Abstract
Blast-induced neurotrauma has received much attention over the past decade. Vascular injury occurs early following blast exposure. Indeed, in animal models that approximate human mild traumatic brain injury or subclinical blast exposure, vascular pathology can occur in the presence of a normal neuropil, suggesting that the vasculature is particularly vulnerable. Brain endothelial cells and their supporting glial and neuronal elements constitute a neurovascular unit (NVU). Blast injury disrupts gliovascular and neurovascular connections in addition to damaging endothelial cells, basal laminae, smooth muscle cells, and pericytes as well as causing extracellular matrix reorganization. Perivascular pathology becomes associated with phospho-tau accumulation and chronic perivascular inflammation. Disruption of the NVU should impact activity-dependent regulation of cerebral blood flow, blood–brain barrier permeability, and glymphatic flow. Here, we review work in an animal model of low-level blast injury that we have been studying for over a decade. We review work supporting the NVU as a locus of low-level blast injury. We integrate our findings with those from other laboratories studying similar models that collectively suggest that damage to astrocytes and other perivascular cells as well as chronic immune activation play a role in the persistent neurobehavioral changes that follow blast injury.
Keywords:
animal models; astrocytes; blast; inflammation; traumatic brain injury; vascular pathology 1. Blast-Induced Neurotrauma in the Military
Public awareness of traumatic brain injury (TBI) in the military greatly increased over the past decade because of the conflicts in Iraq and Afghanistan [1] where estimates are that 10–20% of returning veterans suffered a TBI [2,3]. While military-related TBIs occur for various reasons, certain types of TBIs are relatively unique to military settings, the most prominent being blast-induced neurotrauma (BINT). For service members in Iraq and Afghanistan, exposure to improvised explosive devices (IEDs) caused most TBIs [2,3,4,5].
Initially, focus in the most recent conflicts was on the moderate-to-severe end of the TBI spectrum [6], the type of injury that would be recognized in-theater, and the conflict in Iraq led to the highest number of service-related severe TBIs since the Vietnam era [7]. However, as hostilities continued, what became apparent was that many returning veterans were appearing at Department of Veterans Affairs (VA) medical facilities with symptoms suggesting residual effects of mild TBIs (mTBIs) never recognized during service. Indeed, mTBIs greatly outnumbered moderate-to-severe TBIs during these conflicts [2,3].
In addition to in-theatre exposures, there is increasing concern regarding the effects of subclinical blast exposure [8,9,10]. This type of exposure, now being referred to as military occupational blast exposure, is common for many service members during training and military operations [8]. Whether this type of repetitive, low-level blast exposure causes health problems later in life is unclear [8,11], but recent studies suggest that cumulative low-level blast exposure over a service member’s career is associated with chronically worse brain health, more post-traumatic stress disorder (PTSD)-related symptoms, and a heightened risk for developing symptoms after a later blast injury [12,13,14,15]. Studies in breachers who routinely use explosives in their operations to, e.g., gain entry into buildings, further show chronic structural and functional brain imaging changes as well as altered blood biomarkers suggestive of brain injury [16]. Recent news media reports have brought additional attention to this topic [17].
A history of TBI is frequent among veterans seeking treatment at VA mental health facilities [18]. TBI has been linked to numerous mental health problems including depression, anxiety, poor impulse control, sleep disorders, and suicide [19,20]. A striking feature in the most recent veterans has been the frequent association of blast-related TBI with PTSD [1,21]. The association of BINT and mental health problems is not new and has historical roots dating back to the entity known as shell shock first recognized during World War I [22].
How much of the association of blast-related TBI with mental health problems is driven by blast-related physical trauma vs. psychologically based trauma needs more study [1,21]. However, PTSD and depression are the most prominent drivers of the worsening functional status seen in many veterans who suffered concussive blast injuries in Afghanistan [23]. Supporting a role for blast-related mechanisms, rats exposed to repetitive, low-level blast exposure develop a variety of chronic PTSD-related behavioral traits that are present more than one year after injury [24,25,26,27,28,29].
Over the last 15 years, interest in how blast exposure affects the nervous system has led to a rapid expansion of clinical as well as animal studies [21,30,31]. Here, we review findings from a model of low-level blast exposure in rats that we have been studying for more than 10 years that exhibits an early and selective vascular injury [24,25,26,27,28,29,32,33,34,35,36,37,38,39,40,41,42]. We focus on the neurovascular unit (NVU) as a locus of initial BINT injury and how changes in the perivascular environment might lead to the chronic neurobehavioral disturbances seen in this model in the absence of initial direct neuronal injury. We integrate findings from our own studies with those from others who have been studying the effects of low-level BINT.
2. A Rat Model of Low-Level Blast Injury with Chronic PTSD-Related Behavioral Traits
In 2012, Ahlers et al. [43] described an animal model of BINT developed to mimic a level of blast exposure that would be associated with human mild TBI or subclinical exposure. Initial studies established that exposures up to 74.5 kPa (equivalent to 10.8 psi) caused no post-exposure apnea or mortality [43]. While representing a level of blast transmitted to brain [44], these exposures produced only mild transient behavioral disturbances and no widespread brain histopathology [24,43]. Examination of the lungs showed no hemorrhagic petechiae or other pathology often caused by blast exposure [43]. By contrast, higher exposures (120 kPa, 17.4 psi) led to frank subdural and intraparenchymal hemorrhages and visibly evident histopathology in brain along with pulmonary hemorrhages, effects more consistent with moderate-to-severe BINT in the setting of polytrauma [43].
For most studies in our subsequent work, blast exposures were delivered to male rats at 10 weeks of age, with rats lying prone in line with the long axis of the shock tube and the head closest to the blast tube driver which generates the shock wave. Head motion was restricted during exposure to minimize damage from rotational/acceleration injury, which is in keeping with the limited head movements expected of humans exposed to comparable blast forces [45,46]. To mimic the multiple blast exposures commonly experienced by service members in Iraq and Afghanistan [47], for most studies described in this report, rats were subjected to 74.5 kPa exposures (impulse 175.8 kPa * ms; duration 4.8 ms) [43] delivered once per day for 3 consecutive days (3 × 74.5 kPa).
Rats subjected to 3 × 74.5 kPa repetitive low-level blast exposure develop cognitive and PTSD-related behavioral traits including anxiety, enhanced acoustic startle, impaired recognition memory, and exaggerated fear learning [24,25,26,27]. These traits develop in a delayed manner, being absent in the first eight weeks after blast exposure but consistently present three to four months and longer after exposure. Once established, traits remain present for more than one year after blast exposure [24,25,26,27] and are likely present for the lifetime of the animal. These animals thus model the chronic neurobehavioral syndromes that veterans often suffer following BINT [23,48,49].
3. The Neurovascular Unit
The brain receives the largest blood supply of any organ [50]. However, unlike other organs, CNS blood supply is heavily filtered with substances that enter or leave the CNS regulated [51,52]. Regulation is performed at the level of the blood–brain barrier (BBB) in the context of a complex NVU whose cellular elements include an intraluminal glycocalyx, endothelial cells, perivascular astrocytes, microglia, other mural cells including pericytes, and smooth muscle cells as well as perivascular nerves [51,52]. An extracellular matrix is associated with these structural elements that gives rise to inner and outer basement membranes or basal laminae. An inner basement membrane surrounds the endothelial cells, separating them from the most closely associated cells, the pericytes, while an outer basement membrane lies between the pericytes and the astrocytic endfeet [53,54]. Within the blood vessel lumen exists a complex glycocalyx that additionally regulates blood vessel properties [55].
The NVU regulates cerebral blood flow as well as transport of glucose and other metabolites into the CNS [52,56]. It also controls the movement of solutes and clears metabolic waste out of the CNS [52,56]. Collectively, the NVU is critical for homeostatic maintenance and protection of the CNS from toxins or other biologically active molecules, some of which, while functionally beneficial in the periphery, may disrupt CNS homeostasis.
For blood-borne substances to enter the CNS, they must first transit vascular endothelial cells and their associated basement membranes [56]. The CNS vascular endothelium differs from that found in most peripheral organs by the absence of fenestrations and the presence of tight junctions between endothelial cells, which limit paracellular passage of water-soluble molecules that would otherwise enter most peripheral organs [57]. This matrix of neurovascular endothelial cells, closely knit together by their tight junctions, comprises the main elements of the blood–brain barrier (BBB) that governs what can and cannot cross into the brain [58]. CNS endothelial cells also exhibit less pinocytotic activity and the passage of many substances occurs only through active transport mechanisms specific to individual substrates [57]. The basement membranes, which help to maintain the proper association of elements within the NVU, also serve as a reservoir for growth factors and adhesion proteins that regulate BBB permeability [57,59].
Astrocytes interact with blood vessels through their endfeet, where they directly contact endothelial cells and mural cells [60]. Most of the brain capillary network is generated postnatally [61], at the same time that the astrocytic endfeet start to engage the vasculature [62]. Astrocytic endfeet cover nearly the entire surface of the brain’s blood vessels and contain specialized organelles and local protein translation machinery. Astrocytic endfeet have their own unique molecular composition, including scaffold proteins, anchor channels, transporters, receptors, and enzymes that regulate how astrocytes interact with blood vessels [60]. Astrocytic endfeet are thought to help regulate BBB function and govern regional cerebral blood flow to match metabolic supply and demand via microvascular dilation and constriction [60,63]. By their contribution to perivascular structure, astrocytes also regulate the glymphatic clearance system, which transports solutes out of the brain [64,65].
Pericytes are considered multi-functional cells embedded within the walls of capillaries and post-capillary venules [66]. Various functions have been attributed to them, including regulating cerebral blood flow and BBB integrity [66]. They have a role in vascular development and, in adults, behave like stem cells [66]. Pericytes also regulate neuroinflammatory states and immune cell entry into the CNS [66]. Microglia further contact endothelial cells, astrocytes, and neurons, where they regulate neuroinflammatory states as well as modify synaptic contacts [67,68,69].
Smooth muscle cells are the most abundant cell type found in the vascular wall [70]. Smooth muscle is found on all arteries, arterioles, and veins, but is absent from capillaries and small post-capillary venules. The lymphatics also have a smooth muscle layer. Vascular smooth muscle cells contract or relax to regulate intravascular blood volume and local blood pressure [70]. Smooth muscle contraction also regulates periarterial drainage of soluble metabolites [71].
Finally, neurons, through perivascular nerves, make contact with endothelial cells, astrocytes, and, likely, other mural cells, allowing neuronal activity to regulate vascular tone through effects on endothelial cells, smooth muscle, and astrocytes [52]. Thus, a variety of cellular elements within the NVU could be disturbed by blast exposure and contribute to brain dysfunction after BINT.
4. The Vasculature as a Locus of Injury in Low-Level BINT
Blast injury has long been known to affect the cerebral vasculature [31]. Acutely, higher-level blast exposures of 120 kPa or above show a prominent hemorrhagic component in species including rats, mice, rabbits, ferrets, and goats [43,72,73,74,75,76,77]. Varying degrees of subdural, subarachnoid, and intracerebral hemorrhage are visible grossly and microscopically along with edema and vascular congestion on the brain surface. Effects seem similar whether exposures are under shock tube conditions or exposure to live explosives, with a correlation between injury severity and blast overpressure levels or distance from the detonation of live explosives. While higher-level blast exposures have a prominent vascular component, their association with contusions and intraparenchymal and subdural hemorrhage as well as, likely, direct shock wave damage to neuronal and glial elements, make them a mixture of vascular and non-vascular injuries.
In the model that we have been studying, rats exposed to single or multiple (three) 74.5 kPa blast exposures develop a selective vascular pathology in the absence of a more generalized histopathology. General neuropathological screens, including histology (hematoxylin and eosin/Nissl) and a variety of immunostains, do not reveal any consistent histo- or immunopathology [24,32,42]. These exposures can produce some degree of hemorrhage in the choroid plexus and lateral ventricles. Also observed is a type of likely shear-related lesions, which typically follow the course of penetrating cortical vessels [42]. These lesions may be unique to BINT, but because of their focal nature and infrequency, their contribution to the overall pathological effects is not clear.
In multiple studies using electron microscopy (EM) [34,35,37,38,42], we have noted a generalized microvascular pathology in the setting of an otherwise normal neuropil (Figure 1). What is notable in Figure 1 is that despite the abnormalities in blast-exposed vessels, no abnormalities are present in the neighboring neuropil except at the very margins of severely affected blood vessels. While it is difficult to exclude that there may not be some subtle neuronal injury that is not apparent by standard histology or immunostaining, the striking difference between the vascular pathology and the surrounding neuropil suggests that the brunt of the physical blast injury is on the cerebral vasculature.
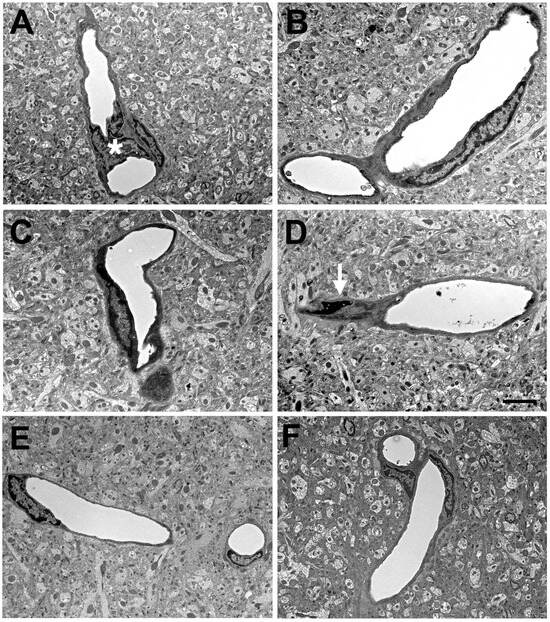
Figure 1.
Blast-induced degenerative changes in cerebral microvessels. In panels (A–D), cerebral microvessels are shown from an animal that received a single 74.5 kPa blast exposure and was sacrificed 24 h later. Panels (E,F) illustrate longitudinally cut cerebral microvessels from non-blast exposed sham controls. All microvessels in panels (A–D) have lost their luminal circularity and the walls are irregular. In panel (A), a dysmorphic endothelial cell nucleus (*) is seen in the vessel lumen. In panel (D), the nucleus of a perivascular cell (arrow) with degenerative changes is indicated. Despite the destruction of the microvessels, the surrounding neuropil appears intact. Scale bar: 1 µm. Figure is reproduced from Gama Sosa et al. [34].
5. Comparisons to Other Models of Low-Level BINT
To compare our results with others, one must first address what low-level BINT is in an animal model. As noted above, initial studies in our model established an overpressure exposure up to 74.5 kPa as compatible with a low-level BINT [21,43]. This consideration was based on these exposure conditions, producing a blast wave that is transmitted to the brain [44], but which does not produce major gross neuropathological or systemic effects nor long-lasting behavioral effects in the immediate recovery period [24,43]. The lack of evidence for coup/contrecoup injuries further supports the relatively mild nature of the brain injury [24,43]. Thus, we later suggested 74.5 kPa as a dividing line between a low-level blast compatible with a low-level BINT as might occur in mTBI or subclinical blast exposure as opposed to moderate-to-severe TBI associated with more widespread tissue damage including subdural and intraparenchymal hemorrhage [21]. Others who have examined this question have come to relatively similar conclusions concerning the magnitude of an overpressure wave consistent with low-level blast exposure [46].
The general requirements for BINT modeling have been discussed in other reviews [45,78,79,80]. Notably, factors other than maximal overpressure of the blast wave contribute to the tissue-damaging potential of a blast wave. The duration and shape of the waveform (i.e., the area under the curve referred to as the impulse) correlate better with tissue damage than raw overpressure. Indeed, pressures as high as 550 kPa generated by live explosives and delivered over brief 0.2 ms durations cause no visible neuronal, axonal, or vascular pathology, as judged by standard histological and immunohistochemical stains [81]. By contrast, 120 kPa exposures delivered in a pressure wave of several ms, which is on the order of the durations produced by most shock tubes, produce extensive tissue damage [43].
Other factors which affect the tissue-damaging potential of a shock wave are body shielding and head restraint. Indeed, it seems that the use of some form of body shielding or selective head exposure has become the norm in many laboratories doing blast research. Body shielding clearly affects CNS vascular injury and is discussed below in the section on direct endothelial injury. Head restraint also affects tissue injury in that the lack of muscle tone in an anesthetized rodent allows for head oscillations and accelerations, creating what has been referred to as a bobblehead effect. These oscillations create the potential for significant rotation/acceleration injuries independent of the effects of the primary blast wave, effects that have been clearly demonstrated by studies in mice [82]. Additionally, some groups expose only one side of the head, which likely increases the potential for a bobblehead effect. While rotation/acceleration injuries from head oscillations are part of most moderate-to-severe blast-related TBIs in humans, they are likely relatively minor factors in subclinical blast exposures and in most blast-related mTBIs.
Despite the rapid expansion of blast research over the last decade, low-level BINT is still relatively little studied. Table 1 summarizes rodent models of blast exposure that have utilized blast overpressures of 80 kPa or less. The studies are hard to compare directly as the use of body shielding and head restraint have been variable. Many studies have described prominent vascular pathology, although some have not. However, those that have not did not examine the vascular ultrastructure with EM. Other studies have described extra-vascular pathology, including neuronal pathology, but it is hard from the published descriptions to judge the relative prominence of vascular vs. non-vascular pathology.

Table 1.
Rodent models of blast exposure utilizing overpressures of 80 kPa or less.
In a mouse model, perhaps the most comparable to ours in rats, Gu and colleagues have studied open-field blasts in mice using a single live explosion [86,87,88,89]. Their protocol generates a 46.6 kPa peak overpressure with an impulse of 60 kPa * ms, and a primary wave duration of 3 ms, parameters somewhat lower than those used in our rat blast model. They report that single-blast injuries do not result in extensive tissue damage at the light microscopy level in brain, although they identified ultrastructural abnormalities in myelin sheaths, mitochondria, and synapses [87,88,89]. In their most recent study [86], multiple ultrastructural changes were described in the NVU, including changes in endothelial cells, pericytes, and basement membranes, as well as swelling of astrocytic endfeet, that seem similar to those seen in our rat model (described below). Clearly, future studies in multiple models seem warranted to clarify the relative selectivity of the vasculature to low-level BINT.
6. Is Direct Damage to Endothelial Cells by BINT a Thoracic Effect?
The endothelial cell is the initial contact point for substances entering the CNS. Direct endothelial cell damage could affect BBB properties as well as disrupt endothelial cell connections to mural cells, the vascular basement membrane, and perivascular astrocytes. Disruption by BINT could thus affect multiple elements of the NVU, contributing to the functional consequences discussed in other sections.
As illustrated in Figure 1, the endothelial cell is damaged by low-level BINT. A question of interest is how this damage occurs. The primary blast wave may affect the brain as it is transmitted through the tissue. BINT may also be associated with acceleration/rotation of the head, imparting mechanical energy to the brain that may cause injuries similar to those seen in non-blast TBI, although this is probably less of an issue with low-level BINT if the head is restrained. A third mechanism that has been suggested is that the blast wave striking the body causes indirect CNS injury through what has been referred to as a thoracic or systemic mechanism [95,96]. Current thinking on this last mechanism conceptualizes the kinetic energy of the shock wave as being transferred into hydraulic energy that is carried through the systemic vasculature causing rapid blood displacement into the lower-pressure intracranial compartment [96,97,98,99]. Such a transmission of energy would be predicted to preferentially damage cellular elements close to cerebral vessels.
The importance of this mechanism has been indirectly assessed by using selective brain exposure or body shielding. While some groups such as ours deliver whole-body exposures, many investigators routinely subject only the head to blast exposure or use various forms of shielding to protect the body. While there are few direct comparisons in the same laboratory, in reviewing studies across many labs, it seems clear that a given blast overpressure exposure produces less intracranial pathology if delivered as a head-only exposure compared to a whole-body exposure (reviewed in [31]). In addition, higher head-only exposures seem necessary to produce the same pathology as lower whole-body exposures [31]. In the few studies that compared head-only vs. whole-body blast exposures in the same experimental system [96,100,101], all found that pathology was significantly less with a head-only exposure.
In a study that addressed this mechanism perhaps the most directly, Simard et al. [102] delivered selective blast exposures to the thorax or jugular veins of rats. They showed acute perivenular inflammatory changes induced by thorax-only exposure that were prevented by ligating the jugular vein and reproduced by selective jugular vein exposure [102]. Taken along with the multiple other studies showing that shielding can reduce or eliminate BINT effects on the brain [96,100,101], the studies of Simard et al. [102] make a compelling case for direct endothelial cell and NVU damage by transmission of a pressure wave through the vasculature.
One study has tried to address the thoracic mechanism in humans [103]. In this study, special weapons and tactics (SWAT) personnel undergoing breacher training were fitted with a jugular vein compression collar. Interestingly, the collar mitigated fMRI (functional magnetic resonance imaging) changes on a working memory task performed during training [103]. More study of this phenomenon seems warranted.
7. Blast Damage to the Endothelial Glycocalyx
The glycocalyx is a carbohydrate-rich layer that coats the luminal surfaces of vascular endothelium [55,104]. It imparts a negative electric charge to endothelial cell surfaces, forming an electrical barrier. Endothelial glycocalyx differs in the CNS from many peripheral tissues in being found over the entire luminal surface of cerebral capillaries while it only partially covers capillaries in organs such as heart and lungs [55]. The glycocalyx in the CNS is also more resistant to stripping from lipopolysaccharide-induced vascular injury, suggesting that CNS glycocalyx has a denser structure [55]. The glycocalyx is thought to play roles in endothelial protection, regulating BBB properties, leukocyte adhesion to endothelial cells and nitric oxide production and as a layer of the vasculature responsible for sensing luminal shear forces [104,105,106,107,108]. Glycocalyx degradation causes BBB dysfunction [105]. The endothelial glycocalyx is thus thought crucial for maintaining brain homeostasis.
While we have not analyzed the glycocalyx in our model of 3 × 74.5 kPa blast exposure, Hall et al. [109,110] studied how repeated low-intensity blast affects the brain capillary glycocalyx using 12 × 40 kPa blast exposures conducted with a minimum of 24 h between blasts. They found that blast exposure reduced the glycocalyx length and density in multiple brain regions. A functional significance to glycocalyx damage was suggested by the observation that administering hyaluronidase, an enzyme that binds to and degrades hyaluronan (a major glycocalyx structural component), prior to blast exposure reduced deficits in water maze performance and induced a thickening of the glycocalyx.
Another study by Chen et al., (this Special Issue) explored the longitudinal effects of a single high-intensity blast (130 kPa, 18.9 PSI) exposure on degradation and recovery of the of glycocalyx, alterations in cerebral blood flow, and the responsiveness of cerebral blood flow to hypercapnia. A significant reduction in the density of the glycocalyx was observed 2–3 h, 1 day, and 3 days after blast exposure. Recovery of the glycocalyx was observed at 28 days post-blast along with an increase in the components of the glycocalyx (heparan sulphate at 14 and 28 days and syndecan-2 and chondroitin sulphate at 28 days post-blast) in the frontal cortex. A significant decrease in cerebral blood flow and an attenuated responsiveness of cerebral blood flow to hypercapnia was observed at 2–3 h, 1, 14, and 28 days after blast exposure.
These findings ([109], Chen et al. this Special Issue) cannot be directly extrapolated to rats that received 3 × 74.5 kPa exposures. However, the effects of altering the glycocalyx on cerebral blood flow and the finding that acute behavioral disturbances could be prevented by pretreatment with hyaluronidase suggests a functional role of glycocalyx disruption following blast injury. Further study seems warranted.
8. Damage to Smooth Muscle Layers by BINT
High-level blast exposure induces prominent vasospasm in humans [111] and animals [112] along with reduced cerebral perfusion and altered contractile properties of large arteries [113,114,115]. The vascular smooth muscle layer is critical in regulating the contractile properties of blood vessels [70] and also affects the periarterial drainage of soluble substances [71]. The vascular smooth muscle layer is affected by blast exposure acutely after injury in our model and remains so chronically. Figure 2 shows examples of disrupted smooth muscle layers soon after injury.
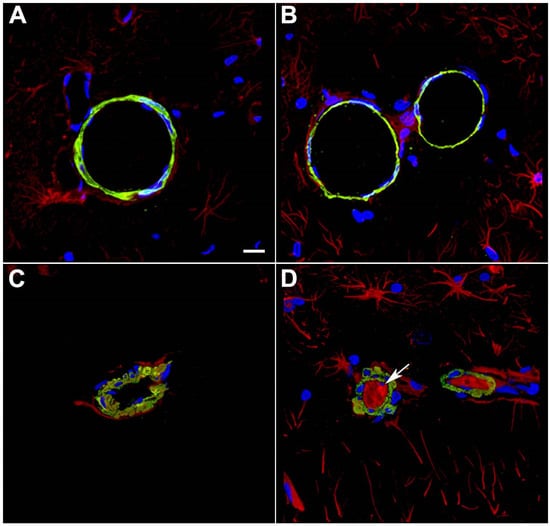
Figure 2.
Disrupted smooth muscle layers and intraluminal astrocytic processes in blood vessels 10 months after blast exposure. Sections from rats euthanized 10 months after the last blast exposure were immunostained for GFAP (red) and α-SMA (green) with a DAPI nuclear counterstain (blue). Panels (A–D) show images from the hippocampal stratum lacunosum moleculare from control (A,B) or blast-exposed (C,D) rats. Note the irregularity and vacuolation in the smooth muscle apparent with α-SMA staining in the blast-exposed animal. An arrow in (D) points to intraluminal accumulation of GFAP. Scale bar, 50 μm. Figure is reproduced from Gama Sosa et al. [35].
While we have not studied the effects of 3 × 74.5 kPa blast exposures on smooth muscle-related physiological functions, Abutarboush et. al. [116], in studying rats after a single 130 kPa (18.9 psi) exposure, found functional alterations in the pial microcirculation that evolved acutely from 2 h through 28 days after injury. They observed delayed and prolonged alterations in cerebrovascular reactivity along with changes in the microarchitecture of the vessel wall and perivascular astrocytes. Impaired arteriolar reactivity appeared only >24 h after blast exposure in the form of reduced hypercapnia-induced vasodilation, increased barium-induced constriction, and reversal of the serotonin effect from constriction to dilation. They also saw a reduction in vascular smooth muscle contractile proteins, as well as a delayed reduction in nitric oxide synthase. Collectively, these results suggest that blast exposure results in an evolving pattern of effects on vascular physiology that would be expected to alter long-term vascular reactivity in the NVU.
Why long-term vascular reactivity would be affected is not well understood, although Alford et al. [117,118] have suggested that blast-induced vasospasm may play a role by initiating a phenotypic switch in vascular smooth muscle cells that has long-term consequences. Using high-velocity stretching of engineered arterial lamellae, they found that one hour after a simulated blast, altered intracellular calcium dynamics were seen as well as hypersensitivity to a contractile stimulus with endothelin-1. Interestingly, Abutarboush et al. [116] reported increased endothelin-1B receptors in astrocytes in their in vivo studies.
Later, Alford et al. [117,118] found that one day after a simulated blast, tissues exhibited a prolonged hypercontraction and changes in vascular smooth muscle cells suggestive of a force-dependent phenotypic switch. They argued that blast-induced cerebral vasospasm causes a mechanically induced switch in vascular smooth muscle that potentiates vascular remodeling, leading to further vasospasm and lumen occlusion. Although this phenomenon has yet to be demonstrated in vivo, blast effects on large cerebral blood vessels acutely are well established at higher levels of blast exposure [112]. Whether similar effects occur under low-level blast conditions has yet to be established. Another mechanism would need to be invoked to explain blast effects on capillaries and post-capillary venules where a smooth muscle layer is lacking.
9. Low-Level Blast Induces a Gliovascular and Neurovascular Disconnection
In a study aimed at uncovering molecular alterations in the vasculature following BINT, we studied purified brain vascular fractions from blast-exposed animals 6 weeks after exposure using two-dimensional differential gel electrophoresis (2D DIGE) and matrix-assisted laser desorption/ionization (MALDI) mass spectrometry (MS) [35]. We were somewhat surprised to find that the protein showing the most altered expression was glial fibrillary acidic protein (GFAP), which was decreased in blast-exposed animals. Western blotting confirmed that GFAP expression was decreased about threefold in isolated brain vascular fractions from blast-exposed rats (Figure 3A).
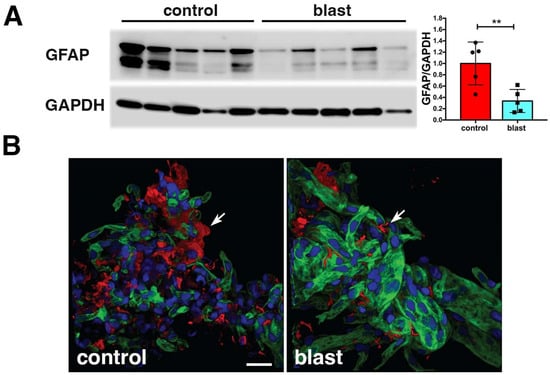
Figure 3.
Reduced GFAP and fewer astroglial attachments in isolated brain vascular fractions following BINT. Panel (A) shows immunoblotting for GFAP (top panel) from five control and five blast-exposed brain-derived vascular fractions. Quantification of the blot with expression normalized to glyceraldehyde 3-phosphate dehydrogenase (GAPDH, lower panel) is shown on the right. All lanes were loaded with 10 μg of protein and contained protein from individual animals. Error bars indicate the SEM (** p < 0.01, unpaired t-test). Panel (B) shows isolated vascular fractions from control and blast-exposed brains immunostained for GFAP (red) and laminin (green) and counterstained with DAPI (blue). Arrows indicate GFAP-immunostained attachments to the isolated vasculature. Note reduced GFAP-labeled attachments in the blast-exposed vessels. All animals were euthanized 6 weeks after blast exposure. Scale bar, 25 μm. Panels (A,B) are from Gama Sosa et al. [35].
GFAP’s presence in isolated vascular fractions, while initially puzzling, seemed to suggest that astrocytic endfeet remain attached to blood vessel walls, with reduced GFAP in blast-exposed samples reflecting a loss of attachments. Indeed, others had previously observed astrocytic endfeet attached to purified mouse brain blood vessels [119,120,121]. Figure 3B shows isolated vascular fractions from control and blast-exposed rats immunostained for GFAP. In controls, GFAP staining was seen in patches, consistent with attached astrocytic endfeet. In blast-exposed rats, perivascular GFAP staining also occurred in patches but was noticeably reduced compared to controls, consistent with astrocytic endfeet being lost. These changes being apparent in Western blots of vascular fractions obtained from whole-brain preparations suggests the widespread nature of the changes.
EM studies showed frequently swollen astrocytic endfeet after blast exposure [35]. Figure 4 shows examples of small arterioles from control and blast-exposed rats at six weeks following blast exposure. Compared to controls, the lumens of blast-exposed vessels appear irregular and thickened. In addition, compared to the tight astrocytic endfeet surrounding the control vessels, perivascular astrocytic endfeet in the blast-exposed vessels are swollen and contain degenerating organelles. Blast-exposed capillaries showed similar changes. Astrocytic changes were more obvious in blood vessels showing the most endothelial cell damage. Although normal-appearing microvessels were apparent in blast-exposed specimens, counts of randomly sampled vessels in the blast-exposed revealed that 18% had clearly swollen astrocytic endfeet [35]. Such changes were not observed in the microvessels of controls.
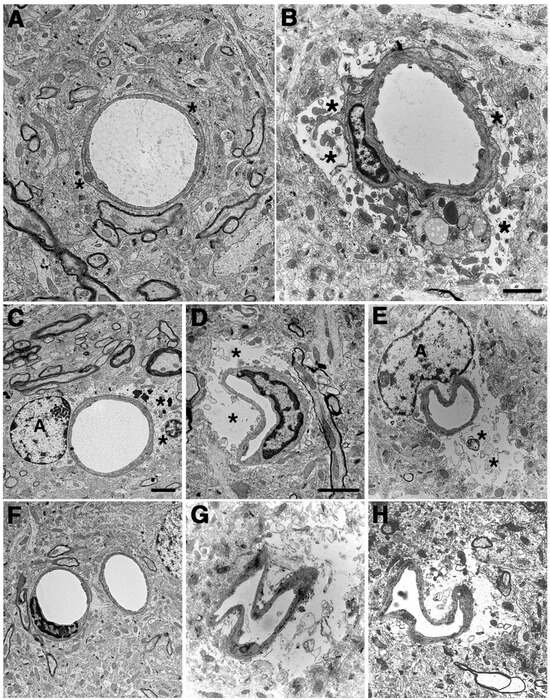
Figure 4.
Blast-induced swelling and degeneration of astrocytic endfeet. Electron micrographs are shown from sections of the frontal motor cortex of control (A,C,F) and blast-exposed (B,D,E,G,H) rats. Animals were euthanized 6 weeks following 3 × 74.5 kPa blast exposures. Asterisks (*) indicate astrocytic endfeet, which are swollen and contain degenerating organelles in all blast-exposed microvessels. The lumens of microvessels in blast-exposed animals also appear irregular and collapsed. Perivascular astrocytes (labeled A) are visible in panels (C,E). Scale bars, 2 μm. Scale bar in panel (B) applies to panels (A,B). Scale bar in panel (C) applies to panels (C–H). Scale bar in panel (D) applies only to panel (D). Figure is reproduced from Gama Sosa et al. [35].
Equally surprising, in addition to GFAP, of the first nine proteins identified with MS, four more were intermediate filament proteins (IFs), but rather than being glial or endothelial, they were neuronal-associated IFs (α-internexin and the light, medium, and heavy chain neurofilaments) [35]. All were decreased in blast-exposed vessels based on 2D DIGE, observations that were confirmed with Western blotting [35]. In each case, as in the proteomic studies, the levels of neuronal IFs were decreased by about threefold in blast-exposed rats.
Neuronal IFs in isolated vascular fractions suggested that neuronal fibers remain tightly associated with purified brain vascular preparations. Others have also observed neuronal IFs in purified preparations of mouse brain vasculature [119,120]. Immunostaining of vascular fractions for heavy neurofilament (NFH) showed focal staining sometimes with fine processes apparent, consistent with neuronal attachments to blood vessels. Their decrease following blast injury suggests that, like gliovascular, neurovascular attachments are being disrupted as a consequence of blast injury.
Interestingly, vascular fractions from animals taken at 8 months post-blast exposure showed normal levels of GFAP with Western blotting suggesting perhaps some attempt at re-establishing astrocytic connections to blood vessels [35]. Yet, a chronic vascular pathology was still visible with EM. A phenomenon frequently observed, however, was GFAP-containing processes within the vascular lumen (Figure 2). We suspect that intraluminal GFAP appears when astrocytic processes regrow towards a damaged vessel but are not able to establish normal connections. Neuronal IFs also become normalized in vascular fractions isolated from rats at 8 months after blast exposure. Collectively, these observations suggest that normalization of GFAP and neuronal IF levels in isolated vascular fractions, while signaling a regrowth of astrocytic and neuronal processes, does not translate into a restoration of a normal vasculature structure [35].
Loss of GFAP in isolated vascular fractions seems to be a general phenomenon in rodents and is not limited to blast-exposed rats. Similar to blast-exposed rats, we have found that GFAP was decreased by about twofold in isolated vascular fractions from blast-exposed mice harvested at four weeks after a repetitive blast exposure protocol [122] (unpublished observations). Thus, blast exposure induces a gliovascular disconnection in rats and mice.
Lost gliovascular connections also appear to occur acutely after injury as Hubbard et al. [92] have shown in male and female rats exposed to single 75.8 kPa (11 psi) peak overpressure exposures. They were further able to show that astrocytic endfoot coverage around blood vessels was decreased in males. Interestingly, by 7 days, GFAP levels had recovered, suggesting that damage following single exposures may recover while multiple repetitive exposures, delivered over-short intervals, as in our model, lead to damage that does not recover [92].
Several studies from other labs have described ultrastructural changes in astrocytic endfeet following BINT [82,123,124]. In a rat model of blast injury that used live explosives (110 kg TNT equivalent), Kaur et al. [123] observed astrocytes in the cerebral and cerebellar cortex with enlarged and swollen endfeet. Lu et al. [124] described enlarged and “watery” astrocytic endfeet in a non-human primate model also utilizing live explosives reported as 80 kPa exposures. These changes, however, occurred in the context of a broader neuronal, glial, and microglial pathology that was absent in our model. Another study described swollen astrocytic endfeet in a mouse model of 77 kPa blast injury, although pathology in this model, which did not restrain the head, occurred in the setting of a broader neuropathology with neuronal tau pathology, axonal changes, and widespread astrogliosis [82].
Supporting the relevance of the animal studies to humans, serum GFAP levels have been found elevated acutely in military personnel after heavy weapons training [125]. Levels remained elevated for at least 3 months in this study [125], although in another study serum, GFAP levels were transiently reduced in military personnel on days 6 and 7 of a 2-week training protocol that involved exposure to moderate blasts [9]. In this second study, GFAP levels negatively correlated with cumulative blasts experienced during training and with duration of military service [9]. Shively et al. [126] have also described a distinctive pattern of interface astroglial scarring at the grey/white matter junctions that may be unique to BINT. The reasons for the variations in time course and the relationship of any of these changes to vascular injury is unclear.
10. Functional Consequences of Lost Gliovascular and Neurovascular Connections on Blood–Brain Barrier Function and Neurovascular Coupling
Perivascular astrocytes play many roles which, by their loss or disruption, could influence the effects of BINT. One of the most important functions of astrocytes is the formation and maintenance of the BBB, a role that may significantly affect the uptake of nutrients, including glucose, hormones, lipids, and amino acids. Astrocytes support BBB integrity by promoting the maintenance of tight junctions between the endothelial cells of blood vessels [60,61,62].
Studies in the 1980s found that in coculture experiments, astrocytes induced BBB properties in brain endothelial cells [60]. Furthermore, when glial progenitors from the CNS were transplanted into peripheral tissues, CNS progenitors induced BBB properties in peripheral vessels that included tight junctions between peripheral endothelial cells which prevented tracer leakage as in the CNS [127]. It would be later shown that an astrocyte-conditioned medium could induce BBB features in endothelial cell cultures, arguing that secreted factors drive BBB formation and maintenance [60]. Astrocytes promote the expression of a number of junctional proteins, enzymatic pathways, and specific transporters, including the glucose transporter-1 (GLUT-1) [128,129,130,131].
Recent studies have attempted to directly assess the role of perivascular astrocytes using laser ablation [60]. Simultaneously ablating adjacent astrocytes caused discontinuous endothelial tight junctions and irregular basement membranes [132]. Astrocyte ablation in the spinal cord reduced expression of the tight-junction protein occludin [133]. However, in these studies, substantial functional BBB damage was not observed based on erythrocyte or fibrinogen extravasation [133,134].
Live in vivo imaging during and after the ablation of single astrocytes or endfeet has shown that these structures have considerable plasticity, with neighboring astrocytes quickly recovering vessel surfaces [60,135,136]. Furthermore, without astrocytic endfoot coverage, vessels showed no leakage of plasma marker dyes such as Evans Blue or dextran-conjugated fluorescein, while when the vascular wall was ablated, Evans Blue extravasation was seen. These studies question how much blast-induced disruption of astrocytic endfeet might be expected to affect BBB integrity, although failure to affect permeability to dyes such as Evans Blue or fluorescein-conjugated dextrans does not preclude that specific transport systems that move smaller solutes including ions, lipids, glucose, and small proteins may be affected.
Astrocytes sense neuronal activity and regulate vascular tone in the context of neurovascular coupling [52]. Glutamate released from neurons acts on astrocytic metabotropic glutamate receptors to evoke the Ca2+-dependent release of vasoactive metabolites from astrocytic endfeet. Under normoxic conditions, astrocytic Ca2+ signaling results in vasodilation, whereas under hyperoxia, vasoconstriction is favored [52].
Cerebral pial vessels are innervated by perivascular nerves from both autonomic and sensory ganglia of sympathetic, parasympathetic, and trigeminal origins [137]. Sympathetic terminals have significant vasoconstrictor effects in large cerebral arteries. Under conditions of elevated hydrostatic pressure, sympathetic vasoconstriction protects against increases in venous pressure, BBB disruption, and edema formation. Stimulation of parasympathetic nerves has potent vasodilator effects on cerebral arteries, resulting in increased blood flow. Thus, BINT-induced alterations in neurovascular connections could disrupt cerebral autoregulation at multiple levels.
11. BINT and Glymphatic Function
The brain produces substances that seem to be best considered as waste products. That material is primarily cleared along cerebrovascular routes, either by direct transport across the BBB into the vascular lumen or via flow along perivascular channels with drainage into the lymphatics [138]. The glymphatic system is a waste-clearance system of perivascular channels formed by astrocytes [65,139]. Cerebrospinal fluid pumped into the brain along penetrating arterioles mixes with interstitial fluid and waste products and is cleared out of the brain along venules [65]. In addition to waste products, the glymphatic system may also help transport non-waste substances including neurotransmitters, glucose, lipids, and amino acids. The glymphatic system has particularly gained attention for its role in transporting potentially neurotoxic waste products involved in neurodegenerative diseases, such as amyloid beta (Aβ) and tau proteins [140,141,142].
An alternate intramural periarterial drainage system is also believed to exist that moves interstitial fluid containing waste products toward the pial surface along the basement membranes of arterioles in the opposite direction of blood flow [71]. Astrocyte endfeet are proposed to drive intramural periarterial drainage by regulating functional hyperemia and cerebrovascular tone [143]. Thus, astrocytes could regulate perivascular flow under either intramural periarterial drainage or glymphatic.
TBI, in general, is a risk factor for the later development of neurodegenerative diseases that may have varied underlying pathologies [144,145,146]. Proteins associated with neurodegenerative diseases, including Aβ, and tau accumulate in brain following TBI [147]. Aβ deposition is a hallmark of Alzheimer’s disease (AD) and epidemiological studies support an association of severe TBI with a later risk for developing AD [146,148]. Acutely, Aβ levels rise rapidly after TBI, with increased levels of soluble Aβ and diffuse cortical deposits present in humans as early as two hours after a severe injury [146,148,149,150].
Amyloid precursor protein (APP) and Aβ elevations occur acutely in brain in many experimental animal models that mimic the type of contusional and rotation/acceleration injuries associated, for example, with motor vehicle accidents or sports injuries [151,152,153,154,155,156,157,158,159,160,161,162]. In these models, there is increased expression of APP, along with BACE-1 (β-site APP-cleaving enzyme-1), the principal α-secretase and the γ-secretase complex responsible for the C-terminal cleavages that together are responsible for generating the 39–43 amino acid peptide Aβ [152,163,164,165,166]. Upregulation of amyloidogenic APP processing, which favors Aβ production over other non-amyloidogenic APP processing [167], has been suggested as an explanation for the epidemiological associations between TBI and AD [146,148].
Widespread disruption of gliovascular connections following blast exposure seems to predict that glymphatic flow would be widely disrupted and reduce Aβ clearance. While this has not been studied in our chronic rat model of repetitive blast exposure, in a study in both rat and mouse models of acute blast exposure, rather than increasing, rodent brain Aβ42 levels decreased in the first week following blast exposure [32]. Interestingly, the effect on Aβ42 was most prominent in rats exposed to lower blast overpressures (36.6 kPa and 74.5 kPa), while there were no effects on Aβ42 at the 116.7-kPa exposure level [32].
In a later study [168], we subjected APP/presenilin 1 transgenic (Tg) mice (APP/PS1 Tg), a transgenic mouse model of AD, to an extended sequence of repetitive low-level blast exposures (34.5 kPa, 5 psi) designed to mimic human subclinical blast exposures. Exposures were administered three times per week over 8 weeks beginning at either 20 or 36 weeks of age, which represent times before and after this line of APP/PS1 Tg mice develop significant plaque burdens [169].
Curiously, repetitive exposures reduced anxiety as well as improved cognition and social interactions in APP/PS1 Tg, returning many behavioral parameters in APP/PS1 Tg mice to the levels of non-transgenic, wild-type mice [168]. Effects were most apparent in APP/PS1 Tg mice that received blast exposures beginning at 20 weeks of age. Results were less robust in mice when blast exposure began at 36 weeks of age, likely reflecting the greater difficulty of reversing behavioral deficits in mice with more extensive amyloid burden [169]. While amyloid plaque loads were unchanged, soluble, insoluble, and oligomeric Aβ42 levels were reduced in the brains of mice exposed to repetitive low-level blast exposure [168].
More recently, Abutarboush et al. [170] explored the mechanistic basis for these effects in rats by examining the effects of a single 37 kPa (5.4 psi) blast exposure on brain Aβ levels, production, and clearance mechanisms in acute (24 h) and delayed (28 days) phases after blast exposure. Aβ, including the neurotoxic detergent soluble Aβ42 form, was reduced at 24 h but not 28 days after blast exposure. Aβ42 reduction was not associated with changes in levels of Aβ oligomers, levels of APP, or in the β- and ϒ-secretases BACE-1 and presenilin-1, which are involved in the amyloidogenic cleavage of APP. Levels of the α-secretase ADAM17 (also known as tumor-necrosis factor α [TNFα]-converting enzyme), which promotes the non-amyloidogenic processing of APP, were decreased. Levels of BACE-1, and the γ-secretase component, presenilin-1, were also unchanged in our prior study of acute blast exposure in rats [32].
In Abutarboush’s study [170], contrasting with the lack of effects on the enzymes involved in the processing of APP into Aβ, at 24 h after blast exposure, brain levels of the endothelial transporter, low-density related protein 1 (LRP1) increased, and there was enhanced aquaporin-4 (AQP4) localization on perivascular astrocytic endfeet. These findings seem to best explain lowered brain Aβ levels after blast exposure as due to enhanced transvascular endothelial clearance of Aβ by LRP1-mediated transcytosis and altered AQP4-aided glymphatic clearance, suggesting that a low-intensity blast alters transvascular and perivascular Aβ clearance. Future studies will be needed to understand whether the same mechanisms apply to APP/PS1 Tg mice or rats after chronic repetitive exposures.
Few other studies have examined Aβ levels after blast exposure. Konan et al. [88], using a mouse low-level blast exposure protocol, found Aβ40 and 42 elevated at 30 days but not at 7 days after exposure. Harper et al. [171] examined the chronic effects of blast TBI on retinal ganglion cells, optic nerve, and brain amyloid load in APP/PS1 Tg mice and non-transgenic littermates exposed to a single 20 psi blast at 2 to 3 months of age. Two months later at ca. 4.5 months, the APP/PS1 Tg mice exposed to the blast had poorer retinal function, thinner retinal ganglion cell layers, and more optic nerve damage than sham-exposed APP/PS1 Tg mice or blast-exposed non-transgenic mice [171]. No Aβ plaque deposits were found in the retinas of APP/PS1 mice, although increased APP/Aβ-immunostaining was found in blast- compared to sham-exposed transgenic mice, particularly in perivascular regions. They found a statistically non-significant trend towards greater cerebral cortical Aβ plaque loads in the blast-exposed mice compared to the sham-exposed transgenic mice [171].
Studies in U.S. military personnel have largely mirrored results in animal studies by showing decreased Aβ following blast exposure. Edwards et al. [172] found that during a 10-day training exercise that involved repeated blast exposure, Aβ42 was lowered in blood at 24 h following blast exposure [172]. Transient reductions in APP and alterations of the APP signaling network in blood were also observed during training exercises that involved a moderate blast exposure [173]. These studies suggest that as in experimental animals, altered APP processing is an effect of acute blast exposure in humans, although another study found elevated serum Aβ42 in military personnel who experienced repeated blast exposures from firing 0.50-caliber rifles in training sessions conducted over multiple days [174]. In addition, chronically elevated serum levels of Aβ40 and Aβ42 have been found in healthy, male, active-duty military and law enforcement personnel exposed to regular military occupational blasts at U.S. Department of Defense and civilian law enforcement training sites [175]. Thus, effects in humans may vary with type and intensity of exposure as well as the temporal relationship to testing. Further studies will be needed to understand these relationships.
12. Perivascular Tau following BINT
Chronic traumatic encephalopathy (CTE) is a progressive cognitive/behavioral syndrome that most often follows repetitive mTBI [144]. A few reports have appeared of pathologically confirmed CTE in military veterans with a history of blast exposure [82,176,177]. Increased plasma tau has been seen in recently deployed veterans with a history of TBI, with correlations between levels and symptom severity [178]. Elevations of neuron-derived tau in extracellular vesicles have also been observed in serum from experienced breachers where it correlated with neurobehavioral symptoms [179]. Three studies have observed increased retention of the PET tau ligand [18F]AV1451 (flortaucipir) either in recent military veterans or Vietnam-era veterans who suffered from chronic cognitive and neurobehavioral syndromes after blast injury [33,180,181]. The prevalence of CTE pathology in veterans with chronic cognitive/behavioral syndromes following blast exposure remains to be determined, although a large post-mortem study found, in general, little association between blast-related TBI and CTE pathology post-mortem [182].
Our model of repetitive low-level blast exposure develops pathological accumulations of hyperphosphorylated tau (p-tau) in neuronal perikarya and perivascular astroglial processes in multiple brain areas [33]. These deposits are present at least as early as six weeks after blast exposure and remain present at one year [33]. Examples of perivascular deposits of p-tau are shown in Figure 5. The accumulations using Western blotting showed a regional pattern in that p-tau was increased in the anterior neocortex and hippocampus but not in the amygdala [33].
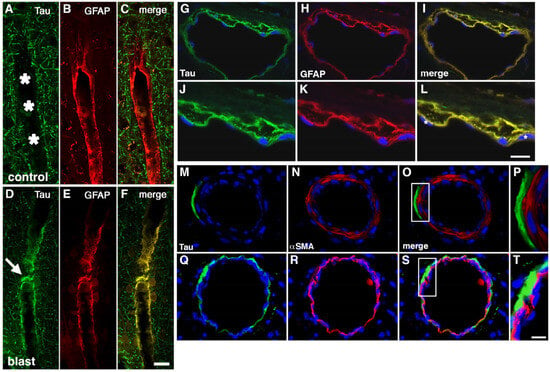
Figure 5.
Perivascular p-tau in astroglial processes 10 months after blast exposure. Penetrating cortical vessels from control (A–C) and blast-exposed rats (D–F) immunostained for AT270 (green, A,D) and GFAP (red, B,E) are shown. Asterisks in panel (A) point to the vascular lumen. The arrow in panel (D) indicates p-tau staining. Scale bar in panel (F): 20 μm. In panels (G–L), a thalamic vessel is shown from a blast-exposed rat sacrificed 10 months after the last blast exposure immunostained with AT270 (green, G,J) and GFAP (red, H,K). Merged images are shown in panels (I,L). DAPI is shown in blue. Panels (J-L) show higher-power images of the vessel shown in panels (G–I). Note the localization of p-tau immunostaining mostly within GFAP immunostained perivascular astroglial processes. Asterisks in panel (L) indicate endothelial cell nuclei identifiable by their elongated appearance. Scale bar in panel (L): 40 μm for panels (G–I); 20 μm for panels (J–L). Panels (M–T) show a lack of p-tau in the vascular smooth muscle layer. Shown is a pial cortical vessel (M–P) or a thalamic vessel (Q–T) from a blast-exposed rat sacrificed 10 months after the last blast exposure. Sections were immunostained with AT270 (green, M,Q) and α-SMA (red, N,R). DAPI is shown in blue. Merged images are shown in panels (O,S). Panels (P,T) show higher-power images of the boxed areas indicated in panels (O,S). Note the lack of co-localization of p-tau immunostaining with the α-SMA stained smooth muscle layer. Scale bar is 40 μm for (M–O,Q–S); 10 μm for (P,T). Figures are reproduced from Dickstein et al. [33].
Tau’s phosphorylation pattern is complex, with over 40 known sites [183,184] and changes in tau phosphorylation following blast exposure were specific to certain p-tau sites [33]. Following blast exposure, we found increased phosphorylation at p-Thr181 [33], which is hyperphosphorylated in human paired helical filaments (PHF) tau [185]. Increased phosphorylation was also seen with the antibody CP13, also raised against PHF tau from human AD brain and recognizing p-Ser202 [33]. No changes were found at three other phosphorylation sites (Thr231, Ser396, and Ser404) that either inhibit microtubule stabilization (Thr231) or promote tau’s self-association properties and oligomerization (Ser396, Ser404) [183,184]. The presence of two phosphor-sites found in human PHF tau (p-Thr181, p-Ser202) following blast exposure suggests a pathological character to the p-tau deposition. Interpreting how the pattern of tau phosphorylation after blast exposure affects tau aggregation and microtubule function more broadly is difficult from the available data.
A puzzling aspect of our rat model is the laterality of blast effects on tau [33,186]. If a brain area was affected, the right side was always involved with the corresponding left side only affected if the right was as well. Particularly striking were the asymmetrical effects on the right and left hippocampus. p-Tau levels further correlated with blast-induced behavioral traits [186]. Elevated p-tau Thr181 in anterior neocortical regions and right hippocampus correlated with anxiety as well as fear learning and novel object localization. By contrast, there were no correlations with levels in the amygdala or posterior neocortical regions and levels did not correlate with hyperarousal. Results were specific to Thr181 in that no correlations were observed for three other sites (Thr231, Ser396, and Ser404) and no consistent correlations were linked with total tau.
Since the blast exposure in our model is delivered as a straight frontal exposure, no systematic variation in the rat’s placement within the blast tube should cause the right hemisphere to be differentially impacted. We can only speculate that this pattern reflects some laterality of hemispheric function in rats, which causes the right side to respond differently to the shock wave or is a feature of the evolution of the injury. Although not as well appreciated as in humans, rats and mice have been known to exhibit paw preference and hemispheric laterality for complex behavioral functions since the 1970s [187]. Hemispheric dominance, in particular, affects spatial memory in rodents [188,189]. Dopaminergic systems, which are disrupted by blast exposure [190] also exhibit lateralization patterns [191,192,193].
Biological models of PTSD postulate that alterations in frontal and limbic structures, including the prefrontal cortex, amygdala, and hippocampus, are involved in generating exaggerated fear responses in humans [194,195,196], which are also seen in blast-exposed rats [26]. Functional neuroimaging in humans is consistent with such models, suggesting that in PTSD, heightened amygdala activity is associated with decreased hippocampal and orbitofrontal activity [194].
p-Tau in our rat model was increased in the anterior cortex and hippocampus, including prelimbic cortex, but not in the amygdala or posterior cortex [33,186]. These correlations are significant in suggesting that p-tau accumulation in anterior neocortical regions and hippocampus may lead to disinhibited amygdala function without p-tau elevation in the amygdala itself. If an evolving tauopathy reduces prefrontal and hippocampal inhibition of the amygdala then, as in biological models of PTSD, this could lead to enhanced fear responses in blast-exposed rats [26] suggestive of amygdala hyperactivity.
Interestingly, in our rat model at six weeks after blast exposure, p-tau was increased in the right anterior neocortex but not in the left [33]. By contrast, at 10 months, it was increased on both sides. This evolution has a relationship to the developing PTSD-related behavioral phenotype, which is absent before 6 weeks but present after 12 weeks [27]. One can speculate that this “spread” of p-tau to both hemispheres marks the critical point at which frontal inhibition of the amygdala fails and onset of the fear- related phenotype begins.
Correlations with fear-related behavior and p-tau in the right hippocampus also suggest a role for hippocampal tau in progression of the behavioral phenotype [186]. Whether in this context p-tau is serving as a marker of neurodegeneration or an actual spread of tau is occurring, as is believed to happen in many neurodegenerative diseases, [197,198,199] is unclear, but seems worthy of further study. Some in vitro evidence supports the notion that astrocytes may be a mediator of cell-to-cell spread of pathological tau [199]. If applicable to blast pathophysiology, this mechanism could have relevance as to how an initial vascular injury with perivascular astrocyte injury leads to a later behavioral phenotype that must have a neuronal basis.
Tau changes might be more broadly involved in the behavioral phenotype of blast- exposed rats in that stress affects phosphorylation at Thr231, Ser262, and Ser396/404 [200]. Tau also seems essential for mediating stress responses [200,201]. This was clearly demonstrated by Lopes et al. [201], who subjected tau-null mutant mice to chronic, unpredictable stress. Atrophy of apical dendrites and spine loss in prefrontal cortical neurons as well as impairments in working memory induced by chronic stress in wild-type mice was absent in tau-null mutant animals [200,201].
p-Tau in perivascular astrocytic processes following blast exposure is of interest in comparing blast pathology to CTE. In CTE, p-tau aggregates occur in neurons and astrocytes in perivascular locations, especially within the depths of the cortical sulci [202,203]. However, recent consensus statements have emphasized the pathognomonic perivascular lesion in CTE as p-tau in perivascular neurons [202,203,204]. p-Tau in astrocytes, while present, is considered a less consistent feature of CTE. Perivascular neuronal p-tau is also apparent in blast-exposed rats [33].
Altered tau processing in the form of increased p-tau has frequently been observed in experimental animal models of blast injury [82,87,88,205,206,207,208,209,210,211,212,213,214,215,216,217,218,219,220,221,222,223,224,225,226]. Two studies reported tau oligomers following blast exposure [218,220]. In mice, p-tau accumulations have been seen in retinal neurons and retinal glia following blast injury [223]. Tau deposits have also been observed in the superficial layers of the mouse cerebral cortex [82].
Meabon et al. [217] described perivascular accumulations of tau in a mouse model of blast injury. These accumulations appeared within hours of injury. Once present in our model, perivascular tau accumulations remain late in the course of the disease [33]. Thus, perivascular tau accumulation seems to be an early and persistent feature of the injury.
Astrocytes influence a diverse variety of animal behaviors through their interactions with neurotransmitters released in perisynaptic regions and their role in regulating calcium currents in the brain [227]. How much of a role tau in perivascular astrocytes plays after blast injury remains to be determined. Brain responses to blast exposure can be conceptualized as a type of stress response, making it of interest to determine how blast responses would be altered in tau-null mutant mice or rats.
13. BINT Damage to Pericytes and the Extracellular Matrix
Pericytes and their normal association with other components of the NVU are disrupted following blast injury in the context of the general disruption of the NVU seen following BINT [37,38]. Damage to pericytes in the walls of capillaries and post-capillary venules could contribute to any number of altered functions that have been attributed to pericytes including BBB maintenance, regulation of immune cell entry into the CNS, and neuroinflammatory states [66]. Neuronal activity may also regulate capillary blood flow through Ca2+-dependent astrocytic mechanisms that actively dilate and constrict pericytes in response to vasoactive agents [228,229]. Pericyte degeneration leads to neurovascular uncoupling and limits oxygen supply to the brain [230]. What specific role pericyte disruption plays in BINT in the context of a more generally disrupted NVU remains to be determined.
The vascular extracellular matrix exists as a complex mixture of proteins, proteoglycans, and other structural proteins that include those in the basement membranes but extend well beyond the immediate perivascular region [231,232]. As a reservoir for growth factors and adhesion proteins that regulate BBB permeability [57,59], disruption of the vascular extracellular matrix could affect a variety of functions within the NVU following blast injury.
In studies investigating whether blast exposure induced changes in the vascular extracellular matrix in our rat model, we performed immunohistochemical staining for vascular extracellular matrix components including collagen IV and laminin [34,37]. We and others have found that efficient immunostaining of collagen IV and laminin in mature adult rodent brain requires a proteolytic epitope unmasking step in perfusion-fixed tissues [233]. In wild-type adult brains, widespread immunostaining for collagen IV and laminin can only be reliably detected following pepsin pretreatment [233]. However, under certain pathological conditions that include vascular degeneration, immunodetection of collagen IV and laminin occurs without antigen retrieval [234].
In blast-exposed animals examined at six weeks after exposure, immunostaining of vascular elements with laminin and collagen IV was observed without pepsin pretreatment [34,37] with areas of enhanced staining that extended broadly across the cortical regions. Pathological staining for collagen IV (Figure 6) and laminin was present at 10 months and longer after blast exposure, suggesting that an altered vascular extracellular matrix is present at subacute stages and a chronic feature of injury [34,37].
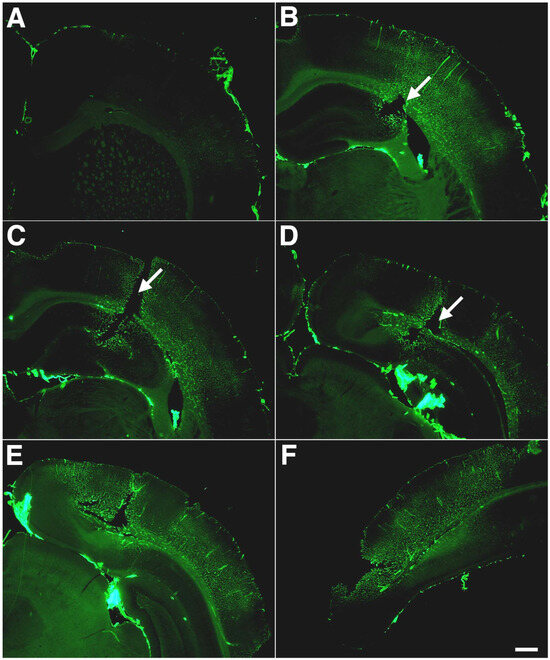
Figure 6.
Altered extracellular matrix immunostaining of blast-exposed animals. Shown is collagen IV immunostaining without pepsin pretreatment of serial sections (A–F) taken 1200 μm apart from a blast-exposed rat that received 3 × 74.5 kPa exposures and was sacrificed 10 months after exposure. A focal cortical lesion is indicated by arrows in panels (B–D). Note the extensive lateral and rostro-caudal extent of altered collagen IV immunostaining without pepsin pretreatment. Scale bar: 750 μm. Figures are reproduced from Gama Sosa et al. [34].
Vascular immunostaining with collagen IV or laminin in the brains of blast-exposed rats without pepsin digestion argues for a reorganization of the vascular extracellular matrix resulting in increased accessibility of these antigens. Increases in matrix metalloproteinases 2 and 9 (MMP-2 and MMP-9) have been observed in isolated vascular fractions at six weeks after blast exposure [37]. Elevation of MMP-2 and MMP-9 suggests that elevation of endogenous degrading proteases may be the basis for these alterations [37]. Others have observed changes in matrix metalloproteinases acutely following blast exposure in animals [235,236,237]. In humans, levels of serum MMP-9 have been found elevated acutely and then for at least 3 months in military personnel during heavy weapons training [125].
Interestingly, the staining of vessels with antigens such as collagen IV without pepsin treatment is typical of immature vessels [233] and seen in vessels in injured areas of the adult brain following mechanical trauma [238], as well as in some models of neurodegeneration [234]. The physiological basis for these differing patterns of staining is incompletely understood, but seems to correlate with the tightness of gliovascular junctions (reviewed in [233]). Indeed, one major structural difference between immature and mature microvessels in the brain is the large perivascular space present in immature microvessels that separates the outer glial basal lamina and the inner vascular one [238,239].
The tightness of gliovascular junctions correlates with the intensity of laminin immunostaining following mechanical injury [238]. Thus, by analogy, altered collagen IV and laminin staining following blast exposure may reflect the loss of normal tightness of gliovascular junctions after blast exposure [35] and could be interpreted as evidence of increased vascular remodeling. Others have observed decreases in the vascular junctional proteins occludin, claudin-5, and ZO-1 acutely following blast exposure [92,122,240,241,242,243,244]. In humans, levels of occludin and claudin-5 are elevated acutely and then for at least 3 months in military personnel during heavy weapons training [125], changes that suggest a loss of vascular tight junctions in human low-level BINT.
Extracellular matrix proteins play many functional roles in the brain, but among their potentially most far-reaching is their role in perineuronal nets (PNNs) [245,246]. PNNs are specialized structures responsible for synaptic stabilization in the adult brain. PNN development plays a critical role in the closure of critical developmental periods associated with high plasticity [245,246]. The digestion of PNNs in adult tissue can restore critical period-like synaptic plasticity [245,246]. Key molecular components of PNNs are chondroitin sulfate proteoglycans (CSPGs), hyaluronan, hyaluronan- and proteoglycan-link proteins (HAPLNs), and the glycoprotein tenascin-R, some of which, as discussed above, are also found in the vascular glycocalyx, which is affected acutely by BINT [109]. PNNs form reticular meshes that surround the perikarya and proximal dendrites of subsets of neurons. PNNs have been most studied in relation to parvalbumin-expressing inhibitory neurons. However, projection neurons, including some hippocampal pyramidal cells, possess PNNs [247,248].
Evidence suggesting that PNNs regulate synaptic plasticity in the adult has come from experiments showing that ocular dominance plasticity could be restored after treatment with chondroitinase ABC (ChABC), an enzyme that degrades CSPGs [249]. Other studies have suggested a role for PNNs in memory retention [245] and PNNs have been implicated in regulating fear-related memory and anxiety [250,251,252,253,254,255]. While initial attention focused on PNNs in the amygdala [251], PNN disruption in the hippocampus affects fear learning in rats [253]. Disruption of PNNs in the medial prefrontal cortex impairs some forms of fear memory as well [253] and intrahippocampal injection of hyaluronidase has been reported to impair contextual fear memory in mice [254]. Given the alterations seen in the extracellular matrix, PNNs seem to warrant exploration in the context of BINT.
Collectively, these observations suggest that blast exposure induces degradation and remodeling of the extracellular matrix. Why vascular remodeling would continue many months after blast exposure, as suggested by our findings in rats, is unknown. Further examination of the molecular changes occurring, particularly in the chronic phase, will be needed to understand the pathophysiological basis of this injury. Functional studies will also be needed to determine whether BBB function is disrupted as well.
14. Vascular-Associated Neuroinflammation following BINT
Microglia have macrophage-like properties and represent 10% of cells in the brain [256]. While not formally part of the NVU, microglia are found near and sometimes contacting blood vessels, as well as synapses, dendritic spines, and processes of perisynaptic and perivascular astrocytes. Microglia support brain health by removing tissue debris and pathogens, as well as remodeling synapses [257].
After injury, microglia are activated, becoming proliferative and undergoing morphological and physiological changes that are associated with increased phagocytic activity and proinflammatory cytokine secretion [256]. This results in the propagation of inflammatory responses that may, in some cases, contribute to brain pathology, although microglia also play roles in maintaining vascular integrity and vascular repair [258].
Microglial morphologies can be correlated with different activation states [259,260,261,262,263,264]. Functionally, microglial activation can be divided into separate states [265]. A pro-inflammatory M1 state is associated with neurotoxicity and extracellular matrix damage. By contrast, the M2 state is associated with phagocytic, anti-inflammatory, and neuroprotective effects that promote processes such as wound healing [265].
We have examined inflammation in our model of low-level blast exposure in several previous studies [36,37,38]. In a study that focused on changes 6 weeks after blast exposure, we found no clear morphological evidence for inflammation in the brain [36]. In this same study, an analysis of 27 cytokines and growth factors in plasma and four brain regions, including the right and left anterior and posterior neocortex, hippocampus, and amygdala, was conducted at 6 and 40 weeks after blast exposure. While a handful of cytokines were decreased in the plasma at 6 weeks (fractalkine, interleukin-1β, lipopolysaccharide-inducible CXC chemokine, macrophage inflammatory protein 1α, and vascular endothelial growth factor), none of the changes in plasma were reflected in brain. No differences in plasma cytokine levels were detected between blast-exposed and control rats at 40 weeks after blast exposure. In the brain, isolated changes were seen in levels of selected cytokines at 6 weeks following blast exposure, but none of these changes were found in both hemispheres or at 40 weeks after blast exposure [36]. The conclusion of this study was that inflammation is not a prominent feature in our model at 6 weeks after blast exposure.
In a later study, we focused on inflammatory changes at one year and longer after blast exposure [38]. This study found patchy neuroinflammatory changes in areas where there were vascular alterations. At 13 months after blast exposure, small perivascular patches of activated microglia expressing high levels of major histocompatibility type II antigens (MHCII) were often observed (Figure 7), a feature characteristic of M1 microglia (the proinflammatory type associated with tissue damage). Morphologically, the microglia were predominantly the more pro-inflammatory types 3 and 4. Some perivascular microglia were visibly undergoing apoptosis and evidence for perivascular microglial phagocytosis of astrocytes was found in the form of intracellular GFAP-immunostained material within perivascular microglia. A variety of EM changes were seen in perivascular microglia, including dilated endoplasmic reticulum, mitochondrial alterations, and cholesterol crystals in lysosomal vacuoles, all suggestive of phagocytic activity [38].
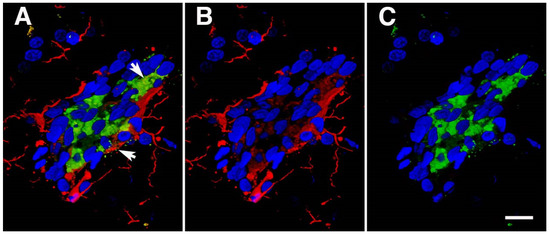
Figure 7.
Activated perivascular microglia. Shown is a luminal view of a patch of perivascular activated M1 microglia expressing MHCII. Panel (A) shows merged images; panel (B), Iba1 immunostaining (microglia, red); and panel (C), MHCII immunostaining (activated M1 microglia, green). Arrows in panel (A) show an apoptotic-activated microglial cell (green) and a degenerating perivascular microglial cell (red). Scale, 20 μm. Figure is reproduced from Gama Sosa et al. [38].
The results of our early [36] and late studies [38] suggest that perivascular neuroinflammation develops after 6 weeks following blast exposure. However, once established, it likely persists into the chronic phase. This time frame correlates roughly with the appearance of the blast-related behavioral phenotype, which is absent before 8 weeks and present 12 weeks and longer after blast exposure [27]. The patchiness of the morphologically visible neuroinflammatory response, however, limits direct correlations [38]. While in individual animals, focal patches of inflammation seemed to bleed into more widespread strips of inflammation that were associated with neuronal and synaptic loss, this was not widely found in the brain [38].
A more general suggestion of a blast-associated inflammatory response was found in a transcriptomic study that examined blast-induced alterations in the anterior neocortex, hippocampus, amygdala, and cerebellum across the time frame over which the PTSD-related behavioral phenotype develops [40]. This study found a group of differentially expressed genes (DEGs) regulated by tumor necrosis factor α (TNFα) whose expression was altered in a direction consistent with TNFα activation in the amygdala and anterior cortex between 6 weeks and 12 months after blast exposure [40].
A group of 6 DEGs (Hapln1, Grm2, P2ry12, Ccr5, Pbld1, and Cdh23) were identified in blast-exposed animals that were common to the anterior neocortex, amygdala, and hippocampus. Of these, the purinergic receptor P2ry12 is of particular interest, since it is highly expressed in quiescent non-inflammatory M2 microglia, but its expression is downregulated in activated M1 inflammatory microglia associated with neuroinflammation [266]. P2ry12 has been identified as one of a set of genes highly enriched in microglia that can be used for phenotyping microglia [267]. The widespread downregulation of P2ry12 in multiple brain regions from studies conducted on whole-tissue extracts suggests a broader inflammatory signal after blast exposure that may not be associated with morphological evidence of inflammation.
Inflammation has long been suspected of playing a role in BINT [31]. Inflammatory changes have been studied mostly following acute blast exposure [31]. In animal models, blast exposure causes increases in many markers of inflammation within blood and plasma [31]. Microglial activation has been reported in many studies [31,42,82,85,123,206,268,269,270,271,272,273]. A recent study by Stone et al. [274] demonstrated increased brain inflammation in special forces personnel using positron emission tomography (PET) to detect uptake of the translocator protein (TSPO) ligand DPA-714, which recognizes activated microglia. Increases in brain inflammation were associated with decreased cortical thickness and volume changes and were positively associated with lifetime repetitive blast exposure.
In other animal studies, elevations of multiple pro-inflammatory cytokines and chemokines including TNFα have been observed in multiple brain regions at 5 days after a single 70 kPa low-level blast exposure in rats [84]. TNFα has been elevated in many studies following blast injury in rodents [84,94,275,276,277,278,279], although in some of them, elevations were transient returning to baseline within 48 h of injury [275,276]. Simard et al. [102] observed perivascular induction of TNFα in their studies of selective vascular exposure.
Interestingly, TNFα in the brain has biological activities independent of its role in inflammation, being implicated in PTSD-related hyperarousal states and regulating fear learning [280,281,282,283,284]. For example, increased TNFα blocks the retrieval and reconsolidation of contextual fear memories [280], while intracerebroventricular injection of TNFα impairs fear extinction [282]. TNFα signaling in the hippocampus has also been implicated as mediating enhanced fear learning during drug withdrawal [281] and maternal TNFα production is suggested to program innate fear responses in offspring [284]. The cellular basis for these effects is unclear, although microglial production of TNFα has been implicated in sustaining fear memories [283]. Thus, a variety of evidence supports a potential role for TNFα signaling in the neurobiological basis of blast effects on behavior through inflammatory or possibly non-inflammatory mechanisms.
Human studies in experienced breachers exposed to high numbers of career blast exposures also show the dysregulation of genes associated with chronic inflammation and immune response in blood [285] and have identified elevated TNFα levels in neuronal-derived extracellular vesicles isolated from serum [286]. TNFα is a therapeutic target in many human inflammatory conditions and has revolutionized the treatment of some [287,288]. Given its role as a key mediator of brain damage in many conditions, including brain trauma [289,290], exploration of its role in BINT is warranted.
15. Linking Neurovascular and Chronic Neurobehavioral Effects That Follow BINT
At the levels of blast exposure we are studying, the initial injury appears largely vascular. The question then becomes, could an initial vascular injury contribute to a delayed and chronic neurobehavioral phenotype that must have a neuronal basis? It is possible that some neuronal injury hidden from sight initiates a strictly neuronal sequence of events leading to the behavioral phenotype. All we can say at this point is that if there is, it is not yet inapparent, while an early vascular pathology is readily apparent.
In this review, we have highlighted the various injuries to the NVU that might play a role in a vascular hypothesis suggesting that an initial vascular injury could initiate a sequence of pathophysiological events leading to a later behavioral disturbance. These changes are summarized and presented in relationship to the late behavioral phenotype’s appearance in Table 2 and Figure 8. Broadly, this model suggests that an early and selective vascular pathology occurs with direct endothelial injury. Associated with endothelial injury there is damage to, and loss of, perivascular astrocytes, altered vascular extracellular matrix proteins, and damage to the smooth muscle layer and other mural cells. Perivascular tau accumulation is also an early feature of the injury, and each of these factors potentially has different contributions to the resulting NVU pathology (Table 2).
Table 2.
Pathophysiological events following blast injury and their relationship to later behavioral disturbance.
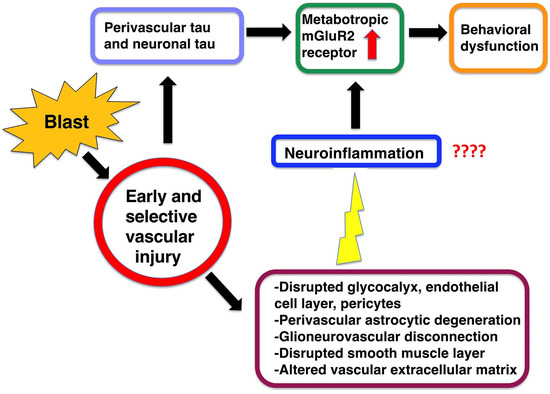
Figure 8.
Hypothetical scenario for how early vascular injury leads to a delayed and chronic neurobehavioral phenotype. Details are discussed in the text.
Effects at the level of the glycocalyx, endothelial cells, perivascular astrocytes, and nerves, as well as the pericyte and smooth muscle layer, could all contribute to a disturbed NVU affecting a variety of vascular-related functions, including BBB permeability, neurovascular coupling, and glymphatic flow. A perivascular tauopathy might have consequences directly on vascular function or indirectly, if it served as a seed for subsequent neuronal spread of tau.
Of the primary elements within the NVU that are disrupted by BINT, perivascular astrocytes, given their diverse effects on the NVU, emerge as particularly good candidates for playing an important role. Their loss or disruption could influence BBB properties, nutrient uptake, neurovascular coupling, and immune cell entry into the CNS. Astrocytes are also attractive because of their broader role in regulating neuronal activity as part of the tripartite synapse [227]. Astrocytes are activated by and clear synaptically released neurotransmitters, particularly glutamate, and intracellular Ca2+ signaling in astrocytes regulates neuronal activity at distant sites [227]. With their broad anatomic reach and well-known effects on behavior, astrocytes are attractive as mediators of an initial vascular injury to distant sites.
Among the secondary factors that a vascular injury might promote, neuroinflammation is attractive as a linking mechanism between vascular injury and a chronic behavioral phenotype. Substantial evidence over many years has established chronic, low-grade inflammation as a feature of many mental health disorders including depression and PTSD as well as a pathophysiological driver of symptoms [291,292,293,294,295,296].
Haroon et al. [297] suggested an interesting model proposing that in mood disorders, initial perivascular inflammation is a seed that leads to later glutamatergic dysfunction and mood disorders. In this model, perivascular inflammation is postulated to convert the synaptic environment from a homeostatic one into a toxic inflammatory one, leading to a hyperglutamatergic and excitotoxic environment [297]. While in mood disorders there is no established vascular lesion that might serve as a seed to trigger such a cascade, this model seems attractive for low-level blast injury where evidence for an initial vascular injury is well established.
Supporting an early, potentially hyperglutamatergic, state following blast injury, a number of studies have shown increased evoked synaptic NMDA receptor activity in hippocampal slices taken from blast-exposed rats in vivo [298,299] or in vitro [300], with one study [299] showing increases still present six weeks after blast exposure. Collectively, a vascular injury followed by perivascular astrocyte injury and neuroinflammation might explain how an injury that starts in the NVU spreads to involve neuronal circuits and produces a behavioral phenotype that must have a neuronal basis.
While the neurobiological basis for the behavioral traits in our model are not fully understood, our most recent work suggests that the late behavioral traits seem to be caused by abnormalities in the group II metabotropic receptor mGluR2 [301]. mGluR2 is elevated after blast exposure in a time course that correlates with the appearance of the behavioral phenotype [27,301]. A causative role for mGluR2 elevation is supported by the prevention [28] or reversal [301] of the behavioral phenotype by mGluR2/3 antagonists. It is also supported by (2R, 6R) hydroxynorketamine’s rescue of the phenotype [39] since (2R, 6R) hydroxynorketamine acts principally through mGluR2-related mechanisms [302,303].
Late increased mGluR2 can also be seen as consistent with the hypothesis discussed above that chronic blast injury induces a hypoglutamatergic state. Presynaptic mGluR2/3 receptors act as autoreceptors that, when stimulated, inhibit glutamate release [288]. Thus, by analogy, increased mGluR2 levels would be predicted to be a marker of a hypoglutamatergic state. This analogy assumes that a pharmacological blockade has the same physiological consequences as reducing receptor overexpression. Support for this argument can be seen in our previous findings that treatment with mGluR2/3 antagonists reverses blast-related anxiety and fear-related behaviors as well as long-term recognition memory [28,288].
16. Future Studies
While we have some understanding of the neurochemical basis of late behavioral effects, much work remains to understand the early molecular evolution of the disease. Chiefly, a better understanding of the molecular evolution of changes at the NVU level and beyond is needed. Although some important clues concerning the role of inflammation in this model have come from regional transcriptomic studies conducted on whole-tissue RNA [40], the difficulty of examining single cell-type effects may be limiting. Indeed, single-nuclei RNA sequencing has revealed a heterogeneity to microglia and astrocytes not appreciated at the morphological level [267,304]. For example, one study identified a set of about 100 genes highly enriched in microglia that have been proposed to represent a microglial “tool set” for sensing the brain milieu [267]. Single-nuclei RNA sequencing has, in addition, identified five transcriptionally, distinct types of astrocytes, which differ in their expression of inflammatory mediators [304]. A recent single-nucleus, multi-region transcriptomic analysis of the brain vasculature in AD further demonstrated the power of single-cell approaches applied to the vasculature [305]. Future studies at the single-cell transcriptomic level seem warranted in our model of low-level BINT.
Finally, a proximate cause for the late neurobehavioral abnormalities seems to lie in the elevated expression of the group II metabotropic receptor mGluR2 [301]. However, the molecular basis for why mGluR2 elevation occurs and its relationship to an early vascular insult are unclear. In addition to developing strategies to address the late neurobehavioral consequences, strategies aimed at preventing or reversing vascular damage or modulating immune responses may improve chronic neuropsychiatric symptoms associated with BINT in humans.
Author Contributions
G.A.E.: conceptualization; data curation; formal analysis; funding acquisition; project administration; supervision; visualization; writing—original draft. M.A.G.S.: conceptualization; data curation; formal analysis; investigation; project administration; writing—review and editing. R.D.G.: conceptualization; data curation; formal analysis; investigation; project administration; visualization; writing—review and editing. G.P.G.: conceptualization; data curation; formal analysis; investigation; visualization; writing—review and editing. G.M.P.: investigation; writing—review and editing. R.A.: investigation; writing—review and editing. U.K.: investigation; methodology; writing—review and editing. C.W.Z.: formal analysis; writing—review and editing. W.G.M.J.: investigation, visualization; writing—review and editing. J.R.S.: conceptualization; writing—review and editing. P.R.H.: conceptualization; visualization; writing—review and editing. D.G.C.: conceptualization; funding acquisition; visualization; writing—review and editing. S.T.A.: conceptualization; methodology; funding acquisition; writing—review and editing. All authors have read and agreed to the published version of the manuscript.
Funding
This work was supported by the Department of Veterans Affairs, Veterans Health Administration, Rehabilitation Research and Development Service awards 1I01RX002660 (G.A.E.), 1I01RX003846 (G.A.E.), and the Department of Veterans Affairs Office of Research and Development Medical Research Service awards 1I01BX004067 (G.A.E.), 1I01BX005882 (G.A.E.), and 1I01BX002311 (D.G.C.), Department of Defense work unit number 0000B999.0000.000.A1503 (S.T.A.) and NIA P30 AG066514 (P.R.H., C.W.Z.).
Data Availability Statement
Data underlying the basis of this report is available from the authors.
Acknowledgments
The views expressed in this article reflect the results of research conducted by the authors and do not necessarily reflect the official policy or position of the of the Uniformed Services University of the Health Sciences (USUHS), the Henry M. Jackson Foundation for the Advancement of Military Medicine Inc., the Department of the Navy, or the Department of Defense (DoD). The mention of trade names, commercial products, or organizations does not imply endorsement by the U.S. government. All study protocols that involved the use of animals were reviewed and approved by the Walter Reed Army Institute of Research/Naval Medical Research Command Institutional Animal Care and Use Committee in compliance with all applicable federal regulations governing the protection of animals in research. The experiments reported herein were conducted in compliance with the Animal Welfare Act and per the principles set forth in the Guide for Care and Use of Laboratory Animals, Institute of Laboratory Animals Resources, National Research Council, National Academy Press, 2011. Some of the authors are military service members or federal/contracted employees of the United States Government. This work was prepared as part of their official duties. Title 17 U.S.C. § 105 provides that ‘Copyright protection under this title is not available for any work of the United States Government’. Title 17 U.S.C. § 101 defines a U.S. government work as a work prepared by a military service member or employee of the U.S. government as part of that person’s official duties.
Conflicts of Interest
The authors declare no conflicts of interest.
References
- Elder, G.A.; Ehrlich, M.E.; Gandy, S. Relationship of traumatic brain injury to chronic mental health problems and dementia in military veterans. Neurosci. Lett. 2019, 707, 134294. [Google Scholar] [CrossRef]
- Hoge, C.W.; McGurk, D.; Thomas, J.L.; Cox, A.L.; Engel, C.C.; Castro, C.A. Mild traumatic brain injury in U.S. Soldiers returning from Iraq. N. Engl. J. Med. 2008, 358, 453–463. [Google Scholar] [CrossRef] [PubMed]
- Tanielian, T.; Jaycox, L.H. Invisible Wounds of War: Psychological and Cognitive Injuries, Their Consequences, and Services to Assist Recovery; Rand Corporation: Santa Monica, CA, USA, 2008. [Google Scholar]
- DePalma, R.G. Combat TBI: History, Epidemiology and Injury Modes. In Brain Neurotrauma: Molecular, Neuropsychological, and Rehabilitation Aspects; Kobeissy, F., Ed.; CRC Press: Boca Raton, FL, USA, 2015; pp. 5–14. [Google Scholar]
- Elder, G.A. Update on TBI and Cognitive Impairment in Military Veterans. Curr. Neurol. Neurosci. Rep. 2015, 15, 68. [Google Scholar] [CrossRef] [PubMed]
- Warden, D.L.; Ryan, L.; Helmick, K.; Schwab, K.; French, L.; Lu, W.; Lux, W.; Ling, G.; Ecklund, J. War neurotrauma: The defense and veterans brain injury center (DVBIC) experience at the Walter Reed Army Medical Center. J. Neurotrauma 2005, 22, 1178. [Google Scholar]
- Bell, R.S.; Vo, A.H.; Neal, C.J.; Tigno, J.; Roberts, R.; Mossop, C.; Dunne, J.R.; Armonda, R.A. Military traumatic brain and spinal column injury: A 5-year study of the impact blast and other military grade weaponry on the central nervous system. J. Trauma. 2009, 66 (Suppl. 4), S104–S111. [Google Scholar] [CrossRef] [PubMed]
- Engel, C.; Hoch, E.; Simmons, M. The Neurological Effects of Repeated Exposure to Military Occupational Blast: Implications for Prevention and Health, Proceedings of the Findings, and Expert Recommendations from the Seventh Department of Defense State-of-the-Science Meeting, Arlington, VA, USA, 12–14 March 2018; Rand Corporation: Arlington VA USA, 2019. [Google Scholar]
- Tschiffely, A.E.; Statz, J.K.; Edwards, K.A.; Goforth, C.; Ahlers, S.T.; Carr, W.S.; Gill, J.M. Assessing a Blast-Related Biomarker in an Operational Community: Glial Fibrillary Acidic Protein in Experienced Breachers. J. Neurotrauma 2020, 37, 1091–1096. [Google Scholar] [CrossRef]
- Wang, Z.; Wilson, C.M.; Mendelev, N.; Ge, Y.; Galfalvy, H.; Elder, G.; Ahlers, S.; Yarnell, A.M.; LoPresti, M.L.; Kamimori, G.H.; et al. Acute and Chronic Molecular Signatures and Associated Symptoms of Blast Exposure in Military Breachers. J. Neurotrauma 2020, 37, 1221–1232. [Google Scholar] [CrossRef]
- Pagulayan, K.F.; Rau, H.; Madathil, R.; Werhane, M.; Millard, S.P.; Petrie, E.C.; Parmenter, B.; Peterson, S.; Sorg, S.; Hendrickson, R.; et al. Retrospective and Prospective Memory Among OEF/OIF/OND Veterans With a Self-Reported History of Blast-Related mTBI. J. Int. Neuropsychol. Soc. 2018, 24, 324–334. [Google Scholar] [CrossRef]
- Bailie, J.; Lippa, S.; Hungerford, L.; French, L.M.; Brickell, T.A.; Lange, R.T. Cumulative Blast Exposure During a Military Career Negatively Impacts Recovery from Traumatic Brain Injury. J. Neurotrauma 2023. [Google Scholar] [CrossRef]
- Lange, R.T.; French, L.M.; Lippa, S.M.; Gillow, K.; Tippett, C.E.; Barnhart, E.A.; Glazer, M.E.; Bailie, J.M.; Hungerford, L.; Brickell, T.A. High Lifetime Blast Exposure Using the Blast Exposure Threshold Survey Is Associated With Worse Warfighter Brain Health Following Mild Traumatic Brain Injury. J. Neurotrauma 2024, 41, 186–198. [Google Scholar] [CrossRef]
- Belding, J.N.; Englert, R.; Bonkowski, J.; Thomsen, C.J. Occupational Risk of Low-Level Blast Exposure and TBI-Related Medical Diagnoses: A Population-Based Epidemiological Investigation (2005–2015). Int. J. Environ. Res. Public. Health 2021, 18, 12925. [Google Scholar] [CrossRef] [PubMed]
- Belding, J.N.; Fitzmaurice, S.; Englert, R.M.; Lee, I.; Kowitz, B.; Highfill-McRoy, R.M.; Thomsen, C.J.; da Silva, U. Blast Exposure and Risk of Recurrent Occupational Overpressure Exposure Predict Deployment TBIs. Mil. Med. 2020, 185, e538–e544. [Google Scholar] [CrossRef]
- Stone, J.R.; Avants, B.B.; Tustison, N.J.; Wassermann, E.M.; Gill, J.; Polejaeva, E.; Dell, K.C.; Carr, W.; Yarnell, A.M.; LoPresti, M.L.; et al. Functional and Structural Neuroimaging Correlates of Repetitive Low-Level Blast Exposure in Career Breachers. J. Neurotrauma 2020, 37, 2468–2481. [Google Scholar] [CrossRef] [PubMed]
- Phillips, D.U.S. Troops Still Train on Weapons With Known Risk of Brain Injury. New York Times, 26 November 2023. [Google Scholar]
- Brenner, L.A.; Homaifar, B.Y.; Olson-Madden, J.H.; Nagamoto, H.T.; Huggins, J.; Schneider, A.L.; Forster, J.E.; Matarazzo, B.; Corrigan, J.D. Prevalence and screening of traumatic brain injury among veterans seeking mental health services. J. Head. Trauma. Rehabil. 2013, 28, 21–30. [Google Scholar] [CrossRef] [PubMed]
- Jorge, R.E.; Arciniegas, D.B. Mood disorders after TBI. Psychiatr. Clin. North. Am. 2014, 37, 13–29. [Google Scholar] [CrossRef]
- Dreer, L.E.; Tang, X.; Nakase-Richardson, R.; Pugh, M.J.; Cox, M.K.; Bailey, E.K.; Finn, J.A.; Zafonte, R.; Brenner, L.A. Suicide and traumatic brain injury: A review by clinical researchers from the National Institute for Disability and Independent Living Rehabilitation Research (NIDILRR) and Veterans Health Administration Traumatic Brain Injury Model Systems. Curr. Opin. Psychol. 2018, 22, 73–78. [Google Scholar] [CrossRef]
- Elder, G.A.; Stone, J.R.; Ahlers, S.T. Effects of Low-Level Blast Exposure on the Nervous System: Is There Really a Controversy? Front. Neurol. 2014, 5, 269. [Google Scholar] [CrossRef]
- Jones, E.; Fear, N.T.; Wessely, S. Shell shock and mild traumatic brain injury: A historical review. Am. J. Psychiatry 2007, 164, 1641–1645. [Google Scholar] [CrossRef]
- Mac Donald, C.L.; Barber, J.; Jordan, M.; Johnson, A.M.; Dikmen, S.; Fann, J.R.; Temkin, N. Early Clinical Predictors of 5-Year Outcome After Concussive Blast Traumatic Brain Injury. JAMA Neurol. 2017, 74, 821–829. [Google Scholar] [CrossRef]
- Elder, G.A.; Dorr, N.P.; De Gasperi, R.; Gama Sosa, M.A.; Shaughness, M.C.; Maudlin-Jeronimo, E.; Hall, A.A.; McCarron, R.M.; Ahlers, S.T. Blast exposure induces post-traumatic stress disorder-related traits in a rat model of mild traumatic brain injury. J. Neurotrauma 2012, 29, 2564–2575. [Google Scholar] [CrossRef]
- Perez-Garcia, G.; Gama Sosa, M.A.; De Gasperi, R.; Lashof-Sullivan, M.; Maudlin-Jeronimo, E.; Stone, J.R.; Haghighi, F.; Ahlers, S.T.; Elder, G.A. Chronic post-traumatic stress disorder-related traits in a rat model of low-level blast exposure. Behav. Brain Res. 2018, 340, 117–125. [Google Scholar] [CrossRef] [PubMed]
- Perez-Garcia, G.; Gama Sosa, M.A.; De Gasperi, R.; Tschiffely, A.E.; McCarron, R.M.; Hof, P.R.; Gandy, S.; Ahlers, S.T.; Elder, G.A. Blast-induced “PTSD”: Evidence from an animal model. Neuropharmacology 2019, 145 Pt B, 220–229. [Google Scholar] [CrossRef]
- Perez Garcia, G.; Perez, G.M.; De Gasperi, R.; Gama Sosa, M.A.; Otero-Pagan, A.; Pryor, D.; Abutarboush, R.; Kawoos, U.; Hof, P.R.; Cook, D.G.; et al. Progressive Cognitive and Post-Traumatic Stress Disorder-Related Behavioral Traits in Rats Exposed to Repetitive Low-Level Blast. J. Neurotrauma 2021, 38, 2030–2045. [Google Scholar] [CrossRef] [PubMed]
- Perez-Garcia, G.; De Gasperi, R.; Gama Sosa, M.A.; Perez, G.M.; Otero-Pagan, A.; Tschiffely, A.; McCarron, R.M.; Ahlers, S.T.; Elder, G.A.; Gandy, S. PTSD-Related Behavioral Traits in a Rat Model of Blast-Induced mTBI Are Reversed by the mGluR2/3 Receptor Antagonist BCI-838. eNeuro 2018, 5. [Google Scholar] [CrossRef] [PubMed]
- Perez-Garcia, G.; Gama Sosa, M.A.; De Gasperi, R.; Lashof-Sullivan, M.; Maudlin-Jeronimo, E.; Stone, J.R.; Haghighi, F.; Ahlers, S.T.; Elder, G.A. Exposure to a Predator Scent Induces Chronic Behavioral Changes in Rats Previously Exposed to Low-level Blast: Implications for the Relationship of Blast-Related TBI to PTSD. Front. Neurol. 2016, 7, 176. [Google Scholar] [CrossRef] [PubMed]
- Kobeissy, F.; Mondello, S.; Tumer, N.; Toklu, H.Z.; Whidden, M.A.; Kirichenko, N.; Zhang, Z.; Prima, V.; Yassin, W.; Anagli, J.; et al. Assessing Neuro-Systemic & Behavioral Components in the Pathophysiology of Blast-Related Brain Injury. Front. Neurol. 2013, 4, 186. [Google Scholar]
- Elder, G.A.; Gama Sosa, M.A.; De Gasperi, R.; Stone, J.R.; Dickstein, D.L.; Haghighi, F.; Hof, P.R.; Ahlers, S.T. Vascular and inflammatory factors in the pathophysiology of blast-induced brain injury. Front. Neurol. 2015, 6, 48. [Google Scholar] [CrossRef]
- De Gasperi, R.; Gama Sosa, M.A.; Kim, S.H.; Steele, J.W.; Shaughness, M.C.; Maudlin-Jeronimo, E.; Hall, A.A.; Dekosky, S.T.; McCarron, R.M.; Nambiar, M.P.; et al. Acute blast injury reduces brain abeta in two rodent species. Front. Neurol. 2012, 3, 177. [Google Scholar] [CrossRef]
- Dickstein, D.L.; De Gasperi, R.; Gama Sosa, M.A.; Perez-Garcia, G.; Short, J.A.; Sosa, H.; Perez, G.M.; Tschiffely, A.E.; Dams-O’Connor, K.; Pullman, M.Y.; et al. Brain and blood biomarkers of tauopathy and neuronal injury in humans and rats with neurobehavioral syndromes following blast exposure. Mol. Psychiatry 2021, 26, 5940–5954. [Google Scholar] [CrossRef]
- Gama Sosa, M.A.; De Gasperi, R.; Janssen, P.L.; Yuk, F.J.; Anazodo, P.C.; Pricop, P.E.; Paulino, A.J.; Wicinski, B.; Shaughness, M.C.; Maudlin-Jeronimo, E.; et al. Selective vulnerability of the cerebral vasculature to blast injury in a rat model of mild traumatic brain injury. Acta Neuropathol. Commun. 2014, 2, 67. [Google Scholar] [CrossRef]
- Gama Sosa, M.A.; De Gasperi, R.; Perez Garcia, G.S.; Perez, G.M.; Searcy, C.; Vargas, D.; Spencer, A.; Janssen, P.L.; Tschiffely, A.E.; McCarron, R.M.; et al. Low-level blast exposure disrupts gliovascular and neurovascular connections and induces a chronic vascular pathology in rat brain. Acta Neuropathol. Commun. 2019, 7, 6. [Google Scholar] [CrossRef] [PubMed]
- Gama Sosa, M.A.; De Gasperi, R.; Perez Garcia, G.S.; Sosa, H.; Searcy, C.; Vargas, D.; Janssen, P.L.; Perez, G.M.; Tschiffely, A.E.; Janssen, W.G.; et al. Lack of chronic neuroinflammation in the absence of focal hemorrhage in a rat model of low-energy blast-induced TBI. Acta Neuropathol. Commun. 2017, 5, 80. [Google Scholar] [CrossRef]
- Gama Sosa, M.A.; De Gasperi, R.; Pryor, D.; Perez Garcia, G.S.; Perez, G.M.; Abutarboush, R.; Kawoos, U.; Hogg, S.; Ache, B.; Janssen, W.G.; et al. Low-level blast exposure induces chronic vascular remodeling, perivascular astrocytic degeneration and vascular-associated neuroinflammation. Acta Neuropathol. Commun. 2021, 9, 167. [Google Scholar] [CrossRef]
- Gama Sosa, M.A.; De Gasperi, R.; Pryor, D.; Perez Garcia, G.S.; Perez, G.M.; Abutarboush, R.; Kawoos, U.; Hogg, S.; Ache, B.; Sowa, A.; et al. Late chronic local inflammation, synaptic alterations, vascular remodeling and arteriovenous malformations in the brains of male rats exposed to repetitive low-level blast overpressures. Acta Neuropathol. Commun. 2023, 11, 81. [Google Scholar] [CrossRef] [PubMed]
- Garcia, G.P.; Perez, G.M.; Gasperi, R.; Sosa, M.A.G.; Otero-Pagan, A.; Abutarboush, R.; Kawoos, U.; Statz, J.K.; Patterson, J.; Zhu, C.W.; et al. (2R,6R)-Hydroxynorketamine Treatment of Rats Exposed to Repetitive Low-Level Blast Injury. Neurotrauma Rep. 2023, 4, 197–217. [Google Scholar] [CrossRef] [PubMed]
- De Gasperi, R.; Gama Sosa, M.A.; Perez Garcia, G.S.; Perez, G.M.; Abutarboush, R.; Kawoos, U.; Statz, J.K.; Patterson, J.; Hof, P.R.; Katsel, P.; et al. Progressive Transcriptional Changes in the Amygdala Implicate Neuroinflammation in the Effects of Repetitive Low-Level Blast Exposure in Male Rats. J. Neurotrauma 2023, 40, 561–577. [Google Scholar] [CrossRef]
- Haghighi, F.; Ge, Y.; Chen, S.; Xin, Y.; Umali, M.U.; De Gasperi, R.; Gama Sosa, M.A.; Ahlers, S.T.; Elder, G.A. Neuronal DNA Methylation Profiling of Blast-Related Traumatic Brain Injury. J. Neurotrauma 2015, 32, 1200–1209. [Google Scholar] [CrossRef]
- Gama Sosa, M.A.; De Gasperi, R.; Paulino, A.J.; Pricop, P.E.; Shaughness, M.C.; Maudlin-Jeronimo, E.; Hall, A.A.; Janssen, W.G.; Yuk, F.J.; Dorr, N.P.; et al. Blast overpressure induces shear-related injuries in the brain of rats exposed to a mild traumatic brain injury. Acta Neuropathol. Commun. 2013, 1, 51. [Google Scholar] [CrossRef]
- Ahlers, S.T.; Vasserman-Stokes, E.; Shaughness, M.C.; Hall, A.A.; Shear, D.A.; Chavko, M.; McCarron, R.M.; Stone, J.R. Assessment of the effects of acute and repeated exposure to blast overpressure in rodents: Toward a greater understanding of blast and the potential ramifications for injury in humans exposed to blast. Front. Neurol. 2012, 3, 32. [Google Scholar] [CrossRef]
- Chavko, M.; Koller, W.A.; Prusaczyk, W.K.; McCarron, R.M. Measurement of blast wave by a miniature fiber optic pressure transducer in the rat brain. J. Neurosci. Methods 2007, 159, 277–281. [Google Scholar] [CrossRef]
- Cernak, I.; Stein, D.G.; Elder, G.A.; Ahlers, S.; Curley, K.; DePalma, R.G.; Duda, J.; Ikonomovic, M.; Iverson, G.L.; Kobeissy, F.; et al. Preclinical modelling of militarily relevant traumatic brain injuries: Challenges and recommendations for future directions. Brain Inj. 2017, 31, 1168–1176. [Google Scholar] [CrossRef] [PubMed]
- Song, H.; Cui, J.; Simonyi, A.; Johnson, C.E.; Hubler, G.K.; DePalma, R.G.; Gu, Z. Linking blast physics to biological outcomes in mild traumatic brain injury: Narrative review and preliminary report of an open-field blast model. Behav. Brain Res. 2018, 340, 147–158. [Google Scholar] [CrossRef] [PubMed]
- Elder, G.A.; Mitsis, E.M.; Ahlers, S.T.; Cristian, A. Blast-induced mild traumatic brain injury. Psychiatr. Clin. North. Am. 2010, 33, 757–781. [Google Scholar] [CrossRef]
- Lange, R.T.; Brickell, T.A.; Ivins, B.; Vanderploeg, R.D.; French, L.M. Variable, not always persistent, postconcussion symptoms after mild TBI in U.S. military service members: A five-year cross-sectional outcome study. J. Neurotrauma 2013, 30, 958–969. [Google Scholar] [CrossRef]
- Mac Donald, C.L.; Barber, J.; Patterson, J.; Johnson, A.M.; Dikmen, S.; Fann, J.R.; Temkin, N. Association Between 5-Year Clinical Outcome in Patients With Nonmedically Evacuated Mild Blast Traumatic Brain Injury and Clinical Measures Collected Within 7 Days Postinjury in Combat. JAMA Netw. Open 2019, 2, e186676. [Google Scholar] [CrossRef] [PubMed]
- Xing, C.Y.; Tarumi, T.; Liu, J.; Zhang, Y.; Turner, M.; Riley, J.; Tinajero, C.D.; Yuan, L.J.; Zhang, R. Distribution of cardiac output to the brain across the adult lifespan. J. Cereb. Blood Flow. Metab. 2017, 37, 2848–2856. [Google Scholar] [CrossRef]
- Siegenthaler, J.A.; Sohet, F.; Daneman, R. ‘Sealing off the CNS’: Cellular and molecular regulation of blood-brain barriergenesis. Curr. Opin. Neurobiol. 2013, 23, 1057–1064. [Google Scholar] [CrossRef]
- Iadecola, C. The Neurovascular Unit Coming of Age: A Journey through Neurovascular Coupling in Health and Disease. Neuron 2017, 96, 17–42. [Google Scholar] [CrossRef]
- Del Zoppo, G.J.; Milner, R.; Mabuchi, T.; Hung, S.; Wang, X.; Koziol, J.A. Vascular matrix adhesion and the blood-brain barrier. Biochem. Soc. Trans. 2006, 34, 1261–1266. [Google Scholar] [CrossRef]
- Sorokin, L. The impact of the extracellular matrix on inflammation. Nat. Rev. Immunol. 2010, 10, 712–723. [Google Scholar] [CrossRef]
- Ando, Y.; Okada, H.; Takemura, G.; Suzuki, K.; Takada, C.; Tomita, H.; Zaikokuji, R.; Hotta, Y.; Miyazaki, N.; Yano, H.; et al. Brain-Specific Ultrastructure of Capillary Endothelial Glycocalyx and Its Possible Contribution for Blood Brain Barrier. Sci. Rep. 2018, 8, 17523. [Google Scholar] [CrossRef] [PubMed]
- Strazielle, N.; Ghersi-Egea, J.F. Physiology of blood-brain interfaces in relation to brain disposition of small compounds and macromolecules. Mol. Pharm. 2013, 10, 1473–1491. [Google Scholar] [CrossRef] [PubMed]
- Daneman, R.; Prat, A. The blood-brain barrier. Cold Spring Harb. Perspect. Biol. 2015, 7, a020412. [Google Scholar] [CrossRef] [PubMed]
- Banks, W.A. From blood-brain barrier to blood-brain interface: New opportunities for CNS drug delivery. Nat. Rev. Drug Discov. 2016, 15, 275–292. [Google Scholar] [CrossRef] [PubMed]
- Yurchenco, P.D.; Patton, B.L. Developmental and pathogenic mechanisms of basement membrane assembly. Curr. Pharm. Des. 2009, 15, 1277–1294. [Google Scholar] [CrossRef] [PubMed]
- Diaz-Castro, B.; Robel, S.; Mishra, A. Astrocyte Endfeet in Brain Function and Pathology: Open Questions. Annu. Rev. Neurosci. 2023, 46, 101–121. [Google Scholar] [CrossRef]
- Coelho-Santos, V.; Berthiaume, A.A.; Ornelas, S.; Stuhlmann, H.; Shih, A.Y. Imaging the construction of capillary networks in the neonatal mouse brain. Proc. Natl. Acad. Sci. USA 2021, 118, e2100866118. [Google Scholar] [CrossRef]
- Bushong, E.A.; Martone, M.E.; Ellisman, M.H. Maturation of astrocyte morphology and the establishment of astrocyte domains during postnatal hippocampal development. Int. J. Dev. Neurosci. 2004, 22, 73–86. [Google Scholar] [CrossRef]
- Voutsinos-Porche, B.; Bonvento, G.; Tanaka, K.; Steiner, P.; Welker, E.; Chatton, J.Y.; Magistretti, P.J.; Pellerin, L. Glial glutamate transporters mediate a functional metabolic crosstalk between neurons and astrocytes in the mouse developing cortex. Neuron 2003, 37, 275–286. [Google Scholar] [CrossRef]
- Nedergaard, M.; Ransom, B.; Goldman, S.A. New roles for astrocytes: Redefining the functional architecture of the brain. Trends Neurosci. 2003, 26, 523–530. [Google Scholar] [CrossRef]
- Jessen, N.A.; Munk, A.S.; Lundgaard, I.; Nedergaard, M. The Glymphatic System: A Beginner’s Guide. Neurochem. Res. 2015, 40, 2583–2599. [Google Scholar] [CrossRef]
- Brown, L.S.; Foster, C.G.; Courtney, J.M.; King, N.E.; Howells, D.W.; Sutherland, B.A. Pericytes and Neurovascular Function in the Healthy and Diseased Brain. Front. Cell Neurosci. 2019, 13, 282. [Google Scholar] [CrossRef] [PubMed]
- Haruwaka, K.; Ikegami, A.; Tachibana, Y.; Ohno, N.; Konishi, H.; Hashimoto, A.; Matsumoto, M.; Kato, D.; Ono, R.; Kiyama, H.; et al. Dual microglia effects on blood brain barrier permeability induced by systemic inflammation. Nat. Commun. 2019, 10, 5816. [Google Scholar] [CrossRef]
- Ikegami, A.; Haruwaka, K.; Wake, H. Microglia: Lifelong modulator of neural circuits. Neuropathology 2019, 39, 173–180. [Google Scholar] [CrossRef] [PubMed]
- Gullotta, G.S.; Costantino, G.; Sortino, M.A.; Spampinato, S.F. Microglia and the Blood-Brain Barrier: An External Player in Acute and Chronic Neuroinflammatory Conditions. Int. J. Mol. Sci. 2023, 24, 9144. [Google Scholar] [CrossRef]
- Sorokin, V.; Vickneson, K.; Kofidis, T.; Woo, C.C.; Lin, X.Y.; Foo, R.; Shanahan, C.M. Role of Vascular Smooth Muscle Cell Plasticity and Interactions in Vessel Wall Inflammation. Front. Immunol. 2020, 11, 599415. [Google Scholar] [CrossRef] [PubMed]
- Aldea, R.; Weller, R.O.; Wilcock, D.M.; Carare, R.O.; Richardson, G. Cerebrovascular Smooth Muscle Cells as the Drivers of Intramural Periarterial Drainage of the Brain. Front. Aging Neurosci. 2019, 11, 1. [Google Scholar] [CrossRef]
- Cheng, J.; Gu, J.; Ma, Y.; Yang, T.; Kuang, Y.; Li, B.; Kang, J. Development of a rat model for studying blast-induced traumatic brain injury. J. Neurol. Sci. 2010, 294, 23–28. [Google Scholar] [CrossRef]
- Reneer, D.V.; Hisel, R.D.; Hoffman, J.M.; Kryscio, R.J.; Lusk, B.T.; Geddes, J.W. A multi-mode shock tube for investigation of blast-induced traumatic brain injury. J. Neurotrauma 2011, 28, 95–104. [Google Scholar] [CrossRef]
- Rafaels, K.A.; Bass, C.R.; Panzer, M.B.; Salzar, R.S.; Woods, W.A.; Feldman, S.H.; Walilko, T.; Kent, R.W.; Capehart, B.P.; Foster, J.B.; et al. Brain injury risk from primary blast. J. Trauma. Acute Care Surg. 2012, 73, 895–901. [Google Scholar] [CrossRef]
- Li, B.C.; Li, Y.; Xu, C.; Wang, J.; Chen, Z.; Li, G.; Zhang, J.; Hu, S.; Wang, L.; Feng, H. Blast-induced traumatic brain injury of goats in confined space. Neurol. Res. 2014, 36, 974–982. [Google Scholar] [CrossRef]
- Kuehn, R.; Simard, P.F.; Driscoll, I.; Keledjian, K.; Ivanova, S.; Tosun, C.; Williams, A.; Bochicchio, G.; Gerzanich, V.; Simard, J.M. Rodent model of direct cranial blast injury. J. Neurotrauma 2011, 28, 2155–2169. [Google Scholar] [CrossRef]
- Wang, Y.; Wei, Y.; Oguntayo, S.; Wilkins, W.; Arun, P.; Valiyaveettil, M.; Song, J.; Long, J.B.; Nambiar, M.P. Tightly coupled repetitive blast-induced traumatic brain injury: Development and characterization in mice. J. Neurotrauma 2011, 28, 2171–2183. [Google Scholar] [CrossRef] [PubMed]
- Cernak, I. Blast-induced neurotrauma models and their requirements. Front. Neurol. 2014, 5, 128. [Google Scholar] [CrossRef]
- Josey, T.; Ouellet, S.; Bieler, D.; Cernak, I.; Franke, A.; Gupta, R.; Kirkman, E.; Leggieri, M.J., Jr.; Orru, H.; Philippens, M.; et al. Guidelines for reproducing blast exposures in the laboratory. J. R. Army Med. Corps 2019, 165, 10–14. [Google Scholar] [CrossRef] [PubMed]
- Watts, S.; Kirkman, E.; Bieler, D.; Bjarnason, S.; Franke, A.; Gupta, R.; Leggieri, M.J., Jr.; Orru, H.; Ouellet, S.; Philippens, M.; et al. Guidelines for using animal models in blast injury research. J. R. Army Med. Corps 2019, 165, 38–40. [Google Scholar] [CrossRef] [PubMed]
- Kawa, L.; Arborelius, U.; Yoshitake, T.; Kehr, J.; Hokfelt, T.; Risling, M.; Agoston, D.V. Neurotransmitter systems in a mild blast traumatic brain injury model: Catecholamines and serotonin. J. Neurotrauma 2015, 32, 1190–1199. [Google Scholar] [CrossRef]
- Goldstein, L.E.; Fisher, A.M.; Tagge, C.A.; Zhang, X.L.; Velisek, L.; Sullivan, J.A.; Upreti, C.; Kracht, J.M.; Ericsson, M.; Wojnarowicz, M.W.; et al. Chronic traumatic encephalopathy in blast-exposed military veterans and a blast neurotrauma mouse model. Sci. Transl. Med. 2012, 4, 134ra60. [Google Scholar]
- Rubovitch, V.; Ten-Bosch, M.; Zohar, O.; Harrison, C.R.; Tempel-Brami, C.; Stein, E.; Hoffer, B.J.; Balaban, C.D.; Schreiber, S.; Chiu, W.T.; et al. A mouse model of blast-induced mild traumatic brain injury. Exp. Neurol. 2011, 232, 280–289. [Google Scholar] [CrossRef]
- Li, Y.; Yang, Z.; Liu, B.; Valdez, C.; Chavko, M.; Cancio, L.C. Low-Level Primary Blast Induces Neuroinflammation and Neurodegeneration in Rats. Mil. Med. 2019, 184 (Suppl. 1), 265–272. [Google Scholar] [CrossRef] [PubMed]
- Ravula, A.R.; Rodriguez, J.; Younger, D.; Perumal, V.; Shao, N.; Rama Rao, K.V.; Pfister, B.; Chandra, N. Animal model of repeated low-level blast traumatic brain injury displays acute and chronic neurobehavioral and neuropathological changes. Exp. Neurol. 2022, 349, 113938. [Google Scholar] [CrossRef]
- Li, C.; Chen, S.; Siedhoff, H.R.; Grant, D.; Liu, P.; Balderrama, A.; Jackson, M.; Zuckerman, A.; Greenlief, C.M.; Kobeissy, F.; et al. Low-intensity open-field blast exposure effects on neurovascular unit ultrastructure in mice. Acta Neuropathol. Commun. 2023, 11, 144. [Google Scholar] [CrossRef] [PubMed]
- Chen, M.; Song, H.; Cui, J.; Johnson, C.E.; Hubler, G.K.; DePalma, R.G.; Gu, Z.; Xia, W. Proteomic Profiling of Mouse Brains Exposed to Blast-Induced Mild Traumatic Brain Injury Reveals Changes in Axonal Proteins and Phosphorylated Tau. J. Alzheimers Dis. 2018, 66, 751–773. [Google Scholar] [CrossRef] [PubMed]
- Konan, L.M.; Song, H.; Pentecost, G.; Fogwe, D.; Ndam, T.; Cui, J.; Johnson, C.E.; Grant, D.; White, T.; Chen, M.; et al. Multi-Focal Neuronal Ultrastructural Abnormalities and Synaptic Alterations in Mice after Low-Intensity Blast Exposure. J. Neurotrauma 2019, 36, 2117–2128. [Google Scholar] [CrossRef] [PubMed]
- Song, H.; Konan, L.M.; Cui, J.; Johnson, C.E.; Langenderfer, M.; Grant, D.; Ndam, T.; Simonyi, A.; White, T.; Demirci, U.; et al. Ultrastructural brain abnormalities and associated behavioral changes in mice after low-intensity blast exposure. Behav. Brain Res. 2018, 347, 148–157. [Google Scholar] [CrossRef] [PubMed]
- Nishii, K.; Satoh, Y.; Higashi, T.; Matsui, T.; Ishizuka, T.; Kashitani, M.; Saitoh, D.; Kobayashi, Y. Evans Blue and Fluorescein Isothiocyanate-Dextran Double Labeling Reveals Precise Sequence of Vascular Leakage and Glial Responses After Exposure to Mild-Level Blast-Associated Shock Waves. J. Neurotrauma 2023, 40, 1228–1242. [Google Scholar] [CrossRef]
- Satoh, Y.; Araki, Y.; Kashitani, M.; Nishii, K.; Kobayashi, Y.; Fujita, M.; Suzuki, S.; Morimoto, Y.; Tokuno, S.; Tsumatori, G.; et al. Molecular Hydrogen Prevents Social Deficits and Depression-Like Behaviors Induced by Low-Intensity Blast in Mice. J. Neuropathol. Exp. Neurol. 2018, 77, 827–836. [Google Scholar] [CrossRef]
- Hubbard, W.B.; Velmurugan, G.V.; Brown, E.P.; Sullivan, P.G. Resilience of females to acute blood-brain barrier damage and anxiety behavior following mild blast traumatic brain injury. Acta Neuropathol. Commun. 2022, 10, 93. [Google Scholar] [CrossRef]
- Hubbard, W.B.; Vekaria, H.J.; Velmurugan, G.V.; Kalimon, O.J.; Prajapati, P.; Brown, E.; Geisler, J.G.; Sullivan, P.G. Mitochondrial Dysfunction after Repeated Mild Blast Traumatic Brain Injury Is Attenuated by a Mild Mitochondrial Uncoupling Prodrug. J. Neurotrauma 2023, 40, 2396–2409. [Google Scholar] [CrossRef]
- Heyburn, L.; Batuure, A.; Wilder, D.; Long, J.; Sajja, V.S. Neuroinflammation Profiling of Brain Cytokines Following Repeated Blast Exposure. Int. J. Mol. Sci. 2023, 24, 12564. [Google Scholar] [CrossRef]
- Courtney, A.C.; Courtney, M.W. A thoracic mechanism of mild traumatic brain injury due to blast pressure waves. Med. Hypotheses 2009, 72, 76–83. [Google Scholar] [CrossRef] [PubMed]
- Cernak, I. The importance of systemic response in the pathobiology of blast-induced neurotrauma. Front. Neurol. 2010, 1, 151. [Google Scholar] [CrossRef] [PubMed]
- Bhattacharjee, Y. Neuroscience. Shell shock revisited: Solving the puzzle of blast trauma. Science 2008, 319, 406–408. [Google Scholar] [CrossRef]
- Chen, Y.; Huang, W.; Constantini, S. Concepts and strategies for clinical management of blast-induced traumatic brain injury and posttraumatic stress disorder. J. Neuropsychiatry Clin. Neurosci. 2013, 25, 103–110. [Google Scholar] [CrossRef] [PubMed]
- Rubio, J.E.; Skotak, M.; Alay, E.; Sundaramurthy, A.; Subramaniam, D.R.; Kote, V.B.; Yeoh, S.; Monson, K.; Chandra, N.; Unnikrishnan, G.; et al. Does Blast Exposure to the Torso Cause a Blood Surge to the Brain? Front. Bioeng. Biotechnol. 2020, 8, 573647. [Google Scholar] [CrossRef] [PubMed]
- Long, J.B.; Bentley, T.L.; Wessner, K.A.; Cerone, C.; Sweeney, S.; Bauman, R.A. Blast overpressure in rats: Recreating a battlefield injury in the laboratory. J. Neurotrauma 2009, 26, 827–840. [Google Scholar] [CrossRef] [PubMed]
- Koliatsos, V.E.; Cernak, I.; Xu, L.; Song, Y.; Savonenko, A.; Crain, B.J.; Eberhart, C.G.; Frangakis, C.E.; Melnikova, T.; Kim, H.; et al. A mouse model of blast injury to brain: Initial pathological, neuropathological, and behavioral characterization. J. Neuropathol. Exp. Neurol. 2011, 70, 399–416. [Google Scholar] [CrossRef]
- Simard, J.M.; Pampori, A.; Keledjian, K.; Tosun, C.; Schwartzbauer, G.; Ivanova, S.; Gerzanich, V. Exposure of the thorax to a sublethal blast wave causes a hydrodynamic pulse that leads to perivenular inflammation in the brain. J. Neurotrauma 2014, 31, 1292–1304. [Google Scholar] [CrossRef]
- Yuan, W.; Barber Foss, K.D.; Dudley, J.; Thomas, S.; Galloway, R.; DiCesare, C.; Leach, J.; Scheifele, P.; Farina, M.; Valencia, G.; et al. Impact of Low-Level Blast Exposure on Brain Function after a One-Day Tactile Training and the Ameliorating Effect of a Jugular Vein Compression Neck Collar Device. J. Neurotrauma 2019, 36, 721–734. [Google Scholar] [CrossRef]
- Reitsma, S.; Slaaf, D.W.; Vink, H.; van Zandvoort, M.A.; oude Egbrink, M.G. The endothelial glycocalyx: Composition, functions, and visualization. Pflugers Arch. 2007, 454, 345–359. [Google Scholar] [CrossRef]
- Zhu, J.; Li, X.; Yin, J.; Hu, Y.; Gu, Y.; Pan, S. Glycocalyx degradation leads to blood-brain barrier dysfunction and brain edema after asphyxia cardiac arrest in rats. J. Cereb. Blood Flow. Metab. 2018, 38, 1979–1992. [Google Scholar] [CrossRef]
- Curry, F.E.; Adamson, R.H. Endothelial glycocalyx: Permeability barrier and mechanosensor. Ann. Biomed. Eng. 2012, 40, 828–839. [Google Scholar] [CrossRef] [PubMed]
- Florian, J.A.; Kosky, J.R.; Ainslie, K.; Pang, Z.; Dull, R.O.; Tarbell, J.M. Heparan sulfate proteoglycan is a mechanosensor on endothelial cells. Circ. Res. 2003, 93, e136–e142. [Google Scholar] [CrossRef] [PubMed]
- Schmidt, E.P.; Yang, Y.; Janssen, W.J.; Gandjeva, A.; Perez, M.J.; Barthel, L.; Zemans, R.L.; Bowman, J.C.; Koyanagi, D.E.; Yunt, Z.X.; et al. The pulmonary endothelial glycocalyx regulates neutrophil adhesion and lung injury during experimental sepsis. Nat. Med. 2012, 18, 1217–1223. [Google Scholar] [CrossRef]
- Hall, A.A.; Mendoza, M.I.; Zhou, H.; Shaughness, M.; Maudlin-Jeronimo, E.; McCarron, R.M.; Ahlers, S.T. Repeated Low Intensity Blast Exposure Is Associated with Damaged Endothelial Glycocalyx and Downstream Behavioral Deficits. Front. Behav. Neurosci. 2017, 11, 104. [Google Scholar] [CrossRef] [PubMed]
- Hall, A.A.; Mendoza, M.I.; Zhou, H.; Shaughness, M.; McCarron, R.M.; Ahlers, S.T. Corrigendum: Repeated Low Intensity Blast Exposure Is Associated With Damaged Endothelial Glycocalyx and Downstream Behavioral Deficits. Front. Behav. Neurosci. 2019, 13, 251. [Google Scholar] [CrossRef] [PubMed]
- Armonda, R.A.; Bell, R.S.; Vo, A.H.; Ling, G.; DeGraba, T.J.; Crandall, B.; Ecklund, J.; Campbell, W.W. Wartime traumatic cerebral vasospasm: Recent review of combat casualties. Neurosurgery 2006, 59, 1215–1225. [Google Scholar] [CrossRef] [PubMed]
- Bauman, R.A.; Ling, G.; Tong, L.; Januszkiewicz, A.; Agoston, D.; Delanerolle, N.; Kim, Y.; Ritzel, D.; Bell, R.; Ecklund, J.; et al. An introductory characterization of a combat-casualty-care relevant swine model of closed head injury resulting from exposure to explosive blast. J. Neurotrauma 2009, 26, 841–860. [Google Scholar] [CrossRef]
- Bir, C.; Vandevord, P.; Shen, Y.; Raza, W.; Haacke, E.M. Effects of variable blast pressures on blood flow and oxygen saturation in rat brain as evidenced using MRI. Magn. Reson. Imaging 2012, 30, 527–534. [Google Scholar] [CrossRef]
- Rodriguez, U.A.; Zeng, Y.; Deyo, D.; Parsley, M.A.; Hawkins, B.E.; Prough, D.S.; DeWitt, D.S. Effects of Mild Blast Traumatic Brain Injury on Cerebral Vascular, Histopathological, and Behavioral Outcomes in Rats. J. Neurotrauma 2018, 35, 375–392. [Google Scholar] [CrossRef]
- Toklu, H.Z.; Yang, Z.; Oktay, S.; Sakarya, Y.; Kirichenko, N.; Matheny, M.K.; Muller-Delp, J.; Strang, K.; Scarpace, P.J.; Wang, K.K.W.; et al. Overpressure blast injury-induced oxidative stress and neuroinflammation response in rat frontal cortex and cerebellum. Behav. Brain Res. 2018, 340, 14–22. [Google Scholar] [CrossRef] [PubMed]
- Abutarboush, R.; Gu, M.; Kawoos, U.; Mullah, S.H.; Chen, Y.; Goodrich, S.Y.; Lashof-Sullivan, M.; McCarron, R.M.; Statz, J.K.; Bell, R.S.; et al. Exposure to Blast Overpressure Impairs Cerebral Microvascular Responses and Alters Vascular and Astrocytic Structure. J. Neurotrauma 2019, 36, 3138–3157. [Google Scholar] [CrossRef]
- Alford, P.W.; Dabiri, B.E.; Goss, J.A.; Hemphill, M.A.; Brigham, M.D.; Parker, K.K. Blast-induced phenotypic switching in cerebral vasospasm. Proc. Natl. Acad. Sci. USA 2011, 108, 12705–12710. [Google Scholar] [CrossRef]
- Hald, E.S.; Alford, P.W. Smooth muscle phenotype switching in blast traumatic brain injury-induced cerebral vasospasm. Transl. Stroke Res. 2014, 5, 385–393. [Google Scholar] [CrossRef] [PubMed]
- Boulay, A.C.; Saubamea, B.; Decleves, X.; Cohen-Salmon, M. Purification of Mouse Brain Vessels. J. Vis. Exp. 2015, 105, e53208. [Google Scholar]
- Chun, H.B.; Scott, M.; Niessen, S.; Hoover, H.; Baird, A.; Yates, J., 3rd; Torbett, B.E.; Eliceiri, B.P. The proteome of mouse brain microvessel membranes and basal lamina. J. Cereb. Blood Flow. Metab. 2011, 31, 2267–2281. [Google Scholar] [CrossRef]
- Searcy, J.L.; Le Bihan, T.; Salvadores, N.; McCulloch, J.; Horsburgh, K. Impact of age on the cerebrovascular proteomes of wild-type and Tg-SwDI mice. PLoS ONE 2014, 9, e89970. [Google Scholar] [CrossRef]
- Logsdon, A.F.; Meabon, J.S.; Cline, M.M.; Bullock, K.M.; Raskind, M.A.; Peskind, E.R.; Banks, W.A.; Cook, D.G. Blast exposure elicits blood-brain barrier disruption and repair mediated by tight junction integrity and nitric oxide dependent processes. Sci. Rep. 2018, 8, 11344. [Google Scholar] [CrossRef]
- Kaur, C.; Singh, J.; Lim, M.K.; Ng, B.L.; Ling, E.A. Macrophages/microglia as ‘sensors’ of injury in the pineal gland of rats following a non-penetrative blast. Neurosci. Res. 1997, 27, 317–322. [Google Scholar] [CrossRef]
- Lu, J.; Ng, K.C.; Ling, G.; Wu, J.; Poon, D.J.; Kan, E.M.; Tan, M.H.; Wu, Y.J.; Li, P.; Moochhala, S.; et al. Effect of blast exposure on the brain structure and cognition in Macaca fascicularis. J. Neurotrauma 2012, 29, 1434–1454. [Google Scholar] [CrossRef]
- Agoston, D.V.; McCullough, J.; Aniceto, R.; Lin, I.H.; Kamnaksh, A.; Eklund, M.; Graves, W.M., 3rd; Dunbar, C.; Engall, J.; Schneider, E.B.; et al. Blood-Based Biomarkers of Repetitive, Subconcussive Blast Overpressure Exposure in the Training Environment: A Pilot Study. Neurotrauma Rep. 2022, 3, 479–490. [Google Scholar] [CrossRef] [PubMed]
- Shively, S.B.; Horkayne-Szakaly, I.; Jones, R.V.; Kelly, J.P.; Armstrong, R.C.; Perl, D.P. Characterisation of interface astroglial scarring in the human brain after blast exposure: A post-mortem case series. Lancet Neurol. 2016, 15, 944–953. [Google Scholar] [CrossRef] [PubMed]
- Janzer, R.C.; Raff, M.C. Astrocytes induce blood-brain barrier properties in endothelial cells. Nature 1987, 325, 253–257. [Google Scholar] [CrossRef]
- Obermeier, B.; Verma, A.; Ransohoff, R.M. The blood-brain barrier. Handb. Clin. Neurol. 2016, 133, 39–59. [Google Scholar]
- Spampinato, S.F.; Bortolotto, V.; Canonico, P.L.; Sortino, M.A.; Grilli, M. Astrocyte-Derived Paracrine Signals: Relevance for Neurogenic Niche Regulation and Blood-Brain Barrier Integrity. Front. Pharmacol. 2019, 10, 1346. [Google Scholar] [CrossRef] [PubMed]
- Lecuyer, M.A.; Kebir, H.; Prat, A. Glial influences on BBB functions and molecular players in immune cell trafficking. Biochim. Biophys. Acta 2016, 1862, 472–482. [Google Scholar] [CrossRef] [PubMed]
- Kadry, H.; Noorani, B.; Cucullo, L. A blood-brain barrier overview on structure, function, impairment, and biomarkers of integrity. Fluids Barriers CNS 2020, 17, 69. [Google Scholar] [CrossRef]
- Heithoff, B.P.; George, K.K.; Phares, A.N.; Zuidhoek, I.A.; Munoz-Ballester, C.; Robel, S. Astrocytes are necessary for blood-brain barrier maintenance in the adult mouse brain. Glia 2021, 69, 436–472. [Google Scholar] [CrossRef]
- Schreiner, B.; Romanelli, E.; Liberski, P.; Ingold-Heppner, B.; Sobottka-Brillout, B.; Hartwig, T.; Chandrasekar, V.; Johannssen, H.; Zeilhofer, H.U.; Aguzzi, A.; et al. Astrocyte Depletion Impairs Redox Homeostasis and Triggers Neuronal Loss in the Adult CNS. Cell Rep. 2015, 12, 1377–1384. [Google Scholar] [CrossRef]
- Tsai, H.H.; Li, H.; Fuentealba, L.C.; Molofsky, A.V.; Taveira-Marques, R.; Zhuang, H.; Tenney, A.; Murnen, A.T.; Fancy, S.P.; Merkle, F.; et al. Regional astrocyte allocation regulates CNS synaptogenesis and repair. Science 2012, 337, 358–362. [Google Scholar] [CrossRef]
- Kubotera, H.; Ikeshima-Kataoka, H.; Hatashita, Y.; Allegra Mascaro, A.L.; Pavone, F.S.; Inoue, T. Astrocytic endfeet re-cover blood vessels after removal by laser ablation. Sci. Rep. 2019, 9, 1263. [Google Scholar] [CrossRef]
- Mills, W.A., 3rd; Woo, A.M.; Jiang, S.; Martin, J.; Surendran, D.; Bergstresser, M.; Kimbrough, I.F.; Eyo, U.B.; Sofroniew, M.V.; Sontheimer, H. Astrocyte plasticity in mice ensures continued endfoot coverage of cerebral blood vessels following injury and declines with age. Nat. Commun. 2022, 13, 1794. [Google Scholar] [CrossRef]
- Hamel, E. Perivascular nerves and the regulation of cerebrovascular tone. J Appl Physiol 2006, 100, 1059–1064. [Google Scholar] [CrossRef] [PubMed]
- Wardlaw, J.M.; Benveniste, H.; Nedergaard, M.; Zlokovic, B.V.; Mestre, H.; Lee, H.; Doubal, F.N.; Brown, R.; Ramirez, J.; MacIntosh, B.J.; et al. Perivascular spaces in the brain: Anatomy, physiology and pathology. Nat. Rev. Neurol. 2020, 16, 137–153. [Google Scholar] [CrossRef] [PubMed]
- Rasmussen, M.K.; Mestre, H.; Nedergaard, M. The glymphatic pathway in neurological disorders. Lancet Neurol. 2018, 17, 1016–1024. [Google Scholar] [CrossRef]
- Chakraborty, S.; ThimmaReddygari, J.; Selvaraj, D. G-lymphatic, vascular and immune pathways for Abeta clearance cascade and therapeutic targets for Alzheimer’s disease. Comb. Chem. High. Throughput Screen. 2020, 24, 1083–1092. [Google Scholar] [CrossRef] [PubMed]
- Iliff, J.J.; Chen, M.J.; Plog, B.A.; Zeppenfeld, D.M.; Soltero, M.; Yang, L.; Singh, I.; Deane, R.; Nedergaard, M. Impairment of glymphatic pathway function promotes tau pathology after traumatic brain injury. J. Neurosci. 2014, 34, 16180–16193. [Google Scholar] [CrossRef]
- Peng, W.; Achariyar, T.M.; Li, B.; Liao, Y.; Mestre, H.; Hitomi, E.; Regan, S.; Kasper, T.; Peng, S.; Ding, F.; et al. Suppression of glymphatic fluid transport in a mouse model of Alzheimer’s disease. Neurobiol. Dis. 2016, 93, 215–225. [Google Scholar] [CrossRef]
- Diem, A.K.; Carare, R.O.; Weller, R.O.; Bressloff, N.W. A control mechanism for intra-mural peri-arterial drainage via astrocytes: How neuronal activity could improve waste clearance from the brain. PLoS ONE 2018, 13, e0205276. [Google Scholar] [CrossRef]
- Gandy, S.; Ikonomovic, M.D.; Mitsis, E.; Elder, G.; Ahlers, S.T.; Barth, J.; Stone, J.R.; DeKosky, S.T. Chronic traumatic encephalopathy: Clinical-biomarker correlations and current concepts in pathogenesis. Mol. Neurodegener. 2014, 9, 37. [Google Scholar] [CrossRef]
- DeKosky, S.T.; Blennow, K.; Ikonomovic, M.D.; Gandy, S. Acute and chronic traumatic encephalopathies: Pathogenesis and biomarkers. Nat. Rev. Neurol. 2013, 9, 192–200. [Google Scholar] [CrossRef]
- DeKosky, S.T.; Ikonomovic, M.D.; Gandy, S. Traumatic brain injury: Football, warfare, and long-term effects. N. Engl. J. Med. 2010, 363, 1293–1296. [Google Scholar] [CrossRef] [PubMed]
- Uryu, K.; Chen, X.H.; Martinez, D.; Browne, K.D.; Johnson, V.E.; Graham, D.I.; Lee, V.M.; Trojanowski, J.Q.; Smith, D.H. Multiple proteins implicated in neurodegenerative diseases accumulate in axons after brain trauma in humans. Exp. Neurol. 2007, 208, 185–192. [Google Scholar] [CrossRef] [PubMed]
- Johnson, V.E.; Stewart, W.; Smith, D.H. Traumatic brain injury and amyloid-beta pathology: A link to Alzheimer’s disease? Nat. Rev. Neurosci. 2010, 11, 361–370. [Google Scholar] [CrossRef] [PubMed]
- Ikonomovic, M.D.; Uryu, K.; Abrahamson, E.E.; Ciallella, J.R.; Trojanowski, J.Q.; Lee, V.M.; Clark, R.S.; Marion, D.W.; Wisniewski, S.R.; DeKosky, S.T. Alzheimer’s pathology in human temporal cortex surgically excised after severe brain injury. Exp. Neurol. 2004, 190, 192–203. [Google Scholar] [CrossRef]
- DeKosky, S.T.; Abrahamson, E.E.; Ciallella, J.R.; Paljug, W.R.; Wisniewski, S.R.; Clark, R.S.; Ikonomovic, M.D. Association of increased cortical soluble abeta42 levels with diffuse plaques after severe brain injury in humans. Arch. Neurol. 2007, 64, 541–544. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Q.G.; Laird, M.D.; Han, D.; Nguyen, K.; Scott, E.; Dong, Y.; Dhandapani, K.M.; Brann, D.W. Critical role of NADPH oxidase in neuronal oxidative damage and microglia activation following traumatic brain injury. PLoS ONE 2012, 7, e34504. [Google Scholar] [CrossRef]
- Loane, D.J.; Pocivavsek, A.; Moussa, C.E.; Thompson, R.; Matsuoka, Y.; Faden, A.I.; Rebeck, G.W.; Burns, M.P. Amyloid precursor protein secretases as therapeutic targets for traumatic brain injury. Nat. Med. 2009, 15, 377–379. [Google Scholar] [CrossRef]
- Loane, D.J.; Washington, P.M.; Vardanian, L.; Pocivavsek, A.; Hoe, H.S.; Duff, K.E.; Cernak, I.; Rebeck, G.W.; Faden, A.I.; Burns, M.P. Modulation of ABCA1 by an LXR agonist reduces beta-amyloid levels and improves outcome after traumatic brain injury. J. Neurotrauma 2011, 28, 225–236. [Google Scholar] [CrossRef]
- Yu, F.; Zhang, Y.; Chuang, D.M. Lithium reduces BACE1 overexpression, beta amyloid accumulation, and spatial learning deficits in mice with traumatic brain injury. J. Neurotrauma 2012, 29, 2342–2351. [Google Scholar] [CrossRef]
- Tian, L.; Guo, R.; Yue, X.; Lv, Q.; Ye, X.; Wang, Z.; Chen, Z.; Wu, B.; Xu, G.; Liu, X. Intranasal administration of nerve growth factor ameliorate beta-amyloid deposition after traumatic brain injury in rats. Brain Res. 2012, 1440, 47–55. [Google Scholar] [CrossRef] [PubMed]
- Smith, D.H.; Nakamura, M.; McIntosh, T.K.; Wang, J.; Rodriguez, A.; Chen, X.H.; Raghupathi, R.; Saatman, K.E.; Clemens, J.; Schmidt, M.L.; et al. Brain trauma induces massive hippocampal neuron death linked to a surge in beta-amyloid levels in mice overexpressing mutant amyloid precursor protein. Am. J. Pathol. 1998, 153, 1005–1010. [Google Scholar] [CrossRef] [PubMed]
- Tajiri, N.; Kellogg, S.L.; Shimizu, T.; Arendash, G.W.; Borlongan, C.V. Traumatic brain injury precipitates cognitive impairment and extracellular Abeta aggregation in Alzheimer’s disease transgenic mice. PLoS ONE 2013, 8, e78851. [Google Scholar] [CrossRef] [PubMed]
- Tran, H.T.; LaFerla, F.M.; Holtzman, D.M.; Brody, D.L. Controlled cortical impact traumatic brain injury in 3xTg-AD mice causes acute intra-axonal amyloid-beta accumulation and independently accelerates the development of tau abnormalities. J. Neurosci. 2011, 31, 9513–9525. [Google Scholar] [CrossRef]
- Tran, H.T.; Sanchez, L.; Esparza, T.J.; Brody, D.L. Distinct temporal and anatomical distributions of amyloid-beta and tau abnormalities following controlled cortical impact in transgenic mice. PLoS ONE 2011, 6, e25475. [Google Scholar] [CrossRef] [PubMed]
- Uryu, K.; Laurer, H.; McIntosh, T.; Pratico, D.; Martinez, D.; Leight, S.; Lee, V.M.; Trojanowski, J.Q. Repetitive mild brain trauma accelerates Abeta deposition, lipid peroxidation, and cognitive impairment in a transgenic mouse model of Alzheimer amyloidosis. J. Neurosci. 2002, 22, 446–454. [Google Scholar] [CrossRef]
- Washington, P.M.; Morffy, N.; Parsadanian, M.; Zapple, D.N.; Burns, M.P. Experimental traumatic brain injury induces rapid aggregation and oligomerization of amyloid-beta in an Alzheimer’s disease mouse model. J. Neurotrauma 2014, 31, 125–134. [Google Scholar] [CrossRef]
- Webster, S.J.; Van Eldik, L.J.; Watterson, D.M.; Bachstetter, A.D. Closed head injury in an age-related Alzheimer mouse model leads to an altered neuroinflammatory response and persistent cognitive impairment. J. Neurosci. 2015, 35, 6554–6569. [Google Scholar] [CrossRef]
- Zohar, O.; Lavy, R.; Zi, X.; Nelson, T.J.; Hongpaisan, J.; Pick, C.G.; Alkon, D.L. PKC activator therapeutic for mild traumatic brain injury in mice. Neurobiol. Dis. 2011, 41, 329–337. [Google Scholar] [CrossRef]
- Blasko, I.; Beer, R.; Bigl, M.; Apelt, J.; Franz, G.; Rudzki, D.; Ransmayr, G.; Kampfl, A.; Schliebs, R. Experimental traumatic brain injury in rats stimulates the expression, production and activity of Alzheimer’s disease beta-secretase (BACE-1). J. Neural Transm. 2004, 111, 523–536. [Google Scholar] [CrossRef]
- Nadler, Y.; Alexandrovich, A.; Grigoriadis, N.; Hartmann, T.; Rao, K.S.; Shohami, E.; Stein, R. Increased expression of the gamma-secretase components presenilin-1 and nicastrin in activated astrocytes and microglia following traumatic brain injury. Glia 2008, 56, 552–567. [Google Scholar] [CrossRef]
- Chen, X.H.; Siman, R.; Iwata, A.; Meaney, D.F.; Trojanowski, J.Q.; Smith, D.H. Long-term accumulation of amyloid-beta, beta-secretase, presenilin-1, and caspase-3 in damaged axons following brain trauma. Am. J. Pathol. 2004, 165, 357–371. [Google Scholar] [CrossRef] [PubMed]
- Haass, C.; Hung, A.Y.; Schlossmacher, M.G.; Teplow, D.B.; Selkoe, D.J. Beta-Amyloid peptide and a 3-kDa fragment are derived by distinct cellular mechanisms. J. Biol. Chem. 1993, 268, 3021–3024. [Google Scholar] [CrossRef]
- Perez Garcia, G.; De Gasperi, R.; Tschiffely, A.E.; Gama Sosa, M.A.; Abutarboush, R.; Kawoos, U.; Statz, J.K.; Ciarlone, S.; Reed, E.; Jeyarajah, T.; et al. Repetitive Low-Level Blast Exposure Improves Behavioral Deficits and Chronically Lowers Abeta42 in an Alzheimer Disease Transgenic Mouse Model. J. Neurotrauma 2021, 38, 3146–3173. [Google Scholar] [CrossRef] [PubMed]
- Jankowsky, J.L.; Fadale, D.J.; Anderson, J.; Xu, G.M.; Gonzales, V.; Jenkins, N.A.; Copeland, N.G.; Lee, M.K.; Younkin, L.H.; Wagner, S.L.; et al. Mutant presenilins specifically elevate the levels of the 42 residue beta-amyloid peptide in vivo: Evidence for augmentation of a 42-specific gamma secretase. Hum. Mol. Genet. 2004, 13, 159–170. [Google Scholar] [CrossRef] [PubMed]
- Abutarboush, R.; Reed, E.; Chen, Y.; Gu, M.; Cameron, W.; Kawoos, U.; Statz, J.; Tschiffely, A.; Ciarlone, S.; Perez-Garcia, G.; et al. Exposure to low-intensity blast increases clearance of brain Aβ. J. Neurotruama 2023, Published online. [CrossRef]
- Harper, M.M.; Hedberg-Buenz, A.; Herlein, J.; Abrahamson, E.E.; Anderson, M.G.; Kuehn, M.H.; Kardon, R.H.; Poolman, P.; Ikonomovic, M.D. Blast-Mediated Traumatic Brain Injury Exacerbates Retinal Damage and Amyloidosis in the APPswePSENd19e Mouse Model of Alzheimer’s Disease. Invest. Ophthalmol. Vis. Sci. 2019, 60, 2716–2725. [Google Scholar] [CrossRef]
- Edwards, K.A.; Leete, J.J.; Tschiffely, A.E.; Moore, C.Y.; Dell, K.C.; Statz, J.K.; Carr, W.; Walker, P.B.; LoPresti, M.L.; Ahlers, S.T.; et al. Blast exposure results in tau and neurofilament light chain changes in peripheral blood. Brain Inj. 2020, 34, 1213–1221. [Google Scholar] [CrossRef]
- Gill, J.; Cashion, A.; Osier, N.; Arcurio, L.; Motamedi, V.; Dell, K.C.; Carr, W.; Kim, H.S.; Yun, S.; Walker, P.; et al. Moderate blast exposure alters gene expression and levels of amyloid precursor protein. Neurol. Genet. 2017, 3, e186. [Google Scholar] [CrossRef]
- Thangavelu, B.; LaValle, C.R.; Egnoto, M.J.; Nemes, J.; Boutte, A.M.; Kamimori, G.H. Overpressure Exposure from.50-Caliber Rifle Training Is Associated with Increased Amyloid Beta Peptides in Serum. Front. Neurol. 2020, 11, 620. [Google Scholar] [CrossRef]
- Boutte, A.M.; Thangavelu, B.; Nemes, J.; LaValle, C.R.; Egnoto, M.; Carr, W.; Kamimori, G.H. Neurotrauma Biomarker Levels and Adverse Symptoms Among Military and Law Enforcement Personnel Exposed to Occupational Overpressure Without Diagnosed Traumatic Brain Injury. JAMA Netw. Open 2021, 4, e216445. [Google Scholar] [CrossRef] [PubMed]
- McKee, A.C.; Robinson, M.E. Military-related traumatic brain injury and neurodegeneration. Alzheimers Dement. 2014, 10 (Suppl. 3), S242–S253. [Google Scholar] [CrossRef] [PubMed]
- Omalu, B.; Hammers, J.L.; Bailes, J.; Hamilton, R.L.; Kamboh, M.I.; Webster, G.; Fitzsimmons, R.P. Chronic traumatic encephalopathy in an Iraqi war veteran with posttraumatic stress disorder who committed suicide. Neurosurg. Focus. 2011, 31, E3. [Google Scholar] [CrossRef] [PubMed]
- Olivera, A.; Lejbman, N.; Jeromin, A.; French, L.M.; Kim, H.S.; Cashion, A.; Mysliwiec, V.; Diaz-Arrastia, R.; Gill, J. Peripheral Total Tau in Military Personnel Who Sustain Traumatic Brain Injuries During Deployment. JAMA Neurol. 2015, 72, 1109–1116. [Google Scholar] [CrossRef] [PubMed]
- Edwards, K.A.; Greer, K.; Leete, J.; Lai, C.; Devoto, C.; Qu, B.X.; Yarnell, A.M.; Polejaeva, E.; Dell, K.C.; LoPresti, M.L.; et al. Neuronally-derived tau is increased in experienced breachers and is associated with neurobehavioral symptoms. Sci. Rep. 2021, 11, 19527. [Google Scholar] [CrossRef] [PubMed]
- Mohamed, A.Z.; Cumming, P.; Gotz, J.; Nasrallah, F.; Department of Defense Alzheimer’s Disease Neuroimaging Initiative. Tauopathy in veterans with long-term posttraumatic stress disorder and traumatic brain injury. Eur. J. Nucl. Med. Mol. Imaging 2019, 46, 1139–1151. [Google Scholar] [CrossRef] [PubMed]
- Robinson, M.E.; McKee, A.C.; Salat, D.H.; Rasmusson, A.M.; Radigan, L.J.; Catana, C.; Milberg, W.P.; McGlinchey, R.E. Positron emission tomography of tau in Iraq and Afghanistan Veterans with blast neurotrauma. Neuroimage Clin. 2019, 21, 101651. [Google Scholar] [CrossRef]
- Priemer, D.S.; Iacono, D.; Rhodes, C.H.; Olsen, C.H.; Perl, D.P. Chronic Traumatic Encephalopathy in the Brains of Military Personnel. N. Engl. J. Med. 2022, 386, 2169–2177. [Google Scholar] [CrossRef]
- Johnson, G.V.; Stoothoff, W.H. Tau phosphorylation in neuronal cell function and dysfunction. J. Cell Sci. 2004, 117 Pt 24, 5721–5729. [Google Scholar] [CrossRef]
- Wang, Y.; Mandelkow, E. Tau in physiology and pathology. Nat. Rev. Neurosci. 2016, 17, 5–21. [Google Scholar] [CrossRef]
- Goedert, M.; Jakes, R.; Crowther, R.A.; Cohen, P.; Vanmechelen, E.; Vandermeeren, M.; Cras, P. Epitope mapping of monoclonal antibodies to the paired helical filaments of Alzheimer’s disease: Identification of phosphorylation sites in tau protein. Biochem. J. 1994, 301 Pt 3, 871–877. [Google Scholar] [CrossRef] [PubMed]
- Perez Garcia, G.; De Gasperi, R.; Gama Sosa, M.A.; Perez, G.M.; Otero-Pagan, A.; Pryor, D.; Abutarboush, R.; Kawoos, U.; Hof, P.R.; Dickstein, D.L.; et al. Laterality and region-specific tau phosphorylation correlate with PTSD-related behavioral traits in rats exposed to repetitive low-level blast. Acta Neuropathol. Commun. 2021, 9, 33. [Google Scholar] [CrossRef] [PubMed]
- Denenberg, V. Hemispheric laterality in animals and the effects of early experience. Behav. Brain Sci. 1981, 4, 1–49. [Google Scholar] [CrossRef]
- Klur, S.; Muller, C.; Pereira de Vasconcelos, A.; Ballard, T.; Lopez, J.; Galani, R.; Certa, U.; Cassel, J.C. Hippocampal-dependent spatial memory functions might be lateralized in rats: An approach combining gene expression profiling and reversible inactivation. Hippocampus 2009, 19, 800–816. [Google Scholar] [CrossRef] [PubMed]
- Shinohara, Y.; Hosoya, A.; Yamasaki, N.; Ahmed, H.; Hattori, S.; Eguchi, M.; Yamaguchi, S.; Miyakawa, T.; Hirase, H.; Shigemoto, R. Right-hemispheric dominance of spatial memory in split-brain mice. Hippocampus 2012, 22, 117–121. [Google Scholar] [CrossRef] [PubMed]
- Schindler, A.G.; Meabon, J.S.; Pagulayan, K.F.; Hendrickson, R.C.; Meeker, K.D.; Cline, M.; Li, G.; Sikkema, C.; Wilkinson, C.W.; Perl, D.P.; et al. Blast-related disinhibition and risk seeking in mice and combat Veterans: Potential role for dysfunctional phasic dopamine release. Neurobiol. Dis. 2017, 106, 23–34. [Google Scholar] [CrossRef] [PubMed]
- Castellano, M.A.; Diaz-Palarea, M.D.; Rodriguez, M.; Barroso, J. Lateralization in male rats and dopaminergic system: Evidence of right-side population bias. Physiol. Behav. 1987, 40, 607–612. [Google Scholar] [CrossRef] [PubMed]
- Berridge, C.; Espana, R.; Stalnaker, T. Stress and coping: Asymmetry of dopamine efferents within the prefrontal cortex. In The Asymmetrical Brain; Hugdahl, K., Davidison, R., Eds.; MIT Press: Cambridge, MA, USA, 2003; pp. 69–103. [Google Scholar]
- Sullivan, R.M.; Dufresne, M.M.; Siontas, D.; Chehab, S.; Townsend, J.; Laplante, F. Mesocortical dopamine depletion and anxiety-related behavior in the rat: Sex and hemisphere differences. Prog. Neuropsychopharmacol. Biol. Psychiatry 2014, 54, 59–66. [Google Scholar] [CrossRef]
- Liberzon, I.; Sripada, C.S. The functional neuroanatomy of PTSD: A critical review. Prog. Brain Res. 2008, 167, 151–169. [Google Scholar]
- Mahan, A.L.; Ressler, K.J. Fear conditioning, synaptic plasticity and the amygdala: Implications for posttraumatic stress disorder. Trends Neurosci. 2012, 35, 24–35. [Google Scholar] [CrossRef]
- Liberzon, I.; Abelson, J.L. Context Processing and the Neurobiology of Post-Traumatic Stress Disorder. Neuron 2016, 92, 14–30. [Google Scholar] [CrossRef] [PubMed]
- Mate De Gerando, A.; Welikovitch, L.A.; Khasnavis, A.; Commins, C.; Glynn, C.; Chun, J.E.; Perbet, R.; Hyman, B.T. Tau seeding and spreading in vivo is supported by both AD-derived fibrillar and oligomeric tau. Acta Neuropathol. 2023, 146, 191–210. [Google Scholar] [CrossRef] [PubMed]
- Stern, A.M.; Selkoe, D.J. Soluble oligomers or insoluble fibrils? Scientific commentary on “Tau seeding and spreading in vivo is supported by both AD-derived fibrillar and oligomeric tau”. Acta Neuropathol. 2023, 146, 861–862. [Google Scholar] [CrossRef]
- Mothes, T.; Portal, B.; Konstantinidis, E.; Eltom, K.; Libard, S.; Streubel-Gallasch, L.; Ingelsson, M.; Rostami, J.; Lindskog, M.; Erlandsson, A. Astrocytic uptake of neuronal corpses promotes cell-to-cell spreading of tau pathology. Acta Neuropathol. Commun. 2023, 11, 97. [Google Scholar] [CrossRef]
- Lopes, S.; Vaz-Silva, J.; Pinto, V.; Dalla, C.; Kokras, N.; Bedenk, B.; Mack, N.; Czisch, M.; Almeida, O.F.; Sousa, N.; et al. Tau protein is essential for stress-induced brain pathology. Proc. Natl. Acad. Sci. USA 2016, 113, E3755–E3763. [Google Scholar] [CrossRef]
- Lopes, S.; Teplytska, L.; Vaz-Silva, J.; Dioli, C.; Trindade, R.; Morais, M.; Webhofer, C.; Maccarrone, G.; Almeida, O.F.; Turck, C.W.; et al. Tau Deletion Prevents Stress-Induced Dendritic Atrophy in Prefrontal Cortex: Role of Synaptic Mitochondria. Cereb. Cortex 2016, 27, 2580–2591. [Google Scholar] [CrossRef]
- McKee, A.C. The Neuropathology of Chronic Traumatic Encephalopathy: The Status of the Literature. Semin. Neurol. 2020, 40, 359–369. [Google Scholar] [CrossRef]
- McKee, A.C.; Cairns, N.J.; Dickson, D.W.; Folkerth, R.D.; Keene, C.D.; Litvan, I.; Perl, D.P.; Stein, T.D.; Vonsattel, J.P.; Stewart, W.; et al. The first NINDS/NIBIB consensus meeting to define neuropathological criteria for the diagnosis of chronic traumatic encephalopathy. Acta Neuropathol. 2016, 131, 75–86. [Google Scholar] [CrossRef]
- Butler, M.; Dixon, E.; Stein, T.D.; Alvarez, V.E.; Huber, B.; Buckland, M.E.; McKee, A.C.; Cherry, J.D. Tau Pathology in Chronic Traumatic Encephalopathy is Primarily Neuronal. J. Neuropathol. Exp. Neurol. 2022, 81, 773–780. [Google Scholar] [CrossRef]
- Huber, B.R.; Meabon, J.S.; Martin, T.J.; Mourad, P.D.; Bennett, R.; Kraemer, B.C.; Cernak, I.; Petrie, E.C.; Emery, M.J.; Swenson, E.R.; et al. Blast exposure causes early and persistent aberrant phospho- and cleaved-tau expression in a murine model of mild blast-induced traumatic brain injury. J. Alzheimers Dis. 2013, 37, 309–323. [Google Scholar] [CrossRef]
- Huber, B.R.; Meabon, J.S.; Hoffer, Z.S.; Zhang, J.; Hoekstra, J.G.; Pagulayan, K.F.; McMillan, P.J.; Mayer, C.L.; Banks, W.A.; Kraemer, B.C.; et al. Blast exposure causes dynamic microglial/macrophage responses and microdomains of brain microvessel dysfunction. Neuroscience 2016, 319, 206–220. [Google Scholar] [CrossRef] [PubMed]
- Kovesdi, E.; Gyorgy, A.B.; Kwon, S.K.; Wingo, D.L.; Kamnaksh, A.; Long, J.B.; Kasper, C.E.; Agoston, D.V. The effect of enriched environment on the outcome of traumatic brain injury; a behavioral, proteomics, and histological study. Front. Neurosci. 2011, 5, 42. [Google Scholar] [CrossRef]
- Arun, P.; Abu-Taleb, R.; Oguntayo, S.; Tanaka, M.; Wang, Y.; Valiyaveettil, M.; Long, J.B.; Zhang, Y.; Nambiar, M.P. Distinct patterns of expression of traumatic brain injury biomarkers after blast exposure: Role of compromised cell membrane integrity. Neurosci. Lett. 2013, 552, 87–91. [Google Scholar] [CrossRef]
- Arun, P.; Oguntayo, S.; Albert, S.V.; Gist, I.; Wang, Y.; Nambiar, M.P.; Long, J.B. Acute decrease in alkaline phosphatase after brain injury: A potential mechanism for tauopathy. Neurosci. Lett. 2015, 609, 152–158. [Google Scholar] [CrossRef] [PubMed]
- Ahmed, F.; Plantman, S.; Cernak, I.; Agoston, D.V. The Temporal Pattern of Changes in Serum Biomarker Levels Reveals Complex and Dynamically Changing Pathologies after Exposure to a Single Low-Intensity Blast in Mice. Front. Neurol. 2015, 6, 114. [Google Scholar] [CrossRef]
- Perez-Polo, J.R.; Rea, H.C.; Johnson, K.M.; Parsley, M.A.; Unabia, G.C.; Xu, G.Y.; Prough, D.; DeWitt, D.S.; Spratt, H.; Hulsebosch, C.E. A rodent model of mild traumatic brain blast injury. J. Neurosci. Res. 2015, 93, 549–561. [Google Scholar] [CrossRef] [PubMed]
- Sajja, V.S.; Hubbard, W.B.; Hall, C.S.; Ghoddoussi, F.; Galloway, M.P.; VandeVord, P.J. Enduring deficits in memory and neuronal pathology after blast-induced traumatic brain injury. Sci. Rep. 2015, 5, 15075. [Google Scholar] [CrossRef]
- Liu, M.D.; Luo, P.; Wang, Z.J.; Fei, Z. Changes of serum Tau, GFAP, TNF-alpha and malonaldehyde after blast-related traumatic brain injury. Chin. J. Traumatol. 2014, 17, 317–322. [Google Scholar]
- Lucke-Wold, B.P.; Naser, Z.J.; Logsdon, A.F.; Turner, R.C.; Smith, K.E.; Robson, M.J.; Bailes, J.E.; Lee, J.M.; Rosen, C.L.; Huber, J.D. Amelioration of nicotinamide adenine dinucleotide phosphate-oxidase mediated stress reduces cell death after blast-induced traumatic brain injury. Transl. Res. 2015, 166, 509–528.e1. [Google Scholar] [CrossRef]
- Lucke-Wold, B.P.; Turner, R.C.; Logsdon, A.F.; Nguyen, L.; Bailes, J.E.; Lee, J.M.; Robson, M.J.; Omalu, B.I.; Huber, J.D.; Rosen, C.L. Endoplasmic reticulum stress implicated in chronic traumatic encephalopathy. J. Neurosurg. 2016, 124, 687–702. [Google Scholar] [CrossRef]
- Du, X.; West, M.B.; Cheng, W.; Ewert, D.L.; Li, W.; Saunders, D.; Towner, R.A.; Floyd, R.A.; Kopke, R.D. Ameliorative Effects of Antioxidants on the Hippocampal Accumulation of Pathologic Tau in a Rat Model of Blast-Induced Traumatic Brain Injury. Oxid. Med. Cell Longev. 2016, 2016, 4159357. [Google Scholar] [CrossRef] [PubMed]
- Meabon, J.S.; Huber, B.R.; Cross, D.J.; Richards, T.L.; Minoshima, S.; Pagulayan, K.F.; Li, G.; Meeker, K.D.; Kraemer, B.C.; Petrie, E.C.; et al. Repetitive blast exposure in mice and combat veterans causes persistent cerebellar dysfunction. Sci. Transl. Med. 2016, 8, 321ra6. [Google Scholar] [CrossRef]
- Gerson, J.; Castillo-Carranza, D.L.; Sengupta, U.; Bodani, R.; Prough, D.S.; DeWitt, D.S.; Hawkins, B.E.; Kayed, R. Tau Oligomers Derived from Traumatic Brain Injury Cause Cognitive Impairment and Accelerate Onset of Pathology in Htau Mice. J. Neurotrauma 2016, 33, 2034–2043. [Google Scholar] [CrossRef] [PubMed]
- Cao, J.; Gaamouch, F.E.; Meabon, J.S.; Meeker, K.D.; Zhu, L.; Zhong, M.B.; Bendik, J.; Elder, G.; Jing, P.; Xia, J.; et al. ApoE4-associated phospholipid dysregulation contributes to development of Tau hyper-phosphorylation after traumatic brain injury. Sci. Rep. 2017, 7, 11372. [Google Scholar] [CrossRef] [PubMed]
- Logsdon, A.F.; Lucke-Wold, B.P.; Turner, R.C.; Li, X.; Adkins, C.E.; Mohammad, A.S.; Huber, J.D.; Rosen, C.L.; Lockman, P.R. A mouse Model of Focal Vascular Injury Induces Astrocyte Reactivity, Tau Oligomers, and Aberrant Behavior. Arch. Neurosci. 2017, 4, e44254. [Google Scholar] [CrossRef]
- Du, X.; West, M.B.; Cai, Q.; Cheng, W.; Ewert, D.L.; Li, W.; Floyd, R.A.; Kopke, R.D. Antioxidants reduce neurodegeneration and accumulation of pathologic Tau proteins in the auditory system after blast exposure. Free Radic. Biol. Med. 2017, 108, 627–643. [Google Scholar] [CrossRef] [PubMed]
- Lucke-Wold, B.P.; Logsdon, A.F.; Turner, R.C.; Huber, J.D.; Rosen, C.L. Endoplasmic Reticulum Stress Modulation as a Target for Ameliorating Effects of Blast Induced Traumatic Brain Injury. J. Neurotrauma 2017, 34 (Suppl. 1), S62–S70. [Google Scholar] [CrossRef]
- Mammadova, N.; Ghaisas, S.; Zenitsky, G.; Sakaguchi, D.S.; Kanthasamy, A.G.; Greenlee, J.J.; West Greenlee, M.H. Lasting Retinal Injury in a Mouse Model of Blast-Induced Trauma. Am. J. Pathol. 2017, 187, 1459–1472. [Google Scholar] [CrossRef]
- Shi, Q.X.; Chen, B.; Nie, C.; Zhao, Z.P.; Zhang, J.H.; Si, S.Y.; Cui, S.J.; Gu, J.W. A novel model of blast induced traumatic brain injury caused by compressed gas produced sustained cognitive deficits in rats: Involvement of phosphorylation of tau at the Thr205 epitope. Brain Res. Bull. 2020, 157, 149–161. [Google Scholar] [CrossRef]
- Bugay, V.; Bozdemir, E.; Vigil, F.A.; Chun, S.H.; Holstein, D.M.; Elliott, W.R.; Sprague, C.J.; Cavazos, J.E.; Zamora, D.O.; Rule, G.; et al. A Mouse Model of Repetitive Blast Traumatic Brain Injury Reveals Post-Trauma Seizures and Increased Neuronal Excitability. J. Neurotrauma 2020, 37, 248–261. [Google Scholar] [CrossRef]
- Murphy, E.K.; Iacono, D.; Pan, H.; Grimes, J.B.; Parks, S.; Raiciulescu, S.; Leonessa, F.; Perl, D.P. Explosive-driven double-blast exposure: Molecular, histopathological, and behavioral consequences. Sci. Rep. 2020, 10, 17446. [Google Scholar] [CrossRef] [PubMed]
- Kofuji, P.; Araque, A. Astrocytes and Behavior. Annu. Rev. Neurosci. 2021, 44, 49–67. [Google Scholar] [CrossRef]
- Biesecker, K.R.; Srienc, A.I.; Shimoda, A.M.; Agarwal, A.; Bergles, D.E.; Kofuji, P.; Newman, E.A. Glial Cell Calcium Signaling Mediates Capillary Regulation of Blood Flow in the Retina. J. Neurosci. 2016, 36, 9435–9445. [Google Scholar] [CrossRef]
- Mishra, A.; Reynolds, J.P.; Chen, Y.; Gourine, A.V.; Rusakov, D.A.; Attwell, D. Astrocytes mediate neurovascular signaling to capillary pericytes but not to arterioles. Nat. Neurosci. 2016, 19, 1619–1627. [Google Scholar] [CrossRef]
- Kisler, K.; Nelson, A.R.; Rege, S.V.; Ramanathan, A.; Wang, Y.; Ahuja, A.; Lazic, D.; Tsai, P.S.; Zhao, Z.; Zhou, Y.; et al. Pericyte degeneration leads to neurovascular uncoupling and limits oxygen supply to brain. Nat. Neurosci. 2017, 20, 406–416. [Google Scholar] [CrossRef] [PubMed]
- Davis, G.E.; Senger, D.R. Endothelial extracellular matrix: Biosynthesis, remodeling, and functions during vascular morphogenesis and neovessel stabilization. Circ. Res. 2005, 97, 1093–1107. [Google Scholar] [CrossRef]
- Kruegel, J.; Miosge, N. Basement membrane components are key players in specialized extracellular matrices. Cell Mol. Life Sci. 2010, 67, 2879–2895. [Google Scholar] [CrossRef] [PubMed]
- Franciosi, S.; De Gasperi, R.; Dickstein, D.L.; English, D.F.; Rocher, A.B.; Janssen, W.G.; Christoffel, D.; Sosa, M.A.; Hof, P.R.; Buxbaum, J.D.; et al. Pepsin pretreatment allows collagen IV immunostaining of blood vessels in adult mouse brain. J. Neurosci. Methods 2007, 163, 76–82. [Google Scholar] [CrossRef]
- Gama Sosa, M.A.; Gasperi, R.D.; Rocher, A.B.; Wang, A.C.; Janssen, W.G.; Flores, T.; Perez, G.M.; Schmeidler, J.; Dickstein, D.L.; Hof, P.R.; et al. Age-related vascular pathology in transgenic mice expressing presenilin 1-associated familial Alzheimer’s disease mutations. Am. J. Pathol. 2010, 176, 353–368. [Google Scholar] [CrossRef]
- Prima, V.; Serebruany, V.L.; Svetlov, A.; Hayes, R.L.; Svetlov, S.I. Impact of moderate blast exposures on thrombin biomarkers assessed by calibrated automated thrombography in rats. J. Neurotrauma 2013, 30, 1881–1887. [Google Scholar] [CrossRef]
- Inui, T.; Hoffer, M.; Balaban, C.D. Mild blast wave exposure produces intensity-dependent changes in MMP2 expression patches in rat brains—Findings from different blast severities. Brain Res. 2021, 1767, 147541. [Google Scholar] [CrossRef] [PubMed]
- Scrimgeour, A.G.; Carrigan, C.T.; Condlin, M.L.; Urso, M.L.; van den Berg, R.M.; van Helden, H.P.M.; Montain, S.J.; Joosen, M.J.A. Dietary Zinc Modulates Matrix Metalloproteinases in Traumatic Brain Injury. J. Neurotrauma 2018, 35, 2495–2506. [Google Scholar] [CrossRef]
- Szabo, A.; Kalman, M. Disappearance of the post-lesional laminin immunopositivity of brain vessels is parallel with the formation of gliovascular junctions and common basal lamina. A double-labelling immunohistochemical study. Neuropathol. Appl. Neurobiol. 2004, 30, 169–177. [Google Scholar] [CrossRef]
- Caley, D.W.; Maxwell, D.S. Development of the blood vessels and extracellular spaces during postnatal maturation of rat cerebral cortex. J. Comp. Neurol. 1970, 138, 31–47. [Google Scholar] [CrossRef]
- Abdul-Muneer, P.M.; Schuetz, H.; Wang, F.; Skotak, M.; Jones, J.; Gorantla, S.; Zimmerman, M.C.; Chandra, N.; Haorah, J. Induction of oxidative and nitrosative damage leads to cerebrovascular inflammation in an animal model of mild traumatic brain injury induced by primary blast. Free Radic. Biol. Med. 2013, 60, 282–291. [Google Scholar] [CrossRef] [PubMed]
- Lucke-Wold, B.P.; Logsdon, A.F.; Smith, K.E.; Turner, R.C.; Alkon, D.L.; Tan, Z.; Naser, Z.J.; Knotts, C.M.; Huber, J.D.; Rosen, C.L. Bryostatin-1 Restores Blood Brain Barrier Integrity following Blast-Induced Traumatic Brain Injury. Mol. Neurobiol. 2015, 52, 1119–1134. [Google Scholar] [CrossRef][Green Version]
- Willis, C.L.; Leach, L.; Clarke, G.J.; Nolan, C.C.; Ray, D.E. Reversible disruption of tight junction complexes in the rat blood-brain barrier, following transitory focal astrocyte loss. Glia 2004, 48, 1–13. [Google Scholar] [CrossRef]
- Hue, C.D.; Cao, S.; Bass, C.R.; Meaney, D.F.; Morrison, B., 3rd. Repeated Primary Blast Injury Causes Delayed Recovery, but not Additive Disruption, in an In Vitro Blood-Brain Barrier Model. J. Neurotrauma 2014, 31, 951–960. [Google Scholar] [CrossRef]
- Heyburn, L.; Abutarboush, R.; Goodrich, S.; Urioste, R.; Batuure, A.; Statz, J.; Wilder, D.; Ahlers, S.T.; Long, J.B.; Sajja, V. Repeated Low-Level Blast Overpressure Leads to Endovascular Disruption and Alterations in TDP-43 and Piezo2 in a Rat Model of Blast TBI. Front. Neurol. 2019, 10, 766. [Google Scholar] [CrossRef]
- De Luca, C.; Papa, M. Looking Inside the Matrix: Perineuronal Nets in Plasticity, Maladaptive Plasticity and Neurological Disorders. Neurochem. Res. 2016, 41, 1507–1515. [Google Scholar] [CrossRef]
- John, U.; Patro, N.; Patro, I. Perineuronal nets: Cruise from a honeycomb to the safety nets. Brain Res. Bull. 2022, 190, 179–194. [Google Scholar] [CrossRef]
- Bruckner, G.; Grosche, J.; Hartlage-Rubsamen, M.; Schmidt, S.; Schachner, M. Region and lamina-specific distribution of extracellular matrix proteoglycans, hyaluronan and tenascin-R in the mouse hippocampal formation. J. Chem. Neuroanat. 2003, 26, 37–50. [Google Scholar] [CrossRef] [PubMed]
- Carstens, K.E.; Phillips, M.L.; Pozzo-Miller, L.; Weinberg, R.J.; Dudek, S.M. Perineuronal Nets Suppress Plasticity of Excitatory Synapses on CA2 Pyramidal Neurons. J. Neurosci. 2016, 36, 6312–6320. [Google Scholar] [CrossRef] [PubMed]
- Pizzorusso, T.; Medini, P.; Berardi, N.; Chierzi, S.; Fawcett, J.W.; Maffei, L. Reactivation of ocular dominance plasticity in the adult visual cortex. Science 2002, 298, 1248–1251. [Google Scholar] [CrossRef]
- Callaghan, B.L.; Graham, B.M.; Li, S.; Richardson, R. From resilience to vulnerability: Mechanistic insights into the effects of stress on transitions in critical period plasticity. Front. Psychiatry 2013, 4, 90. [Google Scholar] [CrossRef]
- Gogolla, N.; Caroni, P.; Luthi, A.; Herry, C. Perineuronal nets protect fear memories from erasure. Science 2009, 325, 1258–1261. [Google Scholar] [CrossRef]
- Quirk, G.J.; Pare, D.; Richardson, R.; Herry, C.; Monfils, M.H.; Schiller, D.; Vicentic, A. Erasing fear memories with extinction training. J. Neurosci. 2010, 30, 14993–14997. [Google Scholar] [CrossRef] [PubMed]
- Hylin, M.J.; Orsi, S.A.; Moore, A.N.; Dash, P.K. Disruption of the perineuronal net in the hippocampus or medial prefrontal cortex impairs fear conditioning. Learn. Mem. 2013, 20, 267–273. [Google Scholar] [CrossRef]
- Kochlamazashvili, G.; Henneberger, C.; Bukalo, O.; Dvoretskova, E.; Senkov, O.; Lievens, P.M.; Westenbroek, R.; Engel, A.K.; Catterall, W.A.; Rusakov, D.A.; et al. The extracellular matrix molecule hyaluronic acid regulates hippocampal synaptic plasticity by modulating postsynaptic L-type Ca2+ channels. Neuron 2010, 67, 116–128. [Google Scholar] [CrossRef]
- Li, X.; Ren, D.; Luo, B.; Liu, Z.; Li, N.; Zhou, T.; Fei, E. Perineuronal Nets Alterations Contribute to Stress-Induced Anxiety-Like Behavior. Mol. Neurobiol. 2023, 1–12. [Google Scholar] [CrossRef]
- Prinz, M.; Masuda, T.; Wheeler, M.A.; Quintana, F.J. Microglia and Central Nervous System-Associated Macrophages-From Origin to Disease Modulation. Annu. Rev. Immunol. 2021, 39, 251–277. [Google Scholar] [CrossRef]
- Bisht, K.; Sharma, K.P.; Lecours, C.; Sanchez, M.G.; El Hajj, H.; Milior, G.; Olmos-Alonso, A.; Gomez-Nicola, D.; Luheshi, G.; Vallieres, L.; et al. Dark microglia: A new phenotype predominantly associated with pathological states. Glia 2016, 64, 826–839. [Google Scholar] [CrossRef]
- Halder, S.K.; Milner, R. A critical role for microglia in maintaining vascular integrity in the hypoxic spinal cord. Proc. Natl. Acad. Sci. USA 2019, 116, 26029–26037. [Google Scholar] [CrossRef]
- Kettenmann, H.; Hanisch, U.K.; Noda, M.; Verkhratsky, A. Physiology of microglia. Physiol. Rev. 2011, 91, 461–553. [Google Scholar] [CrossRef]
- Kreutzberg, G.W. Microglia: A sensor for pathological events in the CNS. Trends Neurosci. 1996, 19, 312–318. [Google Scholar] [CrossRef] [PubMed]
- Sheng, J.G.; Mrak, R.E.; Griffin, W.S. Neuritic plaque evolution in Alzheimer’s disease is accompanied by transition of activated microglia from primed to enlarged to phagocytic forms. Acta Neuropathol. 1997, 94, 1–5. [Google Scholar] [CrossRef]
- Soltys, Z.; Ziaja, M.; Pawlinski, R.; Setkowicz, Z.; Janeczko, K. Morphology of reactive microglia in the injured cerebral cortex. Fractal analysis and complementary quantitative methods. J. Neurosci. Res. 2001, 63, 90–97. [Google Scholar] [CrossRef] [PubMed]
- Stence, N.; Waite, M.; Dailey, M.E. Dynamics of microglial activation: A confocal time-lapse analysis in hippocampal slices. Glia 2001, 33, 256–266. [Google Scholar] [CrossRef]
- Torres-Platas, S.G.; Comeau, S.; Rachalski, A.; Bo, G.D.; Cruceanu, C.; Turecki, G.; Giros, B.; Mechawar, N. Morphometric characterization of microglial phenotypes in human cerebral cortex. J. Neuroinflammation 2014, 11, 12. [Google Scholar] [CrossRef]
- Varnum, M.M.; Ikezu, T. The classification of microglial activation phenotypes on neurodegeneration and regeneration in Alzheimer’s disease brain. Arch Immunol Ther Exp (Warsz) 2012, 60, 251–266. [Google Scholar] [CrossRef] [PubMed]
- Lin, S.S.; Tang, Y.; Illes, P.; Verkhratsky, A. The Safeguarding Microglia: Central Role for P2Y(12) Receptors. Front. Pharmacol. 2020, 11, 627760. [Google Scholar] [CrossRef]
- Crotti, A.; Ransohoff, R.M. Microglial Physiology and Pathophysiology: Insights from Genome-wide Transcriptional Profiling. Immunity 2016, 44, 505–515. [Google Scholar] [CrossRef]
- Readnower, R.D.; Chavko, M.; Adeeb, S.; Conroy, M.D.; Pauly, J.R.; McCarron, R.M.; Sullivan, P.G. Increase in blood-brain barrier permeability, oxidative stress, and activated microglia in a rat model of blast-induced traumatic brain injury. J. Neurosci. Res. 2010, 88, 3530–3539. [Google Scholar] [CrossRef]
- Turner, R.C.; Naser, Z.J.; Logsdon, A.F.; DiPasquale, K.H.; Jackson, G.J.; Robson, M.J.; Gettens, R.T.; Matsumoto, R.R.; Huber, J.D.; Rosen, C.L. Modeling clinically relevant blast parameters based on scaling principles produces functional & histological deficits in rats. Exp. Neurol. 2013, 248, 520–529. [Google Scholar]
- Cho, H.J.; Sajja, V.S.; Vandevord, P.J.; Lee, Y.W. Blast induces oxidative stress, inflammation, neuronal loss and subsequent short-term memory impairment in rats. Neuroscience 2013, 253, 9–20. [Google Scholar] [CrossRef]
- Sajja, V.S.; Ereifej, E.S.; VandeVord, P.J. Hippocampal vulnerability and subacute response following varied blast magnitudes. Neurosci. Lett. 2014, 570, 33–37. [Google Scholar] [CrossRef]
- Kamnaksh, A.; Ahmed, F.; Kovedsi, E.; Barry, E.; Grunberg, N.; Long, J.; Agoston, D. Molecular mechanisms of increased cerebral vulnerability after repeated mild blast-induced traumatic brain injury. Translational Proteomics 2014, 3, 22–37. [Google Scholar] [CrossRef]
- Valiyaveettil, M.; Alamneh, Y.A.; Miller, S.A.; Hammamieh, R.; Arun, P.; Wang, Y.; Wei, Y.; Oguntayo, S.; Long, J.B.; Nambiar, M.P. Modulation of cholinergic pathways and inflammatory mediators in blast-induced traumatic brain injury. Chem. Biol. Interact. 2013, 203, 371–375. [Google Scholar] [CrossRef]
- Stone, J.R.; Avants, B.B.; Tustison, N.; Gill, J.; Wilde, E.A.; Neumann, K.D.; Gladney, L.A.; Kilgore, M.O.; Bowling, F.; Wilson, C.M.; et al. Neurological effects of repeated blast exposure in Special Operations personnel. J. Neurotrauma 2024. [Google Scholar] [CrossRef]
- Dalle Lucca, J.J.; Chavko, M.; Dubick, M.A.; Adeeb, S.; Falabella, M.J.; Slack, J.L.; McCarron, R.; Li, Y. Blast-induced moderate neurotrauma (BINT) elicits early complement activation and tumor necrosis factor alpha (TNFalpha) release in a rat brain. J. Neurol. Sci. 2012, 318, 146–154. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Chavko, M.; Slack, J.L.; Liu, B.; McCarron, R.M.; Ross, J.D.; Dalle Lucca, J.J. Protective effects of decay-accelerating factor on blast-induced neurotrauma in rats. Acta Neuropathol. Commun. 2013, 1, 52. [Google Scholar] [CrossRef]
- Hamacher, J.; Hadizamani, Y.; Huwer, H.; Moehrlen, U.; Bally, L.; Stammberger, U.; Wendel, A.; Lucas, R. Characteristics of inflammatory response and repair after experimental blast lung injury in rats. PLoS ONE 2023, 18, e0281446. [Google Scholar] [CrossRef]
- Meng, X.Y.; Lu, Q.Y.; Zhang, J.F.; Li, J.F.; Shi, M.Y.; Huang, S.Y.; Yu, S.F.; Zhao, Y.M.; Fan, H.J. A novel animal model of primary blast lung injury and its pathological changes in mice. J. Trauma. Acute Care Surg. 2022, 93, 530–537. [Google Scholar] [CrossRef]
- Pierce, M.E.; Hayes, J.; Huber, B.R.; Jeromin, A.; Fortier, C.B.; Fonda, J.R.; Lasseter, H.; Chaby, L.; McGlinchey, R.; Milberg, W. Plasma biomarkers associated with deployment trauma and its consequences in post-9/11 era veterans: Initial findings from the TRACTS longitudinal cohort. Transl. Psychiatry 2022, 12, 80. [Google Scholar] [CrossRef]
- Takahashi, S.; Fukushima, H.; Yu, Z.; Tomita, H.; Kida, S. Tumor necrosis factor alpha negatively regulates the retrieval and reconsolidation of hippocampus-dependent memory. Brain Behav. Immun. 2021, 94, 79–88. [Google Scholar] [CrossRef]
- Parekh, S.V.; Paniccia, J.E.; Adams, L.O.; Lysle, D.T. Hippocampal TNF-alpha Signaling Mediates Heroin Withdrawal-Enhanced Fear Learning and Withdrawal-Induced Weight Loss. Mol. Neurobiol. 2021, 58, 2963–2973. [Google Scholar] [CrossRef]
- Zhang, Z.; Song, Z.; Shen, F.; Xie, P.; Wang, J.; Zhu, A.S.; Zhu, G. Ginsenoside Rg1 Prevents PTSD-Like Behaviors in Mice Through Promoting Synaptic Proteins, Reducing Kir4.1 and TNF-alpha in the Hippocampus. Mol. Neurobiol. 2021, 58, 1550–1563. [Google Scholar] [CrossRef]
- Yu, Z.; Fukushima, H.; Ono, C.; Sakai, M.; Kasahara, Y.; Kikuchi, Y.; Gunawansa, N.; Takahashi, Y.; Matsuoka, H.; Kida, S.; et al. Microglial production of TNF-alpha is a key element of sustained fear memory. Brain Behav. Immun. 2017, 59, 313–321. [Google Scholar] [CrossRef]
- Zupan, B.; Liu, B.; Taki, F.; Toth, J.G.; Toth, M. Maternal Brain TNF-alpha Programs Innate Fear in the Offspring. Curr. Biol. 2017, 27, 3859–3863.e3. [Google Scholar] [CrossRef]
- Vorn, R.; Edwards, K.A.; Hentig, J.; Yun, S.; Kim, H.S.; Lai, C.; Devoto, C.; Yarnell, A.M.; Polejaeva, E.; Dell, K.C.; et al. A Pilot Study of Whole-Blood Transcriptomic Analysis to Identify Genes Associated with Repetitive Low-Level Blast Exposure in Career Breachers. Biomedicines 2022, 10, 690. [Google Scholar] [CrossRef]
- Edwards, K.A.; Leete, J.J.; Smith, E.G.; Quick, A.; Modica, C.M.; Wassermann, E.M.; Polejaeva, E.; Dell, K.C.; LoPresti, M.; Walker, P.; et al. Elevations in Tumor Necrosis Factor Alpha and Interleukin 6 From Neuronal-Derived Extracellular Vesicles in Repeated Low-Level Blast Exposed Personnel. Front. Neurol. 2022, 13, 723923. [Google Scholar] [CrossRef]
- Muth, K.N.; Rech, J.; Losch, F.O.; Hoerning, A. Reversing the Inflammatory Process-25 Years of Tumor Necrosis Factor-alpha Inhibitors. J. Clin. Med. 2023, 12, 5039. [Google Scholar] [CrossRef]
- Souza, R.F.; Caetano, M.A.F.; Magalhaes, H.I.R.; Castelucci, P. Study of tumor necrosis factor receptor in the inflammatory bowel disease. World J. Gastroenterol. 2023, 29, 2733–2746. [Google Scholar] [CrossRef]
- Tuttolomondo, A.; Pecoraro, R.; Pinto, A. Studies of selective TNF inhibitors in the treatment of brain injury from stroke and trauma: A review of the evidence to date. Drug Des. Devel Ther. 2014, 8, 2221–2238. [Google Scholar] [CrossRef]
- Gonzalez Caldito, N. Role of tumor necrosis factor-alpha in the central nervous system: A focus on autoimmune disorders. Front. Immunol. 2023, 14, 1213448. [Google Scholar] [CrossRef]
- Muller, N. Immunology of major depression. Neuroimmunomodulation 2014, 21, 123–130. [Google Scholar] [CrossRef]
- Wieck, A.; Grassi-Oliveira, R.; Hartmann do Prado, C.; Teixeira, A.L.; Bauer, M.E. Neuroimmunoendocrine interactions in post-traumatic stress disorder: Focus on long-term implications of childhood maltreatment. Neuroimmunomodulation 2014, 21, 145–151. [Google Scholar] [CrossRef]
- Lee, D.H.; Lee, J.Y.; Hong, D.Y.; Lee, E.C.; Park, S.W.; Lee, M.R.; Oh, J.S. Neuroinflammation in Post-Traumatic Stress Disorder. Biomedicines 2022, 10, 953. [Google Scholar] [CrossRef]
- Sun, Y.; Qu, Y.; Zhu, J. The Relationship Between Inflammation and Post-traumatic Stress Disorder. Front. Psychiatry 2021, 12, 707543. [Google Scholar] [CrossRef]
- Hassamal, S. Chronic stress, neuroinflammation, and depression: An overview of pathophysiological mechanisms and emerging anti-inflammatories. Front. Psychiatry 2023, 14, 1130989. [Google Scholar] [CrossRef]
- Lee, C.H.; Giuliani, F. The Role of Inflammation in Depression and Fatigue. Front. Immunol. 2019, 10, 1696. [Google Scholar] [CrossRef] [PubMed]
- Haroon, E.; Miller, A.H.; Sanacora, G. Inflammation, Glutamate, and Glia: A Trio of Trouble in Mood Disorders. Neuropsychopharmacology 2017, 42, 193–215. [Google Scholar] [CrossRef]
- Chen, S.; Siedhoff, H.R.; Zhang, H.; Liu, P.; Balderrama, A.; Li, R.; Johnson, C.; Greenlief, C.M.; Koopmans, B.; Hoffman, T.; et al. Low-intensity blast induces acute glutamatergic hyperexcitability in mouse hippocampus leading to long-term learning deficits and altered expression of proteins involved in synaptic plasticity and serine protease inhibitors. Neurobiol. Dis. 2022, 165, 105634. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Sawyer, T.W.; Tse, Y.C.; Fan, C.; Hennes, G.; Barnes, J.; Josey, T.; Weiss, T.; Nelson, P.; Wong, T.P. Primary Blast-Induced Changes in Akt and GSK3beta Phosphorylation in Rat Hippocampus. Front. Neurol. 2017, 8, 413. [Google Scholar] [CrossRef] [PubMed]
- Smith, M.; Piehler, T.; Benjamin, R.; Farizatto, K.L.; Pait, M.C.; Almeida, M.F.; Ghukasyan, V.V.; Bahr, B.A. Blast waves from detonated military explosive reduce GluR1 and synaptophysin levels in hippocampal slice cultures. Exp. Neurol. 2016, 286, 107–115. [Google Scholar] [CrossRef]
- De Gasperi, R.; Gama Sosa, M.; Perez Garcia, G.; Perez, G.; Pryor, D.; Morrison, C.L.-A.; Lind, R.; Abutarboush, R.; Kawoos, U.; Statz, J.; et al. Metabotropic glutamate receptor 2 expression is chronically elevated in male rats with post-traumatic stress disorder related behavioral traits following repetitive low-level blast exposure. J. Neurotrauma 2023. Published online. [Google Scholar] [CrossRef]
- Highland, J.N.; Zanos, P.; Riggs, L.M.; Georgiou, P.; Clark, S.M.; Morris, P.J.; Moaddel, R.; Thomas, C.J.; Zarate, C.A., Jr.; Pereira, E.F.R.; et al. Hydroxynorketamines: Pharmacology and Potential Therapeutic Applications. Pharmacol. Rev. 2021, 73, 763–791. [Google Scholar] [CrossRef] [PubMed]
- Zanos, P.; Highland, J.N.; Stewart, B.W.; Georgiou, P.; Jenne, C.E.; Lovett, J.; Morris, P.J.; Thomas, C.J.; Moaddel, R.; Zarate, C.A., Jr.; et al. (2R,6R)-hydroxynorketamine exerts mGlu2 receptor-dependent antidepressant actions. Proc. Natl. Acad. Sci. USA 2019, 116, 6441–6450. [Google Scholar] [CrossRef]
- Batiuk, M.Y.; Martirosyan, A.; Wahis, J.; de Vin, F.; Marneffe, C.; Kusserow, C.; Koeppen, J.; Viana, J.F.; Oliveira, J.F.; Voet, T.; et al. Identification of region-specific astrocyte subtypes at single cell resolution. Nat. Commun. 2020, 11, 1220. [Google Scholar] [CrossRef]
- Sun, N.; Akay, L.A.; Murdock, M.H.; Park, Y.; Galiana-Melendez, F.; Bubnys, A.; Galani, K.; Mathys, H.; Jiang, X.; Ng, A.P.; et al. Single-nucleus multiregion transcriptomic analysis of brain vasculature in Alzheimer’s disease. Nat. Neurosci. 2023, 26, 970–982. [Google Scholar] [CrossRef]
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).