
The Role of Glutamine Homeostasis in Emotional and Cognitive Functions


{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction: Despite Being a Non-Essential Amino Acid, Glutamine Is Essential for Maintaining the Glutamate–Glutamine Cycle
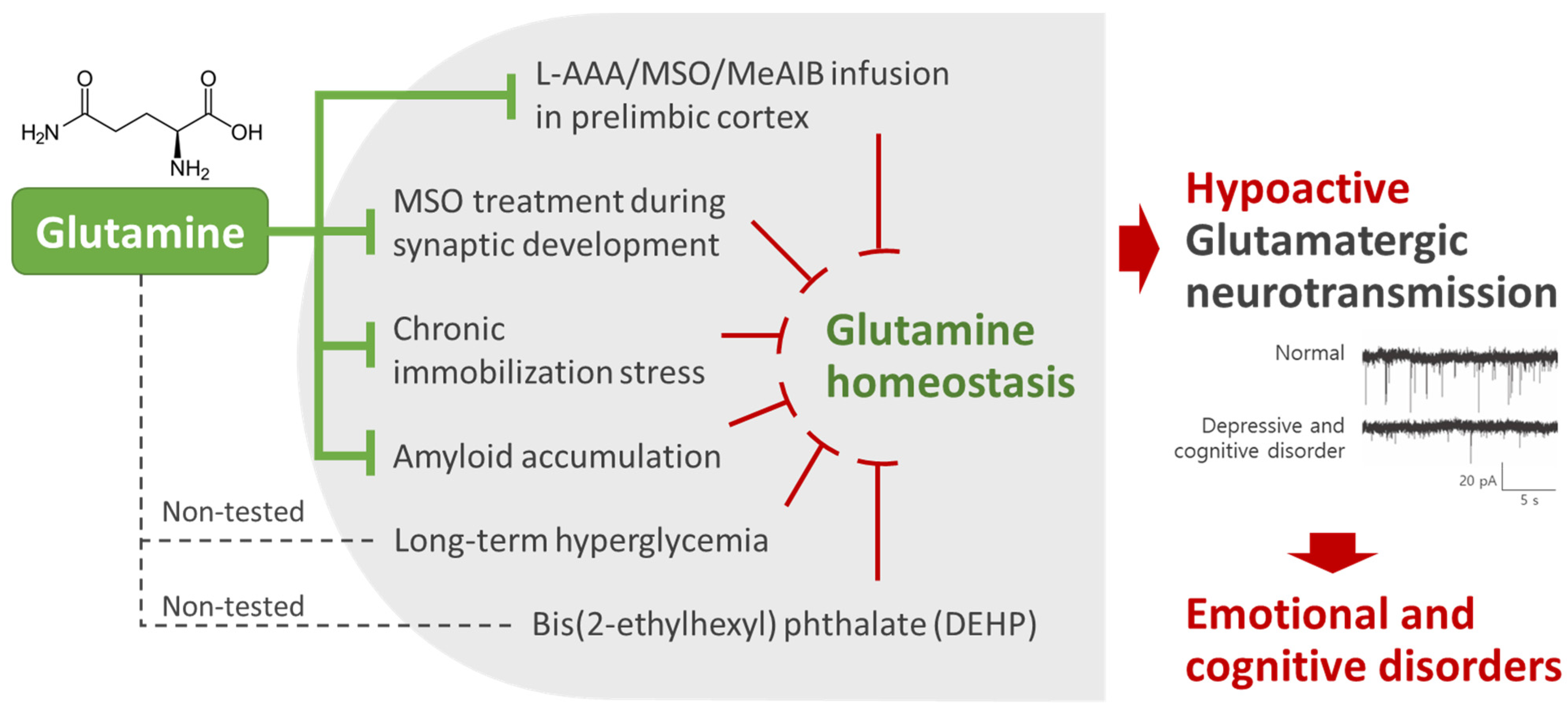
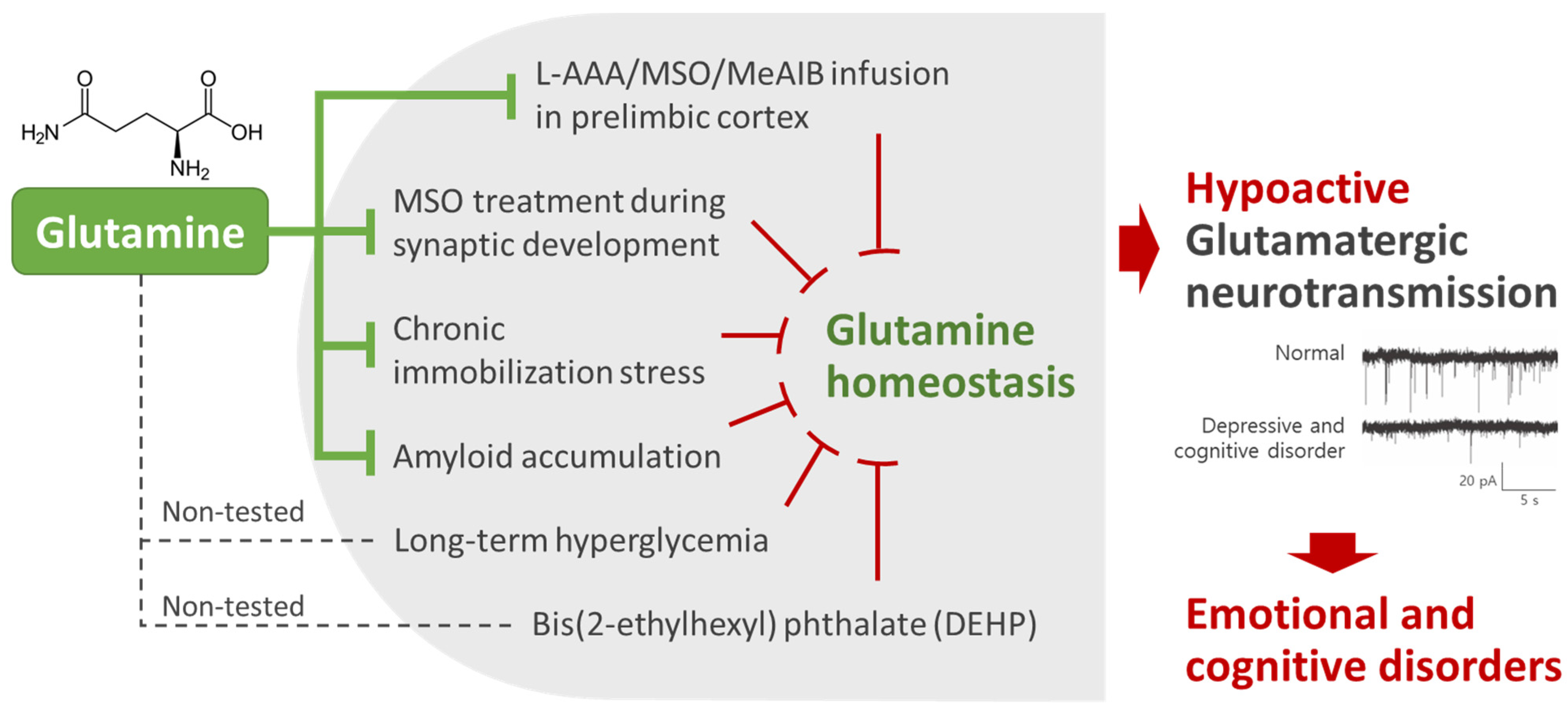
2. Glutamine Homeostasis Is Closely Related to Emotional and Cognitive Functions
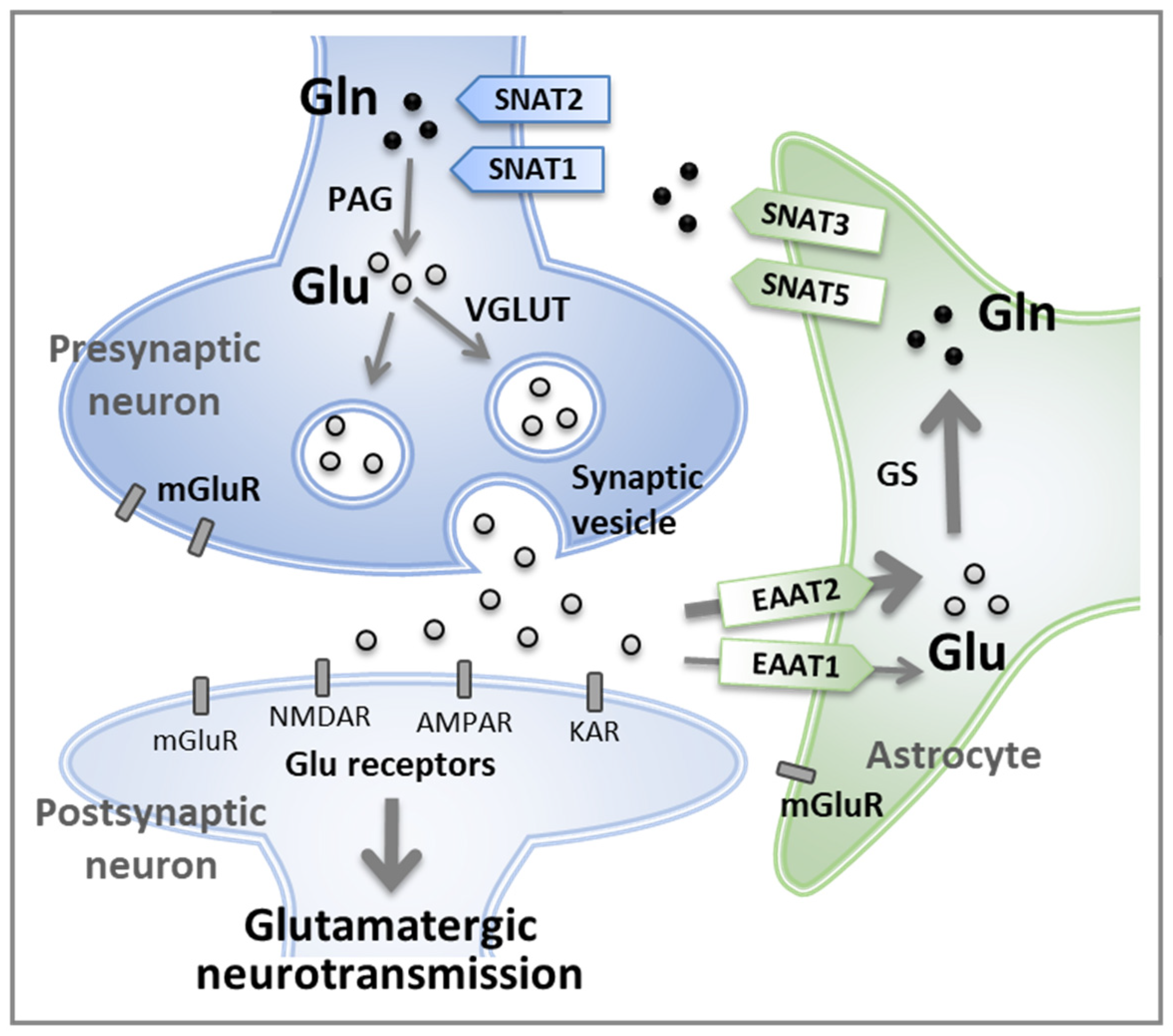
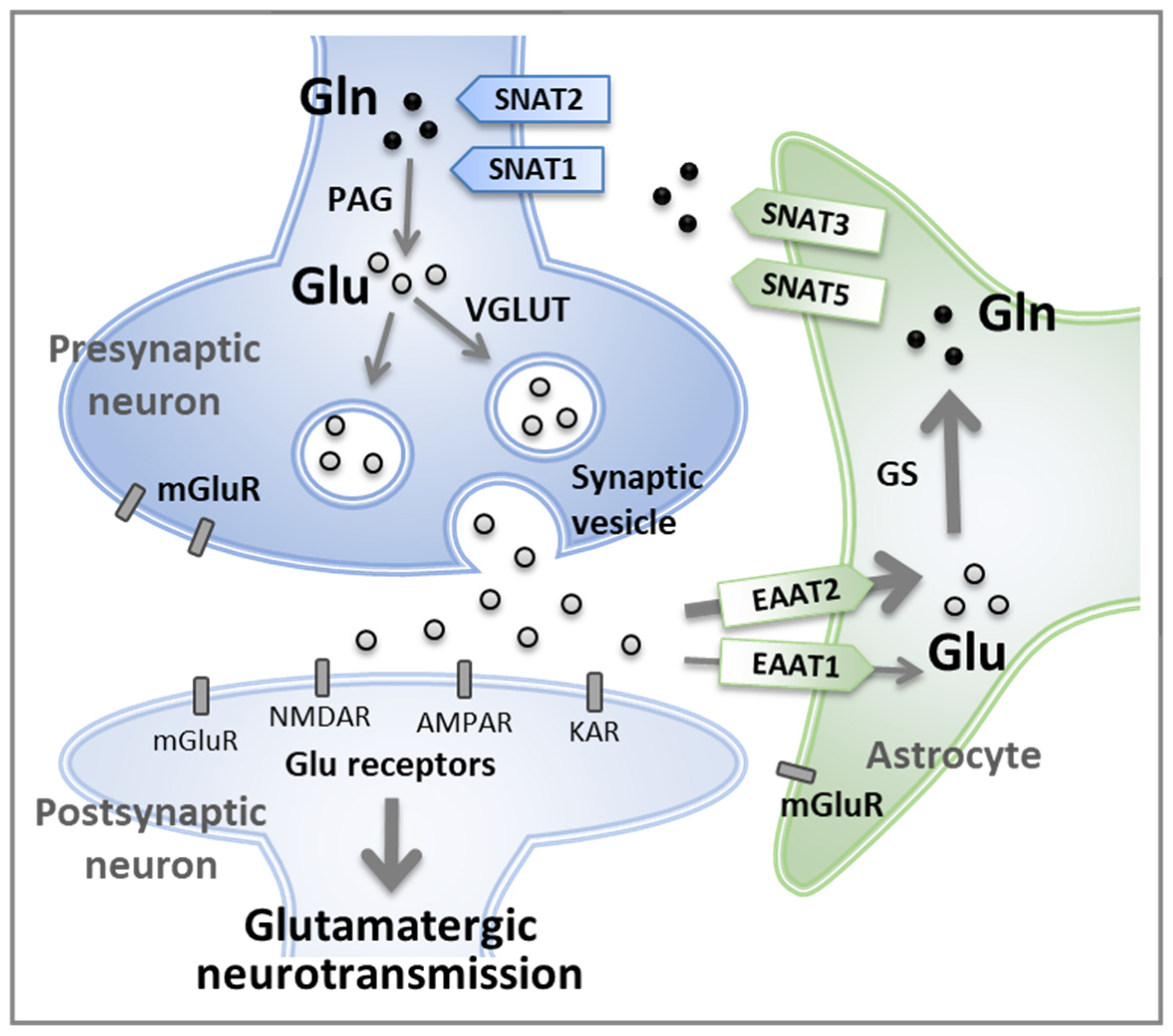
3. Regulation of Glutamine Concentration in the Central Nervous System
4. Clinical Studies of the Effects of Glutamine on Emotional and Cognitive Functions
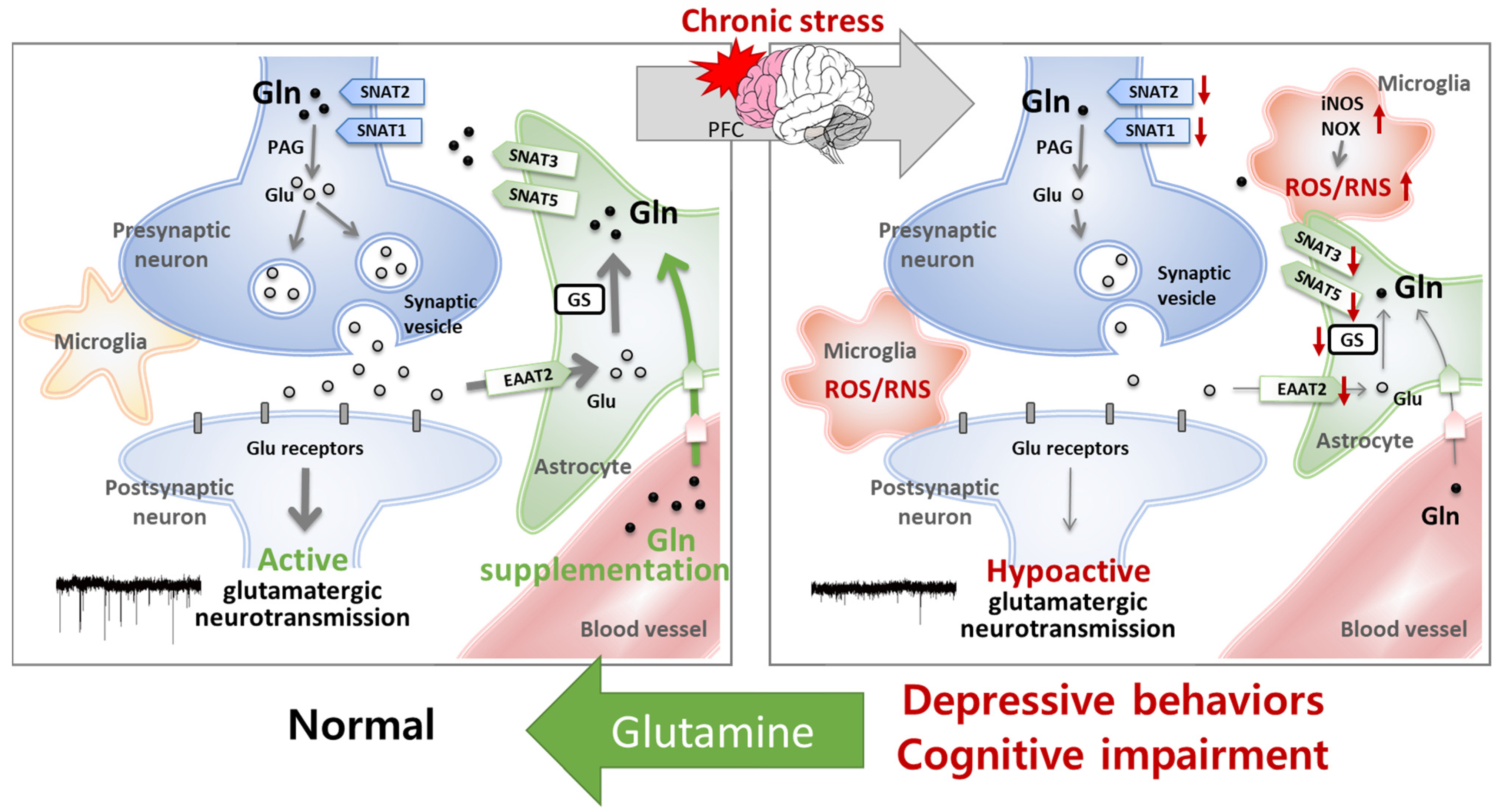
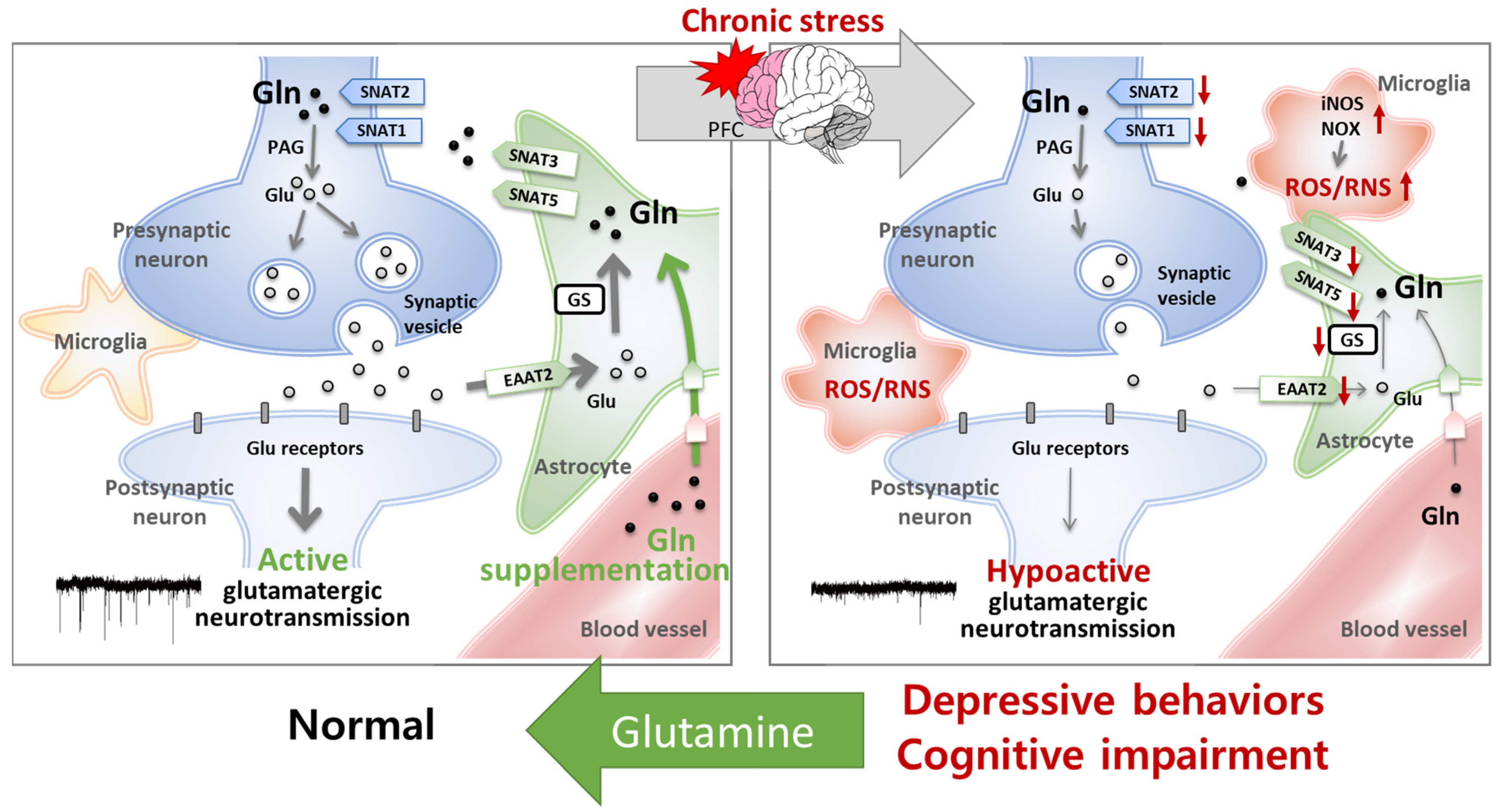
5. Mechanism Studies of the Effects of Glutamine on Emotional and Cognitive Function
5.1. The Homeostasis of Glutamatergic Neurotransmission Is Essential for Normal Behaviors
5.2. Glutamine Has Protective Effects on the Glu–Gln Cycle and Glutamatergic Neuronal Activity
5.3. Glutamine Has Antioxidant and Anti-Inflammatory Effects
6. Glutamine Supplementation Dosage
7. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Abbreviations
| 1H-MRS | proton magnetic resonance spectroscopy |
| 3xTg | triple-transgenic |
| AD | Alzheimer’s disease |
| AMPAR | α-amino-3-hydroxyl-5-methyl-4-isoxazole-propionate receptor |
| BBB | blood–brain barrier |
| CIS | chronic immobilization stress |
| CNS | central nervous system |
| CSF | cerebrospinal fluid |
| DEHP | bis(2-ethylhexyl) phthalate |
| EAAT | excitatory amino acid transporter |
| ECF | extracellular fluid |
| ECT | electroconvulsive therapy |
| GABA | γ-aminobutyric acid |
| Gln | glutamine |
| Glu | glutamate |
| Glx | Glu + Gln |
| GS | glutamine synthetase |
| iNOS | inducible nitric oxide synthase |
| KAR | kainate receptor |
| L-AAA | L-α-aminoadipic acid |
| MCI | mild cognitive impairment |
| MDD | major depressive disorder |
| MeAIB | α-methyl-amino-isobutyric acid |
| mGluR | metabotropic glutamate receptor |
| mPFC | medial prefrontal cortex |
| MSO | methionine sulfoximine |
| NMDAR | N-methyl-D-aspartate receptor |
| NMR | nuclear magnetic resonance |
| NOX | NADPH oxidase |
| PAG | phosphate-activated glutaminase |
| PFC | prefrontal cortex |
| PLC | prelimbic cortex |
| ROS/RNS | reactive oxygen/nitrogen species |
| sEPSC | spontaneous excitatory postsynaptic current |
| SNAT | sodium-coupled neutral amino acid transporter |
| VGLUT | vesicular glutamate transporter |
References
- Albrecht, J.; Sidoryk-Wegrzynowicz, M.; Zielinska, M.; Aschner, M. Roles of glutamine in neurotransmission. Neuron Glia Biol. 2010, 6, 263–276. [Google Scholar] [CrossRef] [PubMed]
- Watford, M. Glutamine and glutamate: Nonessential or essential amino acids? Anim. Nutr. 2015, 1, 119–122. [Google Scholar] [CrossRef] [PubMed]
- Fernández-Montoya, J.; Avendaño, C.; Negredo, P. The glutamatergic system in primary somatosensory neurons and its involvement in sensory input-dependent plasticity. Int. J. Mol. Sci. 2017, 19, 69. [Google Scholar] [CrossRef] [PubMed]
- Gasiorowska, A.; Wydrych, M.; Drapich, P.; Zadrozny, M.; Steczkowska, M.; Niewiadomski, W.; Niewiadomska, G. The biology and pathobiology of glutamatergic, cholinergic, and dopaminergic signaling in the aging brain. Front. Aging Neurosci. 2021, 13, 654931. [Google Scholar] [CrossRef] [PubMed]
- Attwell, D.; Laughlin, S.B. An energy budget for signaling in the grey matter of the brain. J. Cereb. Blood Flow. Metab. 2001, 21, 1133–1145. [Google Scholar] [CrossRef] [PubMed]
- Wernerman, J. Glutamine supplementation. Ann. Intensive Care 2011, 1, 25. [Google Scholar] [CrossRef] [PubMed]
- Popoli, M.; Yan, Z.; McEwen, B.S.; Sanacora, G. The stressed synapse: The impact of stress and glucocorticoids on glutamate transmission. Nat. Rev. Neurosci. 2012, 13, 22–37. [Google Scholar] [CrossRef]
- Bauminger, H.; Gaisler-Salomon, I. Beyond NMDA receptors: Homeostasis at the glutamate tripartite synapse and its contributions to cognitive dysfunction in schizophrenia. Int. J. Mol. Sci. 2022, 23, 8617. [Google Scholar] [CrossRef]
- Sarawagi, A.; Soni, N.D.; Patel, A.B. Glutamate and GABA homeostasis and neurometabolism in major depressive disorder. Front. Psychiatry 2021, 12, 637863. [Google Scholar] [CrossRef]
- Reiner, A.; Levitz, J. Glutamatergic signaling in the central nervous system: Ionotropic and metabotropic receptors in concert. Neuron 2018, 98, 1080–1098. [Google Scholar] [CrossRef]
- Zhou, Y.; Danbolt, N.C. Glutamate as a neurotransmitter in the healthy brain. J. Neural. Transm. (Vienna) 2014, 121, 799–817. [Google Scholar] [CrossRef] [PubMed]
- Boulland, J.L.; Osen, K.K.; Levy, L.M.; Danbolt, N.C.; Edwards, R.H.; Storm-Mathisen, J.; Chaudhry, F.A. Cell-specific expression of the glutamine transporter SN1 suggests differences in dependence on the glutamine cycle. Eur. J. Neurosci. 2002, 15, 1615–1631. [Google Scholar] [CrossRef] [PubMed]
- Cubelos, B.; Gonzalez-Gonzalez, I.M.; Gimenez, C.; Zafra, F. Amino acid transporter SNAT5 localizes to glial cells in the rat brain. Glia 2005, 49, 230–244. [Google Scholar] [CrossRef]
- González-González, I.M.; Cubelos, B.; Giménez, C.; Zafra, F. Immunohistochemical localization of the amino acid transporter SNAT2 in the rat brain. Neuroscience 2005, 130, 61–73. [Google Scholar] [CrossRef] [PubMed]
- Jenstad, M.; Quazi, A.Z.; Zilberter, M.; Haglerod, C.; Berghuis, P.; Saddique, N.; Goiny, M.; Buntup, D.; Davanger, S.; Haug, F.M.S.; et al. System a transporter SAT2 mediates replenishment of dendritic glutamate pools controlling retrograde signaling by glutamate. Cerebral Cortex 2009, 19, 1092–1106. [Google Scholar] [CrossRef]
- Mackenzie, B.; Schäfer, M.K.; Erickson, J.D.; Hediger, M.A.; Weihe, E.; Varoqui, H. Functional properties and cellular distribution of the system a glutamine transporter SNAT1 support specialized roles in central neurons. J. Biol. Chem. 2003, 278, 23720–23730. [Google Scholar] [CrossRef] [PubMed]
- Kanamori, K.; Ross, B.D.; Kondrat, R.W. Glial uptake of neurotransmitter glutamate from the extracellular fluid studied in vivo by microdialysis and 13C NMR. J. Neurochem. 2002, 83, 682–695. [Google Scholar] [CrossRef]
- Lebon, V.; Petersen, K.F.; Cline, G.W.; Shen, J.; Mason, G.F.; Dufour, S.; Behar, K.L.; Shulman, G.I.; Rothman, D.L. Astroglial contribution to brain energy metabolism in humans revealed by 13C nuclear magnetic resonance spectroscopy: Elucidation of the dominant pathway for neurotransmitter glutamate repletion and measurement of astrocytic oxidative metabolism. J. Neurosci. 2002, 22, 1523–1531. [Google Scholar] [CrossRef]
- Sibson, N.R.; Dhankhar, A.; Mason, G.F.; Behar, K.L.; Rothman, D.L.; Shulman, R.G. In vivo 13C NMR measurements of cerebral glutamine synthesis as evidence for glutamate-glutamine cycling. Proc. Natl. Acad. Sci. USA 1997, 94, 2699–2704. [Google Scholar] [CrossRef]
- Son, H.; Baek, J.H.; Go, B.S.; Jung, D.H.; Sontakke, S.B.; Chung, H.J.; Lee, D.H.; Roh, G.S.; Kang, S.S.; Cho, G.J.; et al. Glutamine has antidepressive effects through increments of glutamate and glutamine levels and glutamatergic activity in the medial prefrontal cortex. Neuropharmacology 2018, 143, 143–152. [Google Scholar] [CrossRef]
- Merritt, K.; McGuire, P.; Egerton, A. Relationship between glutamate dysfunction and symptoms and cognitive function in psychosis. Front. Psychiatry 2013, 4, 151. [Google Scholar] [CrossRef]
- Son, H.; Kim, S.; Jung, D.H.; Baek, J.H.; Lee, D.H.; Roh, G.S.; Kang, S.S.; Cho, G.J.; Choi, W.S.; Lee, D.K.; et al. Insufficient glutamine synthetase activity during synaptogenesis causes spatial memory impairment in adult mice. Sci. Rep. 2019, 9, 252. [Google Scholar] [CrossRef]
- Cocchi, R. Antidepressive properties of I-glutamine. Prelim. Report. Acta Psychiatr. Belg. 1976, 76, 658–666. [Google Scholar]
- Dos Santos Quaresma, M.; Souza, W.; Lemos, V.A.; Caris, A.V.; Thomatieli-Santos, R.V. The possible importance of glutamine supplementation to mood and cognition in hypoxia from high altitude. Nutrients 2020, 12, 3627. [Google Scholar] [CrossRef]
- Ravel, J.M.; Felsing, B.; Lansford, E.M., Jr.; Trubey, R.H.; Shive, W. Reversal of alcohol toxicity by glutamine. J. Biol. Chem. 1955, 214, 497–501. [Google Scholar] [CrossRef]
- Rogers, L.L.; Pelton, R.B. Glutamine in the treatment of alcoholism; a preliminary report. Q. J. Stud. Alcohol. 1957, 18, 581–587. [Google Scholar] [CrossRef]
- Rogers, L.L.; Pelton, R.B. Effect of glutamine in IQ scores of mentally deficient children. Tex. Rep. Biol. Med. 1957, 15, 84–90. [Google Scholar]
- Young, L.S.; Bye, R.; Scheltinga, M.; Ziegler, T.R.; Jacobs, D.O.; Wilmore, D.W. Patients receiving glutamine-supplemented intravenous feedings report an improvement in mood. J. Parenter. Enteral Nutr. 1993, 17, 422–427. [Google Scholar] [CrossRef]
- Baek, J.H.; Jung, S.; Son, H.; Kang, J.S.; Kim, H.J. Glutamine supplementation prevents chronic stress-induced mild cognitive impairment. Nutrients 2020, 12, 910. [Google Scholar] [CrossRef]
- Baek, J.H.; Vignesh, A.; Son, H.; Lee, D.H.; Roh, G.S.; Kang, S.S.; Cho, G.J.; Choi, W.S.; Kim, H.J. Glutamine supplementation ameliorates chronic stress-induced reductions in glutamate and glutamine transporters in the mouse prefrontal cortex. Exp. Neurobiol. 2019, 28, 270–278. [Google Scholar] [CrossRef]
- Lee, Y.; Son, H.; Kim, G.; Kim, S.; Lee, D.H.; Roh, G.S.; Kang, S.S.; Cho, G.J.; Choi, W.S.; Kim, H.J. Glutamine deficiency in the prefrontal cortex increases depressive-like behaviours in male mice. J. Psychiatry Neurosci. 2013, 38, 183–191. [Google Scholar] [CrossRef]
- Baek, J.H.; Kang, J.S.; Song, M.; Lee, D.K.; Kim, H.J. Glutamine supplementation preserves glutamatergic neuronal activity in the infralimbic cortex, which delays the onset of mild cognitive impairment in 3xTg-AD female mice. Nutrients 2023, 15, 2794. [Google Scholar] [CrossRef]
- Albrecht, J.; Sonnewald, U.; Waagepetersen, H.S.; Schousboe, A. Glutamine in the central nervous system: Function and dysfunction. Front. Biosci. 2007, 12, 332–343. [Google Scholar] [CrossRef]
- Bukke, V.N.; Archana, M.; Villani, R.; Romano, A.D.; Wawrzyniak, A.; Balawender, K.; Orkisz, S.; Beggiato, S.; Serviddio, G.; Cassano, T. The dual role of glutamatergic neurotransmission in Alzheimer’s disease: From pathophysiology to pharmacotherapy. Int. J. Mol. Sci. 2020, 21, 7452. [Google Scholar] [CrossRef]
- Auer, D.P.; Putz, B.; Kraft, E.; Lipinski, B.; Schill, J.; Holsboer, F. Reduced glutamate in the anterior cingulate cortex in depression: An in vivo proton magnetic resonance spectroscopy study. Biol. Psychiatry 2000, 47, 305–313. [Google Scholar] [CrossRef]
- Hasler, G.; van der Veen, J.W.; Tumonis, T.; Meyers, N.; Shen, J.; Drevets, W.C. Reduced prefrontal glutamate/glutamine and gamma-aminobutyric acid levels in major depression determined using proton magnetic resonance spectroscopy. Arch. Gen. Psychiatry 2007, 64, 193–200. [Google Scholar] [CrossRef]
- Michael, N.; Erfurth, A.; Ohrmann, P.; Arolt, V.; Heindel, W.; Pfleiderer, B. Metabolic changes within the left dorsolateral prefrontal cortex occurring with electroconvulsive therapy in patients with treatment resistant unipolar depression. Psychol. Med. 2003, 33, 1277–1284. [Google Scholar] [CrossRef]
- Pfleiderer, B.; Michael, N.; Erfurth, A.; Ohrmann, P.; Hohmann, U.; Wolgast, M.; Fiebich, M.; Arolt, V.; Heindel, W. Effective electroconvulsive therapy reverses glutamate/glutamine deficit in the left anterior cingulum of unipolar depressed patients. Psychiatry Res. 2003, 122, 185–192. [Google Scholar] [CrossRef]
- Madeira, C.; Alheira, F.V.; Calcia, M.A.; Silva, T.C.S.; Tannos, F.M.; Vargas-Lopes, C.; Fisher, M.; Goldenstein, N.; Brasil, M.A.; Vinogradov, S.; et al. Blood levels of glutamate and glutamine in recent onset and chronic schizophrenia. Front. Psychiatry 2018, 9, 713. [Google Scholar] [CrossRef]
- Shen, J.; Tomar, J.S. Elevated brain glutamate levels in bipolar disorder and pyruvate carboxylase-mediated anaplerosis. Front. Psychiatry 2021, 12, 640977. [Google Scholar] [CrossRef]
- Chen, L.P.; Dai, H.Y.; Dai, Z.Z.; Xu, C.T.; Wu, R.H. Anterior cingulate cortex and cerebellar hemisphere neurometabolite changes in depression treatment: A 1H magnetic resonance spectroscopy study. Psychiatry Clin. Neurosci. 2014, 68, 357–364. [Google Scholar] [CrossRef]
- Njau, S.; Joshi, S.H.; Espinoza, R.; Leaver, A.M.; Vasavada, M.; Marquina, A.; Woods, R.P.; Narr, K.L. Neurochemical correlates of rapid treatment response to electroconvulsive therapy in patients with major depression. J. Psychiatry Neurosci. 2017, 42, 6–16. [Google Scholar] [CrossRef]
- Moghaddam, B.; Adams, B.; Verma, A.; Daly, D. Activation of glutamatergic neurotransmission by ketamine: A novel step in the pathway from NMDA receptor blockade to dopaminergic and cognitive disruptions associated with the prefrontal cortex. J. Neurosci. 1997, 17, 2921–2927. [Google Scholar] [CrossRef]
- Zanos, P.; Moaddel, R.; Morris, P.J.; Georgiou, P.; Fischell, J.; Elmer, G.I.; Alkondon, M.; Yuan, P.; Pribut, H.J.; Singh, N.S.; et al. NMDAR inhibition-independent antidepressant actions of ketamine metabolites. Nature 2016, 533, 481–486. [Google Scholar] [CrossRef]
- Rajkowska, G.; Miguel-Hidalgo, J.J. Gliogenesis and glial pathology in depression. CNS Neurol. Disord. Drug Targets 2007, 6, 219–233. [Google Scholar] [CrossRef]
- Sanacora, G.; Banasr, M. From pathophysiology to novel antidepressant drugs: Glial contributions to the pathology and treatment of mood disorders. Biol. Psychiatry 2013, 73, 1172–1179. [Google Scholar] [CrossRef]
- Choudary, P.V.; Molnar, M.; Evans, S.J.; Tomita, H.; Li, J.Z.; Vawter, M.P.; Myers, R.M.; Bunney, W.E.; Akil, H.; Watson, S.J.; et al. Altered cortical glutamatergic and GABAergic signal transmission with glial involvement in depression. Proc. Natl. Acad. Sci. USA 2005, 102, 15653–15658. [Google Scholar] [CrossRef]
- Boksha, I.S.; Tereshkina, E.B.; Savushkina, O.K.; Prokhorova, T.A.; Vorobyeva, E.A.; Burbaeva, G.S. Comparative studies of glutamine synthetase levels in the brains of patients with schizophrenia and mentally healthy people. Neurochem. J. 2018, 12, 95–101. [Google Scholar] [CrossRef]
- Kantrowitz, J.T.; Dong, Z.; Milak, M.S.; Rashid, R.; Kegeles, L.S.; Javitt, D.C.; Lieberman, J.A.; John Mann, J. Ventromedial prefrontal cortex/anterior cingulate cortex Glx, glutamate, and GABA levels in medication-free major depressive disorder. Transl. Psychiatry 2021, 11, 419. [Google Scholar] [CrossRef]
- McEwen, A.M.; Burgess, D.T.; Hanstock, C.C.; Seres, P.; Khalili, P.; Newman, S.C.; Baker, G.B.; Mitchell, N.D.; Khudabux-Der, J.; Allen, P.S.; et al. Increased glutamate levels in the medial prefrontal cortex in patients with postpartum depression. Neuropsychopharmacology 2012, 37, 2428–2435. [Google Scholar] [CrossRef]
- Naylor, B.; Hesam-Shariati, N.; McAuley, J.H.; Boag, S.; Newton-John, T.; Rae, C.D.; Gustin, S.M. Reduced glutamate in the medial prefrontal cortex is associated with emotional and cognitive dysregulation in people with chronic pain. Front. Neurol. 2019, 10, 1110. [Google Scholar] [CrossRef]
- Shirayama, Y.; Takahashi, M.; Osone, F.; Hara, A.; Okubo, T. Myo-inositol, glutamate, and glutamine in the prefrontal cortex, hippocampus, and amygdala in major depression. Biol. Psychiatry Cogn. Neurosci. Neuroimaging 2017, 2, 196–204. [Google Scholar] [CrossRef]
- Moriguchi, S.; Takamiya, A.; Noda, Y.; Horita, N.; Wada, M.; Tsugawa, S.; Plitman, E.; Sano, Y.; Tarumi, R.; ElSalhy, M.; et al. Glutamatergic neurometabolite levels in major depressive disorder: A systematic review and meta-analysis of proton magnetic resonance spectroscopy studies. Mol. Psychiatry 2019, 24, 952–964. [Google Scholar] [CrossRef]
- Baeken, C.; Lefaucheur, J.-P.; Van Schuerbeek, P. The impact of accelerated high frequency rTMS on brain neurochemicals in treatment-resistant depression: Insights from 1H MR spectroscopy. Clin. Neurophysiol. 2017, 128, 1664–1672. [Google Scholar] [CrossRef]
- Narayan, G.A.; Hill, K.R.; Wengler, K.; He, X.; Wang, J.; Yang, J.; Parsey, R.V.; DeLorenzo, C. Does the change in glutamate to GABA ratio correlate with change in depression severity? A randomized, double-blind clinical trial. Mol. Psychiatry 2022, 27, 3833–3841. [Google Scholar] [CrossRef]
- Gabbay, V.; Bradley, K.A.; Mao, X.; Ostrover, R.; Kang, G.; Shungu, D.C. Anterior cingulate cortex γ-aminobutyric acid deficits in youth with depression. Transl. Psychiatry 2017, 7, e1216. [Google Scholar] [CrossRef]
- Reddy-Thootkur, M.; Kraguljac, N.V.; Lahti, A.C. The role of glutamate and GABA in cognitive dysfunction in schizophrenia and mood disorders—A systematic review of magnetic resonance spectroscopy studies. Schizophr. Res. 2022, 249, 74–84. [Google Scholar] [CrossRef]
- Ongür, D.; Prescot, A.P.; Jensen, J.E.; Cohen, B.M.; Renshaw, P.F. Creatine abnormalities in schizophrenia and bipolar disorder. Psychiatry Res. 2009, 172, 44–48. [Google Scholar] [CrossRef]
- Tumati, S.; Martens, S.; Aleman, A. Magnetic resonance spectroscopy in mild cognitive impairment: Systematic review and meta-analysis. Neurosci. Biobehav. Rev. 2013, 37, 2571–2586. [Google Scholar] [CrossRef]
- Ino, H.; Honda, S.; Yamada, K.; Horita, N.; Tsugawa, S.; Yoshida, K.; Noda, Y.; Meyer, J.H.; Mimura, M.; Nakajima, S.; et al. Glutamatergic neurometabolite levels in bipolar disorder: A systematic review and meta-analysis of proton magnetic resonance spectroscopy studies. Biol. Psychiatry Cogn. Neurosci. Neuroimaging 2023, 8, 140–150. [Google Scholar] [CrossRef]
- Bernstein, H.G.; Meyer-Lotz, G.; Dobrowolny, H.; Bannier, J.; Steiner, J.; Walter, M.; Bogerts, B. Reduced density of glutamine synthetase immunoreactive astrocytes in different cortical areas in major depression but not in bipolar I disorder. Front. Cell. Neurosci. 2015, 9, 273. [Google Scholar] [CrossRef]
- Son, H.; Jung, S.; Shin, J.H.; Kang, M.J.; Kim, H.J. Anti-stress and anti-depressive effects of spinach extracts on a chronic stress-induced depression mouse model through lowering blood corticosterone and increasing brain glutamate and glutamine levels. J. Clin. Med. 2018, 7, 406. [Google Scholar] [CrossRef]
- Kang, J.S.; Baek, J.H.; Jung, S.; Chung, H.J.; Lee, D.K.; Kim, H.J. Ingestion of bis(2-ethylhexyl) phthalate (DEHP) during adolescence causes depressive-like behaviors through hypoactive glutamatergic signaling in the medial prefrontal cortex. Environ. Pollut. 2021, 289, 117978. [Google Scholar] [CrossRef]
- Kang, J.S.; Baek, J.H.; Song, M.Y.; Rehman, N.U.; Chung, H.J.; Lee, D.K.; Yoo, D.Y.; Kim, H.J. Long-term exposure changes the environmentally relevant bis(2-ethylhexyl) phthalate to be a neuro-hazardous substance disrupting neural homeostasis in emotional and cognitive functions. Environ. Pollut. 2023, 324, 121387. [Google Scholar] [CrossRef]
- D’Souza, R.; Powell-Tuck, J. Glutamine supplements in the critically ill. J. R. Soc. Med. 2004, 97, 425–427. [Google Scholar] [CrossRef]
- Madeira, C.; Vargas-Lopes, C.; Brandão, C.O.; Reis, T.; Laks, J.; Panizzutti, R.; Ferreira, S.T. Elevated glutamate and glutamine levels in the cerebrospinal fluid of patients with probable Alzheimer’s disease and depression. Front. Psychiatry 2018, 9, 561. [Google Scholar] [CrossRef]
- He, W.; Wu, G. Metabolism of amino acids in the brain and their roles in regulating food intake. Adv. Exp. Med. Biol. 2020, 1265, 167–185. [Google Scholar] [CrossRef]
- Broer, S.; Brookes, N. Transfer of glutamine between astrocytes and neurons. J. Neurochem. 2001, 77, 705–719. [Google Scholar] [CrossRef]
- Hawkins, R.A. The blood-brain barrier and glutamate. Am. J. Clin. Nutr. 2009, 90, 867s–874s. [Google Scholar] [CrossRef]
- Alajangi, H.K.; Kaur, M.; Sharma, A.; Rana, S.; Thakur, S.; Chatterjee, M.; Singla, N.; Jaiswal, P.K.; Singh, G.; Barnwal, R.P. Blood–brain barrier: Emerging trends on transport models and new-age strategies for therapeutics intervention against neurological disorders. Mol. Brain 2022, 15, 49. [Google Scholar] [CrossRef]
- Golden, P.L.; Pollack, G.M. Blood-brain barrier efflux transport. J. Pharm. Sci. 2003, 92, 1739–1753. [Google Scholar] [CrossRef] [PubMed]
- Newsholme, P. Why is L-glutamine metabolism important to cells of the immune system in health, postinjury, surgery or infection? J. Nutr. 2001, 131, 2515S–2522S. [Google Scholar] [CrossRef] [PubMed]
- Peters, A. The selfish brain: Competition for energy resources. Am. J. Hum. Biol. 2011, 23, 29–34. [Google Scholar] [CrossRef] [PubMed]
- Wilmore, D.W.; Shabert, J.K. Role of glutamine in immunologic responses. Nutrition 1998, 14, 618–626. [Google Scholar] [CrossRef] [PubMed]
- Walsh, N.P.; Blannin, A.K.; Bishop, N.C.; Robson, P.J.; Gleeson, M. Effect of oral glutamine supplementation on human neutrophil lipopolysaccharide-stimulated degranulation following prolonged exercise. Int. J. Sport. Nutr. Exerc. Metab. 2000, 10, 39–50. [Google Scholar] [CrossRef] [PubMed]
- Krzywkowski, K.; Petersen, E.W.; Ostrowski, K.; Link-Amster, H.; Boza, J.; Halkjaer-Kristensen, J.; Pedersen, B.K. Effect of glutamine and protein supplementation on exercise-induced decreases in salivary IgA. J. Appl. Physiol. 2001, 91, 832–838. [Google Scholar] [CrossRef] [PubMed]
- Gleeson, M. Dosing and efficacy of glutamine supplementation in human exercise and sport training. J. Nutr. 2008, 138, 2045S–2049S. [Google Scholar] [CrossRef]
- Legault, Z.; Bagnall, N.; Kimmerly, D.S. The influence of oral L-glutamine supplementation on muscle strength recovery and soreness following unilateral knee extension eccentric exercise. Int. J. Sport. Nutr. Exerc. Metab. 2015, 25, 417–426. [Google Scholar] [CrossRef]
- Bailey, D.M.; Castell, L.M.; Newsholme, E.A.; Davies, B. Continuous and intermittent exposure to the hypoxia of altitude: Implications for glutamine metabolism and exercise performance. Br. J. Sports Med. 2000, 34, 210–212. [Google Scholar] [CrossRef]
- Caris, A.V.; Lira, F.S.; de Mello, M.T.; Oyama, L.M.; dos Santos, R.V. Carbohydrate and glutamine supplementation modulates the Th1/Th2 balance after exercise performed at a simulated altitude of 4500 m. Nutrition 2014, 30, 1331–1336. [Google Scholar] [CrossRef]
- Wischmeyer, P.E.; Kahana, M.; Wolfson, R.; Ren, H.; Musch, M.M.; Chang, E.B. Glutamine reduces cytokine release, organ damage, and mortality in a rat model of endotoxemia. Shock 2001, 16, 398–402. [Google Scholar] [CrossRef] [PubMed]
- Pereira, M.F.; Amaral, I.M.; Lopes, C.; Leitão, C.; Madeira, D.; Lopes, J.P.; Gonçalves, F.Q.; Canas, P.M.; Cunha, R.A.; Agostinho, P. L-α-aminoadipate causes astrocyte pathology with negative impact on mouse hippocampal synaptic plasticity and memory. FASEB J. 2021, 35, e21726. [Google Scholar] [CrossRef] [PubMed]
- Nishimura, R.N.; Santos, D.; Fu, S.T.; Dwyer, B.E. Induction of cell death by L-alpha-aminoadipic acid exposure in cultured rat astrocytes: Relationship to protein synthesis. Neurotoxicology 2000, 21, 313–320. [Google Scholar] [PubMed]
- Hamani, C.; Diwan, M.; Macedo, C.E.; Brandao, M.L.; Shumake, J.; Gonzalez-Lima, F.; Raymond, R.; Lozano, A.M.; Fletcher, P.J.; Nobrega, J.N. Antidepressant-like effects of medial prefrontal cortex deep brain stimulation in rats. Biol. Psychiatry 2010, 67, 117–124. [Google Scholar] [CrossRef] [PubMed]
- Hamani, C.; Machado, D.C.; Hipolide, D.C.; Dubiela, F.P.; Suchecki, D.; Macedo, C.E.; Tescarollo, F.; Martins, U.; Covolan, L.; Nobrega, J.N. Deep brain stimulation reverses anhedonic-like behavior in a chronic model of depression: Role of serotonin and brain derived neurotrophic factor. Biol. Psychiatry 2012, 71, 30–35. [Google Scholar] [CrossRef]
- Warden, M.R.; Selimbeyoglu, A.; Mirzabekov, J.J.; Lo, M.; Thompson, K.R.; Kim, S.Y.; Adhikari, A.; Tye, K.M.; Frank, L.M.; Deisseroth, K. A prefrontal cortex-brainstem neuronal projection that controls response to behavioural challenge. Nature 2012, 492, 428–432. [Google Scholar] [CrossRef]
- Fuchikami, M.; Thomas, A.; Liu, R.; Wohleb, E.S.; Land, B.B.; DiLeone, R.J.; Aghajanian, G.K.; Duman, R.S. Optogenetic stimulation of infralimbic pfc reproduces ketamine’s rapid and sustained antidepressant actions. Proc. Natl. Acad. Sci. USA 2015, 112, 8106–8111. [Google Scholar] [CrossRef]
- Baek, J.H.; Son, H.; Kang, J.S.; Yoo, D.Y.; Chung, H.J.; Lee, D.K.; Kim, H.J. Long-term hyperglycemia causes depressive behaviors in mice with hypoactive glutamatergic activity in the medial prefrontal cortex, which is not reversed by insulin treatment. Cells 2022, 11, 4012. [Google Scholar] [CrossRef]
- Chaudhry, F.A.; Lehre, K.P.; van Lookeren Campagne, M.; Ottersen, O.P.; Danbolt, N.C.; Storm-Mathisen, J. Glutamate transporters in glial plasma membranes: Highly differentiated localizations revealed by quantitative ultrastructural immunocytochemistry. Neuron 1995, 15, 711–720. [Google Scholar] [CrossRef]
- Kim, M.; Jung, S.; Son, H.; Kim, H.J. Glutamine-supplement diet maintains growth performance and reduces blood corticosterone level in cage-reared growing chicks. J. Agric. Life Sci. 2018, 52, 91–102. [Google Scholar] [CrossRef]
- Joo, Y.; Choi, K.M.; Lee, Y.H.; Kim, G.; Lee, D.H.; Roh, G.S.; Kang, S.S.; Cho, G.J.; Choi, W.S.; Kim, H.J. Chronic immobilization stress induces anxiety- and depression-like behaviors and decreases transthyretin in the mouse cortex. Neurosci. Lett. 2009, 461, 121–125. [Google Scholar] [CrossRef] [PubMed]
- Son, H.; Yang, J.H.; Kim, H.J.; Lee, D.K. A chronic immobilization stress protocol for inducing depression-like behavior in mice. J. Vis. Exp. 2019, 147, e59546. [Google Scholar] [CrossRef]
- Corbett, N.J.; Gabbott, P.L.; Klementiev, B.; Davies, H.A.; Colyer, F.M.; Novikova, T.; Stewart, M.G. Amyloid-beta induced CA1 pyramidal cell loss in young adult rats is alleviated by systemic treatment with FGL, a neural cell adhesion molecule-derived mimetic peptide. PLoS ONE 2013, 8, e71479. [Google Scholar] [CrossRef] [PubMed]
- West, M.J.; Kawas, C.H.; Martin, L.J.; Troncoso, J.C. The CA1 region of the human hippocampus is a hot spot in Alzheimer’s disease. Ann. N. Y Acad. Sci. 2000, 908, 255–259. [Google Scholar] [CrossRef] [PubMed]
- Di Meo, S.; Reed, T.T.; Venditti, P.; Victor, V.M. Role of ROS and RNS sources in physiological and pathological conditions. Oxidative Med. Cell. Longev. 2016, 2016, 1245049. [Google Scholar] [CrossRef] [PubMed]
- Tejada-Simon, M.V.; Serrano, F.; Villasana, L.E.; Kanterewicz, B.I.; Wu, G.Y.; Quinn, M.T.; Klann, E. Synaptic localization of a functional NADPH oxidase in the mouse hippocampus. Mol. Cell. Neurosci. 2005, 29, 97–106. [Google Scholar] [CrossRef]
- Li, Y.; Yu, Z.; Liu, F.; Tan, L.; Wu, B.; Li, J. Oral glutamine ameliorates chemotherapy-induced changes of intestinal permeability and does not interfere with the antitumor effect of chemotherapy in patients with breast cancer: A prospective randomized trial. Tumori J. 2006, 92, 396–401. [Google Scholar] [CrossRef]
- Topkan, E.; Parlak, C.; Topuk, S.; Pehlivan, B. Influence of oral glutamine supplementation on survival outcomes of patients treated with concurrent chemoradiotherapy for locally advanced non-small cell lung cancer. BMC Cancer 2012, 12, 502. [Google Scholar] [CrossRef]
- Tsujimoto, T.; Wasa, M.; Inohara, H.; Ito, T. L-glutamine and survival of patients with locally advanced head and neck cancer receiving chemoradiotherapy. Nutrients 2023, 15, 4117. [Google Scholar] [CrossRef]
- Nair, A.B.; Jacob, S. A simple practice guide for dose conversion between animals and human. J. Basic. Clin. Pharm. 2016, 7, 27–31. [Google Scholar] [CrossRef]
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Baek, J.H.; Park, H.; Kang, H.; Kim, R.; Kang, J.S.; Kim, H.J. The Role of Glutamine Homeostasis in Emotional and Cognitive Functions. Int. J. Mol. Sci. 2024, 25, 1302. https://doi.org/10.3390/ijms25021302
Baek JH, Park H, Kang H, Kim R, Kang JS, Kim HJ. The Role of Glutamine Homeostasis in Emotional and Cognitive Functions. International Journal of Molecular Sciences. 2024; 25(2):1302. https://doi.org/10.3390/ijms25021302
Chicago/Turabian StyleBaek, Ji Hyeong, Hyeongchan Park, Hyeju Kang, Rankyung Kim, Jae Soon Kang, and Hyun Joon Kim. 2024. "The Role of Glutamine Homeostasis in Emotional and Cognitive Functions" International Journal of Molecular Sciences 25, no. 2: 1302. https://doi.org/10.3390/ijms25021302
APA StyleBaek, J. H., Park, H., Kang, H., Kim, R., Kang, J. S., & Kim, H. J. (2024). The Role of Glutamine Homeostasis in Emotional and Cognitive Functions. International Journal of Molecular Sciences, 25(2), 1302. https://doi.org/10.3390/ijms25021302