


Shotgun Metagenomics Reveals Minor Micro“bee”omes Diversity Defining Differences between Larvae and Pupae Brood Combs


Abstract

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
1.1. Bee Hive as Microbiology Model Object
1.2. Bee Hologenome Parts
2. Results
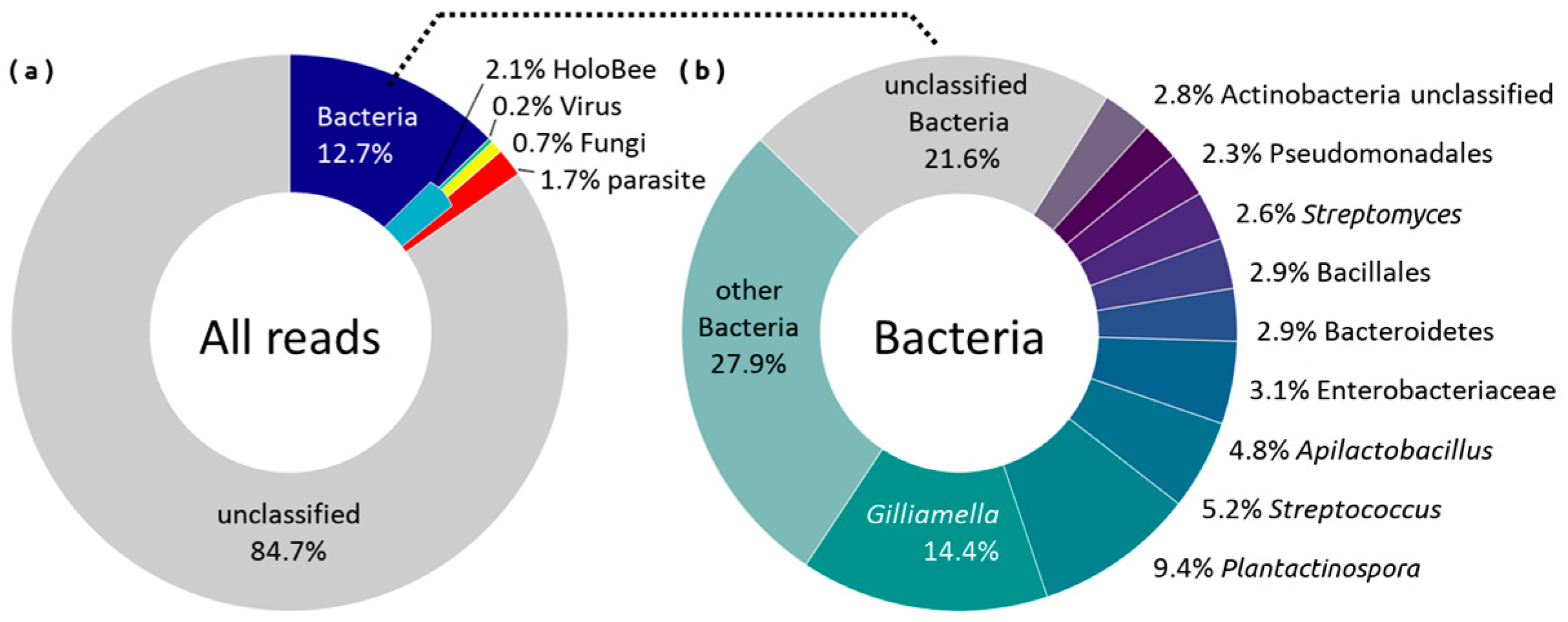
2.1. Taxonomy Annotation
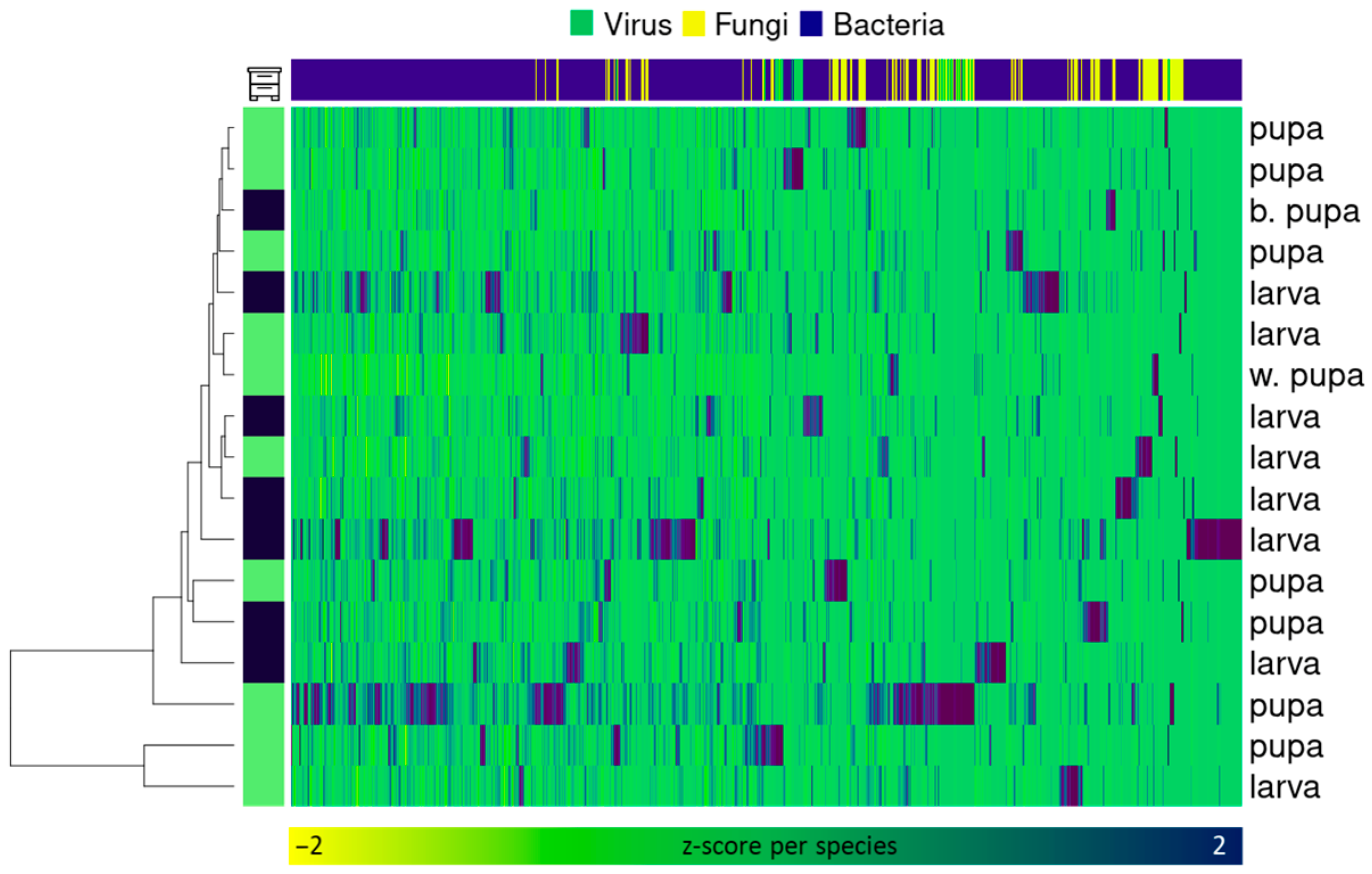
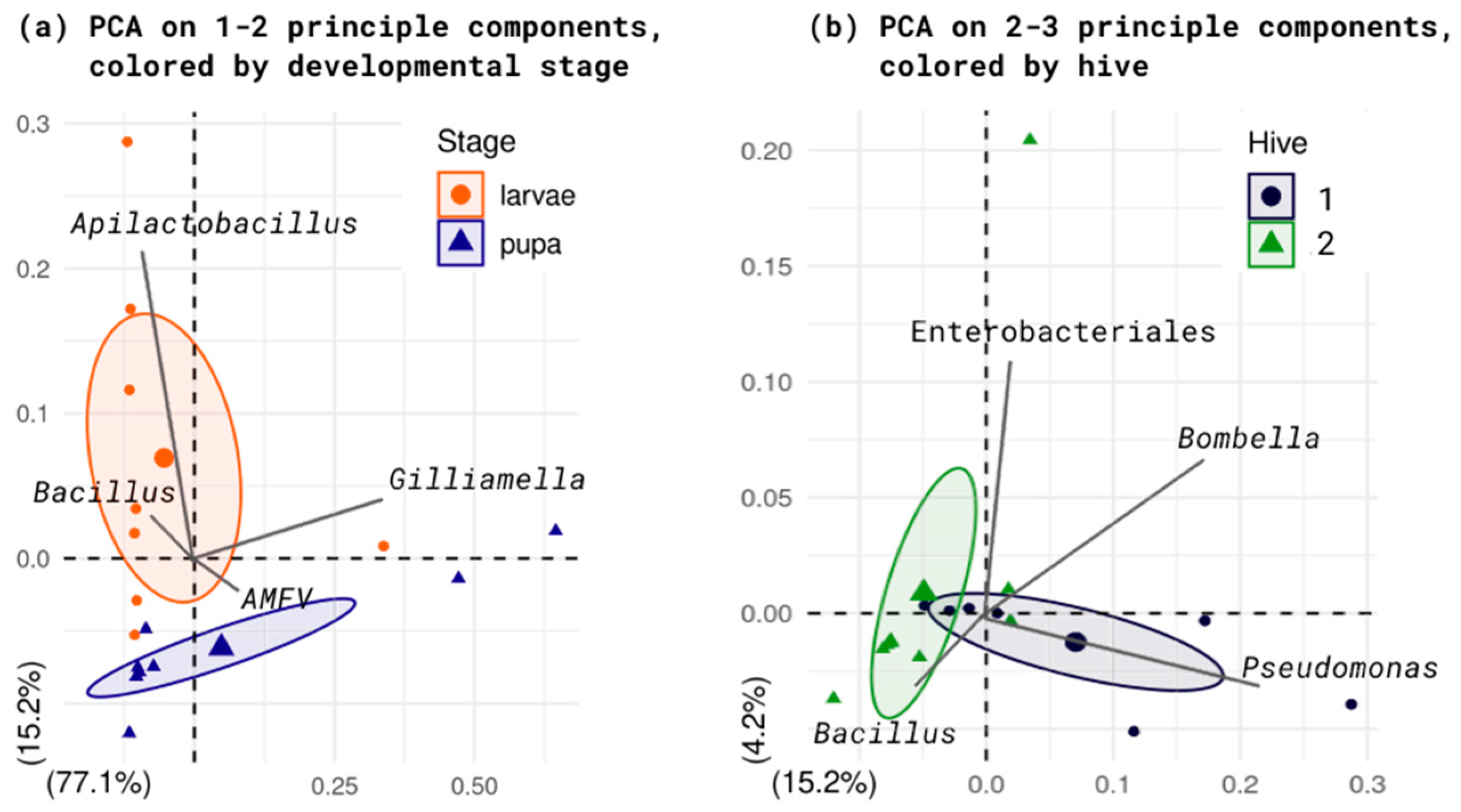
2.2. Diversity Analysis
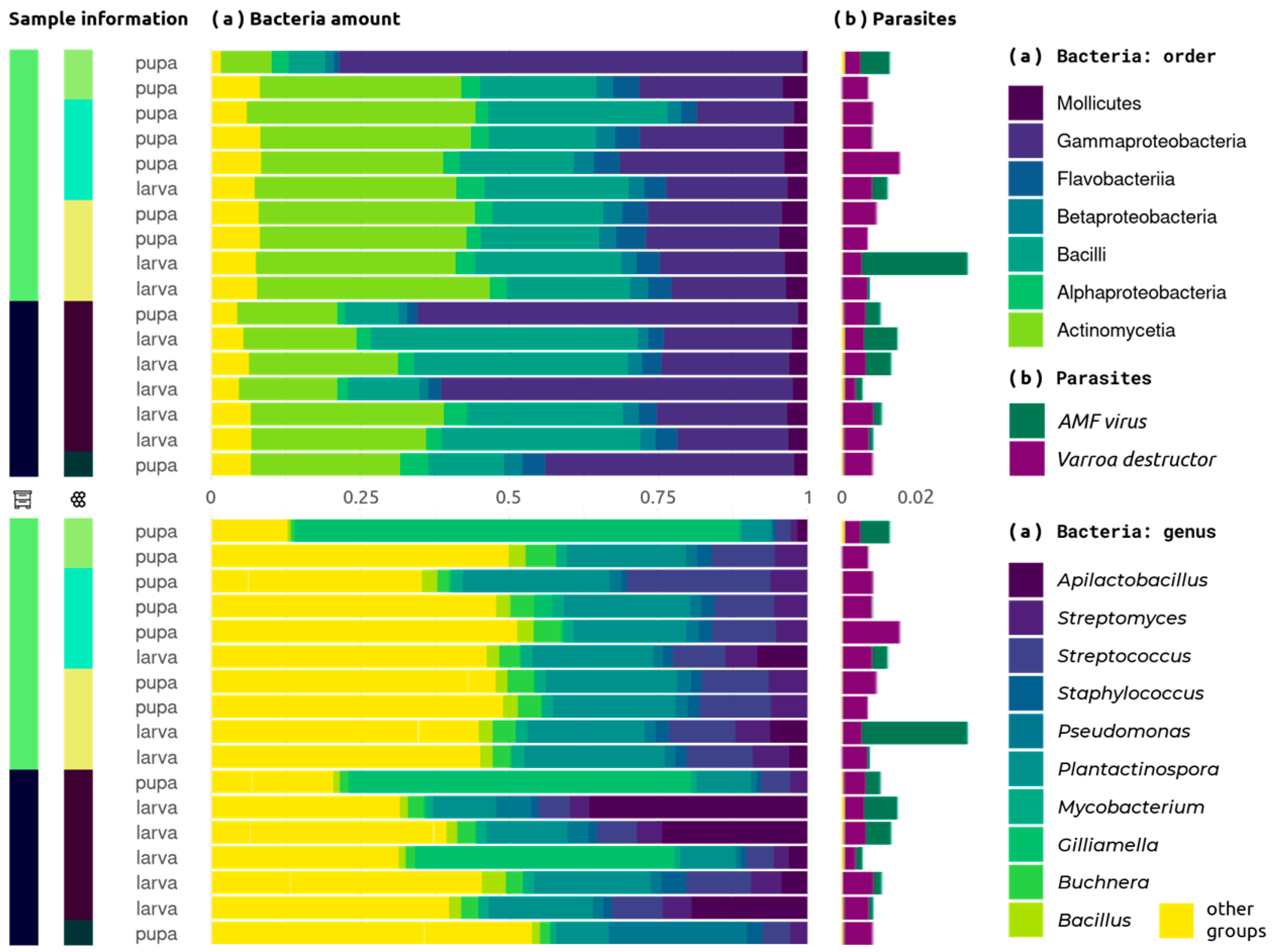
2.3. Composition Analysis
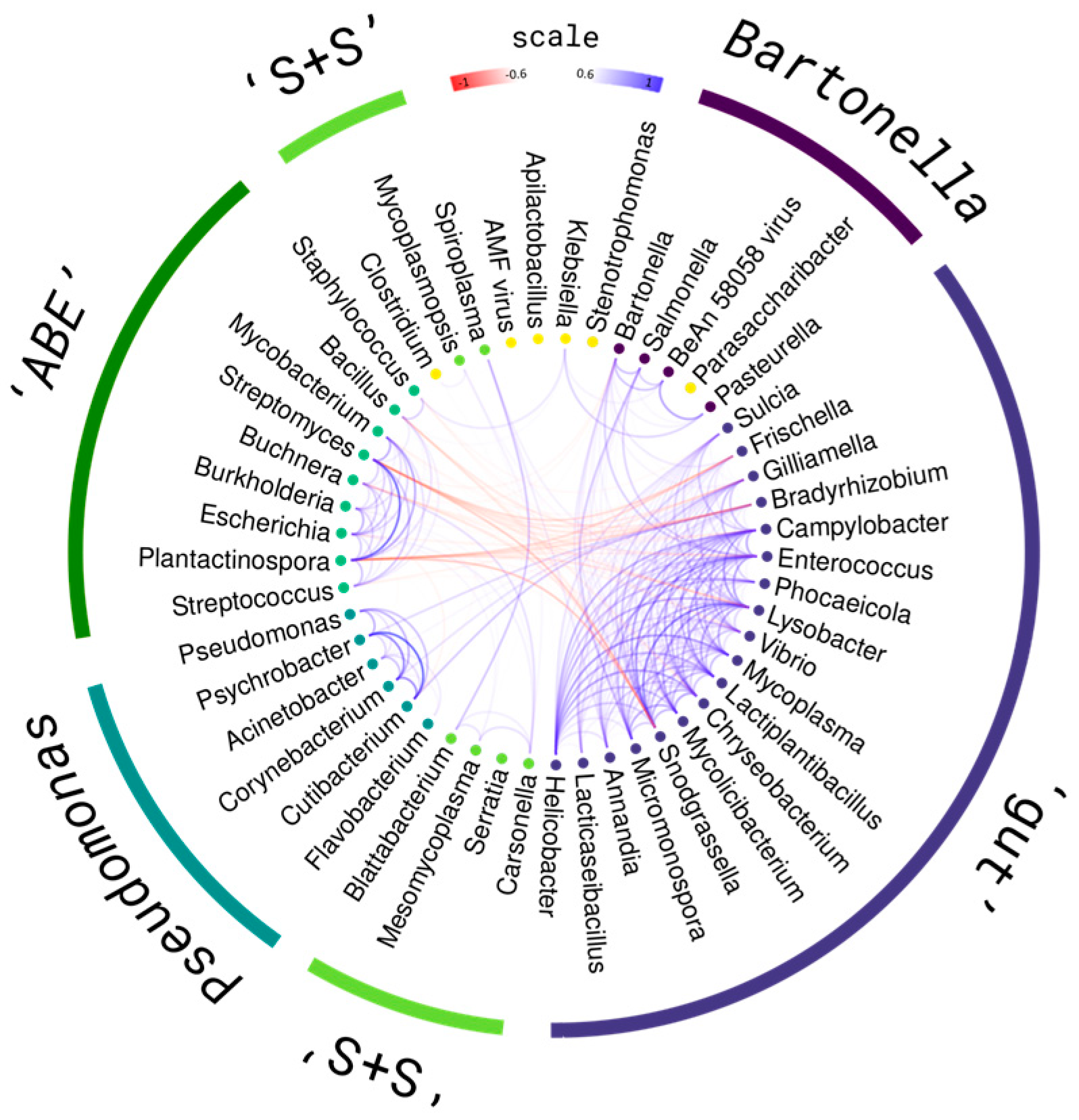
2.4. Composition Associations
2.5. Genome Annotations
3. Discussion
4. Materials and Methods
4.1. Sample Collection
4.2. Sequencing
4.3. Data Processing
4.4. Taxonomic Analysis
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Berg, G.; Rybakova, D.; Fischer, D.; Cernava, T.; Vergès, M.-C.C.; Charles, T.; Chen, X.; Cocolin, L.; Eversole, K.; Corral, G.H.; et al. Microbiome definition re-visited: Old concepts and new challenges. Microbiome 2020, 8, 103. [Google Scholar] [CrossRef]
- Robinson, C.D.; Bohannan, B.J.; Britton, R.A. Scales of persistence: Transmission and the microbiome. Curr. Opin. Microbiol. 2019, 50, 42–49. [Google Scholar] [CrossRef] [PubMed]
- Warren, J.R.; Marshall, B. Unidentified curved bacilli on gastric epithelium in active chronic gastritis. Lancet Lond. Engl. 1983, 1, 1273–1275. [Google Scholar]
- Foley, K.; Fazio, G.; Jensen, A.B.; Hughes, W.O.H. The distribution of Aspergillus spp. opportunistic parasites in hives and their pathogenicity to honey bees. Vet. Microbiol. 2014, 169, 203–210. [Google Scholar] [CrossRef]
- Diggle, S.P.; Whiteley, M. Microbe, Profile: Pseudomonas aeruginosa: Opportunistic pathogen and lab rat. Microbiol. Read. Engl. 2020, 166, 30–33. [Google Scholar] [CrossRef]
- Coyte, K.Z.; Schluter, J.; Foster, K.R. The ecology of the microbiome: Networks, competition, and stability. Science 2015, 350, 663–666. [Google Scholar] [CrossRef] [PubMed]
- Yonatan, Y.; Amit, G.; Friedman, J.; Bashan, A. Complexity-stability trade-off in empirical microbial ecosystems. Nat. Ecol. Evol. 2022, 6, 693–700. [Google Scholar] [CrossRef] [PubMed]
- Shaw, L.P.; Bassam, H.; Barnes, C.P.; Walker, A.S.; Klein, N.; Balloux, F. Modelling microbiome recovery after antibiotics using a stability landscape framework. ISME J. 2019, 13, 1845–1856. [Google Scholar] [CrossRef]
- Watanabe, H.; Nakamura, I.; Mizutani, S.; Kurokawa, Y.; Mori, H.; Kurokawa, K.; Yamada, T. Minor taxa in human skin microbiome contribute to the personal identification. PLoS ONE 2018, 13, e0199947. [Google Scholar] [CrossRef]
- Tabrett, A.; Horton, M.W. The influence of host genetics on the microbiome. F1000Research 2020, 9, 84. [Google Scholar] [CrossRef]
- Barber, C.; Mego, M.; Sabater, C.; Vallejo, F.; Bendezu, R.A.; Masihy, M.; Guarner, F.; Espín, J.C.; Margolles, A.; Azpiroz, F. Differential, Effects of Western and Mediterranean-Type, Diets on Gut, Microbiota: A Metagenomics and Metabolomics, Approach. Nutrients 2021, 13, 2638. [Google Scholar] [CrossRef] [PubMed]
- Kešnerová, L.; Emery, O.; Troilo, M.; Liberti, J.; Erkosar, B.; Engel, P. Gut microbiota structure differs between honeybees in winter and summer. ISME J. 2020, 14, 801–814. [Google Scholar] [CrossRef] [PubMed]
- Kwong, W.K.; Moran, N.A. Gut microbial communities of social bees. Nat. Rev. Microbiol. 2016, 14, 374–384. [Google Scholar] [CrossRef] [PubMed]
- Bovo, S.; Ribani, A.; Utzeri, V.J.; Schiavo, G.; Bertolini, F.; Fontanesi, L. Shotgun metagenomics of honey DNA: Evaluation of a methodological approach to describe a multi-kingdom honey bee derived environmental DNA signature. PLoS ONE 2018, 13, e0205575. [Google Scholar] [CrossRef]
- Khan, K.A.; Al-Ghamdi, A.A.; Ghramh, H.A.; Ansari, M.J.; Ali, H.; Alamri, S.A.; Kahtani, S.N.A.; Adgaba, N.; Qasim, M.; Hafeez, M. Structural diversity and functional variability of gut microbial communities associated with honey bees. Microb. Pathog. 2020, 138, 103793. [Google Scholar] [CrossRef]
- Anderson, K.E.; Sheehan, T.H.; Mott, B.M.; Maes, P.; Snyder, L.; Schwan, M.R.; Walton, A.; Jones, B.M.; Corby-Harris, V. Microbial ecology of the hive and pollination landscape: Bacterial associates from floral nectar, the alimentary tract and stored food of honey bees (Apis mellifera). PLoS ONE 2013, 8, e83125. [Google Scholar] [CrossRef]
- Bonilla-Rosso, G.; Engel, P. Functional roles and metabolic niches in the honey bee gut microbiota. Curr. Opin. Microbiol. 2018, 43, 69–76. [Google Scholar] [CrossRef]
- Copeland, D.C.; Anderson, K.E.; Mott, B.M. Early queen development in honey bees: Social context and queen breeder source affect gut microbiota and associated metabolism. Microbiol. Spectr. 2022, 10, e0038322. [Google Scholar] [CrossRef]
- Maes, P.W.; Rodrigues, P.A.P.; Oliver, R.; Mott, B.M.; Anderson, K.E. Diet-related gut bacterial dysbiosis correlates with impaired development, increased mortality and Nosema disease in the honeybee (Apis mellifera). Mol. Ecol. 2016, 25, 5439–5450. [Google Scholar] [CrossRef]
- Tarpy, D.R.; Mattila, H.R.; Newton, I.L.G. Development of the honey bee gut microbiome throughout the queen-rearing process. Appl. Environ. Microbiol. 2015, 81, 3182–3191. [Google Scholar] [CrossRef]
- Kwong, W.K.; Mancenido, A.L.; Moran, N.A. Immune system stimulation by the native gut microbiota of honey bees. R. Soc. Open Sci. 2017, 4, 170003. [Google Scholar] [CrossRef]
- Leonard, S.P.; Powell, J.E.; Perutka, J.; Geng, P.; Heckmann, L.C.; Horak, R.D.; Davies, B.W.; Ellington, A.D.; Barrick, J.E.; Moran, N.A. Engineered symbionts activate honey bee immunity and limit pathogens. Science 2020, 367, 573–576. [Google Scholar] [CrossRef]
- Anderson, K.E.; Sheehan, T.H.; Eckholm, B.J.; Mott, B.M.; DeGrandi-Hoffman, G. An emerging paradigm of colony health: Microbial balance of the honey bee and hive (Apis mellifera). Insectes Sociaux 2011, 58, 431–444. [Google Scholar] [CrossRef]
- Harwood, G.; Salmela, H.; Freitak, D.; Amdam, G. Social immunity in honey bees: Royal jelly as a vehicle in transferring bacterial pathogen fragments between nestmates. J. Exp. Biol. 2021, 224, jeb231076. [Google Scholar] [CrossRef]
- Liberti, J.; Engel, P. The gut microbiota—Brain axis of insects. Curr. Opin. Insect Sci. 2020, 39, 6–13. [Google Scholar] [CrossRef]
- Huang, Y.; Li, N.; Yang, C.; Lin, Y.; Wen, Y.; Zheng, L.; Zhao, C. Honeybee as a food nutrition analysis model of neural development and gut microbiota. Neurosci. Biobehav. Rev. 2023, 153, 105372. [Google Scholar] [CrossRef]
- Good, A.P.; Gauthier, M.-P.L.; Vannette, R.L.; Fukami, T. Honey bees avoid nectar colonized by three bacterial species, but not by a yeast species, isolated from the bee gut. PLoS ONE 2014, 9, e86494. [Google Scholar] [CrossRef] [PubMed]
- Gaggìa, F.; Jakobsen, R.R.; Alberoni, D.; Baffoni, L.; Cutajar, S.; Mifsud, D.; Nielsen, D.S.; Di Gioia, D. Environment or genetic isolation? An atypical intestinal microbiota in the Maltese honey bee Apis mellifera spp. ruttneri. Front. Microbiol. 2023, 14, 1127717. [Google Scholar] [CrossRef]
- Kwong, W.K.; Moran, N.A. Evolution of host specialization in gut microbes: The bee gut as a model. Gut Microbes 2015, 6, 214–220. [Google Scholar] [CrossRef] [PubMed]
- Zheng, H.; Steele, M.I.; Leonard, S.P.; Motta, E.V.S.; Moran, N.A. Honey bees as models for gut microbiota research. Lab. Anim. 2018, 47, 317–325. [Google Scholar] [CrossRef] [PubMed]
- Smutin, D.; Lebedev, E.; Selitskiy, M.; Panyushev, N.; Adonin, L. Micro”bee”ota: Honey bee normal microbiota as a part of superorganism. Microorganisms 2022, 10, 2359. [Google Scholar] [CrossRef]
- Schwarz, R.S.; Huang, Q.; Evans, J.D. Hologenome theory and the honey bee pathosphere. Curr. Opin. Insect Sci. 2015, 10, 1–7. [Google Scholar] [CrossRef]
- Ghosh, S.; Namin, S.M.; Jung, C. Differential bacterial community of bee bread and bee pollen revealed by 16s rrna high-throughput sequencing. Insects 2022, 13, 863. [Google Scholar] [CrossRef]
- Dharampal, P.S.; Carlson, C.; Currie, C.R.; Steffan, S.A. Pollen-borne microbes shape bee fitness. Proc. Biol. Sci. 2019, 286, 20182894. [Google Scholar] [CrossRef] [PubMed]
- Bovo, S.; Utzeri, V.J.; Ribani, A.; Cabbri, R.; Fontanesi, L. Shotgun sequencing of honey DNA can describe honey bee derived environmental signatures and the honey bee hologenome complexity. Sci. Rep. 2020, 10, 9279. [Google Scholar] [CrossRef] [PubMed]
- Miller, D.L.; Parish, A.J.; Newton, I.L. Transitions and transmission: Behavior and physiology as drivers of honey bee-associated microbial communities. Curr. Opin. Microbiol. 2019, 50, 1–7. [Google Scholar] [CrossRef] [PubMed]
- Anderson, K.E.; Carroll, M.J.; Sheehan, T.; Mott, B.M.; Maes, P.; Corby-Harris, V. Hive-stored pollen of honey bees: Many lines of evidence are consistent with pollen preservation, not nutrient conversion. Mol. Ecol. 2014, 23, 5904–5917. [Google Scholar] [CrossRef] [PubMed]
- Nicolson, S.W.; Neves, S.D.S.D.; Human, H.; Pirk, C.W. Digestibility and nutritional value of fresh and stored pollen for honey bees (Apis mellifera scutellata). J. Insect Physiol. 2018, 107, 302–308. [Google Scholar] [CrossRef] [PubMed]
- Wen, Y.; Wang, L.; Jin, Y.; Zhang, J.; Su, L.; Zhang, X.; Zhou, J.; Li, Y. The microbial community dynamics during the vitex honey ripening process in the honeycomb. Front. Microbiol. 2017, 8, 1649. [Google Scholar] [CrossRef] [PubMed]
- Wirta, H.; Abrego, N.; Miller, K.; Roslin, T.; Vesterinen, E. DNA traces the origin of honey by identifying plants, bacteria and fungi. Sci. Rep. 2021, 11, 4798. [Google Scholar] [CrossRef]
- Anderson, K.E.; Mott, B.M. Ecology of pollen storage in honey bees: Sugar tolerant yeast and the aerobic social microbiota. Insects 2023, 14, 265. [Google Scholar] [CrossRef] [PubMed]
- Donkersley, P.; Rhodes, G.; Pickup, R.W.; Jones, K.C.; Wilson, K. Bacterial communities associated with honeybee food stores are correlated with land use. Ecol. Evol. 2018, 8, 4743–4756. [Google Scholar] [CrossRef] [PubMed]
- Casalone, E.; Cavalieri, D.; Daly, G.; Vitali, F.; Perito, B. Propolis hosts a diversemicrobial community. World J. Microbiol. Biotechnol. 2020, 36, 50. [Google Scholar] [CrossRef] [PubMed]
- Garcia-Mazcorro, J.F.; Kawas, J.R.; Marroquin-Cardona, A.G. Descriptive bacterial and fungal characterization of propolis using ultra-high-throughput marker gene sequencing. Insects 2019, 10, 402. [Google Scholar] [CrossRef] [PubMed]
- Alberoni, D.; Gaggìa, F.; Baffoni, L.; Di Gioia, D. Beneficial microorganisms for honey bees: Problems and progresses. Appl. Microbiol. Biotechnol. 2016, 100, 9469–9482. [Google Scholar] [CrossRef] [PubMed]
- Boukraâ, L.; Sulaiman, S.A. Rediscovering the antibiotics of the hive. Recent Pat. Anti-Infect. Drug Discov. 2009, 4, 206–213. [Google Scholar] [CrossRef] [PubMed]
- Khataybeh, B.; Jaradat, Z.; Ababneh, Q. Anti-bacterial, anti-biofilm and anti-quorum sensing activities of honey: A review. J. Ethnopharmacol. 2023, 317, 116830. [Google Scholar] [CrossRef] [PubMed]
- Saelao, P.; Borba, R.S.; Ricigliano, V.; Spivak, M.; Simone-Finstrom, M. Honeybee microbiome is stabilized in the presence of propolis. Biol. Lett. 2020, 16, 20200003. [Google Scholar] [CrossRef]
- Vásquez, A.; Olofsson, T.C. The lactic acid bacteria involved in the production of bee pollen and bee bread. J. Apic. Res. 2009, 48, 189–195. [Google Scholar] [CrossRef]
- Castelli, L.; Branchiccela, B.; Romero, H.; Zunino, P.; Antúnez, K. Seasonal dynamics of the honey bee gut microbiota in colonies under subtropical climate: Seasonal dynamics of honey bee gut microbiota. Microb. Ecol. 2022, 83, 492–500. [Google Scholar] [CrossRef]
- Prado, A.; Barret, M.; Vaissière, B.E.; Torres-Cortes, G. Honey bees change the microbiota of pollen. bioRxiv 2022, 101, 127–133. [Google Scholar] [CrossRef]
- Ricigliano, V.A.; Fitz, W.; Copeland, D.C.; Mott, B.M.; Maes, P.; Floyd, A.S.; Dockstader, A.; Anderson, K.E. The impact of pollen consumption on honey bee (Apis mellifera) digestive physiology and carbohydrate metabolism. Arch. Insect Biochem. Physiol. 2017, 96, e21406. [Google Scholar] [CrossRef] [PubMed]
- Fernandes, K.E.; Frost, E.A.; Remnant, E.J.; Schell, K.R.; Cokcetin, N.N.; Carter, D.A. The role of honey in the ecology of the hive: Nutrition, detoxification, longevity, and protection against hive pathogens. Front. Nutr. 2022, 9, 954170. [Google Scholar] [CrossRef] [PubMed]
- Grubbs, K.J.; Scott, J.J.; Budsberg, K.J.; Read, H.; Balser, T.C.; Currie, C.R. Unique honey bee (Apis mellifera) hive component-based communities as detected by a hybrid of phospholipid fatty-acid and fatty-acid methyl ester analyses. PLoS ONE 2015, 10, e0121697. [Google Scholar] [CrossRef]
- Piccini, C.; Antúnez, K.; Zunino, P. An approach to the characterization of the honey bee hive bacterial flora. J. Apic. Res. 2004, 43, 101–104. [Google Scholar] [CrossRef]
- Tajabadi, N.; Mardan, M.; Manap, M.Y.A.; Mustafa, S. Molecular identification of Lactobacillus spp. isolated from the honey comb of the honey bee (Apis dorsata) by 16S rRNA gene sequencing. J. Apic. Res. 2013, 52, 235–241. [Google Scholar] [CrossRef]
- Gilliam, M. Identification and roles of non-pathogenic microflora associated with honey bees. FEMS Microbiol. Lett. 1997, 155, 1–10. [Google Scholar] [CrossRef]
- Promnuan, Y.; Kudo, T.; Chantawannakul, P. Actinomycetes isolated from beehives in Thailand. World J. Microbiol. Biotechnol. 2009, 25, 1685–1689. [Google Scholar] [CrossRef]
- Huang, Q.; Lopez, D.; Evans, J.D. Shared and unique microbes between Small hive beetles (Aethina tumida) and their honey bee hosts. MicrobiologyOpen 2019, 8, e899. [Google Scholar] [CrossRef]
- Kešnerová, L.; Moritz, R.; Engel, P. Bartonella apis sp. nov., a honey bee gut symbiont of the class Alphaproteobacteria. Int. J. Syst. Evol. Microbiol. 2016, 66, 414–421. [Google Scholar] [CrossRef]
- Corby-Harris, V.; Snyder, L.A.; Schwan, M.R.; Maes, P.; McFrederick, Q.S.; Anderson, K.E. Origin and effect of Alpha 2.2 Acetobacteraceae in honey bee larvae and description of Parasaccharibacter apium gen. nov., sp. nov. Appl. Environ. Microbiol. 2014, 80, 7460–7472. [Google Scholar] [CrossRef] [PubMed]
- Pattemore, D.E.; Goodwin, R.M.; McBrydie, H.M.; Hoyte, S.M.; Vanneste, J.L. Evidence of the role of honey bees (Apis mellifera) as vectors of the bacterial plant pathogen Pseudomonas syringae. Australas. Plant Pathol. 2014, 43, 571–575. [Google Scholar] [CrossRef]
- Papadopoulou-Karabela, K.; Iliadis, N.; Liakos, V.; Bourdzy-Hatzopoulou, E. Experimental infection of honeybees by Pseudomonas aeruginosa. Apidologie 1992, 23, 393–397. [Google Scholar] [CrossRef]
- Anjum, S.I.; Shah, A.H.; Aurongzeb, M.; Kori, J.; Azim, M.K.; Ansari, M.J.; Bin, L. Characterization of gut bacterial flora of Apis mellifera from north-west Pakistan. Saudi J. Biol. Sci. 2018, 25, 388–392. [Google Scholar] [CrossRef] [PubMed]
- Tsadila, C.; Amoroso, C.; Mossialos, D. Microbial diversity in bee species and bee products: Pseudomonads’ contribution to bee well-being and the biological activity exerted by honey bee products: A narrative review. Diversity 2023, 15, 1088. [Google Scholar] [CrossRef]
- Raymann, K.; Coon, K.L.; Shaffer, Z.; Salisbury, S.; Moran, N.A. Pathogenicity of Serratia marcescens strains in honey bees. mBio 2018, 9, e01649-18. [Google Scholar] [CrossRef] [PubMed]
- Schwarz, R.S.; Teixeira, É.W.; Tauber, J.P.; Birke, J.M.; Martins, M.F.; Fonseca, I.; Evans, J.D. Honey bee colonies act as reservoirs for two Spiroplasma facultative symbionts and incur complex, multiyear infection dynamics. MicrobiologyOpen 2014, 3, 341–355. [Google Scholar] [CrossRef] [PubMed]
- Avalos, J.; Rosero-Mayanquer, H.; Maldonado, G.; Reynaldi, F. Honey bee louse (Braula schmitzi) as a honey bee virus vector? J. Apic. Res. 2019, 58, 427–429. [Google Scholar] [CrossRef]
- Kowallik, V.; Mikheyev, A.S. Honey bee larval and adult microbiome life stages are effectively decoupled with vertical transmission overcoming early life perturbations. mBio 2021, 12, e0296621. [Google Scholar] [CrossRef]
- Vojvodic, S.; Rehan, S.M.; Anderson, K.E. Microbial gut diversity of africanized and european honey bee larval instars. PLoS ONE 2013, 8, e72106. [Google Scholar] [CrossRef]
- Hroncova, Z.; Havlik, J.; Killer, J.; Doskocil, I.; Tyl, J.; Kamler, M.; Titera, D.; Hakl, J.; Mrazek, J.; Bunesova, V.; et al. Variation in honey bee gut microbial diversity affected by ontogenetic stage, age and geographic location. PLoS ONE 2015, 10, e0118707. [Google Scholar] [CrossRef] [PubMed]
- Engel, P.; Kwong, W.K.; McFrederick, Q.; Anderson, K.E.; Barribeau, S.M.; Chandler, J.A.; Cornman, R.S.; Dainat, J.; de Miranda, J.R.; Doublet, V.; et al. The bee microbiome: Impact on bee health and model for evolution and ecology of host-microbe interactions. mBio 2016, 7, e02164-02115. [Google Scholar] [CrossRef] [PubMed]
- Cox-Foster, D.L.; Conlan, S.; Holmes, E.C.; Palacios, G.; Evans, J.D.; Moran, N.A.; Quan, P.-L.; Briese, T.; Hornig, M.; Geiser, D.M.; et al. A metagenomic survey of microbes in honey bee colony collapse disorder. Science 2007, 318, 283–287. [Google Scholar] [CrossRef]
- Regan, T.; Barnett, M.W.; Laetsch, D.R.; Bush, S.J.; Wragg, D.; Budge, G.E.; Highet, F.; Dainat, B.; de Miranda, J.R.; Watson, M.; et al. Characterisation of the British honey bee metagenome. Nat. Commun. 2018, 9, 4995. [Google Scholar] [CrossRef] [PubMed]
- Li, C.; Tang, M.; Li, X.; Zhou, X. Community dynamics in structure and function of honey bee gut bacteria in response to winter dietary shift. mBio 2022, 13, e0113122. [Google Scholar] [CrossRef] [PubMed]
- Crovadore, J.; Gérard, F.; Chablais, R.; Cochard, B.; Bergman Jensen, K.K.; Lefort, F. Deeper, Insight in beehives: Metagenomes of royal jelly, pollen, and honey from lavender, chestnut, and fir honeydew and epiphytic and endophytic microbiota of lavender and rose flowers. Genome Announc. 2017, 5, e00425-17. [Google Scholar] [CrossRef] [PubMed]
- Powell, J.E.; Martinson, V.G.; Urban-Mead, K.; Moran, N.A. Routes of Acquisition of the Gut Microbiota of the Honey Bee Apis mellifera. Appl. Environ. Microbiol. 2014, 80, 7378–7387. [Google Scholar] [CrossRef] [PubMed]
- Neveling, D.P.; Endo, A.; Dicks, L.M.T. Fructophilic, Lactobacillus kunkeei and Lactobacillus brevis isolated from fresh flowers, bees and bee-hives. Curr. Microbiol. 2012, 65, 507–515. [Google Scholar] [CrossRef]
- Hilgarth, M.; Redwitz, J.; Ehrmann, M.A.; Vogel, R.F.; Jakob, F. Bombella favorum sp. nov. and Bombella mellum sp. nov., two novel species isolated from the honeycombs of Apis mellifera. Int. J. Syst. Evol. Microbiol. 2021, 71, 4633. [Google Scholar] [CrossRef]
- Maeno, S.; Tanizawa, Y.; Kanesaki, Y.; Kubota, E.; Kumar, H.; Dicks, L.; Salminen, S.; Nakagawa, J.; Arita, M.; Endo, A. Genomic characterization of a fructophilic bee symbiont Lactobacillus kunkeei reveals its niche-specific adaptation. Syst. Appl. Microbiol. 2016, 39, 516–526. [Google Scholar] [CrossRef]
- Farmer, J.J.; Davis, B.R.; Hickman-Brenner, F.W.; McWhorter, A.; Huntley-Carter, G.P.; Asbury, M.A.; Riddle, C.; Wathen-Grady, H.G.; Elias, C.; Fanning, G.R.; et al. Biochemical identification of new species and biogroups of Enterobacteriaceae isolated from clinical specimens. J. Clin. Microbiol. 1985, 21, 46–76. [Google Scholar] [CrossRef] [PubMed]
- Corby-Harris, V.; Snyder, L.; Meador, C.a.D.; Naldo, R.; Mott, B.; Anderson, K.E. Parasaccharibacter apium, gen. nov., sp. nov., improves honey bee (Hymenoptera: Apidae) resistance to Nosema. J. Econ. Entomol. 2016, 109, 537–543. [Google Scholar] [CrossRef] [PubMed]
- Martinson, V.G.; Danforth, B.N.; Minckley, R.L.; Rueppell, O.; Tingek, S.; Moran, N.A. A simple and distinctive microbiota associated with honey bees and bumble bees. Mol. Ecol. 2011, 20, 619–628. [Google Scholar] [CrossRef] [PubMed]
- Gilliam, M.; Morton, H.L. Enterobacteriaceae isolated from honey bees, Apis mellifera, treated with 2,4-D and antibiotics. J. Invertebr. Pathol. 1974, 23, 42–45. [Google Scholar] [CrossRef]
- de Landa, G.F.; Porrini, M.P.; Revainera, P.; Porrini, D.P.; Farina, J.; Correa-Benítez, A.; Maggi, M.D.; Eguaras, M.J.; Quintana, S. Pathogens detection in the small hive beetle (Aethina tumida (Coleoptera: Nitidulidae)). Neotrop. Entomol. 2021, 50, 312–316. [Google Scholar] [CrossRef]
- Promnuan, Y.; Promsai, S.; Meelai, S. Antimicrobial activity of Streptomyces spp. isolated from Apis dorsata combs against some phytopathogenic bacteria. PeerJ 2020, 8, e10512. [Google Scholar] [CrossRef] [PubMed]
- Brudzynski, K. Honey as an ecological reservoir of antibacterial compounds produced by antagonistic microbial interactions in plant nectars, honey and honey bee. Antibiotics 2021, 10, 551. [Google Scholar] [CrossRef] [PubMed]
- Ellegaard, K.M.; Engel, P. New, Reference Genome, Sequences for 17 Bacterial, Strains of the Honey, Bee Gut, Microbiota. Microbiol. Resour. Announc. 2018, 7, e00834-18. [Google Scholar] [CrossRef]
- Gismondi, A.; De Rossi, S.; Canuti, L.; Novelli, S.; Di Marco, G.; Fattorini, L.; Canini, A. From, Robinia pseudoacacia L. nectar to Acacia monofloral honey: Biochemical changes and variation of biological properties. J. Sci. Food Agric. 2018, 98, 4312–4322. [Google Scholar] [CrossRef]
- Van der Heiden, E.; Delmarcelle, M.; Lebrun, S.; Freichels, R.; Brans, A.; Vastenavond, C.M.; Galleni, M.; Joris, B. A pathway closely related to the (D)-tagatose pathway of gram-negative enterobacteria identified in the gram-positive bacterium Bacillus licheniformis. Appl. Environ. Microbiol. 2013, 79, 3511–3515. [Google Scholar] [CrossRef]
- Rajagopalan, K.V. Molybdopterin–problems and perspectives. BioFactors 1988, 1, 273–278. [Google Scholar] [PubMed]
- Zhang, Z.; Guo, Y.; Yang, F.; Li, J. Pan-genome analysis reveals functional divergences in gut-restricted Gilliamella and Snodgrassella. Bioengineering 2022, 9, 544. [Google Scholar] [CrossRef]
- Hagan, A.K.; Carlson, P.E.; Hanna, P.C. Flying under the radar: The non-canonical biochemistry and molecular biology of petrobactin from Bacillus anthracis. Mol. Microbiol. 2016, 102, 196–206. [Google Scholar] [CrossRef] [PubMed]
- Gilliam, M.; Prest, D.B. Microbiology of feces of the larval honey bee, Apis mellifera. J. Invertebr. Pathol. 1987, 49, 70–75. [Google Scholar] [CrossRef]
- Hroncova, Z.; Killer, J.; Hakl, J.; Titera, D.; Havlik, J. In-hive variation of the gut microbial composition of honey bee larvae and pupae from the same oviposition time. BMC Microbiol. 2019, 19, 110. [Google Scholar] [CrossRef] [PubMed]
- Guo, B.; Tang, J.; Ding, G.; Mashilingi, S.K.; Huang, J.; An, J. Gut microbiota is a potential factor in shaping phenotypic variation in larvae and adults of female bumble bees. Front. Microbiol. 2023, 14, 1117077. [Google Scholar] [CrossRef] [PubMed]
- Al-Ghamdi, A.; Ali Khan, K.; Javed Ansari, M.; Almasaudi, S.B.; Al-Kahtani, S. Effect of gut bacterial isolates from Apis mellifera jemenitica on Paenibacillus larvae infected bee larvae. Saudi J. Biol. Sci. 2018, 25, 383–387. [Google Scholar] [CrossRef] [PubMed]
- Chen, W.-F.; Wang, H.-F.; Wang, Y.; Liu, Z.-G.; Xu, B.-H. Honey bee larval culture in vitro: Gut emptying determines the transition from larva to prepupa and recombinant AccApidaecin improves antibacterial activity. Bull. Entomol. Res. 2023, 113, 368–379. [Google Scholar] [CrossRef]
- Bolger, A.M.; Lohse, M.; Usadel, B. Trimmomatic: A flexible trimmer for Illumina sequence data. Bioinformatics 2014, 30, 2114–2120. [Google Scholar] [CrossRef]
- Langmead, B.; Trapnell, C.; Pop, M.; Salzberg, S.L. Ultrafast and memory-efficient alignment of short DNA sequences to the human genome. Genome Biol. 2009, 10, R25. [Google Scholar] [CrossRef]
- Wood, D.E.; Lu, J.; Langmead, B. Improved metagenomic analysis with Kraken 2. Genome Biol. 2019, 20, 257. [Google Scholar] [CrossRef]
- Menzel, P.; Ng, K.L.; Krogh, A. Fast and sensitive taxonomic classification for metagenomics with Kaiju. Nat. Commun. 2016, 7, 11257. [Google Scholar] [CrossRef] [PubMed]
- R Core Team. R: A Language and Environment for Statistical Computing; R Foundation for Statistical Computing: Vienna, Austria, 2023. [Google Scholar]
- RStudio Team. RStudio: Integrated Development Environment for R; RStudio, P.B.C.: Boston, MA, USA, 2020. [Google Scholar]
- Gu, Z.; Gu, L.; Eils, R.; Schlesner, M.; Brors, B. Circlize implements and enhances circular visualization in R. Bioinformatics 2014, 30, 2811–2812. [Google Scholar] [CrossRef] [PubMed]
- Kassambara, A.; Mundt, F. Factoextra: Extract and Visualize the Results of Multivariate Data Analyses; R package: Vienna, Austria, 2020. [Google Scholar]
- Pedersen, T.L. Ggraph: An. Implementation of Grammar of Graphics for Graphs and Networks; R package: Vienna, Austria, 2022. [Google Scholar]
- Slowikowski, K. Ggrepel: Automatically Position Non-Overlapping Text Labels with “ggplot2”; R package: Vienna, Austria, 2023. [Google Scholar]
- Csárdi, G.; Nepusz, T.; Kirill, M.; Traag, V.; Horvát, S.; Zanini, F.; Noom, D. Igraph: Network, Analysis and Visualization in R; Zenodo: Genève, Switzerland, 2023. [Google Scholar] [CrossRef]
- Wei, T.; Simko, V.R. Package “corrplot”: Visualization of a Correlation Matrix; R package: Vienna, Austria, 2021. [Google Scholar]
- Krijthe, J.H. Rtsne: T-Distributed Stochastic Neighbor Embedding Using Barnes-Hut Implementation; R package: Vienna, Austria, 2015. [Google Scholar]
- Garnier, S.; Ross, N.; Rudis, R.; Filipovic-Pierucci, A.; Galili, T.; timelyportfolio; O’Callaghan, A.; Greenwell, B.; Sievert, C.; Harris, D.J.; et al. Viridis(Lite)—Colorblind-Friendly Color Maps for R; Zenodo: Genève, Switzerland, 2023. [Google Scholar] [CrossRef]
- Wickham, H.; Averick, M.; Bryan, J.; Chang, W.; McGowan, L.D.; François, R.; François, R.; Grolemund, G.; Hayes, A.; Henry, L.; et al. Welcome to the tidyverse. J. Open Source Softw. 2019, 4, 1686. [Google Scholar] [CrossRef]
- Bittinger, K. Abdiv: Alpha and Beta Diversity Measures; R package: Vienna, Austria, 2020. [Google Scholar]
- Peng, Y.; Leung, H.C.M.; Yiu, S.M.; Chin, F.Y.L. IDBA-UD: A de novo assembler for single-cell and metagenomic sequencing data with highly uneven depth. Bioinformatics 2012, 28, 1420–1428. [Google Scholar] [CrossRef]
- Quevillon, E.; Silventoinen, V.; Pillai, S.; Harte, N.; Mulder, N.; Apweiler, R.; Lopez, R. InterProScan: Protein domains identifier. Nucleic Acids Res. 2005, 33, W116–W120. [Google Scholar] [CrossRef] [PubMed]
- Medema, M.H.; Blin, K.; Cimermancic, P.; de Jager, V.; Zakrzewski, P.; Fischbach, M.A.; Weber, T.; Takano, E.; Breitling, R. Antismash: Rapid identification, annotation and analysis of secondary metabolite biosynthesis gene clusters in bacterial and fungal genome sequences. Nucleic Acids Res. 2011, 39, W339–W346. [Google Scholar] [CrossRef] [PubMed]
- Seemann, T. Prokka: Rapid prokaryotic genome annotation. Bioinformatics 2014, 30, 2068–2069. [Google Scholar] [CrossRef] [PubMed]
- Carver, T.; Harris, S.R.; Berriman, M.; Parkhill, J.; McQuillan, J.A. Artemis: An integrated platform for visualization and analysis of high-throughput sequence-based experimental data. Bioinformatics 2012, 28, 464–469. [Google Scholar] [CrossRef]
- Ye, Y.; Doak, T. A Parsimony approach to biological pathway reconstruction/inference for metagenomes. PLoS Comput. Biol. 2009, 5, e1000465. [Google Scholar] [CrossRef]
- Sun, Z.; Huang, S.; Zhang, M.; Zhu, Q.; Haiminen, N.; Carrieri, A.P.; Vázquez-Baeza, Y.; Parida, L.; Kim, H.-C.; Knight, R.; et al. Challenges in Benchmarking Metagenomic Profilers. Nat. Methods 2021, 18, 618–626. [Google Scholar] [CrossRef]
- Lu, J.; Rincon, N.; Wood, D.E.; Breitwieser, F.; Pockrandt, C.; Langmead, B.; Salzberg, S.L.; Steinegger, M. Metagenome analysis using the Kraken software suite. Nat. Protoc. 2022, 17, 2815–2839. [Google Scholar] [CrossRef] [PubMed]
- Johnson, J.; Sun, S.; Fodor, A.A. Systematic classification error profoundly impacts inference in high-depth Whole Genome Shotgun Sequencing datasets. bioRxiv 2022. [Google Scholar] [CrossRef]
- Garrido-Sanz, L.; Senar, M.À.; Piñol, J. Drastic reduction of false positive species in samples of insects by intersecting the default output of two popular metagenomic classifiers. PLoS ONE 2022, 17, e0275790. [Google Scholar] [CrossRef] [PubMed]
- Ye, S.H.; Siddle, K.J.; Park, D.J.; Sabeti, P.C. Benchmarking Metagenomics Tools for Taxonomic Classification. Cell 2019, 178, 779–794. [Google Scholar] [CrossRef] [PubMed]
- Kang, D.D.; Li, F.; Kirton, E.; Thomas, A.; Egan, R.; An, H.; Wang, Z. MetaBAT 2: An adaptive binning algorithm for robust and efficient genome reconstruction from metagenome assemblies. PeerJ 2019, 7, e7359. [Google Scholar] [CrossRef] [PubMed]
- Baumann, P.; Baumann, L.; Lai, C.Y.; Rouhbakhsh, D.; Moran, N.A.; Clark, M.A. Genetics, physiology, and evolutionary relationships of the genus Buchnera: Intracellular symbionts of aphids. Annu. Rev. Microbiol. 1995, 49, 55–94. [Google Scholar] [CrossRef] [PubMed]
- Li, W.; Larsen, A.; Murphy, B.; Fregulia, P. Liver microbial community and associated host transcriptome in calves with feed induced acidosis. Front. Vet. Sci. 2023, 10, 1193473. [Google Scholar] [CrossRef]
- Martyn, C.; Hayes, B.; Lauko, D.; Mithun, E.; Castañeda, G.; Bosco-Lauth, A.; Kistler, A.; Pollard, K.S.; Chou, S. mNGS Investigation of Single Ixodes pacificus Ticks Reveals Diverse Microbes, Viruses, and a Novel mRNA-like Endogenous Viral Elements. bioRxiv 2022. [Google Scholar] [CrossRef]
- Mehta, P.; Soni, J.; Chattopadhyay, P.; Devi, P.; Habyarimana, T.; Tardalkar, K.; Joshi, M.; Pandey, R. Single-cell RNA-Seq reveals intracellular microbial diversity within Immune cells during SARS-CoV-2 Infection and Recovery. iScience 2023, 26, 108357. [Google Scholar] [CrossRef]
- Contreras-Castro, L.; Maldonado, L.A.; Quintana, E.T.; Raggi, L.; Sánchez-Flores, A. Draft Genome Sequence of Two Marine Plantactinospora spp. from the Gulf of California. Genome Announc. 2018, 6, e00436-18. [Google Scholar] [CrossRef]
- Sarmiento-Ramírez, J.M.; van der Voort, M.; Raaijmakers, J.M.; Diéguez-Uribeondo, J. Unravelling the microbiome of eggs of the endangered sea turtle Eretmochelys imbricata identifies bacteria with activity against the emerging pathogen Fusarium falciforme. PLoS ONE 2014, 9, e95206. [Google Scholar] [CrossRef] [PubMed]
- Pascar, J. Characterizing Microbiome Variation in Wild Populations of Mosquito Species. Master’s Thesis, Syracuse University, Syracuse, NY, USA, 2022. [Google Scholar]
- Usié, A.; Leão, C.; Gaspar, D.; Monteiro, H.; Tábuas, L.; Bettencourt, E.; Caetano, P.; Padre, L.; Carolino, N.; Ramos, A.M.; et al. A metagenomics approach to characterize the footrot microbiome in Merino sheep. Vet. Microbiol. 2023, 281, 109745. [Google Scholar] [CrossRef] [PubMed]
- Kim, D.; Yu, Y.; Jung, K.S.; Kim, Y.H.; Kim, J.-J. Tumor Microenvironment Can Predict Chemotherapy Response of Patients with Triple-Negative Breast Cancer Receiving Neoadjuvant Chemotherapy. Cancer Res. Treat. 2023. [Google Scholar] [CrossRef] [PubMed]
- Carstens, C.K.; Salazar, J.K.; Sharma, S.V.; Chan, W.; Darkoh, C. Evaluation of the kitchen microbiome and food safety behaviors of predominantly low-income families. Front. Microbiol. 2022, 13, 987925. [Google Scholar] [CrossRef] [PubMed]
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Smutin, D.; Taldaev, A.; Lebedev, E.; Adonin, L. Shotgun Metagenomics Reveals Minor Micro“bee”omes Diversity Defining Differences between Larvae and Pupae Brood Combs. Int. J. Mol. Sci. 2024, 25, 741. https://doi.org/10.3390/ijms25020741
Smutin D, Taldaev A, Lebedev E, Adonin L. Shotgun Metagenomics Reveals Minor Micro“bee”omes Diversity Defining Differences between Larvae and Pupae Brood Combs. International Journal of Molecular Sciences. 2024; 25(2):741. https://doi.org/10.3390/ijms25020741
Chicago/Turabian StyleSmutin, Daniil, Amir Taldaev, Egor Lebedev, and Leonid Adonin. 2024. "Shotgun Metagenomics Reveals Minor Micro“bee”omes Diversity Defining Differences between Larvae and Pupae Brood Combs" International Journal of Molecular Sciences 25, no. 2: 741. https://doi.org/10.3390/ijms25020741
APA StyleSmutin, D., Taldaev, A., Lebedev, E., & Adonin, L. (2024). Shotgun Metagenomics Reveals Minor Micro“bee”omes Diversity Defining Differences between Larvae and Pupae Brood Combs. International Journal of Molecular Sciences, 25(2), 741. https://doi.org/10.3390/ijms25020741