
Regulation of Benzo[a]pyrene-Induced Hepatic Lipid Accumulation through CYP1B1-Induced mTOR-Mediated Lipophagy

Abstract
1. Introduction
2. Results
2.1. B[a]P-Induced Lipid Accumulation Is Decreased by CYP1B1 Knockdown in Hepatic Cells
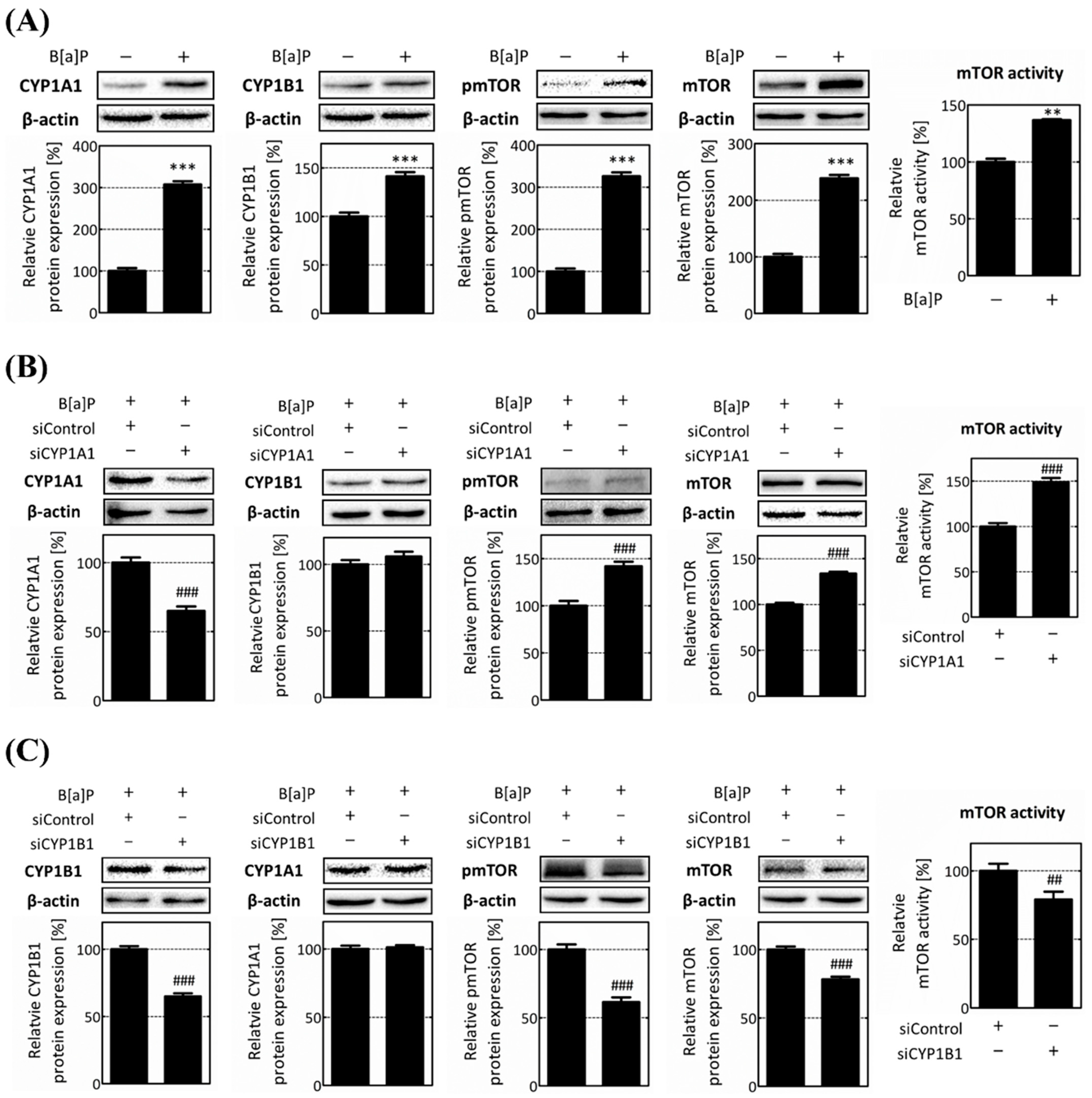
2.2. CYP1B1 Specifically Regulates mTOR and Macroautophagy-Related Genes
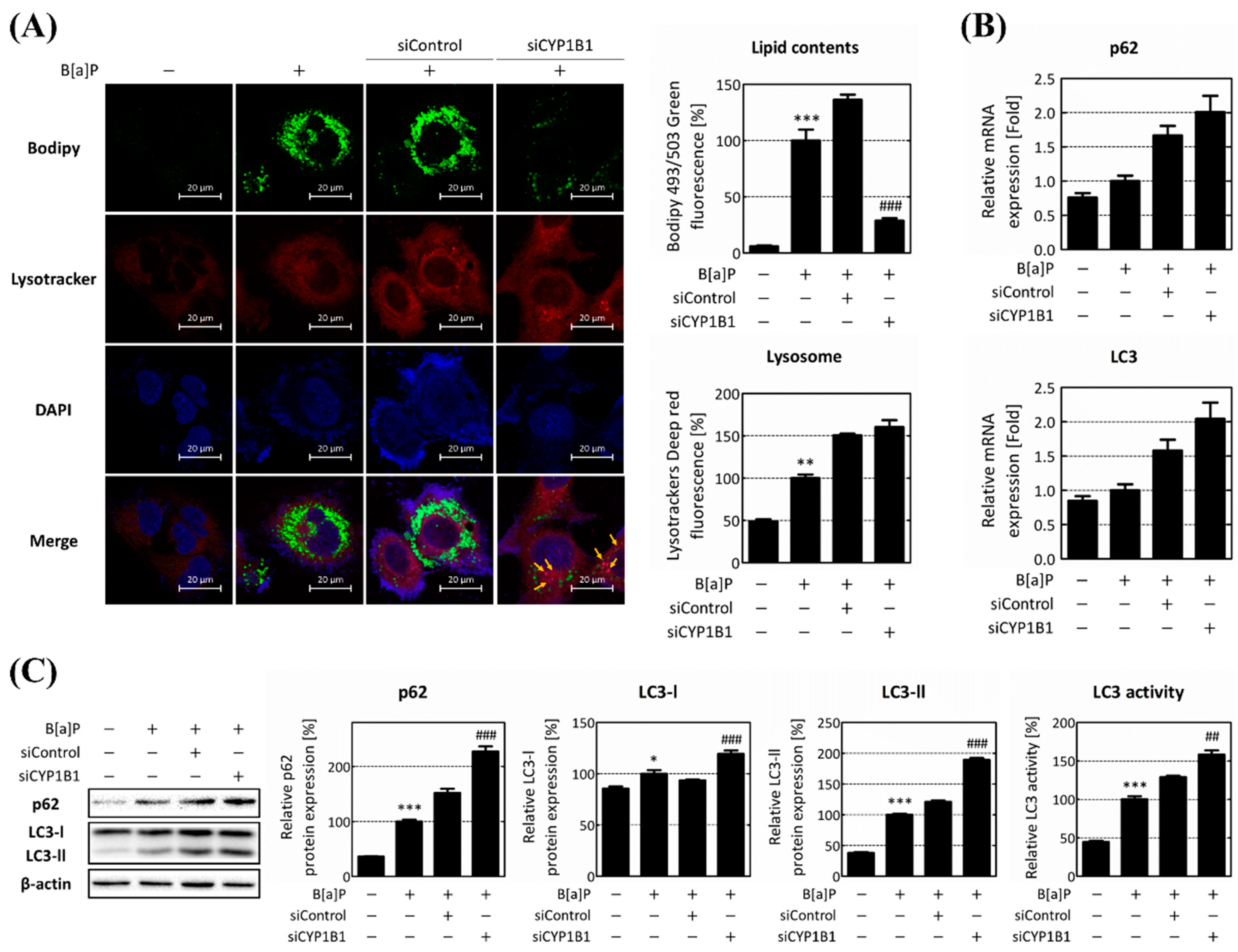
2.3. CYP1B1 Knockdown Mitigates Lipid Accumulation by Enhancing Intracellular Lipophagy, Which Is Reduced by B[a]P
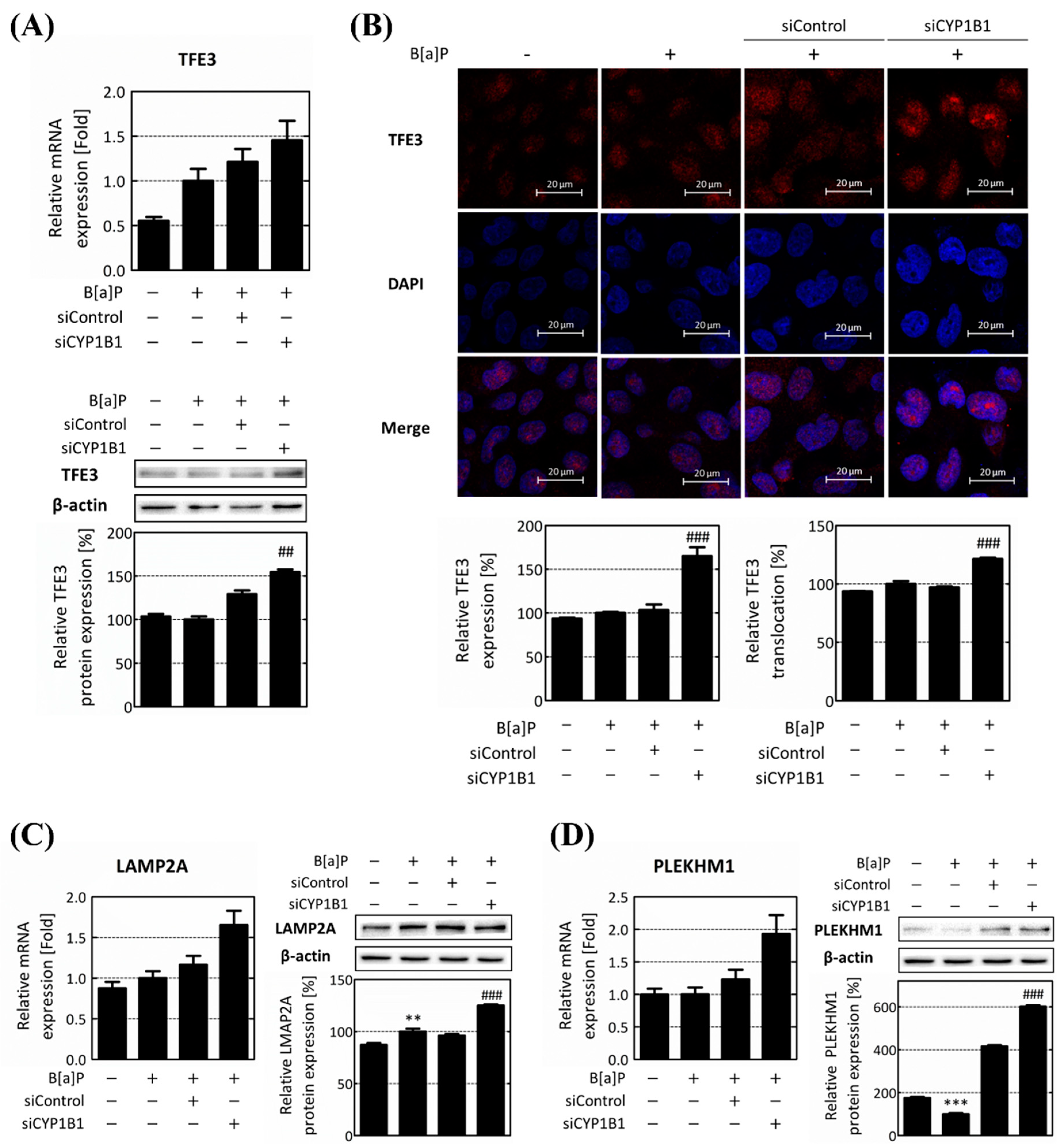
2.4. CYP1B1 Knockdown Enhances Lipophagy via the Induction of TFE3 Translocation
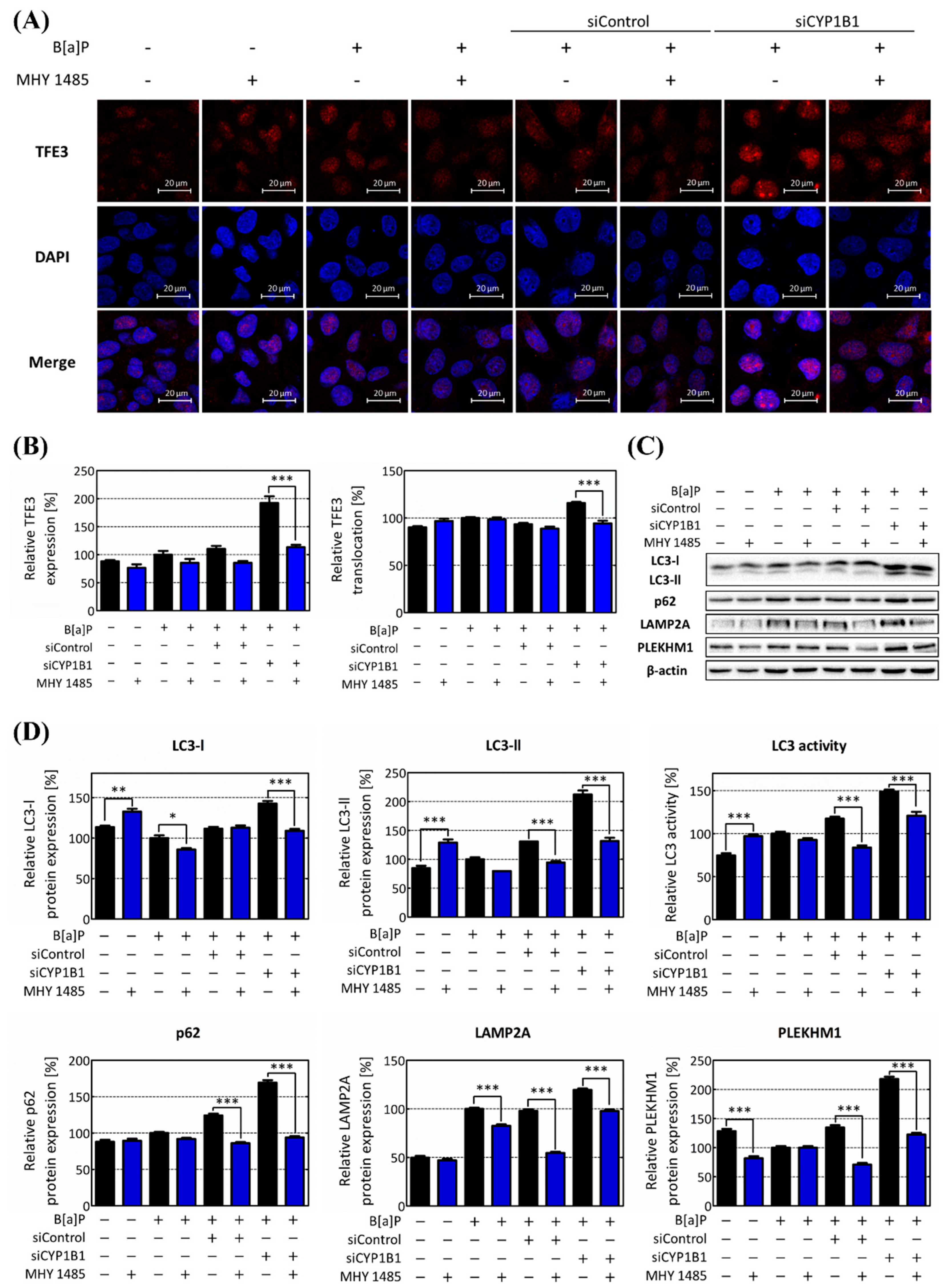
2.5. CYP1B1-Mediated Regulation of mTOR Amplifies Hepatic Lipid Accumulation via Lipophagy Suppression
3. Discussion
4. Materials and Methods
4.1. Reagents
4.2. Cell Culture and Treatment
4.3. siRNA Transfection
4.4. Cell Viability Assay
4.5. Lipid Accumulation Test Using ORO Staining
4.6. mRNA Extraction
4.7. mRNA Quantification Sequencing (Quan-Seq)
4.8. Reverse Transcription-Quantitative Polymerase Chain Reaction (RT-qPCR)
4.9. Western Blot Analysis
4.10. Biogenesis of Phagolysosomes
4.11. Immunofluorescence Analysis
4.12. Statistical Analysis
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Brunt, E.M. Pathology of Nonalcoholic Fatty Liver Disease. Nat. Rev. Gastroenterol. Hepatol. 2010, 7, 195–203. [Google Scholar] [CrossRef] [PubMed]
- Tripodi, A.; Lombardi, R.; Primignani, M.; La Mura, V.; Peyvandi, F.; Fracanzani, A.L. Hypercoagulability in Patients with Non-Alcoholic Fatty Liver Disease (MASLD): Causes and Consequences. Biomedicines 2022, 10, 249. [Google Scholar] [CrossRef]
- Grefhorst, A.; van de Peppel, I.P.; Larsen, L.E.; Jonker, J.W.; Holleboom, A.G. The Role of Lipophagy in the Development and Treatment of Non-Alcoholic Fatty Liver Disease. Front. Endocrinol. 2021, 11, 1627. [Google Scholar] [CrossRef] [PubMed]
- Younossi, Z.M.; Koenig, A.B.; Abdelatif, D.; Fazel, Y.; Henry, L.; Wymer, M. Global Epidemiology of Nonalcoholic Fatty Liver Disease—Meta-analytic Assessment of Prevalence, Incidence, and Outcomes. Hepatology 2016, 64, 73–84. [Google Scholar] [CrossRef] [PubMed]
- Teng, Y.X.; Xie, S.; Guo, P.P.; Deng, Z.J.; Zhang, Z.Y.; Gao, W.; Zhang, W.G.; Zhong, J.H. Hepatocellular Carcinoma in Non-alcoholic Fatty Liver Disease: Current Progresses and Challenges. J. Clin. Transl. Hepatol. 2022, 10, 955–964. [Google Scholar] [CrossRef]
- VanWagner, L.B.; Ning, H.; Lewis, C.E.; Shay, C.M.; Wilkins, J.; Carr, J.J.; Terry, J.G.; Lloyd-Jones, D.M.; Jacobs, D.R.; Carnethon, M.R. Associations between nonalcoholic fatty liver disease and subclinical atherosclerosis in middle-aged adults: The Coronary Artery Risk Development in Young Adults Study. Atherosclerosis 2014, 235, 599–605. [Google Scholar] [CrossRef]
- Zhu, X.; Xiong, T.; Liu, P.; Guo, X.; Xiao, L.; Zhou, F.; Tang, Y.; Yao, P. Quercetin Ameliorates HFD-induced MASLD by Promoting Hepatic VLDL Assembly and Lipophagy via the IRE1a/XBP1s Pathway. Food Chem. Toxicol. 2018, 114, 52–60. [Google Scholar] [CrossRef]
- Baffy, G.; Brunt, E.M.; Caldwell, S.H. Hepatocellular Carcinoma in Non-Alcoholic Fatty Liver Disease: An Emerging Menace. J. Hepatol. 2012, 56, 1384–1391. [Google Scholar] [CrossRef]
- Lindenmeyer, C.C.; McCullough, A.J. The Natural History of Nonalcoholic Fatty Liver Disease—An Evolving View. Clin. Liver Dis. 2018, 22, 11–21. [Google Scholar] [CrossRef]
- Feng, J.; Qiu, S.; Zhou, S.; Tan, Y.; Bai, Y.; Cao, H.; Guo, J.; Su, Z. mTOR: A Potential New Target in Nonalcoholic Fatty Liver Disease. Int. J. Mol. Sci. 2022, 23, 9196. [Google Scholar] [CrossRef]
- Li, Y.; Liang, N.; Tang, T.; Zheng, Z.; Chen, M.; Mo, J.; Zhang, N.; Liao, S.; Lei, Y.; Wu, Y.; et al. Low-Dose Benzo[a]pyrene Exposure Induces Hepatic Lipid Deposition through LCMT1/PP2Ac-Mediated Autophagy Inhibition. Food Chem. Toxicol. 2023, 179, 113986. [Google Scholar] [CrossRef] [PubMed]
- Farhadian, A.; Jinap, S.; Hanifah, H.N.; Zaidul, I.S. Effects of Meat Preheating and Wrapping on the Levels of Polycyclic Aromatic Hydrocarbons in Charcoal-Grilled Meat. Food Chem. 2011, 124, 141–146. [Google Scholar] [CrossRef]
- Ge, Y.; Gu, P.; Wang, W.; Cao, L.; Zhang, L.; Li, J.; Mu, W.; Wang, H. Benzo[a]pyrene Stimulates miR-650 Expression to Promote the Pathogenesis of Fatty Liver Disease and Hepatocellular Carcinoma via SOCS3/JAK/STAT3 Cascades. J. Mol. Cell Biol. 2021, 13, 556–564. [Google Scholar] [CrossRef]
- Uno, S.; Nebert, D.W.; Makishima, M. Cytochrome P450 1A1 (CYP1A1) Protects against Nonalcoholic Fatty Liver Disease Caused by Western Diet Containing Benzo[a]pyrene in Mice. Food Chem. Toxicol. 2018, 113, 73–82. [Google Scholar] [CrossRef] [PubMed]
- Nisbet, I.C.T.; LaGoy, P.K. Toxic Equivalency Factors (TEFs) for Polycyclic Aromatic Hydrocarbons (PAHs). Regul. Toxicol. Pharmacol. 1992, 16, 290–300. [Google Scholar] [CrossRef]
- Lee, S.C.; Jee, S.C.; Kim, M.; Kim, S.; Shin, M.K.; Kim, Y.; Sung, J.S. Curcumin Suppresses the Lipid Accumulation and Oxidative Stress Induced by Benzo[a]pyrene Toxicity in HepG2 Cells. Antioxidants 2021, 10, 1314. [Google Scholar] [CrossRef] [PubMed]
- Barnes, J.L.; Zubair, M.; John, K.; Poirier, M.C.; Martin, F.L. Carcinogens and DNA dDmage. Biochem. Soc. Trans. 2018, 46, 1213–1224. [Google Scholar] [CrossRef]
- Ortiz, L.; Nakamura, B.; Li, X.; Blumberg, B.; Luderer, U. In Utero Exposure to Benzo[a]pyrene Increases Adiposity and Causes Hepatic Steatosis in Female Mice, and Glutathione Deficiency is Protective. Toxicol. Let. 2013, 223, 260–267. [Google Scholar] [CrossRef]
- Lou, W.; Zhang, M.-d.; Chen, Q.; Bai, T.-Y.; Hu, Y.-X.; Gao, F.; Li, J.; Lv, X.-L.; Zhang, Q.; Chang, F.-H. Molecular Mechanism of Benzo[a]Pyrene Regulating Lipid Metabolism via Aryl Hydrocarbon Receptor. Lipids Health Dis. 2022, 21, 13. [Google Scholar] [CrossRef] [PubMed]
- Matsunawa, M.; Amano, Y.; Endo, K.; Uno, S.; Sakaki, T.; Yamada, S.; Makishima, M. The Aryl Hydrocarbon Receptor Activator Benzo[a]pyrene Enhances Vitamin D3 Catabolism in Macrophages. Toxicol. Sci. 2009, 109, 50–58. [Google Scholar] [CrossRef]
- Kim, M.; Jee, S.C.; Kim, K.S.; Kim, H.S.; Yu, K.N.; Sung, J.S. Quercetin and Isorhamnetin Attenuate Benzo[a]pyrene-Induced Toxicity by Modulating Detoxification Enzymes through the AhR and NRF2 Signaling Pathways. Antioxidants 2021, 10, 787. [Google Scholar] [CrossRef] [PubMed]
- Walle, T.; Walle, U.K.; Sedmera, D.; Klausner, M. Benzo[a]pyrene-Induced Oral Carcinogenesis and Chemoprevention: Studies in Bioengineered Human Tissue. Drug Metab. Dispos. 2006, 34, 346–350. [Google Scholar] [CrossRef] [PubMed]
- McDonnell, A.M.; Dang, C.H. Basic Review of the Cytochrome p450 System. J. Adv. Pract. Oncol. 2013, 4, 263–268. [Google Scholar] [CrossRef] [PubMed]
- Fröhlich, E.; Kueznik, T.; Samberger, C.; Roblegg, E.; Wrighton, C.; Pieber, T.R. Size-Dependent Effects of Nanoparticles on the Activity of Cytochrome P450 Isoenzymes. Toxicol. Appl. Pharmacol. 2010, 242, 326–332. [Google Scholar] [CrossRef] [PubMed]
- Zhao, M.; Ma, J.; Li, M.; Zhang, Y.; Jiang, B.; Zhao, X.; Huai, C.; Shen, L.; Zhang, N.; He, L.; et al. Cytochrome P450 Enzymes and Drug Metabolism in Humans. Int. J. Mol. Sci. 2021, 22, 12808. [Google Scholar] [CrossRef]
- Bishop-Bailey, D.; Thomson, S.; Askari, A.; Faulkner, A.; Wheeler-Jones, C. Lipid-Metabolizing CYPs in the Regulation and Dysregulation of Metabolism. Annu. Rev. Nutr. 2014, 34, 261–279. [Google Scholar] [CrossRef]
- Ohashi, H.; Nishioka, K.; Nakajima, S.; Kim, S.; Suzuki, R.; Aizaki, H.; Fukasawa, M.; Kamisuki, S.; Sugawara, F.; Ohtani, N.; et al. The Aryl Hydrocarbon Receptor;Cytochrome P450 1A1 Pathway Controls Lipid Accumulation and Enhances the Permissiveness for Hepatitis C Virus Assembly. J. Biol. Chem. 2018, 293, 19559–19571. [Google Scholar] [CrossRef]
- Zhu, X.-Y.; Xia, H.-G.; Wang, Z.-H.; Li, B.; Jiang, H.-Y.; Li, D.-L.; Jin, R.; Jin, Y. In Vitro and in Vivo Approaches for Identifying the Role of Aryl Hydrocarbon Receptor in the Development of Nonalcoholic Fatty Liver Disease. Toxicol. Let. 2020, 319, 85–94. [Google Scholar] [CrossRef]
- Grabner, G.F.; Xie, H.; Schweiger, M.; Zechner, R. Lipolysis: Cellular Mechanisms for Lipid Mobilization from Fat Stores. Nat. Metab. 2021, 3, 1445–1465. [Google Scholar] [CrossRef]
- Singh, R.; Kaushik, S.; Wang, Y.; Xiang, Y.; Novak, I.; Komatsu, M.; Tanaka, K.; Cuervo, A.M.; Czaja, M.J. Autophagy Regulates Lipid Metabolism. Nature 2009, 458, 1131–1135. [Google Scholar] [CrossRef]
- Carotti, S.; Aquilano, K.; Zalfa, F.; Ruggiero, S.; Valentini, F.; Zingariello, M.; Francesconi, M.; Perrone, G.; Alletto, F.; Antonelli-Incalzi, R.; et al. Lipophagy Impairment Is Associated With Disease Progression in NAFLD. Front. Physiol. 2020, 11, 850. [Google Scholar] [CrossRef] [PubMed]
- Schulze, R.J.; Sathyanarayan, A.; Mashek, D.G. Breaking fat: The Regulation and Mechanisms of Lipophagy. Biochim. Biophys. Acta Mol. Cell Biol. Lipids 2017, 1862, 1178–1187. [Google Scholar] [CrossRef] [PubMed]
- Mao, Y.; Yu, F.; Wang, J.; Guo, C.; Fan, X. Autophagy: A New Target for Nonalcoholic Fatty Liver Disease Therapy. Hepat. Med. 2016, 8, 27–37. [Google Scholar] [CrossRef] [PubMed]
- Ouimet, M.; Franklin, V.; Mak, E.; Liao, X.; Tabas, I.; Marcel, Y.L. Autophagy Regulates Cholesterol Efflux from Macrophage Foam Cells via Lysosomal Acid Lipase. Cell Metab. 2011, 13, 655–667. [Google Scholar] [CrossRef] [PubMed]
- Tan, X.; Huang, X.; Lu, Z.; Chen, L.; Hu, J.; Tian, X.; Qiu, Z. The Essential Effect of mTORC1-Dependent Lipophagy in Non-Alcoholic Fatty Liver Disease. Front. Pharmacol. 2023, 14, 1124003. [Google Scholar] [CrossRef] [PubMed]
- Kim, Y.C.; Guan, K.L. mTOR: A Pharmacologic Target for Autophagy Regulation. J. Clin. Investig. 2015, 125, 25–32. [Google Scholar] [CrossRef]
- Slade, L.; Pulinilkunnil, T. The MiTF/TFE Family of Transcription Factors: Master Regulators of Organelle Signaling, Metabolism, and Stress Adaptation. Mol. Cancer Res. 2017, 15, 1637–1643. [Google Scholar] [CrossRef]
- Raben, N.; Puertollano, R. TFEB and TFE3: Linking Lysosomes to Cellular Adaptation to Stress. Annu. Rev. Cell Dev. Biol. 2016, 32, 255–278. [Google Scholar] [CrossRef]
- Riazi, K.; Azhari, H.; Charette, J.H.; Underwood, F.E.; King, J.A.; Afshar, E.E.; Swain, M.G.; Congly, S.E.; Kaplan, G.G.; Shaheen, A.-A. The Prevalence and Incidence of MASLD Worldwide: A Systematic Review and Meta-Analysis. Lancet Gastroenterol. Hepatol. 2022, 7, 851–861. [Google Scholar] [CrossRef]
- Jee, S.-C.; Kim, M.; Kim, K.S.; Kim, H.-S.; Sung, J.-S. Protective Effects of Myricetin on Benzo[a]pyrene-Induced 8-Hydroxy-2′-Deoxyguanosine and BPDE-DNA Adduct. Antioxidants 2020, 9, 446. [Google Scholar] [CrossRef]
- IARC Working Group on the Evaluation of Carcinogenic Risks to Humans. IARC Working Group on the Evaluation of Carcinogenic Risks to Humans. Chemical Agents and Related Occupations. IARC Monogr. Eval. Carcinog. Risks Hum. 2012, 100, 9–562. [Google Scholar]
- Soret, P.A.; Magusto, J.; Housset, C.; Gautheron, J. In Vitro and In Vivo Models of Non-Alcoholic Fatty Liver Disease: A Critical Appraisal. J. Clin. Med. 2020, 10, 36. [Google Scholar] [CrossRef]
- Messina, A.; Luce, E.; Hussein, M.; Dubart-Kupperschmitt, A. Pluripotent-Stem-Cell-Derived Hepatic Cells: Hepatocytes and Organoids for Liver Therapy and Regeneration. Cells 2020, 9, 420. [Google Scholar] [CrossRef] [PubMed]
- Misaki, K.; Matsui, S.; Matsuda, T. Metabolic Enzyme Induction by HepG2 Cells Exposed to Oxygenated and Nonoxygenated Polycyclic Aromatic Hydrocarbons. Chem. Res. Toxicol. 2007, 20, 277–283. [Google Scholar] [CrossRef] [PubMed]
- Choi, J.M.; Oh, S.J.; Lee, S.Y.; Im, J.H.; Oh, J.M.; Ryu, C.S.; Kwak, H.C.; Lee, J.Y.; Kang, K.W.; Kim, S.K. HepG2 Cells as an in vitro Model for Evaluation of Cytochrome P450 Induction by Xenobiotics. Arch. Pharm. Res. 2015, 38, 691–704. [Google Scholar] [CrossRef]
- Jennen, D.G.J.; Magkoufopoulou, C.; Ketelslegers, H.B.; van Herwijnen, M.H.M.; Kleinjans, J.C.S.; van Delft, J.H.M. Comparison of HepG2 and HepaRG by Whole-Genome Gene Expression Analysis for the Purpose of Chemical Hazard Identification. Toxicol. Sci. 2010, 115, 66–79. [Google Scholar] [CrossRef]
- Tanaka, S.; Hikita, H.; Tatsumi, T.; Sakamori, R.; Nozaki, Y.; Sakane, S.; Shiode, Y.; Nakabori, T.; Saito, Y.; Hiramatsu, N.; et al. Rubicon Inhibits Autophagy and Accelerates Hepatocyte Apoptosis and Lipid Accumulation in Nonalcoholic Fatty Liver Disease in Mice. Hepatology 2016, 64, 1994–2014. [Google Scholar] [CrossRef]
- Cui, W.; Sathyanarayan, A.; Lopresti, M.; Aghajan, M.; Chen, C.; Mashek, D.G. Lipophagy-Derived Fatty Acids Undergo Extracellular Efflux via Lysosomal Exocytosis. Autophagy 2021, 17, 690–705. [Google Scholar] [CrossRef]
- Laplante, M.; Sabatini, D.M. mTOR Signaling in Growth Control and Disease. Cell 2012, 149, 274–293. [Google Scholar] [CrossRef]
- Shroff, A.; Nazarko, T.Y. SQSTM1, Lipid Droplets and Current State of their Lipophagy Affairs. Autophagy 2023, 19, 720–723. [Google Scholar] [CrossRef]
- Ward, C.; Martinez-Lopez, N.; Otten, E.G.; Carroll, B.; Maetzel, D.; Singh, R.; Sarkar, S.; Korolchuk, V.I. Autophagy, Lipophagy and Lysosomal Lipid Storage Disorders. Biochim. Biophys. Acta Mol. Cell Biol. Lipids 2016, 1861, 269–284. [Google Scholar] [CrossRef]
- Mao, Z.; Zhang, W. Role of mTOR in Glucose and Lipid Metabolism. Int. J. Mol. Sci. 2018, 19, 2043. [Google Scholar] [CrossRef]
- Issa, A.R.; Sun, J.; Petitgas, C.; Mesquita, A.; Dulac, A.; Robin, M.; Mollereau, B.; Jenny, A.; Chérif-Zahar, B.; Birman, S. The Lysosomal Membrane Protein LAMP2A Promotes Autophagic Flux and Prevents SNCA-Induced Parkinson Disease-like Symptoms in the Drosophila Brain. Autophagy 2018, 14, 1898–1910. [Google Scholar] [CrossRef] [PubMed]
- McEwan, D.G.; Popovic, D.; Gubas, A.; Terawaki, S.; Suzuki, H.; Stadel, D.; Coxon, F.P.; Miranda de Stegmann, D.; Bhogaraju, S.; Maddi, K.; et al. PLEKHM1 Regulates Autophagosome-Lysosome Fusion through HOPS Complex and LC3/GABARAP proteins. Mol. Cell 2015, 57, 39–54. [Google Scholar] [CrossRef] [PubMed]
- Alfaro, I.E.; Albornoz, A.; Molina, A.; Moreno, J.; Cordero, K.; Criollo, A.; Budini, M. Chaperone Mediated Autophagy in the Crosstalk of Neurodegenerative Diseases and Metabolic Disorders. Front. Endocrinol. 2018, 9, 778. [Google Scholar] [CrossRef] [PubMed]
- He, X.; Xie, Y.; Zheng, Q.; Zhang, Z.; Ma, S.; Li, J.; Li, M.; Huang, Q. TFE3-Mediated Autophagy is Involved in Dopaminergic Neurodegeneration in Parkinson’s Disease. Front. Cell Dev. Biol. 2021, 9, 761773. [Google Scholar] [CrossRef]
- Gao, L.; Lv, G.; Li, R.; Liu, W.T.; Zong, C.; Ye, F.; Li, X.Y.; Yang, X.; Jiang, J.H.; Hou, X.J.; et al. Glycochenodeoxycholate Promotes Hepatocellular Carcinoma Invasion and Migration by AMPK/mTOR Dependent Autophagy Activation. Cancer Lett. 2019, 454, 215–223. [Google Scholar] [CrossRef]
- Choi, Y.J.; Park, Y.J.; Park, J.Y.; Jeong, H.O.; Kim, D.H.; Ha, Y.M.; Kim, J.M.; Song, Y.M.; Heo, H.S.; Yu, B.P.; et al. Inhibitory Dffect of mTOR Activator MHY1485 on Autophagy: Suppression of Lysosomal Fusion. PLoS ONE 2012, 7, e43418. [Google Scholar] [CrossRef]
- Rabanal-Ruiz, Y.; Korolchuk, V.I. mTORC1 and Nutrient Homeostasis: The Central Role of the Lysosome. Int. J. Mol. Sci. 2018, 19, 818. [Google Scholar] [CrossRef]
- Seok, J.K.; Hong, E.-H.; Yang, G.; Lee, H.E.; Kim, S.-E.; Liu, K.-H.; Kang, H.C.; Cho, Y.-Y.; Lee, H.S.; Lee, J.Y. Oxidized Phospholipids in Tumor Microenvironment Stimulate Tumor Metastasis via Regulation of Autophagy. Cells 2021, 10, 558. [Google Scholar] [CrossRef]




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| siRNA | Duplex Sequence | MW | |
|---|---|---|---|
| Control | Sense | CCUCGUGCCGUUCCAUCAGGUAGUU | 7487.7 |
| Antisense | CUACCUGAUGGAACGGCACGAGGUU | 7636.9 | |
| CYP1A1 | Sense | CUGGUAUUCUGGGUAAUCAUU | 6316.1 |
| Antisense | UGAUUACCCAGAAUACCAGUU | 6305.1 | |
| CYP1B1 | Sense | GCAACUUCAGCAACUUCAUUU | 6242.1 |
| Antisense | AUGAAGUUGCUGAAGUUGCUU | 6379.2 | |
| Gene | Sequence | Product Length (bp) | |
|---|---|---|---|
| β-actin | Forward | AACTGGAACGGTGAAGGT | 133 |
| Reverse | CCTGTAACAACGCATCTCATAT | ||
| mTOR | Forward | GAAGAAGGTCACTGAGGAT | 133 |
| Reverse | GGAGATGGAACGGAAGAA | ||
| CYP1A1 | Forward | CACAGCACAACAAGAGAC | 128 |
| Reverse | TCAGGTAGGAACTCAGATG | ||
| CYP1B1 | Forward | TGGAGATGAGGTCAGTTG | 181 |
| Reverse | AAGCACTTAGCACTTAGGA | ||
| LC3 | Forward | GAGCGAGTTGGTCAAGAT | 188 |
| Reverse | CTCAGAAGCCGAAGGTTT | ||
| P62 | Forward | GCAGACCAAGAACTATGAC | 123 |
| Reverse | CACAACTATGAGACAGAAGAG | ||
| TFE3 | Forward | TTGCTCCATCCTTTGTCT | 159 |
| Forward | GTCTCATCCTCACTTCTGT | ||
| LAMP2A | Forward | GCCATCTCCTACTACAACA | 138 |
| Forward | ACTGAAGCAACCTTATCCT | ||
| PLEKHM1 | Forward | CCACAAACACATCATCTCAG | 115 |
| Reverse | CAGGTAGCACTCCATCAG | ||
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Bu, K.-B.; Kim, M.; Shin, M.K.; Lee, S.-H.; Sung, J.-S. Regulation of Benzo[a]pyrene-Induced Hepatic Lipid Accumulation through CYP1B1-Induced mTOR-Mediated Lipophagy. Int. J. Mol. Sci. 2024, 25, 1324. https://doi.org/10.3390/ijms25021324
Bu K-B, Kim M, Shin MK, Lee S-H, Sung J-S. Regulation of Benzo[a]pyrene-Induced Hepatic Lipid Accumulation through CYP1B1-Induced mTOR-Mediated Lipophagy. International Journal of Molecular Sciences. 2024; 25(2):1324. https://doi.org/10.3390/ijms25021324
Chicago/Turabian StyleBu, Kyung-Bin, Min Kim, Min Kyoung Shin, Seung-Ho Lee, and Jung-Suk Sung. 2024. "Regulation of Benzo[a]pyrene-Induced Hepatic Lipid Accumulation through CYP1B1-Induced mTOR-Mediated Lipophagy" International Journal of Molecular Sciences 25, no. 2: 1324. https://doi.org/10.3390/ijms25021324
APA StyleBu, K.-B., Kim, M., Shin, M. K., Lee, S.-H., & Sung, J.-S. (2024). Regulation of Benzo[a]pyrene-Induced Hepatic Lipid Accumulation through CYP1B1-Induced mTOR-Mediated Lipophagy. International Journal of Molecular Sciences, 25(2), 1324. https://doi.org/10.3390/ijms25021324