
Metabolic Flexibility of the Heart: The Role of Fatty Acid Metabolism in Health, Heart Failure, and Cardiometabolic Diseases


Abstract
1. Introduction
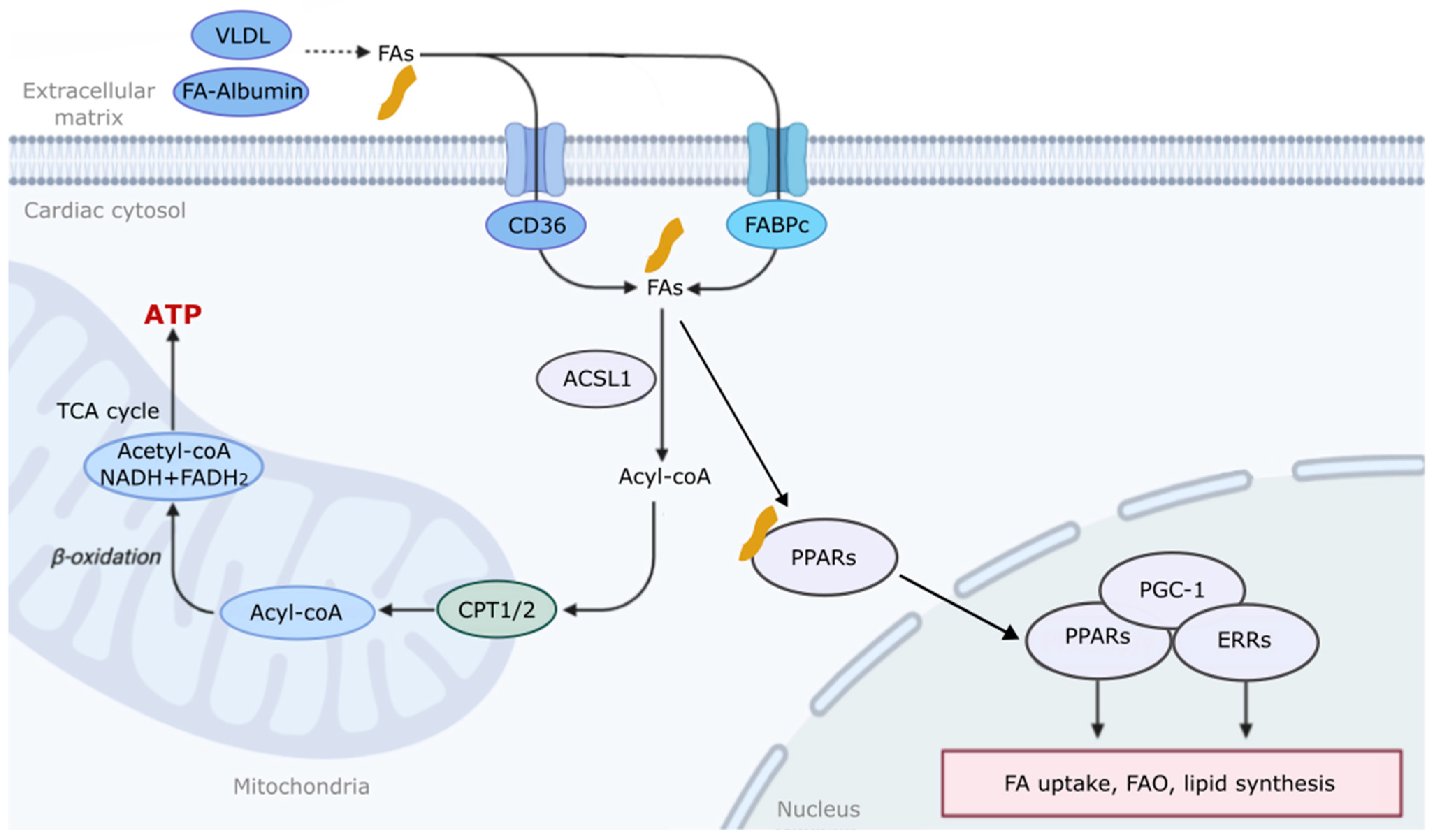
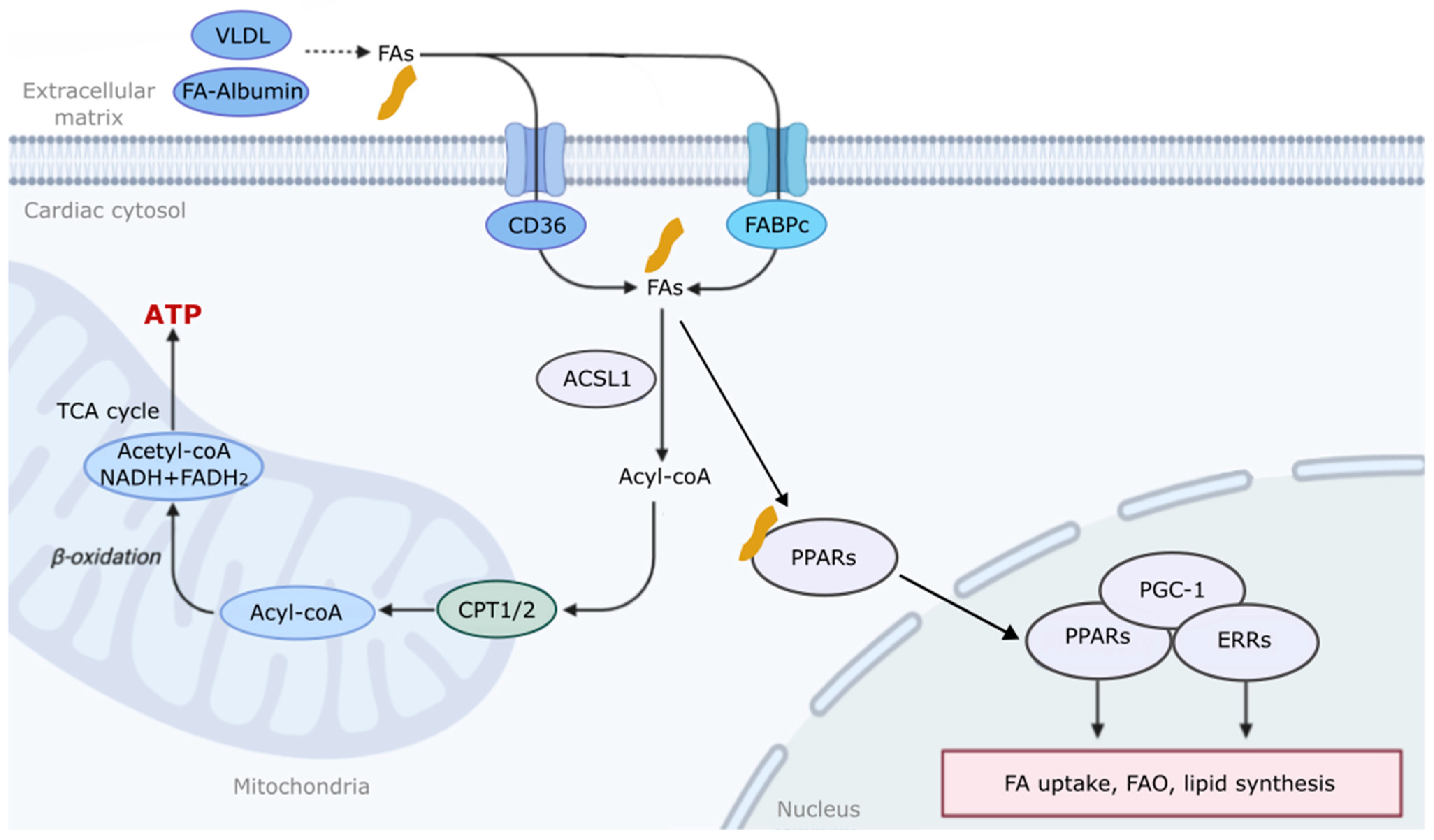
2. The Mechanism of Fatty Acid Metabolism and Regulation in the Heart
3. Fatty Acid Metabolism in Diverse Diseases
4. Fatty Acid Metabolism in Heart Failure with Reduced Ejection Fraction (HFrEF) and Preserved Ejection Fraction (HFpEF)
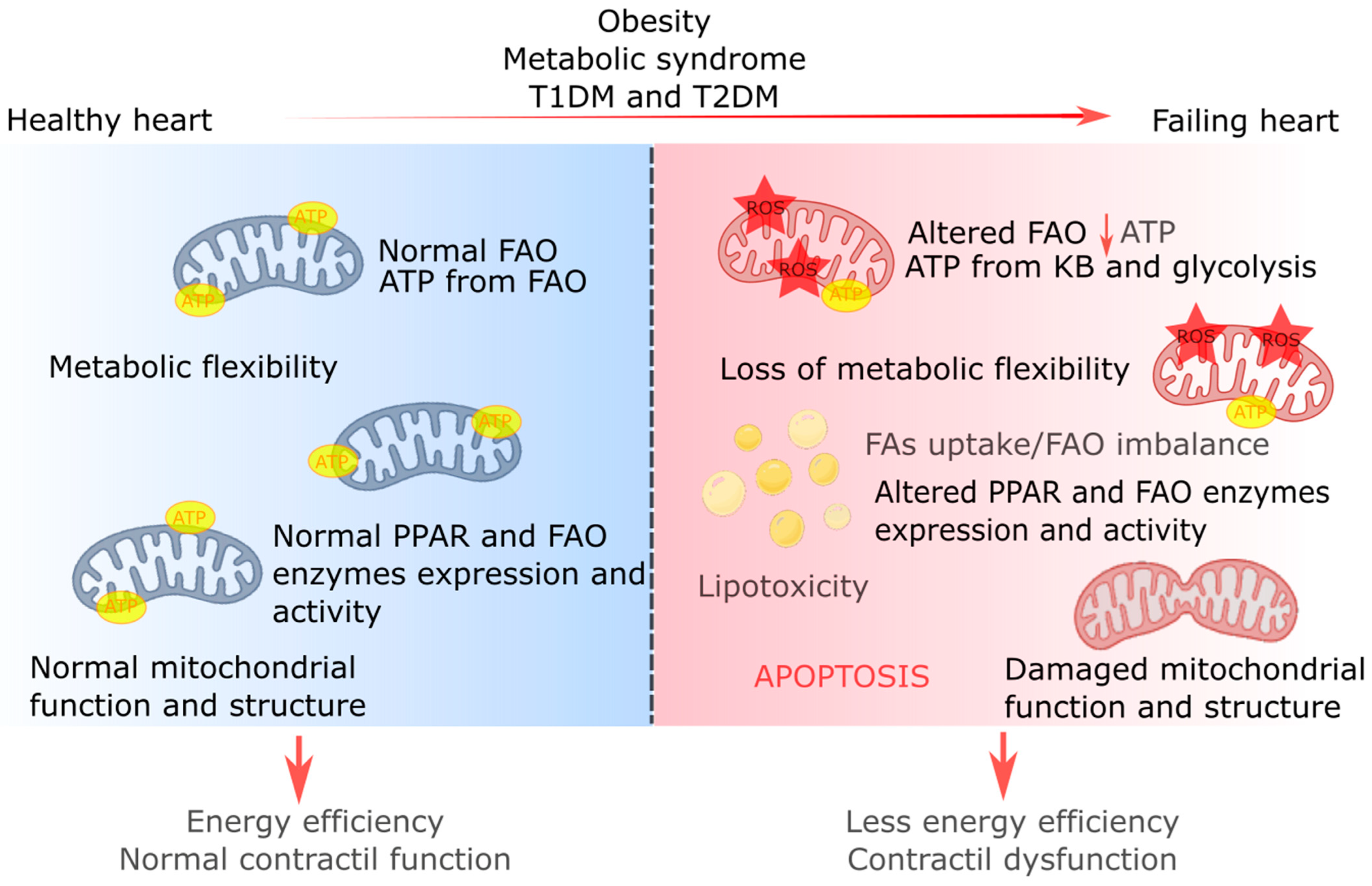
5. Fatty Acid Metabolism in Obesity and Diabetic Cardiomyopathy
6. Modulation of Fatty Acid Metabolism Regulation as Metabolic Therapy for Treating Cardiac Diseases
6.1. Inhibition of FAO
6.2. Stimulation of FAO
7. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Karwi, Q.G.; Uddin, G.M.; Ho, K.L.; Lopaschuk, G.D. Loss of Metabolic Flexibility in the Failing Heart. Front. Cardiovasc. Med. 2018, 5, 68. [Google Scholar] [CrossRef]
- Lopaschuk, G.D.; Ussher, J.R.; Folmes, C.D.L.; Jaswal, J.S.; Stanley, W.C. Myocardial Fatty Acid Metabolism in Health and Disease. Physiol. Rev. 2010, 90, 207–258. [Google Scholar] [CrossRef]
- Lopaschuk, G.D.; Karwi, Q.G.; Tian, R.; Wende, A.R.; Abel, E.D. Cardiac Energy Metabolism in Heart Failure. Circ. Res. 2021, 128, 1487–1513. [Google Scholar] [CrossRef]
- Chen, Z.; Jin, Z.-X.; Cai, J.; Li, R.; Deng, K.-Q.; Ji, Y.-X.; Lei, F.; Li, H.-P.; Lu, Z.; Li, H. Energy substrate metabolism and oxidative stress in metabolic cardiomyopathy. J. Mol. Med. 2022, 100, 1721–1739. [Google Scholar] [CrossRef]
- Nguyen, T.D.; Schulze, P.C. Lipid in the midst of metabolic remodeling—Therapeutic implications for the failing heart. Adv. Drug Deliv. Rev. 2020, 159, 120–132. [Google Scholar] [CrossRef]
- Bertero, E.; Maack, C. Metabolic remodelling in heart failure. Nat. Rev. Cardiol. 2018, 15, 457–470. [Google Scholar] [CrossRef]
- Li, Z.; Lei, H.; Jiang, H.; Fan, Y.; Shi, J.; Li, C.; Chen, F.; Mi, B.; Ma, M.; Lin, J.; et al. Saturated fatty acid biomarkers and risk of cardiometabolic diseases: A meta-analysis of prospective studies. Front. Nutr. 2022, 9, 963471. [Google Scholar] [CrossRef]
- Øie, E.; Ueland, T.; Dahl, C.P.; Bohov, P.; Berge, C.; Yndestad, A.; Gullestad, L.; Aukrust, P.; Berge, R.K. Fatty acid composition in chronic heart failure: Low circulating levels of eicosatetraenoic acid and high levels of vaccenic acid are associated with disease severity and mortality. J. Intern. Med. 2011, 270, 263–272. [Google Scholar] [CrossRef]
- Glatz, J.F.C.; Luiken, J.J.F.P. Dynamic role of the transmembrane glycoprotein CD36 (SR-B2) in cellular fatty acid uptake and utilization. J. Lipid Res. 2018, 59, 1084–1093. [Google Scholar] [CrossRef]
- Goldenberg, J.R.; Wang, X.; Lewandowski, E.D. Acyl CoA synthetase-1 links facilitated long chain fatty acid uptake to intracellular metabolic trafficking differently in hearts of male versus female mice. J. Mol. Cell. Cardiol. 2016, 94, 1–9. [Google Scholar] [CrossRef]
- Schlaepfer, I.R.; Joshi, M. CPT1A-mediated Fat Oxidation, Mechanisms, and Therapeutic Potential. Endocrinology 2020, 161, bqz046. [Google Scholar] [CrossRef]
- Marin, W.; Marin, D.; Ao, X.; Liu, Y. Mitochondria as a therapeutic target for cardiac ischemia-reperfusion injury (Review). Int. J. Mol. Med. 2020, 47, 485–499. [Google Scholar] [CrossRef]
- Ramachandra, C.J.; Hernandez-Resendiz, S.; Crespo-Avilan, G.E.; Lin, Y.-H.; Hausenloy, D.J. Mitochondria in acute myocardial infarction and cardioprotection. EBioMedicine 2020, 57, 102884. [Google Scholar] [CrossRef]
- Wang, W.; Zhang, L.; Battiprolu, P.K.; Fukushima, A.; Nguyen, K.; Milner, K.; Gupta, A.; Altamimi, T.; Byrne, N.; Mori, J.; et al. Malonyl CoA Decarboxylase Inhibition Improves Cardiac Function Post-Myocardial Infarction. JACC Basic Transl. Sci. 2019, 4, 385–400. [Google Scholar] [CrossRef]
- Softic, S.; Meyer, J.G.; Wang, G.-X.; Gupta, M.K.; Batista, T.M.; Lauritzen, H.P.; Fujisaka, S.; Serra, D.; Herrero, L.; Willoughby, J.; et al. Dietary Sugars Alter Hepatic Fatty Acid Oxidation via Transcriptional and Post-translational Modifications of Mitochondrial Proteins. Cell Metab. 2019, 30, 735–753.e4. [Google Scholar] [CrossRef]
- Chi, Z.; Chen, S.; Xu, T.; Zhen, W.; Yu, W.; Jiang, D.; Guo, X.; Wang, Z.; Zhang, K.; Li, M.; et al. Histone Deacetylase 3 Couples Mitochondria to Drive IL-1β-Dependent Inflammation by Configuring Fatty Acid Oxidation. Mol. Cell 2020, 80, 43–58.e7. [Google Scholar] [CrossRef]
- Northam, C.; LeMoine, C.M. Metabolic regulation by the PGC-1α and PGC-1β coactivators in larval zebrafish (Danio rerio). Comp. Biochem. Physiol. Part A Mol. Integr. Physiol. 2019, 234, 60–67. [Google Scholar] [CrossRef]
- Montaigne, D.; Butruille, L.; Staels, B. PPAR control of metabolism and cardiovascular functions. Nat. Rev. Cardiol. 2021, 18, 809–823. [Google Scholar] [CrossRef]
- Monroy-Ramirez, H.C.; Galicia-Moreno, M.; Sandoval-Rodriguez, A.; Meza-Rios, A.; Santos, A.; Armendariz-Borunda, J. PPARs as Metabolic Sensors and Therapeutic Targets in Liver Diseases. Int. J. Mol. Sci. 2021, 22, 8298. [Google Scholar] [CrossRef]
- Montagner, A.; Polizzi, A.; Fouché, E.; Ducheix, S.; Lippi, Y.; Lasserre, F.; Barquissau, V.; Régnier, M.; Lukowicz, C.; Benhamed, F.; et al. Liver PPARα is crucial for whole-body fatty acid homeostasis and is protective against NAFLD. Gut 2016, 65, 1202–1214. [Google Scholar] [CrossRef]
- Badmus, O.O.; Kipp, Z.A.; Bates, E.A.; da Silva, A.A.; Taylor, L.C.; Martinez, G.J.; Lee, W.-H.; Creeden, J.F.; Hinds, T.D.; Stec, D.E. Loss of hepatic PPARα in mice causes hypertension and cardiovascular disease. Am. J. Physiol. Integr. Comp. Physiol. 2023, 325, R81–R95. [Google Scholar] [CrossRef]
- Hinds, T.D.; Kipp, Z.A.; Xu, M.; Yiannikouris, F.B.; Morris, A.J.; Stec, D.F.; Wahli, W.; Stec, D.E. Adipose-Specific PPARα Knockout Mice Have Increased Lipogenesis by PASK–SREBP1 Signaling and a Polarity Shift to Inflammatory Macrophages in White Adipose Tissue. Cells 2021, 11, 4. [Google Scholar] [CrossRef]
- Wang, X.; Zhu, X.-X.; Jiao, S.-Y.; Qi, D.; Yu, B.-Q.; Xie, G.-M.; Liu, Y.; Song, Y.-T.; Xu, Q.-B.; Gonzalez, F.J.; et al. Cardiomyocyte peroxisome proliferator-activated receptor α is essential for energy metabolism and extracellular matrix homeostasis during pressure overload-induced cardiac remodeling. Acta Pharmacol. Sin. 2021, 43, 1231–1242. [Google Scholar] [CrossRef]
- Smeets, P.J.H.; Teunissen, B.E.J.; Willemsen, P.H.M.; van Nieuwenhoven, F.A.; Brouns, A.E.; Janssen, B.J.A.; Cleutjens, J.P.M.; Staels, B.; Van Der Vusse, G.J.; Van Bilsen, M. Cardiac hypertrophy is enhanced in PPARα−/− mice in response to chronic pressure overload. Cardiovasc. Res. 2008, 78, 79–89. [Google Scholar] [CrossRef]
- Tham, Y.K.; Bernardo, B.; Claridge, B.; Woon, L.; Yildiz, G.; Ooi, J.; Matsumoto, A.; Luo, J.; Tai, C.; Harmawan, C.; et al. Estrogen receptor α deficiency in cardiac myocytes reprograms heart-derived extracellular vesicle proteome and induces obesity in female mice. J. Mol. Cell. Cardiol. 2022, 173, S104. [Google Scholar] [CrossRef]
- Kamada, S.; Takeiwa, T.; Ikeda, K.; Horie, K.; Inoue, S. Emerging Roles of COX7RP and Mitochondrial Oxidative Phosphorylation in Breast Cancer. Front. Cell Dev. Biol. 2022, 10, 717881. [Google Scholar] [CrossRef]
- Cho, Y.; Tachibana, S.; Lam, K.; Arita, Y.; Khosrowjerdi, S.; Zhang, O.; Liang, A.; Li, R.; Andreyev, A.; Murphy, A.N.; et al. Perm1 promotes cardiomyocyte mitochondrial biogenesis and protects against hypoxia/reoxygenation-induced damage in mice. J. Biol. Chem. 2021, 297, 100825. [Google Scholar] [CrossRef]
- Rius-Pérez, S.; Torres-Cuevas, I.; Millán, I.; Ortega, Á.L.; Pérez, S. PGC-1α, Inflammation, and Oxidative Stress: An Integrative View in Metabolism. Oxid. Med. Cell. Longev. 2020, 2020, 1452696. [Google Scholar] [CrossRef]
- Liang, H.; Ward, W.F.; Shute, R.J.; Heesch, M.W.; Zak, R.B.; Kreiling, J.L.; Slivka, D.R.; Sun, S.; Li, H.; Chen, J.; et al. PGC-1α: A key regulator of energy metabolism. Adv. Physiol. Educ. 2006, 30, 145–151. [Google Scholar] [CrossRef]
- Arany, Z. PGC-1 coactivators and skeletal muscle adaptations in health and disease. Curr. Opin. Genet. Dev. 2008, 18, 426–434. [Google Scholar] [CrossRef]
- Rowe, G.C.; Jiang, A.; Arany, Z. PGC-1 Coactivators in Cardiac Development and Disease. Circ. Res. 2010, 107, 825–838. [Google Scholar] [CrossRef]
- Essandoh, K.; Philippe, J.M.; Jenkins, P.M.; Brody, M.J. Palmitoylation: A Fatty Regulator of Myocardial Electrophysiology. Front. Physiol. 2020, 11, 108. [Google Scholar] [CrossRef]
- Nakamura, M.; Sadoshima, J. Cardiomyopathy in obesity, insulin resistance and diabetes. J. Physiol. 2019, 598, 2977–2993. [Google Scholar] [CrossRef]
- Da Dalt, L.; Cabodevilla, A.G.; Goldberg, I.J.; Norata, G.D. Cardiac lipid metabolism, mitochondrial function, and heart failure. Cardiovasc. Res. 2023, 119, 1905–1914. [Google Scholar] [CrossRef]
- Morigny, P.; Boucher, J.; Arner, P.; Langin, D. Lipid and glucose metabolism in white adipocytes: Pathways, dysfunction and therapeutics. Nat. Rev. Endocrinol. 2021, 17, 276–295. [Google Scholar] [CrossRef]
- Klein, S.; Gastaldelli, A.; Yki-Järvinen, H.; Scherer, P.E. Why does obesity cause diabetes? Cell Metab. 2022, 34, 11–20. [Google Scholar] [CrossRef] [PubMed]
- Fryk, E.; Olausson, J.; Mossberg, K.; Strindberg, L.; Schmelz, M.; Brogren, H.; Gan, L.-M.; Piazza, S.; Provenzani, A.; Becattini, B.; et al. Hyperinsulinemia and insulin resistance in the obese may develop as part of a homeostatic response to elevated free fatty acids: A mechanistic case-control and a population-based cohort study. EBioMedicine 2021, 65, 103264. [Google Scholar] [CrossRef]
- Berge, R.; Tronstad, K.; Berge, K.; Rost, T.; Wergedahl, H.; Gudbrandsen, O.; Skorve, J. The metabolic syndrome and the hepatic fatty acid drainage hypothesis. Biochimie 2005, 87, 15–20. [Google Scholar] [CrossRef]
- Huang, X.; Su, B.; Li, M.; Zhou, Y.; He, X. Multiomics characterization of fatty acid metabolism for the clinical management of hepatocellular carcinoma. Sci. Rep. 2023, 13, 1–13. [Google Scholar] [CrossRef]
- Mi, Y.; Qi, G.; Vitali, F.; Shang, Y.; Raikes, A.C.; Wang, T.; Jin, Y.; Brinton, R.D.; Gu, H.; Yin, F. Loss of fatty acid degradation by astrocytic mitochondria triggers neuroinflammation and neurodegeneration. Nat. Metab. 2023, 5, 445–465. [Google Scholar] [CrossRef]
- Mustonen, A.-M.; Nieminen, P. Fatty Acids and Oxylipins in Osteoarthritis and Rheumatoid Arthritis—A Complex Field with Significant Potential for Future Treatments. Curr. Rheumatol. Rep. 2021, 23, 41. [Google Scholar] [CrossRef]
- He, J.; Luo, X.; Xin, H.; Lai, Q.; Zhou, Y.; Bai, Y. The Effects of Fatty Acids on Inflammatory Bowel Disease: A Two-Sample Mendelian Randomization Study. Nutrients 2022, 14, 2883. [Google Scholar] [CrossRef]
- Tavasoli, M.; Lahire, S.; Sokolenko, S.; Novorolsky, R.; Reid, S.A.; Lefsay, A.; Otley, M.O.C.; Uaesoontrachoon, K.; Rowsell, J.; Srinivassane, S.; et al. Mechanism of action and therapeutic route for a muscular dystrophy caused by a genetic defect in lipid metabolism. Nat. Commun. 2022, 13, 1559. [Google Scholar] [CrossRef]
- Libby, P. The changing landscape of atherosclerosis. Nature 2021, 592, 524–533. [Google Scholar] [CrossRef] [PubMed]
- Reis, A.H. Acidemia and blood free fatty acids: Analysis of cardiovascular risk factors in a new context. Discov. Med. 2017, 23, 183–188. [Google Scholar]
- Tang, J.-J.; Li, J.-G.; Qi, W.; Qiu, W.-W.; Li, P.-S.; Li, B.-L.; Song, B.-L. Inhibition of SREBP by a Small Molecule, Betulin, Improves Hyperlipidemia and Insulin Resistance and Reduces Atherosclerotic Plaques. Cell Metab. 2011, 13, 44–56. [Google Scholar] [CrossRef]
- Reilly, N.A.; Lutgens, E.; Kuiper, J.; Heijmans, B.T.; Jukema, J.W. Effects of fatty acids on T cell function: Role in atherosclerosis. Nat. Rev. Cardiol. 2021, 18, 824–837. [Google Scholar] [CrossRef]
- Suganami, T.; Tanimoto-Koyama, K.; Nishida, J.; Itoh, M.; Yuan, X.; Mizuarai, S.; Kotani, H.; Yamaoka, S.; Miyake, K.; Aoe, S.; et al. Role of the Toll-like Receptor 4/NF-κB Pathway in Saturated Fatty Acid–Induced Inflammatory Changes in the Interaction Between Adipocytes and Macrophages. Arter. Thromb. Vasc. Biol. 2007, 27, 84–91. [Google Scholar] [CrossRef]
- Chen, Y.; Zhang, J.; Cui, W.; Silverstein, R.L. CD36, a signaling receptor and fatty acid transporter that regulates immune cell metabolism and fate. J. Exp. Med. 2022, 219, e20211314. [Google Scholar] [CrossRef]
- Mozaffarian, D.; Benjamin, E.J.; Go, A.S.; Arnett, D.K.; Blaha, M.J.; Cushman, M.; Das, S.R.; de Ferranti, S.; Despres, J.-P.; Fullerton, H.J.; et al. Stroke Statistics Subcommittee (2016). Heart Disease and Stroke Statistics-2016 Update. Circulation 2016, 133, e38–e60. [Google Scholar] [CrossRef]
- Snipelisky, D.; Chaudhry, S.-P.; Stewart, G.C. The Many Faces of Heart Failure. Card. Electrophysiol. Clin. 2019, 11, 11–20. [Google Scholar] [CrossRef]
- Nguyen, Q.L.; Wang, Y.; Helbling, N.; Simon, M.A.; Shiva, S. Alterations in platelet bioenergetics in Group 2 PH-HFpEF patients. PLoS ONE 2019, 14, e0220490. [Google Scholar] [CrossRef]
- Umbarawan, Y.; Syamsunarno, M.R.A.A.; Koitabashi, N.; Obinata, H.; Yamaguchi, A.; Hanaoka, H.; Hishiki, T.; Hayakawa, N.; Sano, M.; Sunaga, H.; et al. Myocardial fatty acid uptake through CD36 is indispensable for sufficient bioenergetic metabolism to prevent progression of pressure overload-induced heart failure. Sci. Rep. 2018, 8, 12035. [Google Scholar] [CrossRef] [PubMed]
- Zhu, N.; Jiang, W.; Wang, Y.; Wu, Y.; Chen, H.; Zhao, X. Plasma levels of free fatty acid differ in patients with left ventricular preserved, mid-range, and reduced ejection fraction. BMC Cardiovasc. Disord. 2018, 18, 104. [Google Scholar] [CrossRef] [PubMed]
- De Jong, K.A.; Lopaschuk, G.D. Complex Energy Metabolic Changes in Heart Failure with Preserved Ejection Fraction and Heart Failure with Reduced Ejection Fraction. Can. J. Cardiol. 2017, 33, 860–871. [Google Scholar] [CrossRef]
- Voros, G.; Ector, J.; Garweg, C.; Droogne, W.; Van Cleemput, J.; Peersman, N.; Vermeersch, P.; Janssens, S. Increased Cardiac Uptake of Ketone Bodies and Free Fatty Acids in Human Heart Failure and Hypertrophic Left Ventricular Remodeling. Circ. Heart Fail. 2018, 11, e004953. [Google Scholar] [CrossRef]
- Kato, T.; Niizuma, S.; Inuzuka, Y.; Kawashima, T.; Okuda, J.; Tamaki, Y.; Iwanaga, Y.; Narazaki, M.; Matsuda, T.; Soga, T.; et al. Analysis of Metabolic Remodeling in Compensated Left Ventricular Hypertrophy and Heart Failure. Circ. Heart Fail. 2010, 3, 420–430. [Google Scholar] [CrossRef] [PubMed]
- Capone, F.; Sotomayor-Flores, C.; Bode, D.; Wang, R.; Rodolico, D.; Strocchi, S.; Schiattarella, G.G. Cardiac metabolism in HFpEF: From fuel to signalling. Cardiovasc. Res. 2022, 118, 3556–3575. [Google Scholar] [CrossRef]
- Deng, Y.; Xie, M.; Li, Q.; Xu, X.; Ou, W.; Zhang, Y.; Xiao, H.; Yu, H.; Zheng, Y.; Liang, Y.; et al. Targeting Mitochondria-Inflammation Circuit by β-Hydroxybutyrate Mitigates HFpEF. Circ. Res. 2021, 128, 232–245. [Google Scholar] [CrossRef] [PubMed]
- Liu, X.; Zhang, Y.; Deng, Y.; Yang, L.; Ou, W.; Xie, M.; Ding, L.; Jiang, C.; Yu, H.; Li, Q.; et al. Mitochondrial protein hyperacetylation underpins heart failure with preserved ejection fraction in mice. J. Mol. Cell. Cardiol. 2022, 165, 76–85. [Google Scholar] [CrossRef]
- Murashige, D.; Jang, C.; Neinast, M.; Edwards, J.J.; Cowan, A.; Hyman, M.C.; Rabinowitz, J.D.; Frankel, D.S.; Arany, Z. Comprehensive quantification of fuel use by the failing and nonfailing human heart. Science 2020, 370, 364–368. [Google Scholar] [CrossRef] [PubMed]
- Koser, F.; Hobbach, A.J.; Abdellatif, M.; Herbst, V.; Türk, C.; Reinecke, H.; Krüger, M.; Sedej, S.; Linke, W.A. Acetylation and phosphorylation changes to cardiac proteins in experimental HFpEF due to metabolic risk reveal targets for treatment. Life Sci. 2022, 309, 120998. [Google Scholar] [CrossRef]
- Zhang, Z.Y.; Feng, X.Y.; Wang, Z.H.; Huang, Y.Z.; Yang, W.B.; Zhang, W.J.; Zhou, J.; Yuan, Z.Y. [Similarities and differences of myocardial metabolic characteristics between HFpEF and HFrEF mice based on LC-MS/MS metabolomics]. Zhonghua Xin Xue Guan Bing Za Zhi 2023, 51, 722–730. [Google Scholar] [CrossRef]
- Glatz, J.F.; Wang, F.; Nabben, M.; Luiken, J.J. CD36 as a target for metabolic modulation therapy in cardiac disease. Expert Opin. Ther. Targets 2021, 25, 393–400. [Google Scholar] [CrossRef]
- Feng, J.; Zhao, H.; Du, M.; Wu, X. The effect of apelin-13 on pancreatic islet beta cell mass and myocardial fatty acid and glucose metabolism of experimental type 2 diabetic rats. Peptides 2019, 114, 1–7. [Google Scholar] [CrossRef]
- Chen, X.; Wang, Q.; Shao, M.; Ma, L.; Guo, D.; Wu, Y.; Gao, P.; Wang, X.; Li, W.; Li, C.; et al. Ginsenoside Rb3 regulates energy metabolism and apoptosis in cardiomyocytes via activating PPARα pathway. Biomed. Pharmacother. 2019, 120, 109487. [Google Scholar] [CrossRef]
- Lai, Q.; Zhu, X.; Zhang, L.; Kou, J.; Liu, F.; Yu, B.; Li, F. Inhibition of OAT1/3 and CMPF uptake attenuates myocardial ischemia-induced chronic heart failure via decreasing fatty acid oxidation and the therapeutic effects of ruscogenin. Transl. Res. 2023, 261, 1–15. [Google Scholar] [CrossRef]
- Yamamoto, T.; Sano, M. Deranged Myocardial Fatty Acid Metabolism in Heart Failure. Int. J. Mol. Sci. 2022, 23, 996. [Google Scholar] [CrossRef]
- Kolleritsch, S.; Kien, B.; Schoiswohl, G.; Diwoky, C.; Schreiber, R.; Heier, C.; Maresch, L.K.; Schweiger, M.; Eichmann, T.O.; Stryeck, S.; et al. Low cardiac lipolysis reduces mitochondrial fission and prevents lipotoxic heart dysfunction in Perilipin 5 mutant mice. Cardiovasc. Res. 2020, 116, 339–352. [Google Scholar] [CrossRef]
- Da Dalt, L.; Castiglioni, L.; Baragetti, A.; Audano, M.; Svecla, M.; Bonacina, F.; Pedretti, S.; Uboldi, P.; Benzoni, P.; Giannetti, F.; et al. PCSK9 deficiency rewires heart metabolism and drives heart failure with preserved ejection fraction. Eur. Heart J. 2021, 42, 3078–3090. [Google Scholar] [CrossRef]
- Chaanine, A.H.; Joyce, L.D.; Stulak, J.M.; Maltais, S.; Joyce, D.L.; Dearani, J.A.; Klaus, K.; Nair, K.S.; Hajjar, R.J.; Redfield, M.M. Mitochondrial Morphology, Dynamics, and Function in Human Pressure Overload or Ischemic Heart Disease with Preserved or Reduced Ejection Fraction. Circ. Heart Fail. 2019, 12, e005131. [Google Scholar] [CrossRef] [PubMed]
- Schianchi, F.; Glatz, J.F.C.; Gascon, A.N.; Nabben, M.; Neumann, D.; Luiken, J.J.F.P. Putative Role of Protein Palmitoylation in Cardiac Lipid-Induced Insulin Resistance. Int. J. Mol. Sci. 2020, 21, 9438. [Google Scholar] [CrossRef]
- Dato, V.A.; Benitez-Amaro, A.; Garcia, E.; Claudi, L.; Lhoëst, M.T.L.; Iborra, A.; Escola-Gil, J.C.; Guerra, J.M.; Samouillan, V.; Enrich, C.; et al. Targeting cholesteryl ester accumulation in the heart improves cardiac insulin response. Biomed. Pharmacother. 2022, 152, 113270. [Google Scholar] [CrossRef]
- Hamdani, N.; Hervent, A.-S.; Vandekerckhove, L.; Matheeussen, V.; Demolder, M.; Baerts, L.; De Meester, I.; Linke, W.A.; Paulus, W.J.; De Keulenaer, G.W. Left ventricular diastolic dysfunction and myocardial stiffness in diabetic mice is attenuated by inhibition of dipeptidyl peptidase 4. Cardiovasc. Res. 2014, 104, 423–431. [Google Scholar] [CrossRef] [PubMed]
- Koutroumpakis, E.; Jozwik, B.; Aguilar, D.; Taegtmeyer, H. Strategies of Unloading the Failing Heart from Metabolic Stress. Am. J. Med. 2019, 133, 290–296. [Google Scholar] [CrossRef]
- Rawshani, A.; Rawshani, A.; Franzén, S.; Eliasson, B.; Svensson, A.-M.; Miftaraj, M.; McGuire, D.K.; Sattar, N.; Rosengren, A.; Gudbjörnsdottir, S. Mortality and Cardiovascular Disease in Type 1 and Type 2 Diabetes. N. Engl. J. Med. 2017, 376, 1407–1418. [Google Scholar] [CrossRef]
- Piché, M.-E.; Tchernof, A.; Després, J.-P. Obesity Phenotypes, Diabetes, and Cardiovascular Diseases. Circ. Res. 2020, 126, 1477–1500. [Google Scholar] [CrossRef] [PubMed]
- Ren, J.; Wu, N.N.; Wang, S.; Sowers, J.R.; Zhang, Y. Obesity cardiomyopathy: Evidence, mechanisms, and therapeutic implications. Physiol. Rev. 2021, 101, 1745–1807. [Google Scholar] [CrossRef] [PubMed]
- Cortassa, S.; Caceres, V.; Tocchetti, C.G.; Bernier, M.; de Cabo, R.; Paolocci, N.; Sollott, S.J.; Aon, M.A. Metabolic remodelling of glucose, fatty acid and redox pathways in the heart of type 2 diabetic mice. J. Physiol. 2018, 598, 1393–1415. [Google Scholar] [CrossRef]
- Yurista, S.R.; Chen, S.; Welsh, A.; Tang, W.H.W.; Nguyen, C.T. Targeting Myocardial Substrate Metabolism in the Failing Heart: Ready for Prime Time? Curr. Heart Fail. Rep. 2022, 19, 180–190. [Google Scholar] [CrossRef]
- He, L.; Kim, T.; Long, Q.; Liu, J.; Wang, P.; Zhou, Y.; Ding, Y.; Prasain, J.; Wood, P.A.; Yang, Q.; et al. Carnitine Palmitoyltransferase-1b Deficiency Aggravates Pressure Overload–Induced Cardiac Hypertrophy Caused by Lipotoxicity. Circulation 2012, 126, 1705–1716. [Google Scholar] [CrossRef] [PubMed]
- Hu, Q.; Zhang, H.; Cortés, N.G.; Wu, D.; Wang, P.; Zhang, J.; Mattison, J.A.; Smith, E.; Bettcher, L.F.; Wang, M.; et al. Increased Drp1 Acetylation by Lipid Overload Induces Cardiomyocyte Death and Heart Dysfunction. Circ. Res. 2020, 126, 456–470. [Google Scholar] [CrossRef] [PubMed]
- Mouton, A.J.; Li, X.; Hall, M.E.; Hall, J.E. Obesity, Hypertension, and Cardiac Dysfunction. Circ. Res. 2020, 126, 789–806. [Google Scholar] [CrossRef] [PubMed]
- Tsushima, K.; Bugger, H.; Wende, A.R.; Soto, J.; Jenson, G.A.; Tor, A.R.; McGlauflin, R.; Kenny, H.C.; Zhang, Y.; Souvenir, R.; et al. Mitochondrial Reactive Oxygen Species in Lipotoxic Hearts Induce Post-Translational Modifications of AKAP121, DRP1, and OPA1 That Promote Mitochondrial Fission. Circ. Res. 2018, 122, 58–73. [Google Scholar] [CrossRef]
- Fukushima, A.; Lopaschuk, G.D. Cardiac fatty acid oxidation in heart failure associated with obesity and diabetes. Biochim. Biophys. Acta (BBA) Mol. Cell Biol. Lipids 2016, 1861, 1525–1534. [Google Scholar] [CrossRef]
- Dato, V.A.; Benitez-Amaro, A.; de Gonzalo-Calvo, D.; Vazquez, M.; Bonacci, G.; Llorente-Cortés, V.; Chiabrando, G.A. LRP1-Mediated AggLDL Endocytosis Promotes Cholesteryl Ester Accumulation and Impairs Insulin Response in HL-1 Cells. Cells 2020, 9, 182. [Google Scholar] [CrossRef]
- Ritterhoff, J.; Tian, R. Metabolism in cardiomyopathy: Every substrate matters. Cardiovasc. Res. 2017, 113, 411–421. [Google Scholar] [CrossRef] [PubMed]
- Ruiz-Meana, M.; Minguet, M.; Bou-Teen, D.; Miro-Casas, E.; Castans, C.; Castellano, J.; Bonzon-Kulichenko, E.; Igual, A.; Rodriguez-Lecoq, R.; Vázquez, J.; et al. Ryanodine Receptor Glycation Favors Mitochondrial Damage in the Senescent Heart. Circulation 2019, 139, 949–964. [Google Scholar] [CrossRef]
- Knottnerus, S.J.G.; Mengarelli, I.; Wüst, R.C.I.; Baartscheer, A.; Bleeker, J.C.; Coronel, R.; Ferdinandusse, S.; Guan, K.; Ijlst, L.; Li, W.; et al. Electrophysiological Abnormalities in VLCAD Deficient hiPSC-Cardiomyocytes Can Be Improved by Lowering Accumulation of Fatty Acid Oxidation Intermediates. Int. J. Mol. Sci. 2020, 21, 2589. [Google Scholar] [CrossRef] [PubMed]
- Beadle, R.M.; Williams, L.K.; Kuehl, M.; Bowater, S.; Abozguia, K.; Leyva, F.; Yousef, Z.; Wagenmakers, A.J.; Thies, F.; Horowitz, J.; et al. Improvement in Cardiac Energetics by Perhexiline in Heart Failure Due to Dilated Cardiomyopathy. JACC Heart Fail. 2015, 3, 202–211. [Google Scholar] [CrossRef]
- Palm, C.L.; Nijholt, K.T.; Bakker, B.M.; Westenbrink, B.D. Short-Chain Fatty Acids in the Metabolism of Heart Failure—Rethinking the Fat Stigma. Front. Cardiovasc. Med. 2022, 9, 915102. [Google Scholar] [CrossRef]
- Shu, H.; Hang, W.; Peng, Y.; Nie, J.; Wu, L.; Zhang, W.; Wang, D.W.; Zhou, N. Trimetazidine Attenuates Heart Failure by Improving Myocardial Metabolism via AMPK. Front. Pharmacol. 2021, 12, 707399. [Google Scholar] [CrossRef]
- Shu, H.; Peng, Y.; Hang, W.; Zhou, N.; Wang, D.W. Trimetazidine in Heart Failure. Front. Pharmacol. 2021, 11, 569132. [Google Scholar] [CrossRef] [PubMed]
- Marzilli, M.; Vineareanu, D.; Lopaschuk, G.; Chen, Y.; Dalal, J.J.; Danchin, N.; Etriby, E.; Ferrari, R.; Gowdak, L.H.; Lopatin, Y.; et al. Trimetazidine in cardiovascular medicine. Int. J. Cardiol. 2019, 293, 39–44. [Google Scholar] [CrossRef]
- Khuchua, Z.; Glukhov, A.I.; Strauss, A.W.; Javadov, S. Elucidating the Beneficial Role of PPAR Agonists in Cardiac Diseases. Int. J. Mol. Sci. 2018, 19, 3464. [Google Scholar] [CrossRef]
- Kim, N.H.; Kim, S.G. Fibrates Revisited: Potential Role in Cardiovascular Risk Reduction. Diabetes Metab. J. 2020, 44, 213–221. [Google Scholar] [CrossRef] [PubMed]
- Anker, S.D.; Butler, J.; Filippatos, G.; Ferreira, J.P.; Bocchi, E.; Böhm, M.; Brunner–La Rocca, H.-P.; Choi, D.-J.; Chopra, V.; Chuquiure-Valenzuela, E.; et al. Empagliflozin in Heart Failure with a Preserved Ejection Fraction. N. Engl. J. Med. 2021, 385, 1451–1461. [Google Scholar] [CrossRef]
- Verma, S.; Rawat, S.; Ho, K.L.; Wagg, C.S.; Zhang, L.; Teoh, H.; Dyck, J.E.; Uddin, G.M.; Oudit, G.Y.; Mayoux, E.; et al. Empagliflozin Increases Cardiac Energy Production in Diabetes. JACC Basic Transl. Sci. 2018, 3, 575–587. [Google Scholar] [CrossRef] [PubMed]
- Santos-Gallego, C.G.; Requena-Ibanez, J.A.; Antonio, R.S.; Ishikawa, K.; Watanabe, S.; Picatoste, B.; Flores, E.; Garcia-Ropero, A.; Sanz, J.; Hajjar, R.J.; et al. Empagliflozin Ameliorates Adverse Left Ventricular Remodeling in Nondiabetic Heart Failure by Enhancing Myocardial Energetics. J. Am. Coll. Cardiol. 2019, 73, 1931–1944. [Google Scholar] [CrossRef] [PubMed]
- Ferrannini, E.; Mark, M.; Mayoux, E. CV Protection in the EMPA-REG OUTCOME Trial: A “Thrifty Substrate” Hypothesis. Diabetes Care 2016, 39, 1108–1114. [Google Scholar] [CrossRef]
- Mudaliar, S.; Alloju, S.; Henry, R.R. Can a Shift in Fuel Energetics Explain the Beneficial Cardiorenal Outcomes in the EMPA-REG OUTCOME Study? A Unifying Hypothesis. Diabetes Care 2016, 39, 1115–1122. [Google Scholar] [CrossRef] [PubMed]
- Chen, S.; Coronel, R.; Hollmann, M.W.; Weber, N.C.; Zuurbier, C.J. Direct cardiac effects of SGLT2 inhibitors. Cardiovasc. Diabetol. 2022, 21, 45. [Google Scholar] [CrossRef] [PubMed]
- Nabben, M.; Luiken, J.J.F.P.; Glatz, J.F.C. Metabolic remodelling in heart failure revisited. Nat. Rev. Cardiol. 2018, 15, 780. [Google Scholar] [CrossRef]
- Shao, D.; Kolwicz, S.C.; Wang, P.; Roe, N.D.; Villet, O.; Nishi, K.; Hsu, Y.-W.A.; Flint, G.V.; Caudal, A.; Wang, W.; et al. Increasing Fatty Acid Oxidation Prevents High-Fat Diet–Induced Cardiomyopathy Through Regulating Parkin-Mediated Mitophagy. Circulation 2020, 142, 983–997. [Google Scholar] [CrossRef] [PubMed]
- Lee, S.R.; Mukae, M.; Jeong, K.J.; Park, S.H.; Shin, H.J.; Kim, S.W.; Won, Y.S.; Kwun, H.-J.; Baek, I.-J.; Hong, E.-J. PGRMC1 Ablation Protects from Energy-Starved Heart Failure by Promoting Fatty Acid/Pyruvate Oxidation. Cells 2023, 12, 752. [Google Scholar] [CrossRef]
- Nguyen, T.D.; Schenkl, C.; Schlattmann, P.; Heyne, E.; Doenst, T.; Schulze, P.C. P1665 Identifying metabolic treatment strategies for heart failure—A meta-analytic approach. Eur. Heart J. 2019, 40, ehz748.0423. [Google Scholar] [CrossRef]
- Schenkl, C.; Heyne, E.; Doenst, T.; Schulze, P.C.; Nguyen, T.D. Targeting Mitochondrial Metabolism to Save the Failing Heart. Life 2023, 13, 1027. [Google Scholar] [CrossRef]
- Ma, L.; Shao, M.; Cheng, W.; Jiang, J.; Chen, X.; Tan, N.; Ling, G.; Yang, Y.; Wang, Q.; Yang, R.; et al. Neocryptotanshinone ameliorates insufficient energy production in heart failure by targeting retinoid X receptor alpha. Biomed. Pharmacother. 2023, 163, 114868. [Google Scholar] [CrossRef]
- Hahn, V.S.; Petucci, C.; Kim, M.-S.; Bedi, K.C.; Wang, H.; Mishra, S.; Koleini, N.; Yoo, E.J.; Margulies, K.B.; Arany, Z.; et al. Myocardial Metabolomics of Human Heart Failure with Preserved Ejection Fraction. Circulation 2023, 147, 1147–1161. [Google Scholar] [CrossRef]


{kind=link}
{kind=link}
| Metabolic Modulators | Metabolic Mechanism Affected | Cardiac Condition |
|---|---|---|
| CPT1 inhibitors Etomoxir Perhexiline | ↓ FAO ↑ Glucose oxidation | Ischemia–reperfusion injury Chronic HF |
| Trimetazidine (TMZ) | ↓ FAO ↑ AMPK signaling | Pressure overload HF |
| MCD inhibitors | ↑ Malonyl CoA levels ↓ FA uptake ↓ FAO | HF Post-myocardial infarction Ischemia–reperfusion injury |
| ACC2 inhibitors | ↑ FA uptake ↑ FAO ↑ Parkin-mediated mitophagy ↑ Mitochondrial function | Obesity-induced cardiomyopathy |
| PPARα agonists Fibrates | ↑ FA uptake ↑ FAO ↓ Triglycerides ↑ Insulin sensitivity | Ischemia–reperfusion injury T2DM with CVD |
| SGLT2 inhibitors | ↑ Ketone body oxidation ↑ Glucose metabolism ↓ FAO | T2DM with CVD T2DM with HFrEF T2DM with HFpEF |
| RXRα activators Neocryptotanshinone (NCTS) | ↑ CD36 and CPT1 expression ↑ FA uptake ↑ FAO ↑ Mitochondrial biogenesis | HF Post-myocardial infarction |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Actis Dato, V.; Lange, S.; Cho, Y. Metabolic Flexibility of the Heart: The Role of Fatty Acid Metabolism in Health, Heart Failure, and Cardiometabolic Diseases. Int. J. Mol. Sci. 2024, 25, 1211. https://doi.org/10.3390/ijms25021211
Actis Dato V, Lange S, Cho Y. Metabolic Flexibility of the Heart: The Role of Fatty Acid Metabolism in Health, Heart Failure, and Cardiometabolic Diseases. International Journal of Molecular Sciences. 2024; 25(2):1211. https://doi.org/10.3390/ijms25021211
Chicago/Turabian StyleActis Dato, Virginia, Stephan Lange, and Yoshitake Cho. 2024. "Metabolic Flexibility of the Heart: The Role of Fatty Acid Metabolism in Health, Heart Failure, and Cardiometabolic Diseases" International Journal of Molecular Sciences 25, no. 2: 1211. https://doi.org/10.3390/ijms25021211
APA StyleActis Dato, V., Lange, S., & Cho, Y. (2024). Metabolic Flexibility of the Heart: The Role of Fatty Acid Metabolism in Health, Heart Failure, and Cardiometabolic Diseases. International Journal of Molecular Sciences, 25(2), 1211. https://doi.org/10.3390/ijms25021211