
Evaluating the Efficacy of Type 2 Diabetes Polygenic Risk Scores in an Independent European Population

 , , , ,
, , , ,
Abstract
1. Introduction
2. Results
2.1. Cohort Characteristics and Genotype-Based Quality Control
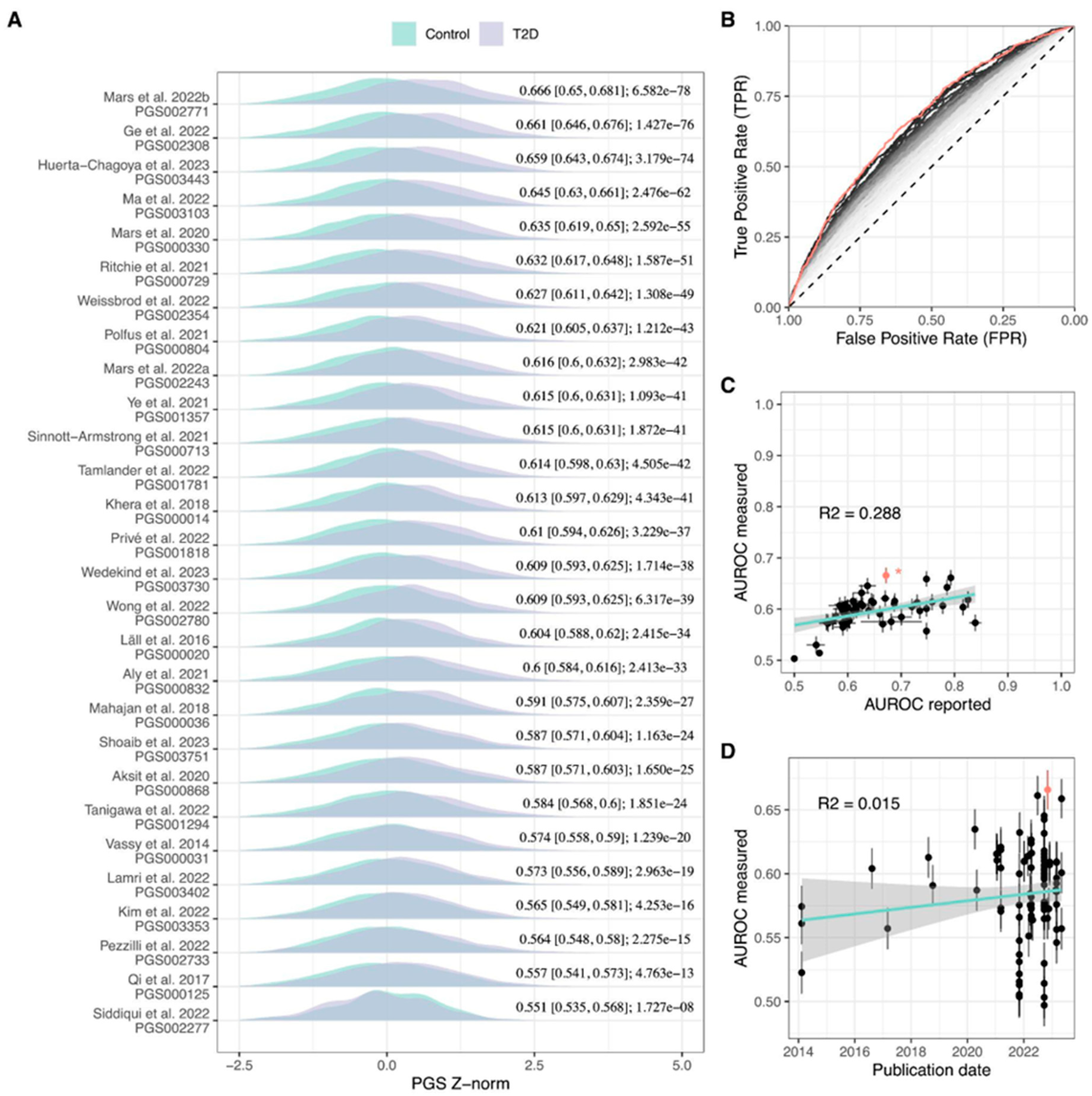
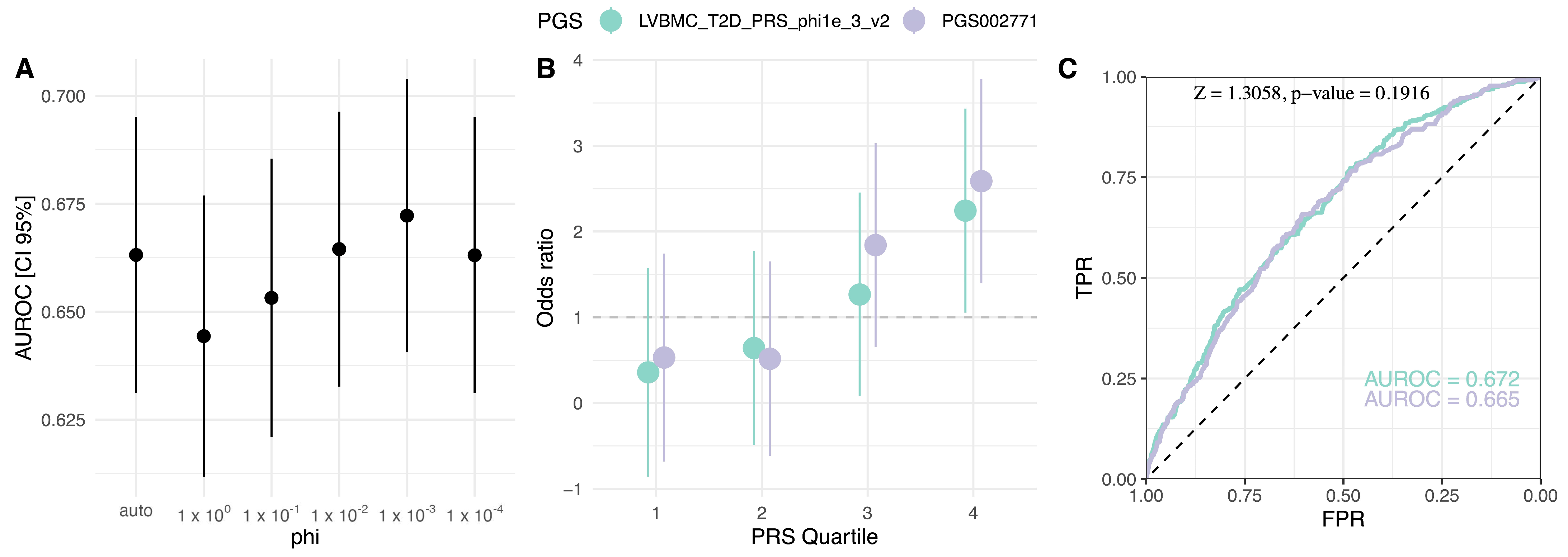
2.2. Evaluation of Published T2D PGS Models
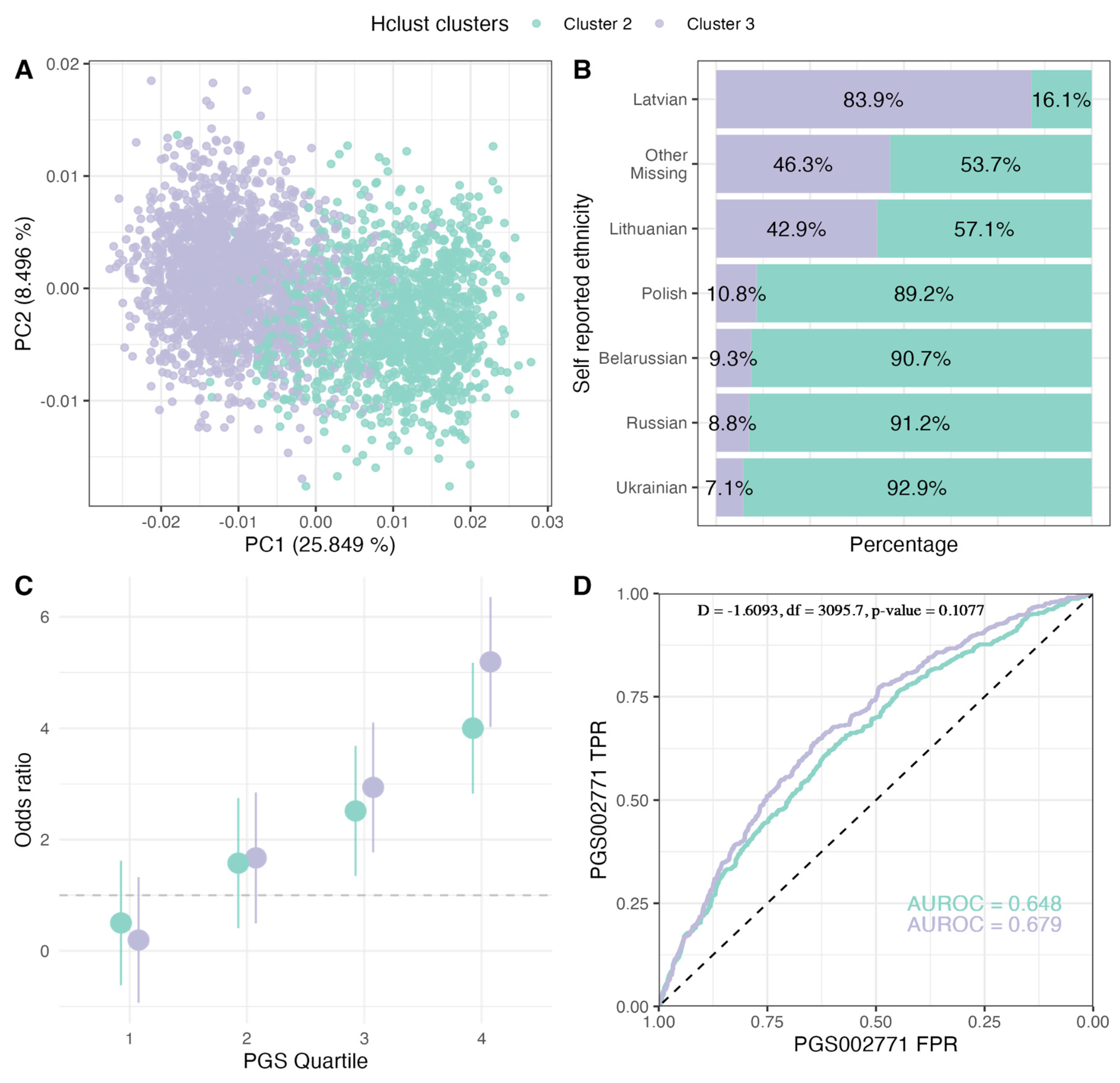
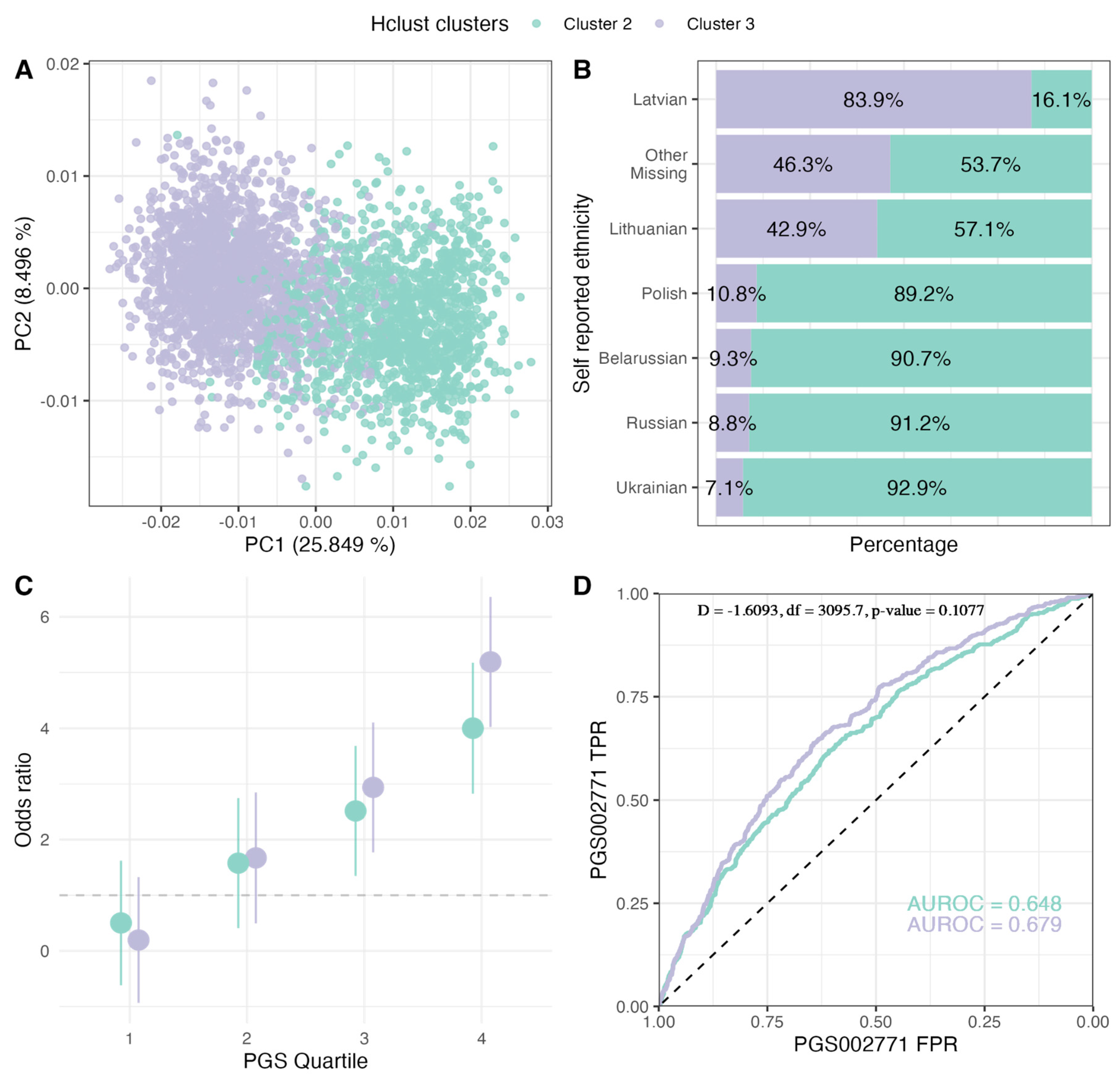
2.3. PGS Performance in Ancestry Clusters
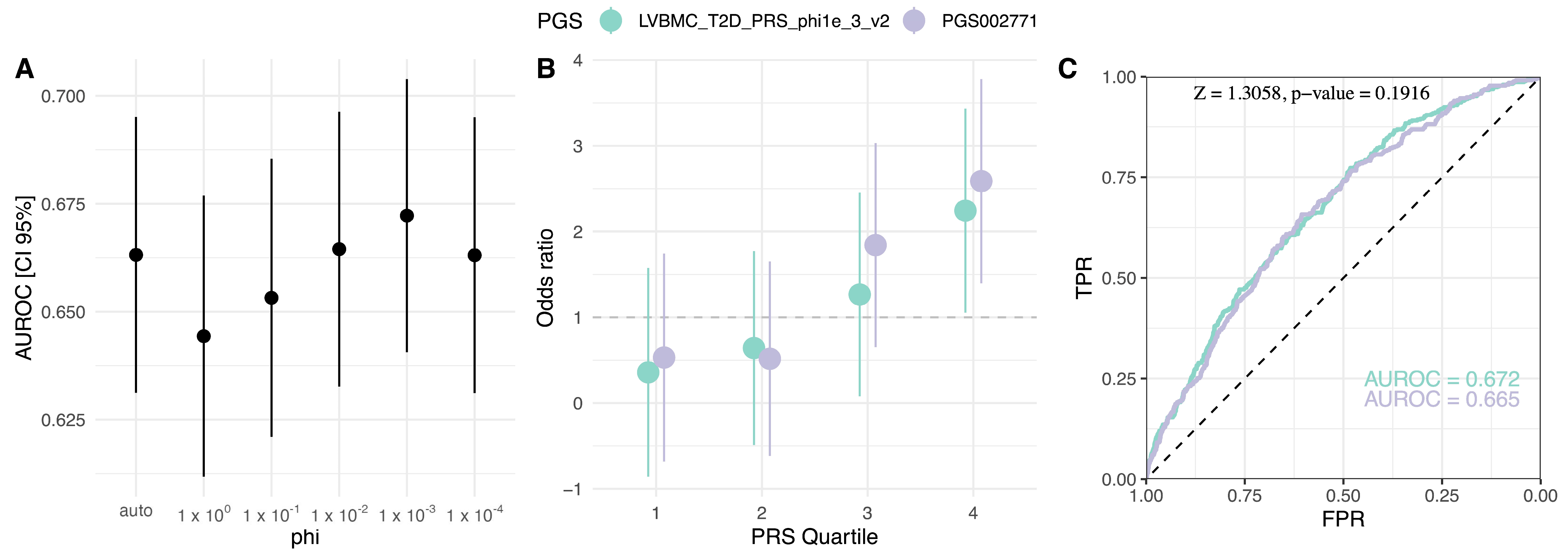
2.4. Population-Specific PGS Development
3. Discussion
4. Methods
4.1. Cohort Description and Data Selection
4.2. Genotype Quality Control
4.3. Polygenic Risk Score Calculation
4.4. PGS Evaluation
4.5. Polygenic Risk Score Model Construction
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Lambert, S.A.; Gil, L.; Jupp, S.; Ritchie, S.C.; Xu, Y.; Buniello, A.; McMahon, A.; Abraham, G.; Chapman, M.; Parkinson, H.; et al. The Polygenic Score Catalog as an Open Database for Reproducibility and Systematic Evaluation. Nat. Genet. 2021, 53, 420–425. [Google Scholar] [CrossRef] [PubMed]
- Albert, E.A.; Kondratieva, O.A.; Baranova, E.E.; Sagaydak, O.V.; Belenikin, M.S.; Zobkova, G.Y.; Kuznetsova, E.S.; Deviatkin, A.A.; Zhurov, A.A.; Karpulevich, E.A.; et al. Transferability of the PRS Estimates for Height and BMI Obtained from the European Ethnic Groups to the Western Russian Populations. Front. Genet. 2023, 14, 1086709. [Google Scholar] [CrossRef] [PubMed]
- Läll, K.; Mägi, R.; Morris, A.; Metspalu, A.; Fischer, K. Personalized Risk Prediction for Type 2 Diabetes: The Potential of Genetic Risk Scores. Genet. Med. 2017, 19, 322–329. [Google Scholar] [CrossRef]
- Weissbrod, O.; Kanai, M.; Shi, H.; Gazal, S.; Peyrot, W.J.; Khera, A.V.; Okada, Y.; Matsuda, K.; Yamanashi, Y.; Furukawa, Y.; et al. Leveraging Fine-Mapping and Multipopulation Training Data to Improve Cross-Population Polygenic Risk Scores. Nat. Genet. 2022, 54, 450–458. [Google Scholar] [CrossRef] [PubMed]
- Scott, R.A.; Scott, L.J.; Mägi, R.; Marullo, L.; Gaulton, K.J.; Kaakinen, M.; Pervjakova, N.; Pers, T.H.; Johnson, A.D.; Eicher, J.D.; et al. An Expanded Genome-Wide Association Study of Type 2 Diabetes in Europeans. Diabetes 2017, 66, 2888–2902. [Google Scholar] [CrossRef] [PubMed]
- Mahajan, A.; Taliun, D.; Thurner, M.; Robertson, N.R.; Torres, J.M.; Rayner, N.W.; Payne, A.J.; Steinthorsdottir, V.; Scott, R.A.; Grarup, N.; et al. Fine-Mapping Type 2 Diabetes Loci to Single-Variant Resolution Using High-Density Imputation and Is-let-Specific Epigenome Maps. Nat. Genet. 2018, 50, 1505–1513. [Google Scholar] [CrossRef] [PubMed]
- Bycroft, C.; Freeman, C.; Petkova, D.; Band, G.; Elliott, L.T.; Sharp, K.; Motyer, A.; Vukcevic, D.; Delaneau, O.; O’Connell, J.; et al. The UK Biobank Resource with Deep Phenotyping and Genomic Data. Nature 2018, 562, 203–209. [Google Scholar] [CrossRef]
- Privé, F.; Aschard, H.; Carmi, S.; Folkersen, L.; Hoggart, C.; O’Reilly, P.F.; Vilhjálmsson, B.J. Portability of 245 Polygenic Scores When Derived from the UK Biobank and Applied to 9 Ancestry Groups from the Same Cohort. Am. J. Hum. Genet. 2022, 109, 12–23. [Google Scholar] [CrossRef]
- Ge, T.; Chen, C.-Y.; Ni, Y.; Feng, Y.-C.A.; Smoller, J.W. Polygenic Prediction via Bayesian Regression and Continuous Shrinkage Priors. Nat. Commun. 2019, 10, 1776. [Google Scholar] [CrossRef]
- Vilhjalmsson, B.; Yang, J.; Finucane, H.K.; Gusev, A.; Lindstrom, S.; Ripke, S.; Genovese, G.; Loh, P.-R.; Bhatia, G.; Do, R.; et al. Modeling Linkage Disequilibrium Increases Accuracy of Polygenic Risk Scores. Am. J. Hum. Genet. 2015, 97, 576–592. [Google Scholar] [CrossRef]
- Aksit, M.A.; Pace, R.G.; Vecchio-Pagán, B.; Ling, H.; Rommens, J.M.; Boelle, P.-Y.; Guillot, L.; Raraigh, K.S.; Pugh, E.; Zhang, P.; et al. Genetic Modifiers of Cystic Fibrosis-Related Diabetes Have Extensive Overlap With Type 2 Diabetes and Related Traits. J. Clin. Endocrinol. Metab. 2020, 105, 1401–1415. [Google Scholar] [CrossRef] [PubMed]
- Oram, R.A.; Sharp, S.A.; Pihoker, C.; Ferrat, L.; Imperatore, G.; Williams, A.; Redondo, M.J.; Wagenknecht, L.; Dolan, L.M.; Lawrence, J.M.; et al. Utility of Diabetes Type–Specific Genetic Risk Scores for the Classification of Diabetes Type Among Multiethnic Youth. Diabetes Care 2022, 45, 1124–1131. [Google Scholar] [CrossRef]
- Rovite, V.; Wolff-Sagi, Y.; Zaharenko, L.; Nikitina-Zake, L.; Grens, E.; Klovins, J. Genome Database of the Latvian Population (LGDB): Design, Goals, and Primary Results. J. Epidemiol. 2018, 28, 353–360. [Google Scholar] [CrossRef] [PubMed]
- Wand, H.; Lambert, S.A.; Tamburro, C.; Iacocca, M.A.; O’Sullivan, J.W.; Sillari, C.; Kullo, I.J.; Rowley, R.; Dron, J.S.; Brockman, D.; et al. Improving Reporting Standards for Polygenic Scores in Risk Prediction Studies. Nature 2021, 591, 211–219. [Google Scholar] [CrossRef] [PubMed]
- Mars, N.; Lindbohm, J.V.; della Briotta Parolo, P.; Widén, E.; Kaprio, J.; Palotie, A.; Ripatti, S. Systematic Comparison of Family History and Polygenic Risk across 24 Common Diseases. Am. J. Hum. Genet. 2022, 109, 2152–2162. [Google Scholar] [CrossRef]
- Ge, T.; Irvin, M.R.; Patki, A.; Srinivasasainagendra, V.; Lin, Y.-F.; Tiwari, H.K.; Armstrong, N.D.; Benoit, B.; Chen, C.-Y.; Choi, K.W.; et al. Development and Validation of a Trans-Ancestry Polygenic Risk Score for Type 2 Diabetes in Diverse Populations. Genome Med. 2022, 14, 70. [Google Scholar] [CrossRef]
- Huerta-Chagoya, A.; Schroeder, P.; Mandla, R.; Deutsch, A.J.; Zhu, W.; Petty, L.; Yi, X.; Cole, J.B.; Udler, M.S.; Dornbos, P.; et al. The Power of TOPMed Imputation for the Discovery of Latino-Enriched Rare Variants Associated with Type 2 Diabetes. Diabetologia 2023, 66, 1273–1288. [Google Scholar] [CrossRef]
- Ma, Y.; Patil, S.; Zhou, X.; Mukherjee, B.; Fritsche, L.G. ExPRSweb: An Online Repository with Polygenic Risk Scores for Common Health-Related Exposures. Am. J. Hum. Genet. 2022, 109, 1742–1760. [Google Scholar] [CrossRef]
- Mars, N.; Koskela, J.T.; Ripatti, P.; Kiiskinen, T.T.J.; Havulinna, A.S.; Lindbohm, J.V.; Ahola-Olli, A.; Kurki, M.; Karjalainen, J.; Palta, P.; et al. Polygenic and Clinical Risk Scores and Their Impact on Age at Onset and Prediction of Cardiometabolic Diseases and Common Cancers. Nat. Med. 2020, 26, 549–557. [Google Scholar] [CrossRef]
- Ritchie, S.C.; Lambert, S.A.; Arnold, M.; Teo, S.M.; Lim, S.; Scepanovic, P.; Marten, J.; Zahid, S.; Chaffin, M.; Liu, Y.; et al. Integrative Analysis of the Plasma Proteome and Polygenic Risk of Cardiometabolic Diseases. Nat. Metab. 2021, 3, 1476–1483. [Google Scholar] [CrossRef]
- Mars, N.; Kerminen, S.; Feng, Y.-C.A.; Kanai, M.; Läll, K.; Thomas, L.F.; Skogholt, A.H.; Della Briotta Parolo, P.; Biobank Japan Project; FinnGen; et al. Genome-Wide Risk Prediction of Common Diseases across Ancestries in One Million People. Cell Genom. 2022, 2, 100118. [Google Scholar] [CrossRef] [PubMed]
- Ye, Y.; Chen, X.; Han, J.; Jiang, W.; Natarajan, P.; Zhao, H. Interactions Between Enhanced Polygenic Risk Scores and Lifestyle for Cardiovascular Disease, Diabetes, and Lipid Levels. Circ. Genom. Precis. Med. 2021, 14, e003128. [Google Scholar] [CrossRef]
- Tamlander, M.; Mars, N.; Pirinen, M.; Palotie, A.; Daly, M.; Riley-Gills, B.; Jacob, H.; Paul, D.; Runz, H.; John, S.; et al. Inte-gration of Questionnaire-Based Risk Factors Improves Polygenic Risk Scores for Human Coronary Heart Disease and Type 2 Diabetes. Commun. Biol. 2022, 5, 158. [Google Scholar] [CrossRef] [PubMed]
- Khera, A.V.; Chaffin, M.; Aragam, K.G.; Haas, M.E.; Roselli, C.; Choi, S.H.; Natarajan, P.; Lander, E.S.; Lubitz, S.A.; Ellinor, P.T.; et al. Genome-Wide Polygenic Scores for Common Diseases Identify Individuals with Risk Equivalent to Monogenic Mutations. Nat. Genet. 2018, 50, 1219–1224. [Google Scholar] [CrossRef] [PubMed]
- Polfus, L.M.; Darst, B.F.; Highland, H.; Sheng, X.; Ng, M.C.Y.; Below, J.E.; Petty, L.; Bien, S.; Sim, X.; Wang, W.; et al. Genetic Discovery and Risk Characterization in Type 2 Diabetes across Diverse Populations. HGG Adv. 2021, 2, 100029. [Google Scholar] [CrossRef] [PubMed]
- Sinnott-Armstrong, N.; Tanigawa, Y.; Amar, D.; Mars, N.; Benner, C.; Aguirre, M.; Venkataraman, G.R.; Wainberg, M.; Ollila, H.M.; Kiiskinen, T.; et al. Genetics of 35 Blood and Urine Biomarkers in the UK Biobank. Nat. Genet. 2021, 53, 185–194. [Google Scholar] [CrossRef]
- Wong, C.K.; Makalic, E.; Dite, G.S.; Whiting, L.; Murphy, N.M.; Hopper, J.L.; Allman, R. Polygenic Risk Scores for Cardiovascular Diseases and Type 2 Diabetes. PLoS ONE 2022, 17, e0278764. [Google Scholar] [CrossRef]
- Wedekind, L.E.; Mahajan, A.; Hsueh, W.-C.; Chen, P.; Olaiya, M.T.; Kobes, S.; Sinha, M.; Baier, L.J.; Knowler, W.C.; McCarthy, M.I.; et al. The Utility of a Type 2 Diabetes Polygenic Score in Addition to Clinical Variables for Prediction of Type 2 Diabetes Incidence in Birth, Youth and Adult Cohorts in an Indigenous Study Population. Diabetologia 2023, 66, 847–860. [Google Scholar] [CrossRef]
- Mansour Aly, D.; Dwivedi, O.P.; Prasad, R.B.; Käräjämäki, A.; Hjort, R.; Thangam, M.; Åkerlund, M.; Mahajan, A.; Udler, M.S.; Florez, J.C.; et al. Genome-Wide Association Analyses Highlight Etiological Differences Underlying Newly Defined Subtypes of Diabetes. Nat. Genet. 2021, 53, 1534–1542. [Google Scholar] [CrossRef]
- Tanigawa, Y.; Qian, J.; Venkataraman, G.; Justesen, J.M.; Li, R.; Tibshirani, R.; Hastie, T.; Rivas, M.A. Significant Sparse Polygenic Risk Scores across 813 Traits in UK Biobank. PLoS Genet. 2022, 18, e1010105. [Google Scholar] [CrossRef]
- Shoaib, M.; Ye, Q.; IglayReger, H.; Tan, M.H.; Boehnke, M.; Burant, C.F.; Soleimanpour, S.A.; Gagliano Taliun, S.A. Evaluation of Polygenic Risk Scores to Differentiate between Type 1 and Type 2 Diabetes. Genet. Epidemiol. 2023, 47, 303–313. [Google Scholar] [CrossRef]
- Vassy, J.L.; Hivert, M.-F.; Porneala, B.; Dauriz, M.; Florez, J.C.; Dupuis, J.; Siscovick, D.S.; Fornage, M.; Rasmussen-Torvik, L.J.; Bouchard, C.; et al. Polygenic Type 2 Diabetes Prediction at the Limit of Common Variant Detection. Diabetes 2014, 63, 2172–2182, Erratum in Diabetes 2018, 67, 1206. [Google Scholar] [CrossRef]
- Lamri, A.; Limbachia, J.; Schulze, K.; Desai, D.; Kelly, B.; de Souza, R.; Paré, G.; Lawlor, D.; Wright, J.; Anand, S.; et al. The Genetic Risk of Gestational Diabetes in South Asian Women. MedRxiv 2022. [Google Scholar] [CrossRef]
- Kim, Y.J.; Moon, S.; Hwang, M.Y.; Han, S.; Jang, H.-M.; Kong, J.; Shin, D.M.; Yoon, K.; Kim, S.M.; Lee, J.-E.; et al. The Con-tribution of Common and Rare Genetic Variants to Variation in Metabolic Traits in 288,137 East Asians. Nat. Commun. 2022, 13, 6642. [Google Scholar] [CrossRef]
- Pezzilli, S.; Tohidirad, M.; Biagini, T.; Scarale, M.G.; Alberico, F.; Mercuri, L.; Mannino, G.C.; Garofolo, M.; Filardi, T.; Tang, Y.; et al. Contribution of Rare Variants in Monogenic Diabetes-Genes to Early-Onset Type 2 Diabetes. Diabetes Metab. 2022, 48, 101353. [Google Scholar] [CrossRef]
- Qi, Q.; Stilp, A.M.; Sofer, T.; Moon, J.-Y.; Hidalgo, B.; Szpiro, A.A.; Wang, T.; Ng, M.C.Y.; Guo, X.; MEta-analysis of type 2 DIabetes in African Americans (MEDIA) Consortium; et al. Genetics of Type 2 Diabetes in U.S. Hispanic/Latino Individuals: Results from the Hispanic Community Health Study/Study of Latinos (HCHS/SOL). Diabetes 2017, 66, 1419–1425. [Google Scholar] [CrossRef] [PubMed]
- Siddiqui, M.K.; Anjana, R.M.; Dawed, A.Y.; Martoeau, C.; Srinivasan, S.; Saravanan, J.; Madanagopal, S.K.; Taylor, A.; Bell, S.; Veluchamy, A.; et al. Correction to: Young-Onset Diabetes in Asian Indians Is Associated with Lower Measured and Genetically Determined Beta Cell Function. Diabetologia 2022, 65, 1237. [Google Scholar] [CrossRef] [PubMed]
- Kerminen, S.; Martin, A.R.; Koskela, J.; Ruotsalainen, S.E.; Havulinna, A.S.; Surakka, I.; Palotie, A.; Perola, M.; Salomaa, V.; Daly, M.J.; et al. Geographic Variation and Bias in the Polygenic Scores of Complex Diseases and Traits in Finland. Am. J. Hum. Genet. 2019, 104, 1169–1181. [Google Scholar] [CrossRef] [PubMed]
- Naret, O.; Kutalik, Z.; Hodel, F.; Xu, Z.M.; Marques-Vidal, P.; Fellay, J. Improving Polygenic Prediction with Genetically Inferred Ancestry. Hum. Genet. Genom. Adv. 2022, 3, 100109. [Google Scholar] [CrossRef]
- Koch, S.; Schmidtke, J.; Krawczak, M.; Caliebe, A. Clinical Utility of Polygenic Risk Scores: A Critical 2023 Appraisal. J. Community Genet. 2023, 14, 471–487. [Google Scholar] [CrossRef]
- Martin, A.R.; Kanai, M.; Kamatani, Y.; Okada, Y.; Neale, B.M.; Daly, M.J. Clinical Use of Current Polygenic Risk Scores May Exacerbate Health Disparities. Nat. Genet. 2019, 51, 584–591. [Google Scholar] [CrossRef]
- Choi, S.W.; García-González, J.; Ruan, Y.; Wu, H.M.; Porras, C.; Johnson, J.; Bipolar Disorder Working group of the Psychiatric Genomics Consortium; Hoggart, C.J.; O’Reilly, P.F. PRSet: Pathway-Based Polygenic Risk Score Analyses and Software. PLoS Genet. 2023, 19, e1010624. [Google Scholar] [CrossRef]
- Taliun, D.; Harris, D.N.; Kessler, M.D.; Carlson, J.; Szpiech, Z.A.; Torres, R.; Taliun, S.A.G.; Corvelo, A.; Gogarten, S.M.; Kang, H.M.; et al. Sequencing of 53,831 Diverse Genomes from the NHLBI TOPMed Program. Nature 2021, 590, 290–299. [Google Scholar] [CrossRef]
- Choi, S.W.; Mak, T.S.-H.; O’Reilly, P.F. Tutorial: A Guide to Performing Polygenic Risk Score Analyses. Nat. Protoc. 2020, 15, 2759–2772. [Google Scholar] [CrossRef]
- Collister, J.A.; Liu, X.; Clifton, L. Calculating Polygenic Risk Scores (PRS) in UK Biobank: A Practical Guide for Epidemiologists. Front. Genet. 2022, 13, 1086709. [Google Scholar] [CrossRef]
- Purcell, S.; Neale, B.; Todd-Brown, K.; Thomas, L.; Ferreira, M.A.R.; Bender, D.; Maller, J.; Sklar, P.; de Bakker, P.I.W.; Daly, M.J.; et al. PLINK: A Tool Set for Whole-Genome Association and Population-Based Linkage Analyses. AJHG 2007, 81, 559–575. [Google Scholar] [CrossRef] [PubMed]
- Danecek, P.; Bonfield, J.K.; Liddle, J.; Marshall, J.; Ohan, V.; Pollard, M.O.; Whitwham, A.; Keane, T.; McCarthy, S.A.; Davies, R.M.; et al. Twelve Years of SAMtools and BCFtools. GigaScience 2021, 10, giab008. [Google Scholar] [CrossRef]
- Durbin, R.M.; Altshuler, D.; Durbin, R.M.; Abecasis, G.R.; Bentley, D.R.; Chakravarti, A.; Clark, A.G.; Collins, F.S.; De La Vega, F.M.; Donnelly, P.; et al. A Map of Human Genome Variation from Population-Scale Sequencing. Nature 2010, 467, 1061–1073. [Google Scholar] [CrossRef]
- Koenig, Z.; Yohannes, M.T.; Nkambule, L.L.; Goodrich, J.K.; Kim, H.A.; Zhao, X.; Wilson, M.W.; Tiao, G.; Hao, S.P.; Sahakian, N.; et al. A Harmonized Public Resource of Deeply Sequenced Diverse Human Genomes. bioRxiv 2023. [Google Scholar] [CrossRef]
- R Core Team. R: A Language and Environment for Statistical Computing; R Foundation for Statistical Computing: Vienna, Austria, 2020. [Google Scholar]
- Robin, X.; Turck, N.; Tiberti, N.; Lisacek, F.; Sanchez, J.-C.; Müller, M.; Siegert, S.; Doering, M.; Billings, Z. pROC: Display and Analyze ROC Curves; R Package Version 1.18.5. 2023. Available online: https://cran.r-project.org/web/packages/pROC/index.html (accessed on 19 September 2023).
- Revelle, W. Psych: Procedures for Psychological, Psychometric, and Personality Research; R Package Version 2.3.9. 2023. Available online: https://CRAN.R-project.org/package=psych (accessed on 19 September 2023).
- Chongsuvivatwong, V. Epidemiological Data Display Package; R Package Version 3.5.0.2. 2022. Available online: https://CRAN.R-project.org/package=epiDisplay (accessed on 19 September 2023).
- Fraley, C.; Raftery, A.E.; Scrucca, L.; Murphy, T.B.; Fop, M. Mclust: Gaussian Mixture Modelling for Model-Based Clustering, Classification, and Density Estimation; R Package Version 6.0.1. 2023. Available online: https://cran.r-project.org/web/packages/mclust/index.html (accessed on 19 September 2023).
- Nakazawa, M. Fmsb: Functions for Medical Statistics Book with Some Demographic Data; R Package Version 0.7.5. 2023. Available online: https://cran.r-project.org/web/packages/fmsb/index.html (accessed on 19 September 2023).
- Ruan, Y.; Lin, Y.-F.; Feng, Y.-C.A.; Chen, C.-Y.; Lam, M.; Guo, Z.; Ahn, Y.M.; Akiyama, K.; Arai, M.; Baek, J.H.; et al. Improving polygenic prediction in ancestrally diverse populations. Nat. Genet. 2022, 54, 573–580. [Google Scholar] [CrossRef]
- Mak, T.S.H.; Porsch, R.M.; Choi, S.W.; Zhou, X.; Sham, P.C. Polygenic scores via penalized regression on summary statistics. Genet. Epidemiol. 2017, 41, 469–480. [Google Scholar] [CrossRef] [PubMed]
- Loh, P.-R.; Tucker, G.; Bulik-Sullivan, B.K.; Vilhjalmsson, B.J.; Finucane, H.K.; Salem, R.M.; Chasman, D.I.; Ridker, P.M.; Neale, B.M.; Berger, B.; et al. Efficient Bayesian mixed model analysis increases association power in large cohorts. Nat. Genet. 2015, 47, 284–290. [Google Scholar] [CrossRef] [PubMed]
- Zheng, Z.; Liu, S.; Sidorenko, J.; Yengo, L.; Turley, P.; Ani, A.; Wang, R.; Nolt, I.; Snieder, H.; Lifelines Cohort Study Yang, J.; et al. Leveraging functional genomic annotations and genome coverage to improve polygenic prediction of complex traits within and between ancestries. bioRxiv 2022. [Google Scholar] [CrossRef]


{kind=link}
{kind=link}
{kind=link}
| PGS Author | GWAS Weight Source Study | PGS Development Method * | PGS Variants (% Match) | AUROC [95% CI] | OR/SD [95% CI] |
|---|---|---|---|---|---|
| PGS002771 Mars et al., 2022b [15] | GCST009379 | PRS-CS | 1,091,608 (97.7) | 0.669 [0.651, 0.686] | 1.883 [1.748, 2.028] |
| PGS002308 Ge et al., 2022 [16] | GCST009379 | PRS-CSx | 1,259,754 (86.9) | 0.665 [0.648, 0.683] | 1.883 [1.747, 2.031] |
| PGS003443 Huerta-Chagoya et al., 2023 [17] | GCST009379 | PRS-CSx | 1,092,496 (98.5) | 0.665 [0.648, 0.683] | 1.899 [1.759, 2.049] |
| PGS003103 Ma et al., 2022 [18] | GCST006867 | PRS-CS | 945,820 (98.9) | 0.651 [0.633, 0.668] | 1.795 [1.663, 1.937] |
| PGS003118 Ma et al., 2022 [18] | GCST006867 | PRS-CS | 945,921 (98.9) | 0.649 [0.631, 0.666] | 1.783 [1.652, 1.923] |
| PGS000330 Mars et al., 2020 [19] | GCST009379 | LDpred | 6,437,380 (87.2) | 0.643 [0.625, 0.661] | 1.743 [1.616, 1.88] |
| PGS003099 Ma et al., 2022 [18] | GCST006867 | lassosum | 555,512 (83.4) | 0.636 [0.618, 0.654] | 1.724 [1.594, 1.864] |
| PGS002354 Weissbrod et al., 2022 [4] | Weissbrod et al., 2022 [4] | BOLT-LMM | 1,109,311 (97.4) | 0.633 [0.615, 0.651] | 1.636 [1.523, 1.757] |
| PGS000729 Ritchie et al., 2021 [20] | GCST007517 | LD thinning | 2,017,388 (80.9) | 0.633 [0.615, 0.651] | 1.611 [1.502, 1.728] |
| PGS002720 Weissbrod et al., 2022 [4] | Weissbrod et al., 2022 [4] | SBayesR | 911,809 (98) | 0.631 [0.613, 0.649] | 1.618 [1.508, 1.736] |
| Cluster 3 | Cluster 2 | |||||
|---|---|---|---|---|---|---|
| Control. | T2D | All | Control | T2D | All | |
| (N = 1317) | (N = 617) | (N = 1934) | (N = 705) | (N = 706) | (N = 1411) | |
| Sex | ||||||
| Female | 808 (61.4%) | 388 (62.9%) | 1196 (61.8%) | 444 (63.0%) | 450 (63.7%) | 894 (63.4%) |
| Male | 469 (35.6%) | 217 (35.2%) | 686 (35.5%) | 237 (33.6%) | 249 (35.3%) | 486 (34.4%) |
| Missing | 40 (3.0%) | 12 (1.9%) | 52 (2.7%) | 24 (3.4%) | 7 (1.0%) | 31 (2.2%) |
| Age | ||||||
| Mean (SD) | 46.9 (14.3) | 56.6 (13.6) | 50.0 (14.8) | 47.3 (14.8) | 58.8 (12.4) | 53.2 (14.8) |
| Median [Min, Max] | 47.0 [5.00, 94.0] | 58.0 [18.0, 85.0] | 50.0 [5.00, 94.0] | 48.0 [9.00, 88.0] | 60.0 [19.0, 89.0] | 55.0 [9.00, 89.0] |
| Missing | 40 (3.0%) | 12 (1.9%) | 52 (2.7%) | 28 (4.0%) | 7 (1.0%) | 35 (2.5%) |
| BMI | ||||||
| Mean (SD) | 26.6 (5.38) | 32.5 (6.72) | 28.5 (6.47) | 27.3 (7.78) | 32.3 (6.36) | 29.8 (7.52) |
| Median [Min, Max] | 25.7 [11.3, 51.0] | 32.3 [18.2, 56.4] | 27.5 [11.3, 56.4] | 26.6 [14.7, 163] | 31.8 [18.7, 61.3] | 29.1 [14.7, 163] |
| Missing | 57 (4.3%) | 19 (3.1%) | 76 (3.9%) | 31 (4.4%) | 15 (2.1%) | 46 (3.3%) |
| Ethnicity | ||||||
| Belarussian | 5 (0.4%) | 5 (0.8%) | 10 (0.5%) | 43 (6.1%) | 55 (7.8%) | 98 (6.9%) |
| Latvian | 1213 (92.1%) | 528 (85.6%) | 1741 (90.0%) | 233 (33.0%) | 100 (14.2%) | 333 (23.6%) |
| Lithuanian | 11 (0.8%) | 7 (1.1%) | 18 (0.9%) | 14 (2.0%) | 10 (1.4%) | 24 (1.7%) |
| Other Missing | 53 (4.0%) | 29 (4.7%) | 82 (4.2%) | 47 (6.7%) | 48 (6.8%) | 95 (6.7%) |
| Polish | 3 (0.2%) | 4 (0.6%) | 7 (0.4%) | 25 (3.5%) | 33 (4.7%) | 58 (4.1%) |
| Russian | 29 (2.2%) | 42 (6.8%) | 71 (3.7%) | 321 (45.5%) | 417 (59.1%) | 738 (52.3%) |
| Ukrainian | 3 (0.2%) | 2 (0.3%) | 5 (0.3%) | 22 (3.1%) | 43 (6.1%) | 65 (4.6%) |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Brīvība, M.; Atava, I.; Pečulis, R.; Elbere, I.; Ansone, L.; Rozenberga, M.; Silamiķelis, I.; Kloviņš, J. Evaluating the Efficacy of Type 2 Diabetes Polygenic Risk Scores in an Independent European Population. Int. J. Mol. Sci. 2024, 25, 1151. https://doi.org/10.3390/ijms25021151
Brīvība M, Atava I, Pečulis R, Elbere I, Ansone L, Rozenberga M, Silamiķelis I, Kloviņš J. Evaluating the Efficacy of Type 2 Diabetes Polygenic Risk Scores in an Independent European Population. International Journal of Molecular Sciences. 2024; 25(2):1151. https://doi.org/10.3390/ijms25021151
Chicago/Turabian StyleBrīvība, Monta, Ivanna Atava, Raitis Pečulis, Ilze Elbere, Laura Ansone, Maija Rozenberga, Ivars Silamiķelis, and Jānis Kloviņš. 2024. "Evaluating the Efficacy of Type 2 Diabetes Polygenic Risk Scores in an Independent European Population" International Journal of Molecular Sciences 25, no. 2: 1151. https://doi.org/10.3390/ijms25021151
APA StyleBrīvība, M., Atava, I., Pečulis, R., Elbere, I., Ansone, L., Rozenberga, M., Silamiķelis, I., & Kloviņš, J. (2024). Evaluating the Efficacy of Type 2 Diabetes Polygenic Risk Scores in an Independent European Population. International Journal of Molecular Sciences, 25(2), 1151. https://doi.org/10.3390/ijms25021151