
The Research Progress of Mitochondrial Transplantation in the Treatment of Mitochondrial Defective Diseases


{kind=link}
{kind=link}
Abstract
1. Introduction
2. Methods of Mitochondrial Transplantation
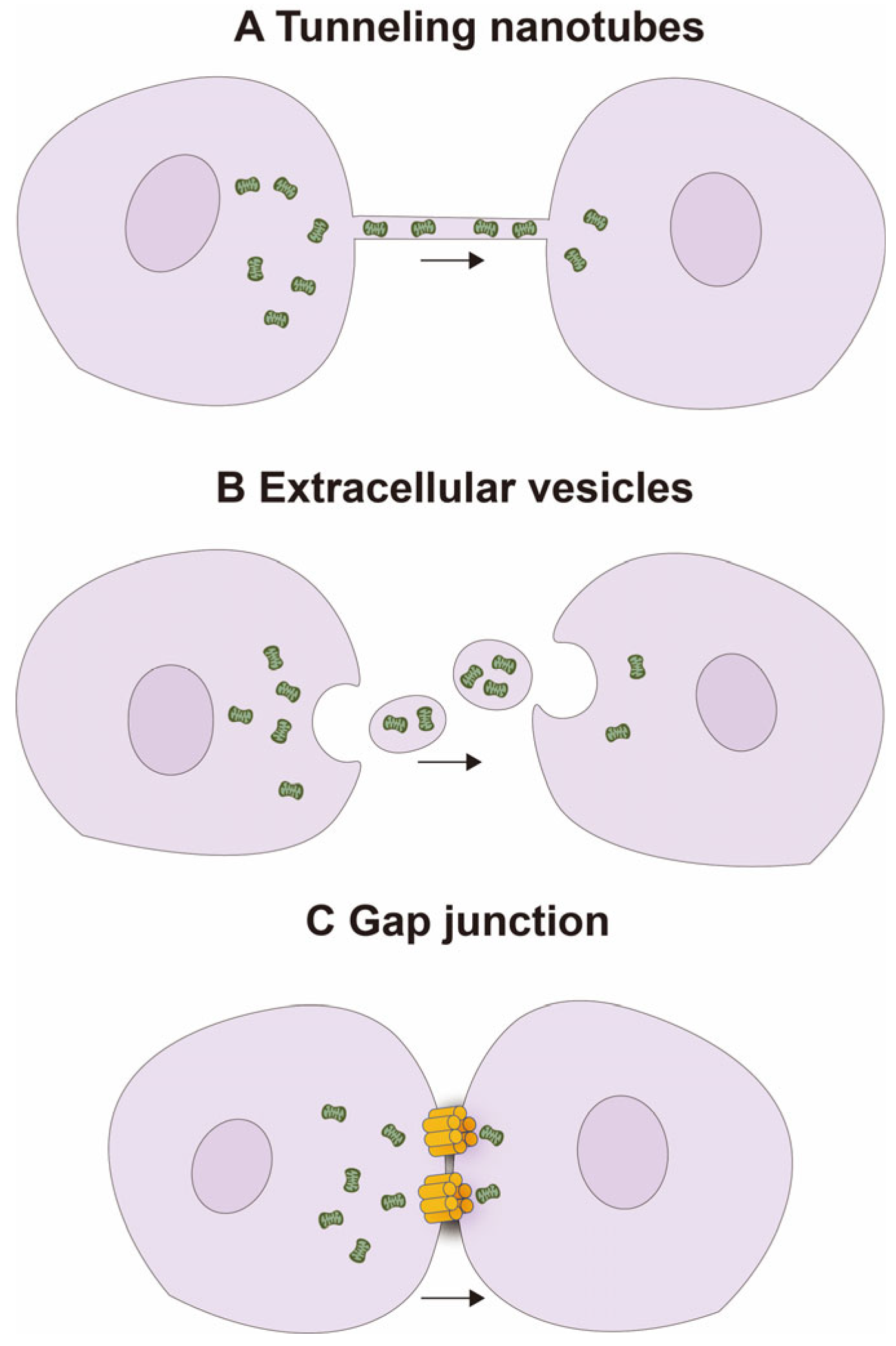
2.1. Natural Mitochondrial Transfer Mechanisms
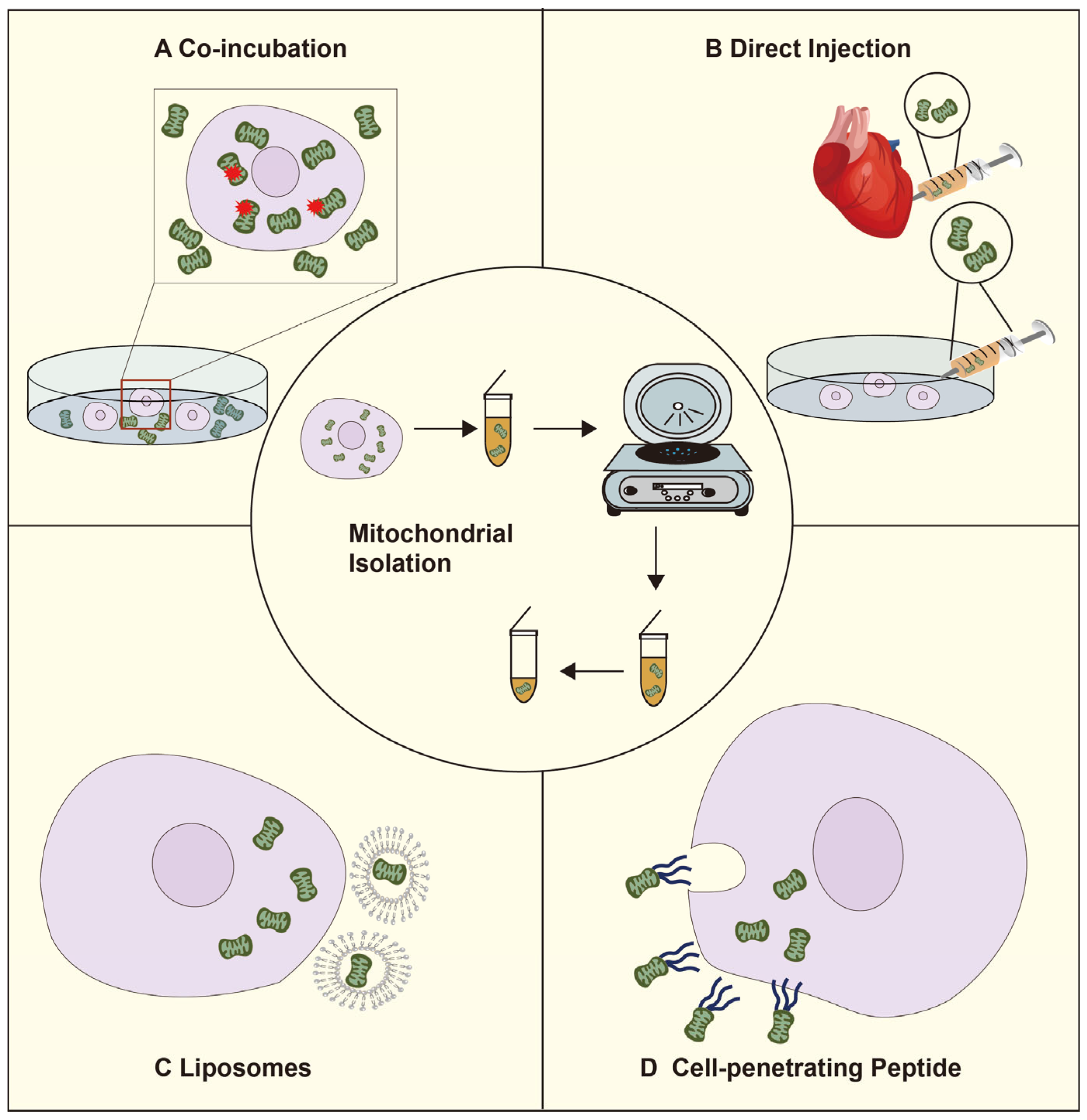
2.2. The Artificial Mitochondrial Transfer Pathway
3. Mitochondrial Transplantation for Mitochondria-Deficient Diseases
3.1. Mitochondrial Transplantation Protects against Neuronal Damage Caused by Cerebral Ischemia
3.2. Mitochondrial Transplantation for Myocardial Ischemia-Reperfusion Injury
3.3. Mitochondrial Transplantation for Tumor Therapy
4. Discussion
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Harris, D.A.; Das, A.M. Control of mitochondrial ATP synthesis in the heart. Biochem. J. 1991, 280 Pt 3, 561–573. [Google Scholar] [CrossRef] [PubMed]
- Rossmann, M.P.; Dubois, S.M.; Agarwal, S.; Zon, L.I. Mitochondrial function in development and disease. Dis. Model. Mech. 2021, 14, dmm048912. [Google Scholar] [CrossRef] [PubMed]
- Srinivasan, S.; Guha, M.; Kashina, A.; Avadhani, N.G. Mitochondrial dysfunction and mitochondrial dynamics-The cancer connection. Biochim. Biophys. Acta Bioenerg. 2017, 1858, 602–614. [Google Scholar] [CrossRef]
- Faria, R.; Boisguérin, P.; Sousa, Â.; Costa, D. Delivery Systems for Mitochondrial Gene Therapy: A Review. Pharmaceutics 2023, 15, 572. [Google Scholar] [CrossRef]
- Huang, T.; Zhang, T.; Gao, J. Targeted mitochondrial delivery: A therapeutic new era for disease treatment. J. Control Release 2022, 343, 89–106. [Google Scholar] [CrossRef] [PubMed]
- Torralba, D.; Baixauli, F.; Sánchez-Madrid, F. Mitochondria Know No Boundaries: Mechanisms and Functions of Intercellular Mitochondrial Transfer. Front. Cell Dev. Biol. 2016, 4, 107. [Google Scholar] [CrossRef] [PubMed]
- Guerra, F.; Arbini, A.A.; Moro, L. Mitochondria and cancer chemoresistance. Biochim. Biophys. Acta Bioenerg. 2017, 1858, 686–699. [Google Scholar] [CrossRef] [PubMed]
- Valenti, D.; Vacca, R.A.; Moro, L.; Atlante, A. Mitochondria Can Cross Cell Boundaries: An Overview of the Biological Relevance, Pathophysiological Implications and Therapeutic Perspectives of Intercellular Mitochondrial Transfer. Int. J. Mol. Sci. 2021, 22, 8312. [Google Scholar] [CrossRef]
- Berridge, M.V.; McConnell, M.J.; Grasso, C.; Bajzikova, M.; Kovarova, J.; Neuzil, J. Horizontal transfer of mitochondria between mammalian cells: Beyond co-culture approaches. Curr. Opin. Genet. Dev. 2016, 38, 75–82. [Google Scholar] [CrossRef]
- Dong, L.F.; Rohlena, J.; Zobalova, R.; Nahacka, Z.; Rodriguez, A.M.; Berridge, M.V.; Neuzil, J. Mitochondria on the move: Horizontal mitochondrial transfer in disease and health. J. Cell Biol. 2023, 222, e202211044. [Google Scholar] [CrossRef]
- Otera, H.; Mihara, K. Molecular mechanisms and physiologic functions of mitochondrial dynamics. J. Biochem. 2011, 149, 241–251. [Google Scholar] [CrossRef] [PubMed]
- Shanmughapriya, S.; Langford, D.; Natarajaseenivasan, K. Inter and Intracellular mitochondrial trafficking in health and disease. Ageing Res. Rev. 2020, 62, 101128. [Google Scholar] [CrossRef] [PubMed]
- Qin, Y.; Jiang, X.; Yang, Q.; Zhao, J.; Zhou, Q.; Zhou, Y. The Functions, Methods, and Mobility of Mitochondrial Transfer Between Cells. Front. Oncol. 2021, 11, 672781. [Google Scholar] [CrossRef] [PubMed]
- Abounit, S.; Zurzolo, C. Wiring through tunneling nanotubes—From electrical signals to organelle transfer. J. Cell Sci. 2012, 125, 1089–1098. [Google Scholar] [CrossRef] [PubMed]
- Mittal, R.; Karhu, E.; Wang, J.S.; Delgado, S.; Zukerman, R.; Mittal, J.; Jhaveri, V.M. Cell communication by tunneling nanotubes: Implications in disease and therapeutic applications. J. Cell Physiol. 2019, 234, 1130–1146. [Google Scholar] [CrossRef] [PubMed]
- Gerdes, H.H.; Bukoreshtliev, N.V.; Barroso, J.F. Tunneling nanotubes: A new route for the exchange of components between animal cells. FEBS Lett. 2007, 581, 2194–2201. [Google Scholar] [CrossRef]
- Zhang, Y.; Yu, Z.; Jiang, D.; Liang, X.; Liao, S.; Zhang, Z.; Yue, W.; Li, X.; Chiu, S.M.; Chai, Y.H.; et al. iPSC-MSCs with High Intrinsic MIRO1 and Sensitivity to TNF-α Yield Efficacious Mitochondrial Transfer to Rescue Anthracycline-Induced Cardiomyopathy. Stem Cell Rep. 2016, 7, 749–763. [Google Scholar] [CrossRef]
- Ahmad, T.; Mukherjee, S.; Pattnaik, B.; Kumar, M.; Singh, S.; Kumar, M.; Rehman, R.; Tiwari, B.K.; Jha, K.A.; Barhanpurkar, A.P.; et al. Miro1 regulates intercellular mitochondrial transport & enhances mesenchymal stem cell rescue efficacy. EMBO J. 2014, 33, 994–1010. [Google Scholar]
- Wang, X.; Gerdes, H.H. Transfer of mitochondria via tunneling nanotubes rescues apoptotic PC12 cells. Cell Death Differ. 2015, 22, 1181–1191. [Google Scholar] [CrossRef]
- Bagheri, H.S.; Bani, F.; Tasoglu, S.; Zarebkohan, A.; Rahbarghazi, R.; Sokullu, E. Mitochondrial donation in translational medicine; from imagination to reality. J. Transl. Med. 2020, 18, 367. [Google Scholar] [CrossRef] [PubMed]
- Aguilar, P.S.; Baylies, M.K.; Fleissner, A.; Helming, L.; Inoue, N.; Podbilewicz, B.; Wang, H.; Wong, M. Genetic basis of cell-cell fusion mechanisms. Trends Genet. 2013, 29, 427–437. [Google Scholar] [CrossRef] [PubMed]
- Acquistapace, A.; Bru, T.; Lesault, P.F.; Figeac, F.; Coudert, A.E.; le Coz, O.; Christov, C.; Baudin, X.; Auber, F.; Yiou, R.; et al. Human mesenchymal stem cells reprogram adult cardiomyocytes toward a progenitor-like state through partial cell fusion and mitochondria transfer. Stem Cells 2011, 29, 812–824. [Google Scholar] [CrossRef] [PubMed]
- Sanz-Ros, J.; Mas-Bargues, C.; Romero-García, N.; Huete-Acevedo, J.; Dromant, M.; Borrás, C. The Potential Use of Mitochondrial Extracellular Vesicles as Biomarkers or Therapeutical Tools. Int. J. Mol. Sci. 2023, 24, 7005. [Google Scholar] [CrossRef] [PubMed]
- Ikeda, G.; Santoso, M.R.; Tada, Y.; Li, A.M.; Vaskova, E.; Jung, J.H.; O’Brien, C.; Egan, E.; Ye, J.; Yang, P.C. Mitochondria-Rich Extracellular Vesicles From Autologous Stem Cell-Derived Cardiomyocytes Restore Energetics of Ischemic Myocardium. J. Am. Coll. Cardiol. 2021, 77, 1073–1088. [Google Scholar] [CrossRef]
- Zhang, Z.; Sheng, H.; Liao, L.; Xu, C.; Zhang, A.; Yang, Y.; Zhao, L.; Duan, L.; Chen, H.; Zhang, B. Mesenchymal Stem Cell-Conditioned Medium Improves Mitochondrial Dysfunction and Suppresses Apoptosis in Okadaic Acid-Treated SH-SY5Y Cells by Extracellular Vesicle Mitochondrial Transfer. J. Alzheimers Dis. 2020, 78, 1161–1176. [Google Scholar] [CrossRef]
- Islam, M.N.; Das, S.R.; Emin, M.T.; Wei, M.; Sun, L.; Westphalen, K.; Rowlands, D.J.; Quadri, S.K.; Bhattacharya, S.; Bhattacharya, J. Mitochondrial transfer from bone-marrow-derived stromal cells to pulmonary alveoli protects against acute lung injury. Nat. Med. 2012, 18, 759–765. [Google Scholar] [CrossRef]
- Yang, J.; Liu, L.; Oda, Y.; Wada, K.; Ago, M.; Matsuda, S.; Hattori, M.; Goto, T.; Ishibashi, S.; Kawashima-Sonoyama, Y.; et al. Extracellular Vesicles and Cx43-Gap Junction Channels Are the Main Routes for Mitochondrial Transfer from Ultra-Purified Mesenchymal Stem Cells, RECs. Int. J. Mol. Sci. 2023, 24, 10294. [Google Scholar] [CrossRef]
- Norris, R.P. Transfer of mitochondria and endosomes between cells by gap junction internalization. Traffic 2021, 22, 174–179. [Google Scholar] [CrossRef]
- Ribeiro-Rodrigues, T.M.; Martins-Marques, T.; Morel, S.; Kwak, B.R.; Girão, H. Role of connexin 43 in different forms of intercellular communication - gap junctions, extracellular vesicles and tunnelling nanotubes. J. Cell Sci. 2017, 130, 3619–3630. [Google Scholar] [CrossRef]
- Li, H.; Wang, C.; He, T.; Zhao, T.; Chen, Y.Y.; Shen, Y.L.; Zhang, X.; Wang, L.L. Mitochondrial Transfer from Bone Marrow Mesenchymal Stem Cells to Motor Neurons in Spinal Cord Injury Rats via Gap Junction. Theranostics 2019, 9, 2017–2035. [Google Scholar] [CrossRef]
- Zhang, T.G.; Miao, C.Y. Mitochondrial transplantation as a promising therapy for mitochondrial diseases. Acta Pharm. Sin. B 2023, 13, 1028–1035. [Google Scholar] [CrossRef] [PubMed]
- McCully, J.D.; Cowan, D.B.; Pacak, C.A.; Toumpoulis, I.K.; Dayalan, H.; Levitsky, S. Injection of isolated mitochondria during early reperfusion for cardioprotection. Am. J. Physiol. Heart Circ. Physiol. 2009, 296, H94–H105. [Google Scholar] [CrossRef] [PubMed]
- Preble, J.M.; Pacak, C.A.; Kondo, H.; MacKay, A.A.; Cowan, D.B.; McCully, J.D. Rapid isolation and purification of mitochondria for transplantation by tissue dissociation and differential filtration. J. Vis. Exp. 2014, 91, e51682. [Google Scholar]
- Berridge, M.V.; Herst, P.M.; Rowe, M.R.; Schneider, R.; McConnell, M.J. Mitochondrial transfer between cells: Methodological constraints in cell culture and animal models. Anal. Biochem. 2018, 552, 75–80. [Google Scholar] [CrossRef] [PubMed]
- Ali Pour, P.; Kenney, M.C.; Kheradvar, A. Bioenergetics Consequences of Mitochondrial Transplantation in Cardiomyocytes. J. Am. Heart Assoc. 2020, 9, e0145012020. [Google Scholar] [CrossRef] [PubMed]
- Bhattacharya, D.; Slavin, M.B.; Hood, D.A. Muscle mitochondrial transplantation can rescue and maintain cellular homeostasis. Am. J. Physiol. Cell Physiol. 2023, 325, C862–C884. [Google Scholar] [CrossRef]
- Kaza, A.K.; Wamala, I.; Friehs, I.; Kuebler, J.D.; Rathod, R.H.; Berra, I.; Ericsson, M.; Yao, R.; Thedsanamoorthy, J.K.; Zurakowski, D.; et al. Myocardial rescue with autologous mitochondrial transplantation in a porcine model of ischemia/reperfusion. J. Thorac. Cardiovasc. Surg. 2017, 153, 934–943. [Google Scholar] [CrossRef]
- Pinkert, C.A.; Irwin, M.H.; Johnson, L.W.; Moffatt, R.J. Mitochondria transfer into mouse ova by microinjection. Transgenic Res. 1997, 6, 379–383. [Google Scholar] [CrossRef]
- Doulamis, I.P.; Guariento, A.; Duignan, T.; Kido, T.; Orfany, A.; Saeed, M.Y.; Weixler, V.H.; Blitzer, D.; Shin, B.; Snay, E.R.; et al. Mitochondrial transplantation by intra-arterial injection for acute kidney injury. Am. J. Physiol. Renal Physiol. 2020, 319, F403–F413. [Google Scholar] [CrossRef]
- Mobarak, H.; Heidarpour, M.; Tsai, P.J.; Rezabakhsh, A.; Rahbarghazi, R.; Nouri, M.; Mahdipour, M. Autologous mitochondrial microinjection; a strategy to improve the oocyte quality and subsequent reproductive outcome during aging. Cell Biosci. 2019, 9, 95. [Google Scholar] [CrossRef]
- Yasuzaki, Y.; Yamada, Y.; Harashima, H. Mitochondrial matrix delivery using MITO-Porter, a liposome-based carrier that specifies fusion with mitochondrial membranes. Biochem. Biophys. Res. Commun. 2010, 397, 181–186. [Google Scholar] [CrossRef] [PubMed]
- Shi, J.; Irwin, M.H.; Pinkert, C.A. Mitochondria transfer into fibroblasts: Liposome-mediated transfer of labeled mitochondria into cultured cells. Ethn. Dis. 2008, 18, s143–s144. [Google Scholar]
- Liu, C.S.; Chang, J.C.; Kuo, S.J.; Liu, K.H.; Lin, T.T.; Cheng, W.L.; Chuang, S.F. Delivering healthy mitochondria for the therapy of mitochondrial diseases and beyond. Int. J. Biochem. Cell Biol. 2014, 53, 141–146. [Google Scholar] [CrossRef] [PubMed]
- Chang, J.C.; Liu, K.H.; Li, Y.C.; Kou, S.J.; Wei, Y.H.; Chuang, C.S.; Hsieh, M.; Liu, C.S. Functional recovery of human cells harbouring the mitochondrial DNA mutation MERRF A8344G via peptide-mediated mitochondrial delivery. Neurosignals 2013, 21, 160–173. [Google Scholar] [CrossRef] [PubMed]
- Kaur, M.M.; Sharma, D.S. Mitochondrial repair as potential pharmacological target in cerebral ischemia. Mitochondrion 2022, 63, 23–31. [Google Scholar] [CrossRef] [PubMed]
- Zhou, Y.; Danbolt, N.C. Glutamate as a neurotransmitter in the healthy brain. J. Neural. Transm. 2014, 121, 799–817. [Google Scholar] [CrossRef]
- Hansen, K.B.; Yi, F.; Perszyk, R.E.; Furukawa, H.; Wollmuth, L.P.; Gibb, A.J.; Traynelis, S.F. Structure, function, and allosteric modulation of NMDA receptors. J. Gen. Physiol. 2018, 150, 1081–1105. [Google Scholar] [CrossRef] [PubMed]
- Kang, J.B.; Park, D.J.; Shah, M.A.; Koh, P.O. Quercetin ameliorates glutamate toxicity-induced neuronal cell death by controlling calcium-binding protein parvalbumin. J. Vet. Sci. 2022, 23, e26. [Google Scholar] [CrossRef]
- Orrenius, S.; Gogvadze, V.; Zhivotovsky, B. Calcium and mitochondria in the regulation of cell death. Biochem. Biophys. Res. Commun. 2015, 460, 72–81. [Google Scholar] [CrossRef]
- Gong, Y.; Lin, J.; Ma, Z.; Yu, M.; Wang, M.; Lai, D.; Fu, G. Mitochondria-associated membrane-modulated Ca(2+) transfer: A potential treatment target in cardiac ischemia reperfusion injury and heart failure. Life Sci. 2021, 278, 119511. [Google Scholar] [CrossRef]
- Hardingham, G.E. Coupling of the NMDA receptor to neuroprotective and neurodestructive events. Biochem. Soc. Trans. 2009, 37, 1147–1160. [Google Scholar] [CrossRef]
- Zorov, D.B.; Juhaszova, M.; Sollott, S.J. Mitochondrial Reactive Oxygen Species (ROS) and ROS-Induced ROS Release. Physiol. Rev. 2014, 94, 909. [Google Scholar] [CrossRef]
- Pourmohammadi-Bejarpasi, Z.; Roushandeh, A.M.; Saberi, A.; Rostami, M.K.; Toosi, S.M.R.; Jahanian-Najafabadi, A.; Tomita, K.; Kuwahara, Y.; Sato, T.; Roudkenar, M.H. Mesenchymal stem cells-derived mitochondria transplantation mitigates I/R-induced injury, abolishes I/R-induced apoptosis, and restores motor function in acute ischemia stroke rat model. Brain Res. Bull. 2020, 165, 70–80. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Z.; Ma, Z.; Yan, C.; Pu, K.; Wu, M.; Bai, J.; Li, Y.; Wang, Q. Muscle-derived autologous mitochondrial transplantation: A novel strategy for treating cerebral ischemic injury. Behav. Brain Res. 2019, 356, 322–331. [Google Scholar] [CrossRef]
- Tseng, N.; Lambie, S.C.; Huynh, C.Q.; Sanford, B.; Patel, M.; Herson, P.S.; Ormond, D.R. Mitochondrial transfer from mesenchymal stem cells improves neuronal metabolism after oxidant injury in vitro: The role of Miro1. J. Cereb. Blood Flow. Metab. 2021, 41, 761–770. [Google Scholar] [CrossRef] [PubMed]
- Hayakawa, K.; Esposito, E.; Wang, X.; Terasaki, Y.; Liu, Y.; Xing, C.; Ji, X.; Lo, E.H. Transfer of mitochondria from astrocytes to neurons after stroke. Nature 2016, 535, 551–555. [Google Scholar] [CrossRef] [PubMed]
- Berridge, M.V.; Neuzil, J. The mobility of mitochondria: Intercellular trafficking in health and disease. Clin. Exp. Pharmacol. Physiol. 2017, 44 (Suppl. 1), 15–20. [Google Scholar] [CrossRef]
- Shin, B.; Cowan, D.B.; Emani, S.M.; Del Nido, P.J.; McCully, J.D. Mitochondrial Transplantation in Myocardial Ischemia and Reperfusion Injury. Adv. Exp. Med. Biol. 2017, 982, 595–619. [Google Scholar] [CrossRef]
- Tian, H.; Zhao, X.; Zhang, Y.; Xia, Z. Abnormalities of glucose and lipid metabolism in myocardial ischemia-reperfusion injury. Biomed. Pharmacother. 2023, 163, 114827. [Google Scholar] [CrossRef]
- Sun, X.; Chen, H.; Gao, R.; Qu, Y.; Huang, Y.; Zhang, N.; Hu, S.; Fan, F.; Zou, Y.; Hu, K.; et al. Intravenous Transplantation of an Ischemic-specific Peptide-TPP-mitochondrial Compound Alleviates Myocardial Ischemic Reperfusion Injury. ACS Nano 2023, 17, 896–909. [Google Scholar] [CrossRef]
- Masuzawa, A.; Black, K.M.; Pacak, C.A.; Ericsson, M.; Barnett, R.J.; Drumm, C.; Seth, P.; Bloch, D.B.; Levitsky, S.; Cowan, D.B.; et al. Transplantation of autologously derived mitochondria protects the heart from ischemia-reperfusion injury. Am. J. Physiol. Heart Circ. Physiol. 2013, 304, H966–H982. [Google Scholar] [CrossRef] [PubMed]
- McCully, J.D.; Levitsky, S.; Del Nido, P.J.; Cowan, D.B. Mitochondrial transplantation for therapeutic use. Clin. Transl. Med. 2016, 5, 16. [Google Scholar] [CrossRef] [PubMed]
- Roche, S.; D’Ippolito, G.; Gomez, L.A.; Bouckenooghe, T.; Lehmann, S.; Montero-Menei, C.N.; Schiller, P.C. Comparative analysis of protein expression of three stem cell populations: Models of cytokine delivery system in vivo. Int. J. Pharm. 2013, 440, 72–82. [Google Scholar] [CrossRef] [PubMed]
- Shen, Y.; Liu, X.; Shi, J.; Wu, X. Involvement of Nrf2 in myocardial ischemia and reperfusion injury. Int. J. Biol. Macromol. 2019, 125, 496–502. [Google Scholar] [CrossRef]
- Somers, P.; Cornelissen, R.; Thierens, H.; Van Nooten, G. An optimized growth factor cocktail for ovine mesenchymal stem cells. Growth Factors 2012, 30, 37–48. [Google Scholar] [CrossRef] [PubMed]
- De Miranda, M.C.; Rodrigues, M.A.; de Angelis Campos, A.C.; Faria, J.; Kunrath-Lima, M.; Mignery, G.A.; Schechtman, D.; Goes, A.M.; Nathanson, M.H.; Gomes, D.A. Epidermal growth factor (EGF) triggers nuclear calcium signaling through the intranuclear phospholipase Cδ-4 (PLCδ4). J. Biol. Chem. 2019, 294, 16650–16662. [Google Scholar] [CrossRef] [PubMed]
- Schenk, S.; Mal, N.; Finan, A.; Zhang, M.; Kiedrowski, M.; Popovic, Z.; McCarthy, P.M.; Penn, M.S. Monocyte chemotactic protein-3 is a myocardial mesenchymal stem cell homing factor. Stem Cells 2007, 25, 245–251. [Google Scholar] [CrossRef]
- Pacak, C.A.; Preble, J.M.; Kondo, H.; Seibel, P.; Levitsky, S.; Del Nido, P.J.; Cowan, D.B.; McCully, J.D. Actin-dependent mitochondrial internalization in cardiomyocytes: Evidence for rescue of mitochondrial function. Biol. Open 2015, 4, 622–626. [Google Scholar] [CrossRef]
- Emani, S.M.; McCully, J.D. Mitochondrial transplantation: Applications for pediatric patients with congenital heart disease. Transl. Pediatr. 2018, 7, 169–175. [Google Scholar] [CrossRef]
- Chen, K.; Lu, P.; Beeraka, N.M.; Sukocheva, O.A.; Madhunapantula, S.V.; Liu, J.; Sinelnikov, M.Y.; Nikolenko, V.N.; Bulygin, K.V.; Mikhaleva, L.M.; et al. Mitochondrial mutations and mitoepigenetics: Focus on regulation of oxidative stress-induced responses in breast cancers. Semin. Cancer Biol. 2022, 83, 556–569. [Google Scholar] [CrossRef]
- Sies, H.; Jones, D.P. Reactive oxygen species (ROS) as pleiotropic physiological signalling agents. Nat. Rev. Mol. Cell Biol. 2020, 21, 363–383. [Google Scholar] [CrossRef] [PubMed]
- Yang, Y.; Karakhanova, S.; Hartwig, W.; D’Haese, J.G.; Philippov, P.P.; Werner, J.; Bazhin, A.V. Mitochondria and Mitochondrial ROS in Cancer: Novel Targets for Anticancer Therapy. J. Cell Physiol. 2016, 231, 2570–2581. [Google Scholar] [CrossRef] [PubMed]
- Yakes, F.M.; Van Houten, B. Mitochondrial DNA damage is more extensive and persists longer than nuclear DNA damage in human cells following oxidative stress. Proc. Natl. Acad. Sci. USA 1997, 94, 514–519. [Google Scholar] [CrossRef] [PubMed]
- Zampieri, L.X.; Silva-Almeida, C.; Rondeau, J.D.; Sonveaux, P. Mitochondrial Transfer in Cancer: A Comprehensive Review. Int. J. Mol. Sci. 2021, 22, 3245. [Google Scholar] [CrossRef]
- Wu, Z.; Wu, J.; Zhao, Q.; Fu, S.; Jin, J. Emerging roles of aerobic glycolysis in breast cancer. Clin. Transl. Oncol. 2020, 22, 631–646. [Google Scholar] [CrossRef] [PubMed]
- Hanahan, D.; Weinberg, R.A. Hallmarks of cancer: The next generation. Cell 2011, 144, 646–674. [Google Scholar] [CrossRef] [PubMed]
- Nile, D.L.; Rae, C.; Walker, D.J.; Waddington, J.C.; Vincent, I.; Burgess, K.; Gaze, M.N.; Mairs, R.J.; Chalmers, A.J. Inhibition of glycolysis and mitochondrial respiration promotes radiosensitisation of neuroblastoma and glioma cells. Cancer Metab. 2021, 9, 24. [Google Scholar] [CrossRef]
- Fang, J.; Ma, Y.; Li, Y.; Li, J.; Zhang, X.; Han, X.; Ma, S.; Guan, F. CCT4 knockdown enhances the sensitivity of cisplatin by inhibiting glycolysis in human esophageal squamous cell carcinomas. Mol. Carcinog. 2022, 61, 1043–1055. [Google Scholar] [CrossRef]
- Zhou, W.; Zhao, Z.; Yu, Z.; Hou, Y.; Keerthiga, R.; Fu, A. Mitochondrial transplantation therapy inhibits the proliferation of malignant hepatocellular carcinoma and its mechanism. Mitochondrion 2022, 65, 11–22. [Google Scholar] [CrossRef]
- Sun, C.; Liu, X.; Wang, B.; Wang, Z.; Liu, Y.; Di, C.; Si, J.; Li, H.; Wu, Q.; Xu, D.; et al. Endocytosis-mediated mitochondrial transplantation: Transferring normal human astrocytic mitochondria into glioma cells rescues aerobic respiration and enhances radiosensitivity. Theranostics 2019, 9, 3595–3607. [Google Scholar] [CrossRef]
- Astigiano, C.; Benzi, A.; Laugieri, M.E.; Piacente, F.; Sturla, L.; Guida, L.; Bruzzone, S.; De Flora, A. Paracrine ADP Ribosyl Cyclase-Mediated Regulation of Biological Processes. Cells 2022, 11, 2637. [Google Scholar] [CrossRef] [PubMed]
- Li, J.P.; Wei, W.; Li, X.X.; Xu, M. Regulation of NLRP3 inflammasome by CD38 through cADPR-mediated Ca(2+) release in vascular smooth muscle cells in diabetic mice. Life Sci. 2020, 255, 117758. [Google Scholar] [CrossRef] [PubMed]
- Takasawa, S. CD38-Cyclic ADP-Ribose Signal System in Physiology, Biochemistry, and Pathophysiology. Int. J. Mol. Sci. 2022, 23, 4306. [Google Scholar] [CrossRef] [PubMed]
- Elliott, R.L.; Jiang, X.P.; Head, J.F. Mitochondria organelle transplantation: Introduction of normal epithelial mitochondria into human cancer cells inhibits proliferation and increases drug sensitivity. Breast Cancer Res. Treat. 2012, 136, 347–354. [Google Scholar] [CrossRef]
- Chang, J.C.; Chang, H.S.; Wu, Y.C.; Cheng, W.L.; Lin, T.T.; Chang, H.J.; Kuo, S.J.; Chen, S.T.; Liu, C.S. Mitochondrial transplantation regulates antitumour activity, chemoresistance and mitochondrial dynamics in breast cancer. J. Exp. Clin. Cancer Res. 2019, 38, 30. [Google Scholar] [CrossRef] [PubMed]
- Yu, Z.; Hou, Y.; Zhou, W.; Zhao, Z.; Liu, Z.; Fu, A. The effect of mitochondrial transplantation therapy from different gender on inhibiting cell proliferation of malignant melanoma. Int. J. Biol. Sci. 2021, 17, 2021–2033. [Google Scholar] [CrossRef]
- Zhang, X.; Li, Y.; Ma, Y.; Yang, L.; Wang, T.; Meng, X.; Zong, Z.; Sun, X.; Hua, X.; Li, H. Yes-associated protein (YAP) binds to HIF-1α and sustains HIF-1α protein stability to promote hepatocellular carcinoma cell glycolysis under hypoxic stress. J. Exp. Clin. Cancer Res. 2018, 37, 216. [Google Scholar] [CrossRef] [PubMed]
- Tian, H.; Zhu, X.; Lv, Y.; Jiao, Y.; Wang, G. Glucometabolic Reprogramming in the Hepatocellular Carcinoma Microenvironment: Cause and Effect. Cancer Manag. Res. 2020, 12, 5957–5974. [Google Scholar] [CrossRef]
- Spees, J.L.; Olson, S.D.; Whitney, M.J.; Prockop, D.J. Mitochondrial transfer between cells can rescue aerobic respiration. Proc. Natl. Acad. Sci. USA 2006, 103, 1283–1288. [Google Scholar] [CrossRef]
- Patel, P.S.; Castelow, C.; Patel, D.S.; Bhattacharya, S.K.; Kuscu, C.; Kuscu, C.; Makowski, L.; Eason, J.D.; Bajwa, A. Mitochondrial Role in Oncogenesis and Potential Chemotherapeutic Strategy of Mitochondrial Infusion in Breast Cancer. Int. J. Mol. Sci. 2022, 23, 12993. [Google Scholar] [CrossRef]
- Prasuhn, J.; Davis, R.L.; Kumar, K.R. Targeting Mitochondrial Impairment in Parkinson’s Disease: Challenges and Opportunities. Front. Cell Dev. Biol. 2020, 8, 615461. [Google Scholar] [CrossRef]
- Rey, F.; Ottolenghi, S.; Zuccotti, G.V.; Samaja, M.; Carelli, S. Mitochondrial dysfunctions in neurodegenerative diseases: Role in disease pathogenesis, strategies for analysis and therapeutic prospects. Neural Regen. Res. 2022, 17, 754–758. [Google Scholar] [PubMed]
- Park, A.; Oh, M.; Lee, S.J.; Oh, K.J.; Lee, E.W.; Lee, S.C.; Bae, K.H.; Han, B.S.; Kim, W.K. Mitochondrial Transplantation as a Novel Therapeutic Strategy for Mitochondrial Diseases. Int. J. Mol. Sci. 2021, 22, 4793. [Google Scholar] [CrossRef] [PubMed]
- Wang, S.; Zhao, H.; Lin, S.; Lv, Y.; Lin, Y.; Liu, Y.; Peng, R.; Jin, H. New therapeutic directions in type II diabetes and its complications: Mitochondrial dynamics. Front. Endocrinol. 2023, 14, 1230168. [Google Scholar] [CrossRef]
- Nasoni, M.G.; Carloni, S.; Canonico, B.; Burattini, S.; Cesarini, E.; Papa, S.; Pagliarini, M.; Ambrogini, P.; Balduini, W.; Luchetti, F. Melatonin reshapes the mitochondrial network and promotes intercellular mitochondrial transfer via tunneling nanotubes after ischemic-like injury in hippocampal HT22 cells. J. Pineal Res. 2021, 71, e12747. [Google Scholar] [CrossRef]
- Tomita, K.; Kuwahara, Y.; Igarashi, K.; Roudkenar, M.H.; Roushandeh, A.M.; Kurimasa, A.; Sato, T. Mitochondrial Dysfunction in Diseases, Longevity, and Treatment Resistance: Tuning Mitochondria Function as a Therapeutic Strategy. Genes 2021, 12, 1348. [Google Scholar] [CrossRef]
- Yamada, Y.; Ito, M.; Arai, M.; Hibino, M.; Tsujioka, T.; Harashima, H. Challenges in Promoting Mitochondrial Transplantation Therapy. Int. J. Mol. Sci. 2020, 21, 6365. [Google Scholar] [CrossRef] [PubMed]
- Lake, N.J.; Compton, A.G.; Rahman, S.; Thorburn, D.R. Leigh syndrome: One disorder, more than 75 monogenic causes. Ann. Neurol. 2016, 79, 190–203. [Google Scholar] [CrossRef] [PubMed]
- Arakawa, K.; Kudo, T.; Ikawa, M.; Morikawa, N.; Kawai, Y.; Sahashi, K.; Lee, J.D.; Kuriyama, M.; Miyamori, I.; Okazawa, H.; et al. Abnormal myocardial energy-production state in mitochondrial cardiomyopathy and acute response to L-arginine infusion. C-11 acetate kinetics revealed by positron emission tomography. Circ. J. 2010, 74, 2702–2711. [Google Scholar] [CrossRef]
- Rawle, M.J.; Larner, A.J. NARP Syndrome: A 20-Year Follow-Up. Case Rep. Neurol. 2013, 5, 204–207. [Google Scholar] [CrossRef]
- Taivassalo, T.; Jensen, T.D.; Kennaway, N.; DiMauro, S.; Vissing, J.; Haller, R.G. The spectrum of exercise tolerance in mitochondrial myopathies: A study of 40 patients. Brain 2003, 126, 413–423. [Google Scholar] [CrossRef] [PubMed]
- Rodan, L.H.; Wells, G.D.; Banks, L.; Thompson, S.; Schneiderman, J.E.; Tein, I. L-Arginine Affects Aerobic Capacity and Muscle Metabolism in MELAS (Mitochondrial Encephalomyopathy, Lactic Acidosis and Stroke-Like Episodes) Syndrome. PLoS ONE 2015, 10, e01270662015. [Google Scholar] [CrossRef] [PubMed]


Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Hu, C.; Shi, Z.; Liu, X.; Sun, C. The Research Progress of Mitochondrial Transplantation in the Treatment of Mitochondrial Defective Diseases. Int. J. Mol. Sci. 2024, 25, 1175. https://doi.org/10.3390/ijms25021175
Hu C, Shi Z, Liu X, Sun C. The Research Progress of Mitochondrial Transplantation in the Treatment of Mitochondrial Defective Diseases. International Journal of Molecular Sciences. 2024; 25(2):1175. https://doi.org/10.3390/ijms25021175
Chicago/Turabian StyleHu, Cuilan, Zheng Shi, Xiongxiong Liu, and Chao Sun. 2024. "The Research Progress of Mitochondrial Transplantation in the Treatment of Mitochondrial Defective Diseases" International Journal of Molecular Sciences 25, no. 2: 1175. https://doi.org/10.3390/ijms25021175
APA StyleHu, C., Shi, Z., Liu, X., & Sun, C. (2024). The Research Progress of Mitochondrial Transplantation in the Treatment of Mitochondrial Defective Diseases. International Journal of Molecular Sciences, 25(2), 1175. https://doi.org/10.3390/ijms25021175