
The Effects of DNA Methylation on Cytoplasmic Male Sterility in Sugar Beet

Abstract
1. Introduction
2. Results
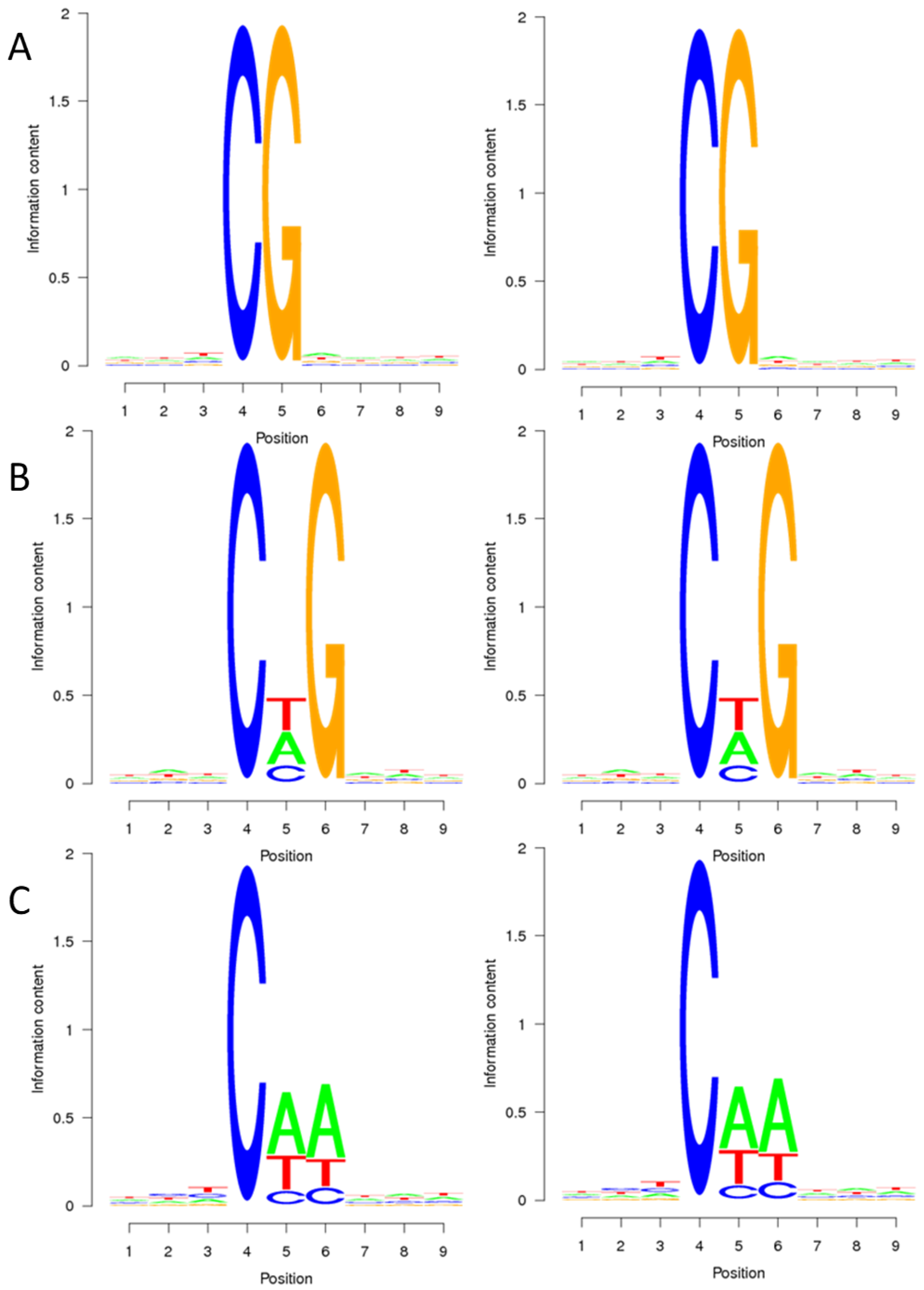
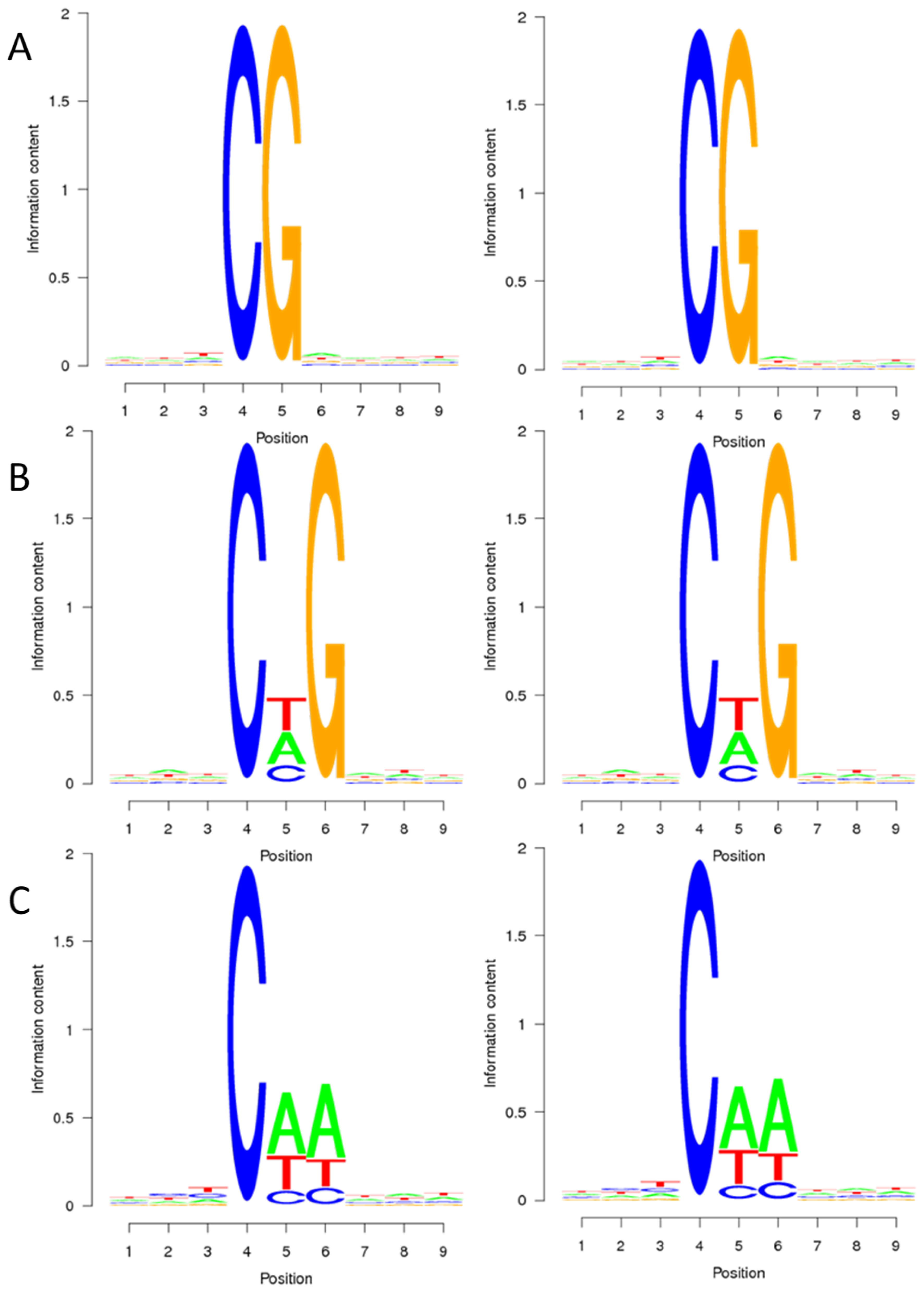
2.1. DNA Methylation Pattern of Sugar Beet
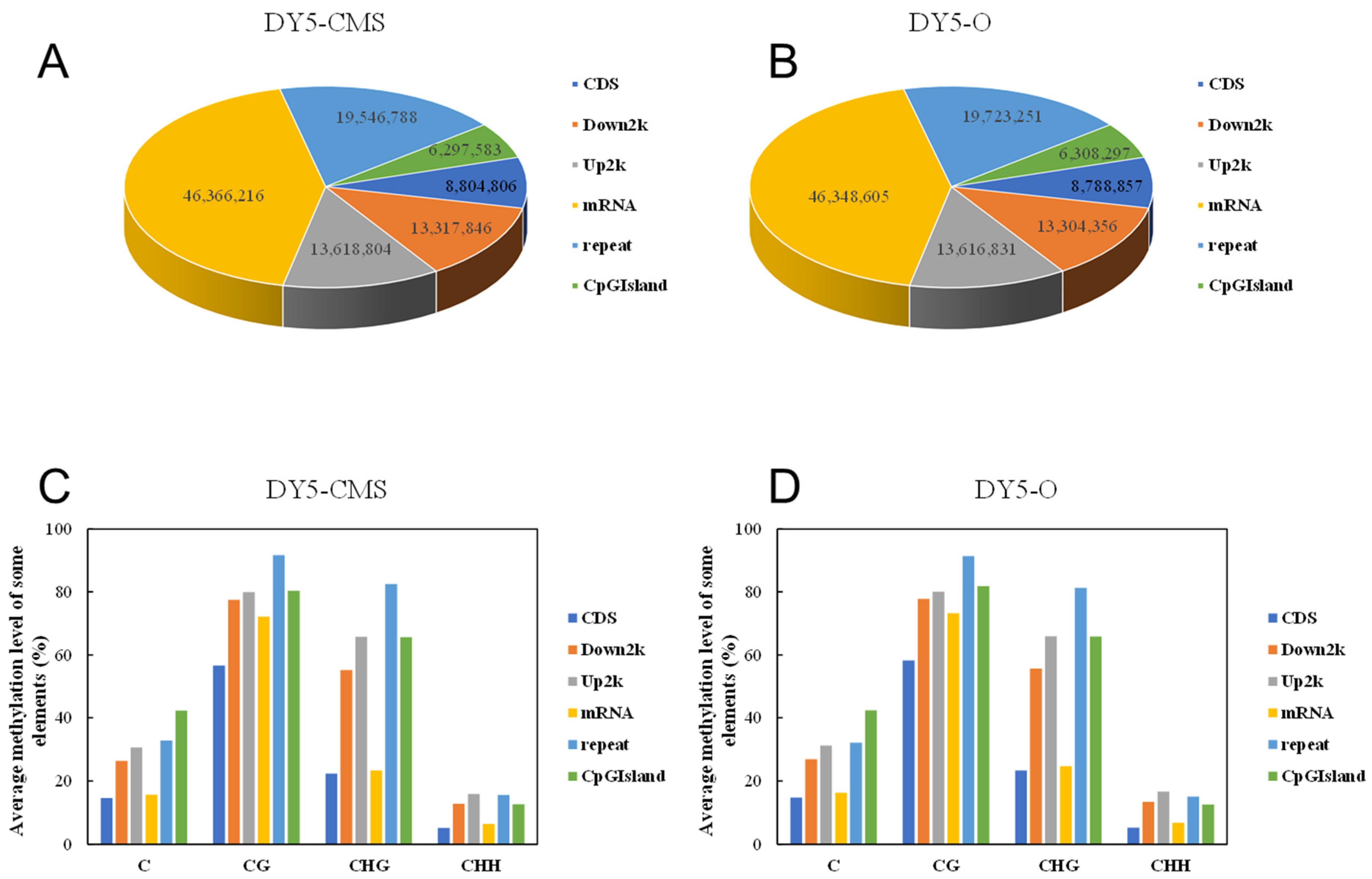
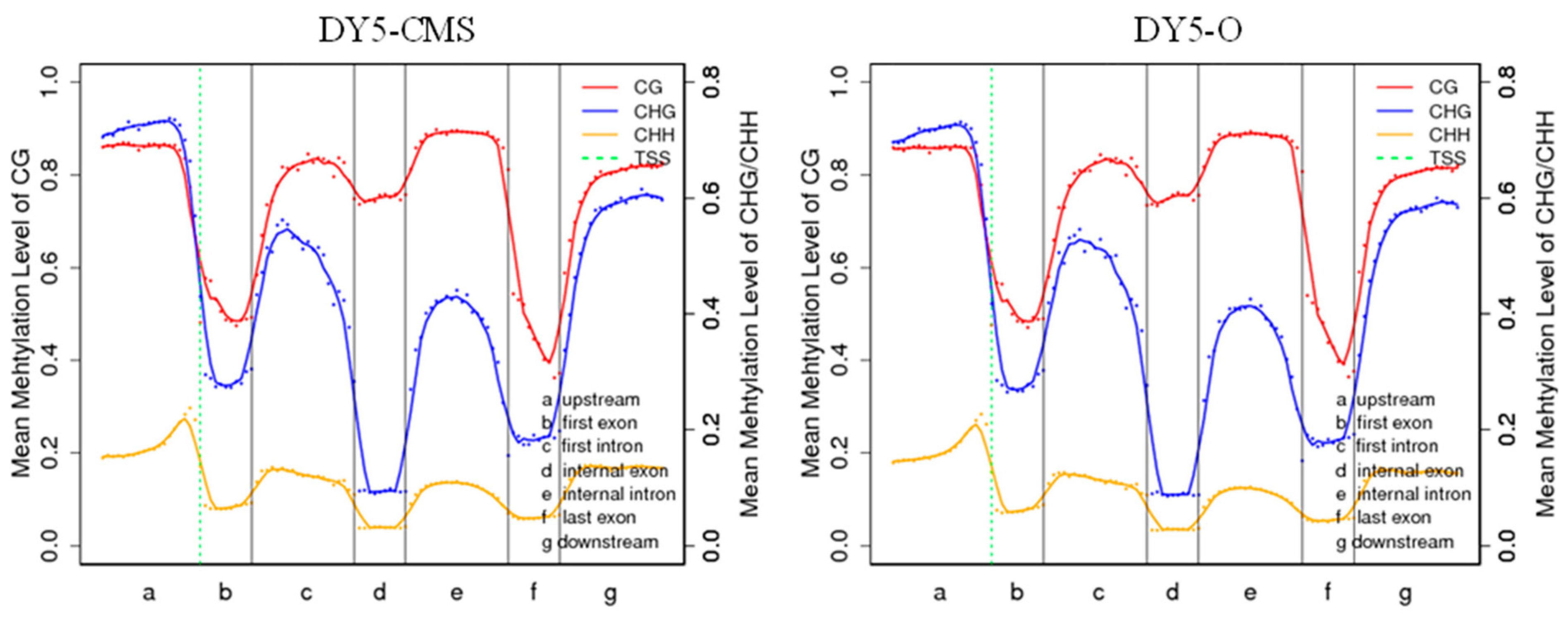
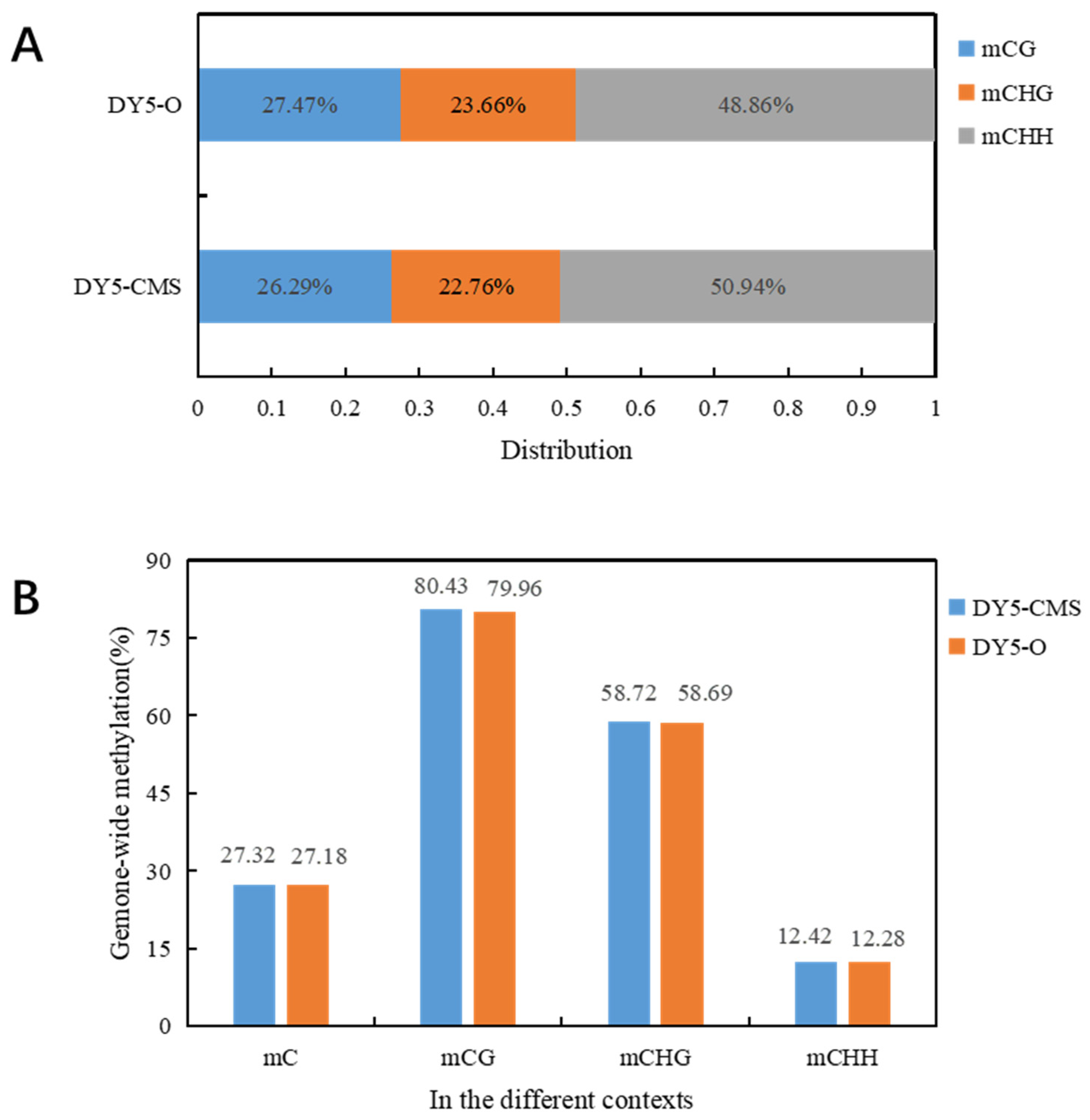
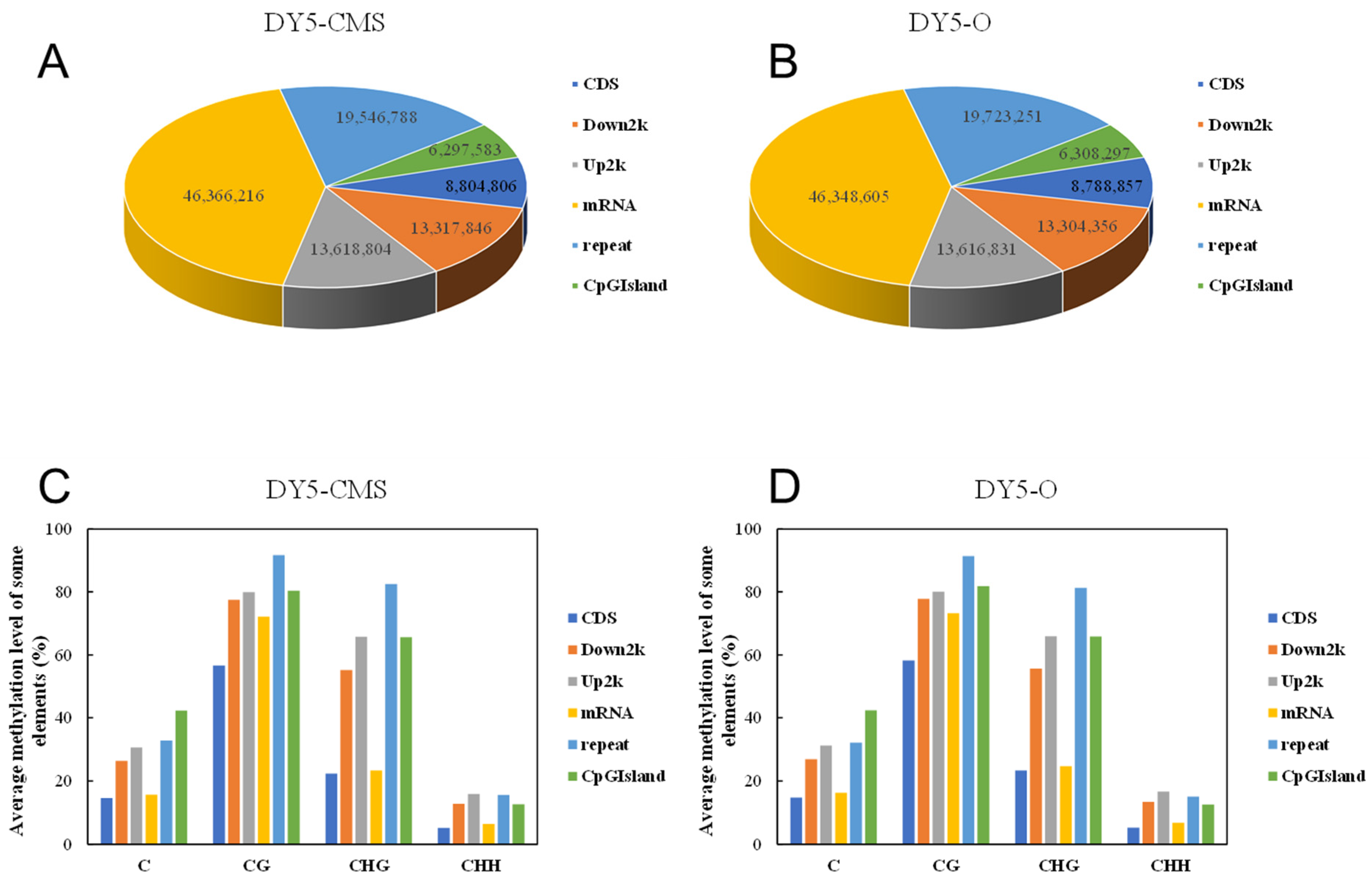
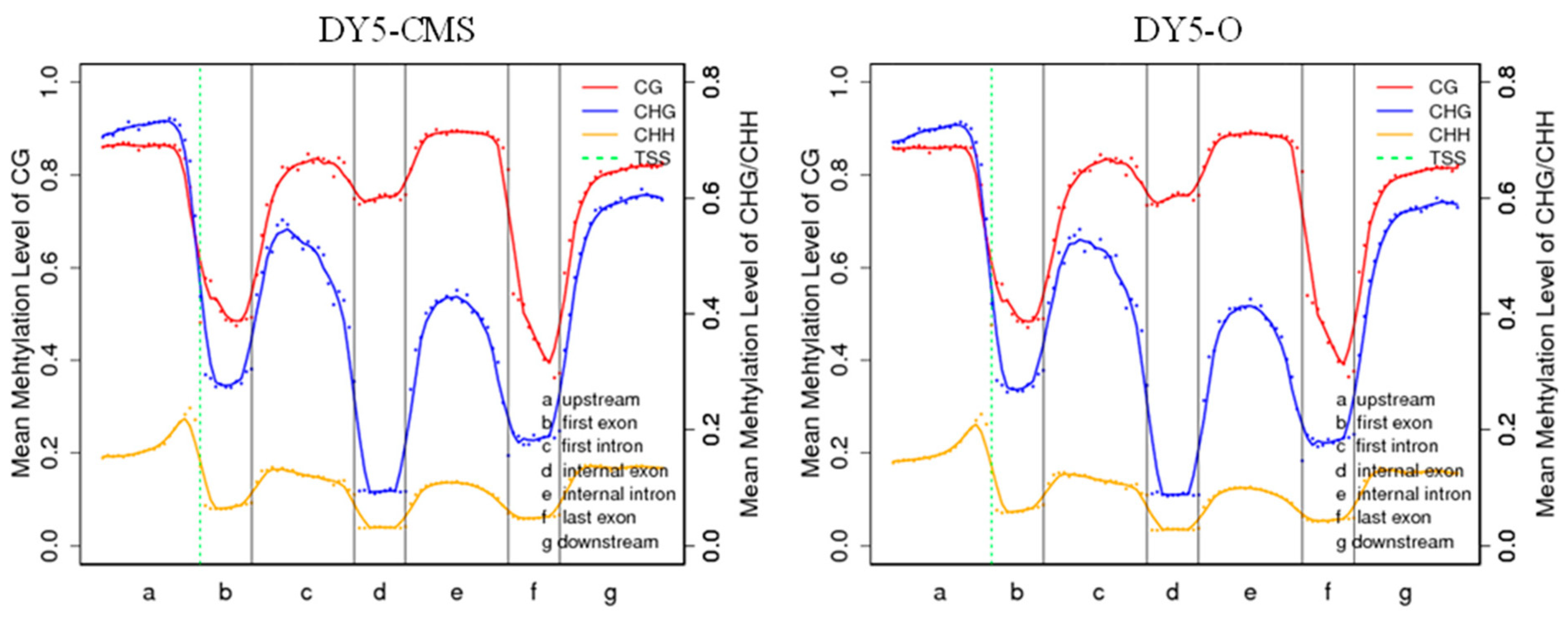
2.2. DNA Methylation Level Analysis of DY5CMS and DY5O
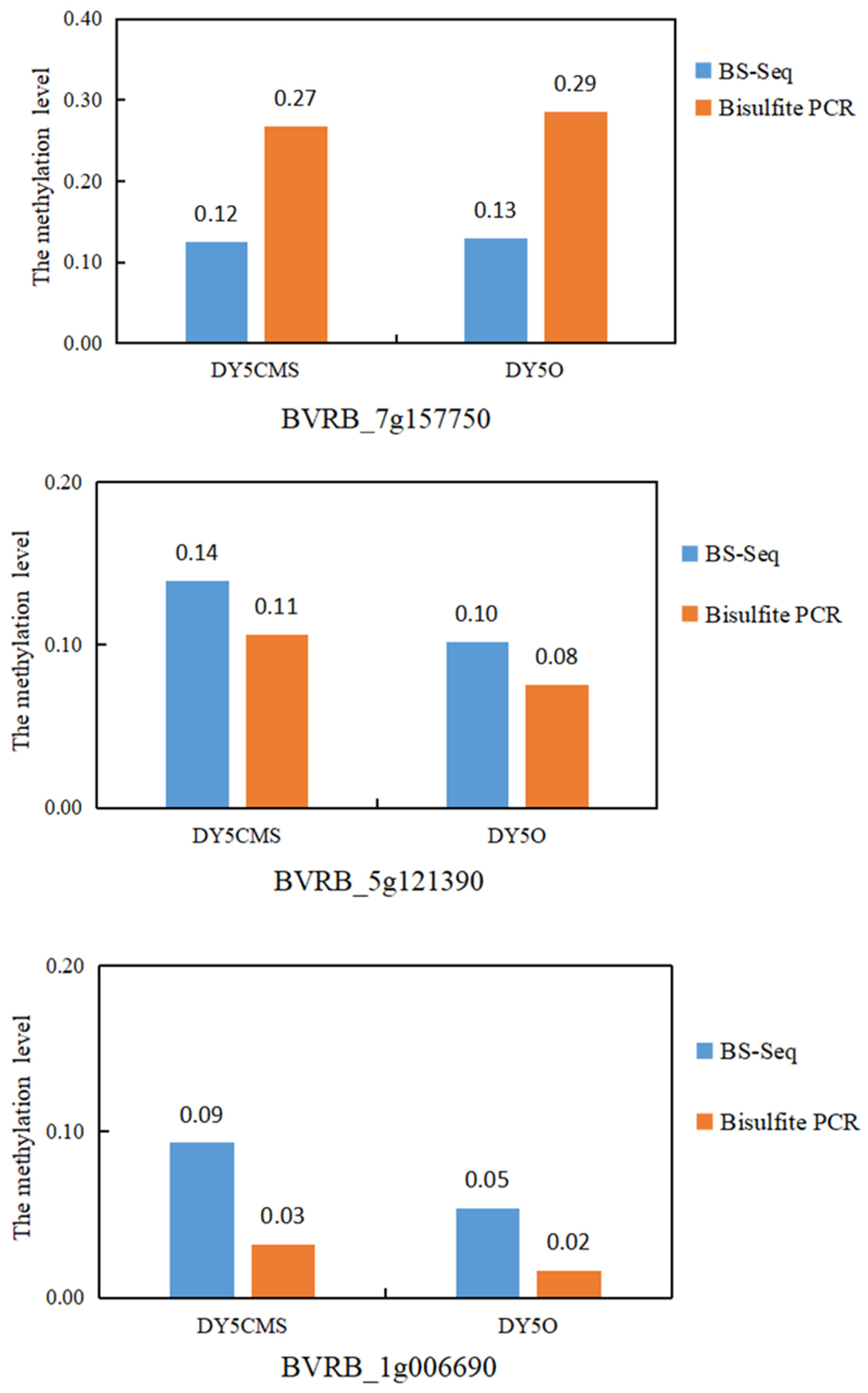
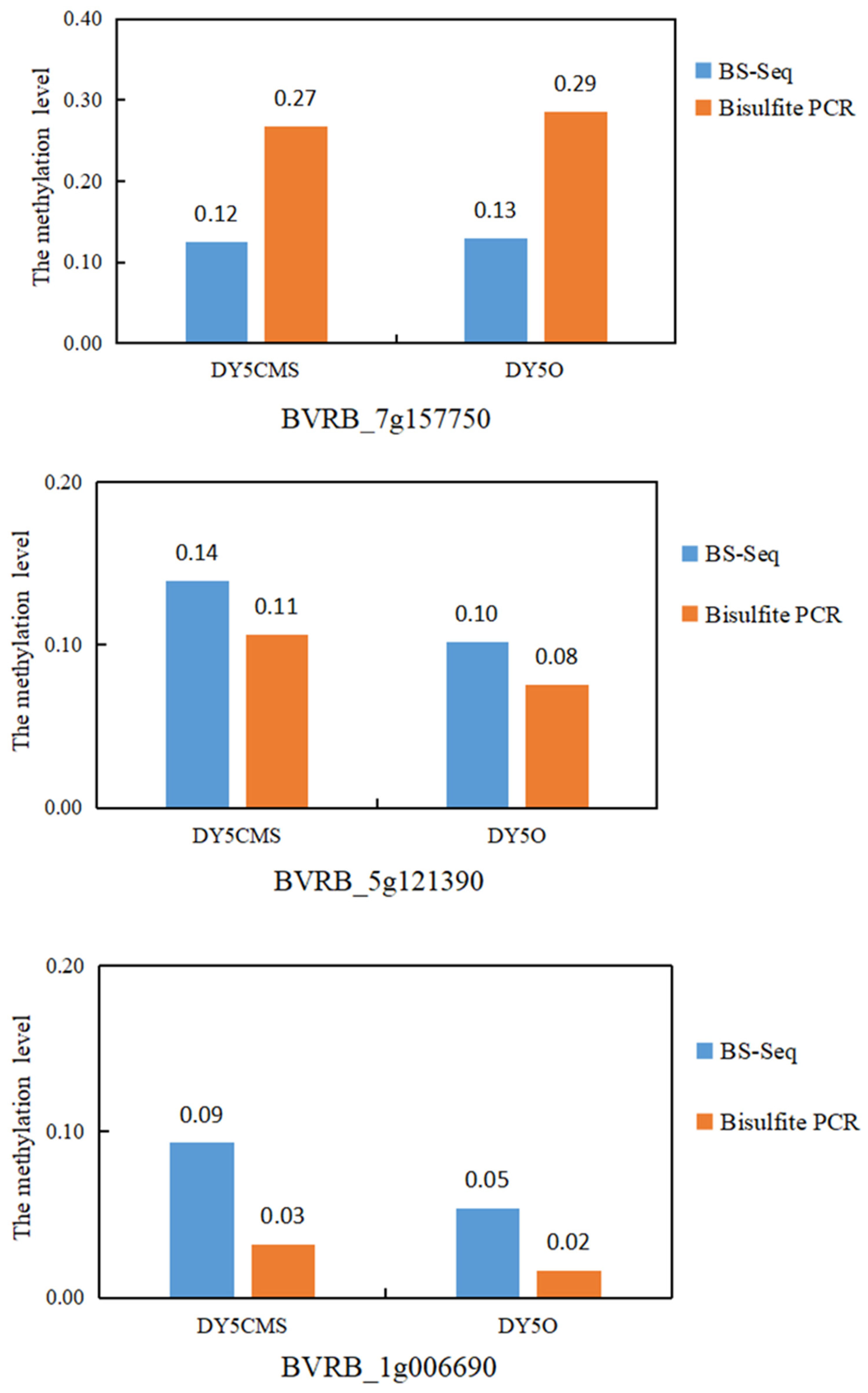
2.3. Verification of WGBS Sequencing Data by Bisulfite Conversion Method
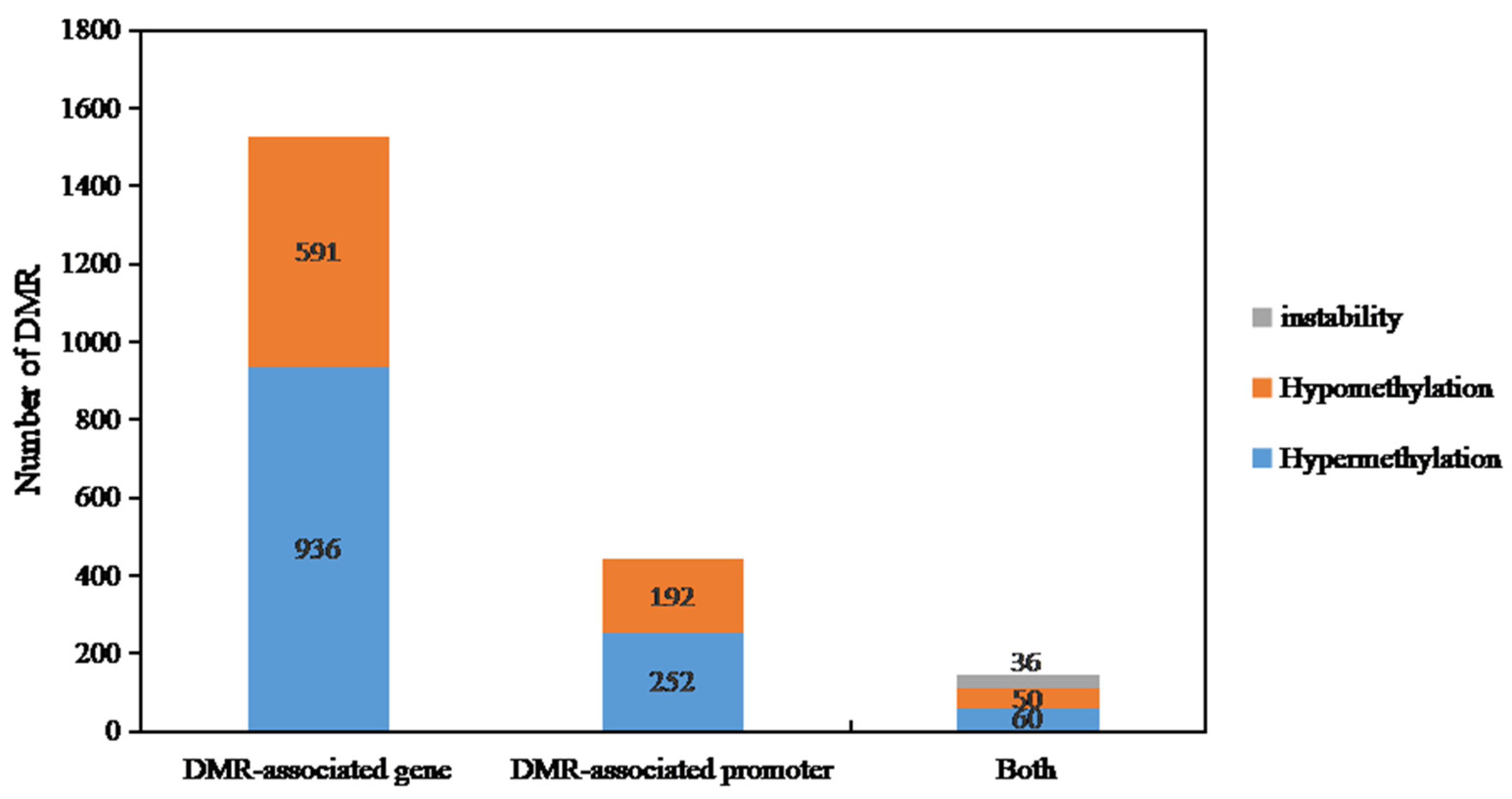
2.4. Differential Methylation Genes (DMGs) Identification of DY5CMS and DY5O
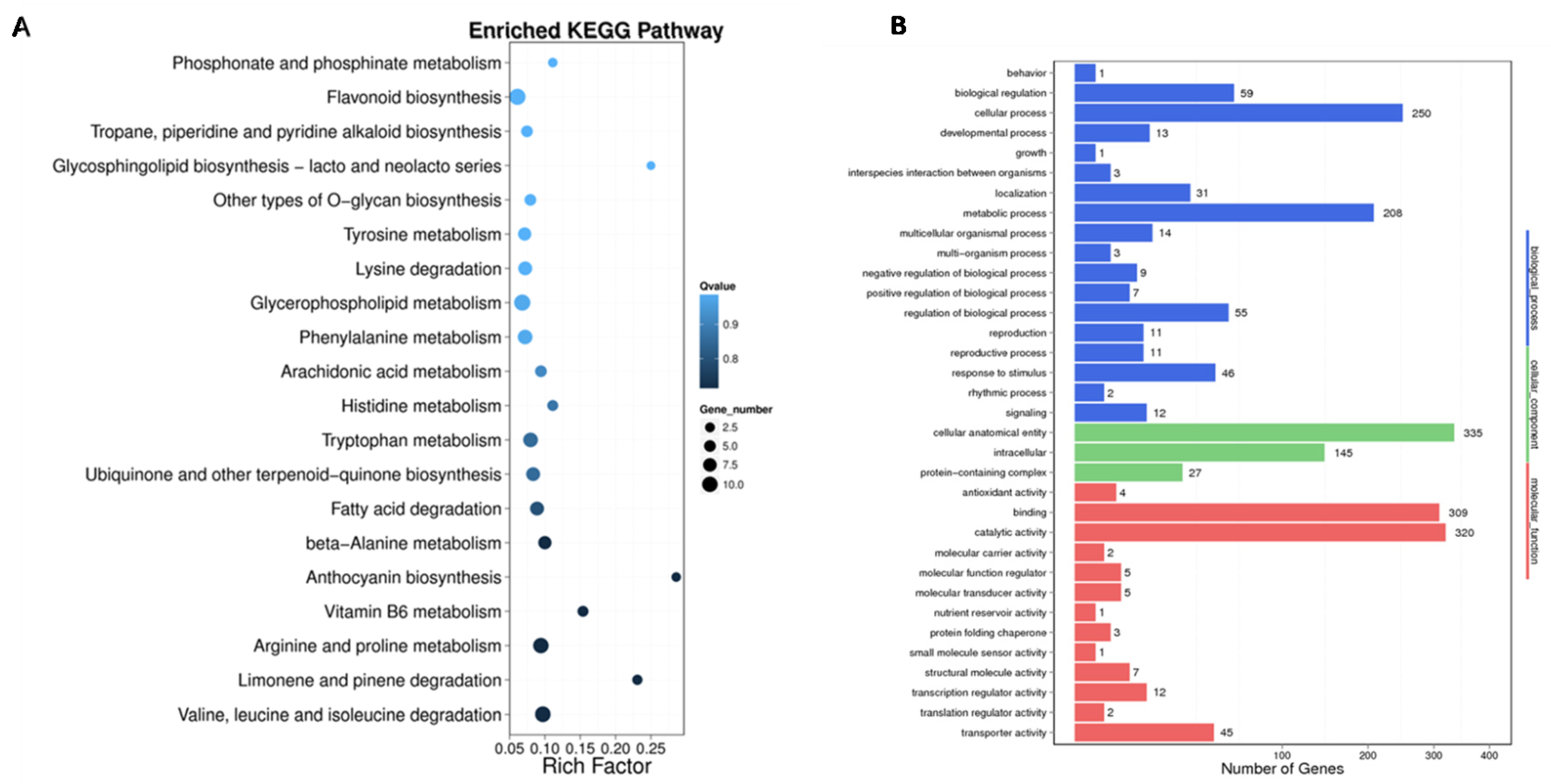
2.5. GO Annotation and KEGG Enrichment Analysis of Sugar Beet DMGs
2.6. Analysis of the Relationship between DMGs’ Methylation and Expression Level’
3. Discussion
3.1. Related DMGs Involved in Carbohydrate and Energy Metabolism
3.2. Related DMGs Involved in Pollen Wall Development
3.3. Related DMGs with Transcriptional Regulation
3.4. DMGs Associated with Endogenous Hormones
4. Materials and Methods
4.1. Plant Materials
4.2. DNA Extraction and WGBS
4.3. WGBS Data Validation
4.4. Evaluation of Methylation Level at Site C
4.5. Differential Methylation Regions (DMRS) and Differential Methylation Genes (DMGS) Were Identified
4.6. GO and KEGG Enrichment Analysis of Differential Methylated Genes
4.7. qRT-PCR Analysis
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Hoffmann, C.M.; Koch, H.; Märländer, B. Chapter 20—Sugar beet. In Crop Physiology Case Histories for Major Crops; Sadras, V.O., Calderini, D.F., Eds.; Academic Press: Cambridge, MA, USA, 2021; pp. 634–672. [Google Scholar]
- Breton, S.; Stewart, D.T.; Brémaud, J.; Havird, J.C.; Smith, C.H.; Hoeh, W.R. Did doubly uniparental inheritance (DUI) of mtDNA originate as a cytoplasmic male sterility (CMS) system? Bioessays 2022, 44, 2100283. [Google Scholar] [CrossRef] [PubMed]
- Meena, O.P.; Dhaliwal, M.S.; Jindal, S.K. Heterosis breeding in chilli pepper by using cytoplasmic male sterile lines for high-yield production with special reference to seed and bioactive compound content under temperature stress regimes. Sci. Hortic. 2020, 262, 109036. [Google Scholar] [CrossRef]
- Xu, F.; Yang, X.; Zhao, N.; Hu, Z.; Mackenzie, S.A.; Zhang, M.; Yang, J. Exploiting sterility and fertility variation in cytoplasmic male sterile vegetable crops. Hortic. Res. 2022, 9, uhab039. [Google Scholar] [CrossRef] [PubMed]
- Chen, L.; Liu, Y. Male Sterility and Fertility Restoration in Crops. Annu. Rev. Plant Biol. 2014, 65, 579–606. [Google Scholar] [CrossRef] [PubMed]
- Sorensen, A.; Kröber, S.; Unte, U.S.; Huijser, P.; Dekker, K.; Saedler, H. The Arabidopsis ABORTED MICROSPORES (AMS) gene encodes a MYC class transcription factor. Plant J. 2003, 33, 413–423. [Google Scholar] [CrossRef] [PubMed]
- Li, J.; Pandeya, D.; Jo, Y.D.; Liu, W.Y.; Kang, B. Reduced activity of ATP synthase in mitochondria causes cytoplasmic male sterility in chili pepper. Planta 2013, 237, 1097–1109. [Google Scholar] [CrossRef] [PubMed]
- Ding, B.; Hao, M.; Mei, D.; Zaman, Q.; Sang, S.; Wang, H.; Wang, W.; Fu, L.; Cheng, H.; Hu, Q. Transcriptome and Hormone Comparison of Three Cytoplasmic Male Sterile Systems in Brassica napus. Int. J. Mol. Sci. 2018, 19, 4022. [Google Scholar] [CrossRef]
- Majewska-Sawka, A.; Rodriguez-Garcia, M.I.; Nakashima, H.; Jassen, B. Ultrastructural expression of cytoplasmic male sterility in sugar beet (Beta vulgaris L.). Sex. Plant Reprod. 1993, 6, 22–32. [Google Scholar] [CrossRef]
- Wesołowski, W.; Szklarczyk, M.; Szalonek, M.; Słowińska, J. Analysis of the mitochondrial proteome in cytoplasmic male-sterile and male-fertile beets. J. Proteom. 2015, 119, 61–74. [Google Scholar] [CrossRef]
- Chase, C.D. Cytoplasmic male sterility: A window to the world of plant mitochondrial–nuclear interactions. Trends Genet. 2007, 23, 81–90. [Google Scholar] [CrossRef]
- Yang, J.; Liu, X.; Yang, X.; Zhang, M. Mitochondrially-targeted expression of a cytoplasmic male sterility-associated orf220 gene causes male sterility in Brassica juncea. BMC Plant Biol. 2010, 10, 231. [Google Scholar] [CrossRef] [PubMed]
- Li, M.; Chen, L.; Tang, D.; Liao, X.; Kong, X.; Li, B.; You, J.; Zhou, R. Discovery of Four Novel ORFs Responsible for Cytoplasmic Male Sterility (CMS) in Cotton (Gossypium hirsutum L.) through Comparative Analysis of the Mitochondrial Genomes of Four Isoplasmic Lines. Agronomy 2020, 10, 765. [Google Scholar] [CrossRef]
- Yamamoto, M.P.; Shinada, H.; Onodera, Y.; Komaki, C.; Mikami, T.; Kubo, T. A male sterility-associated mitochondrial protein in wild beets causes pollen disruption in transgenic plants. Plant J. 2008, 54, 1027–1036. [Google Scholar] [CrossRef] [PubMed]
- Kong, X.; Khan, A.; Li, B.; Zheng, J.; Dawar, F.U.; Li, M.; You, J.; Li, Z.; Zhou, R. Deviant DNA methylation play a key role in the pollen abortion of Gossypium barbadense L. cytoplasmic male sterility. Ind. Crop Prod. 2020, 154, 112622. [Google Scholar] [CrossRef]
- Li, Y.; Ding, X.; Wang, X.; He, T.; Zhang, H.; Yang, L.; Wang, T.; Chen, L.; Gai, J.; Yang, S. Genome-wide comparative analysis of DNA methylation between soybean cytoplasmic male-sterile line NJCMS5A and its maintainer NJCMS5B. BMC Genom. 2017, 18, 596. [Google Scholar] [CrossRef] [PubMed]
- Zhang, M.; Zhang, X.; Guo, L.; Qi, T.; Liu, G.; Feng, J.; Shahzad, K.; Zhang, B.; Li, X.; Wang, H.; et al. Single-base resolution methylome of cotton cytoplasmic male sterility system reveals epigenomic changes in response to high-temperature stress during anther development. J. Exp. Bot. 2020, 71, 951–969. [Google Scholar] [PubMed]
- Li, X.; Zhu, J.; Hu, F.; Ge, S.; Ye, M.; Xiang, H.; Zhang, G.; Zheng, X.; Zhang, H.; Zhang, S.; et al. Single-base resolution maps of cultivated and wild rice methylomes and regulatory roles of DNA methylation in plant gene expression. BMC Genom. 2012, 13, 300. [Google Scholar] [CrossRef]
- Wu, Y.; Sun, Y.; Shen, K.; Sun, S.; Wang, J.; Jiang, T.; Cao, S.; Josiah, S.M.; Pang, J.; Lin, X.; et al. Immediate Genetic and Epigenetic Changes in F1 Hybrids Parented by Species with Divergent Genomes in the Rice Genus (Oryza). PLoS ONE 2015, 10, e132911. [Google Scholar] [CrossRef]
- You, J.; Li, M.; Li, H.; Bai, Y.; Zhu, X.; Kong, X.; Chen, X.; Zhou, R. Integrated Methylome and Transcriptome Analysis Widen the Knowledge of Cytoplasmic Male Sterility in Cotton (Gossypium barbadense L.). Front. Plant Sci. 2022, 13, 770098. [Google Scholar] [CrossRef]
- Han, S.; Li, Y.; Li, J.; Zhang, H.; Ding, X.; He, T.; Gai, J.; Yang, S. Genome-wide analysis of DNA methylation to identify genes and pathways associated with male sterility in soybean. Mol. Breed. 2018, 38, 118. [Google Scholar] [CrossRef]
- Feng, S.; Zhong, Z.; Wang, M.; Jacobsen, S.E. Efficient and accurate determination of genome-wide DNA methylation patterns in Arabidopsis thaliana with enzymatic methyl sequencing. Epigenet. Chromatin 2020, 13, 42. [Google Scholar] [CrossRef] [PubMed]
- Zhang, M.; Guo, L.; Qi, T.; Zhang, X.; Tang, H.; Wang, H.; Qiao, X.; Zhang, B.; Feng, J.; Zuo, Z.; et al. Integrated Methylome and Transcriptome Analysis between the CMS-D2 Line ZBA and Its Maintainer Line ZB in Upland Cotton. Int. J. Mol. Sci. 2019, 20, 6070. [Google Scholar] [CrossRef] [PubMed]
- Li, N.; Zhang, D.; Liu, H.; Yin, C.; Li, X.; Liang, W.; Yuan, Z.; Xu, B.; Chu, H.; Wang, J.; et al. The Rice Tapetum Degeneration Retardation Gene Is Required for Tapetum Degradation and Anther Development. Plant Cell 2006, 18, 2999–3014. [Google Scholar] [CrossRef] [PubMed]
- Ohbayashi, I.; Huang, S.; Fukaki, H.; Song, X.; Sun, S.; Morita, M.T.; Tasaka, M.; Millar, A.H.; Furutani, M. Mitochondrial Pyruvate Dehydrogenase Contributes to Auxin-Regulated Organ Development. Plant Physiol. 2019, 180, 896–909. [Google Scholar] [CrossRef]
- Qin, P.; Tu, B.; Wang, Y.; Deng, L.; Quilichini, T.D.; Li, T.; Wang, H.; Ma, B.; Li, S. ABCG15 Encodes an ABC Transporter Protein, and is Essential for Post-Meiotic Anther and Pollen Exine Development in Rice. Plant Cell Physiol. 2012, 54, 138–154. [Google Scholar] [CrossRef]
- Bacic, A. Breaking an impasse in pectin biosynthesis. Proc. Natl. Acad. Sci. USA 2006, 103, 5639–5640. [Google Scholar] [CrossRef]
- Brown, D.M.; Goubet, F.; Wong, V.W.; Goodacre, R.; Stephens, E.; Dupree, P.; Turner, S.R. Comparison of five xylan synthesis mutants reveals new insight into the mechanisms of xylan synthesis. Plant J. 2007, 52, 1154–1168. [Google Scholar] [CrossRef]
- Ma, Z.; Li, W.; Wang, H.; Yu, D. WRKY transcription factors WRKY12 and WRKY13 interact with SPL10 to modulate age-mediated flowering. J. Integr. Plant Biol. 2020, 62, 1659–1673. [Google Scholar] [CrossRef]
- Fernandez, D.E.; Heck, G.R.; Perry, S.E.; Patterson, S.E.; Bleecker, A.B.; Fang, S. The Embryo MADS Domain Factor AGL15 Acts Postembryonically: Inhibition of Perianth Senescence and Abscission via Constitutive Expression. Plant Cell 2000, 12, 183–197. [Google Scholar] [CrossRef]
- Tang, N.; Liu, W.; Zhang, W.; Tang, D. Integrative analysis of transcriptomic and proteomic changes related to male sterility in Tagetes erecta. Physiol. Mol. Biol. Plants 2020, 26, 2061–2074. [Google Scholar] [CrossRef]
- Shen, S.; Li, B.; Deng, T.; Xiao, Z.; Chen, X.; Hu, H.; Zhang, B.; Wu, G.; Li, F.; Zhao, X.; et al. The equilibrium between sugars and ethylene is involved in shading- and drought-induced kernel abortion in maize. Plant Growth Regul. 2020, 91, 101–111. [Google Scholar] [CrossRef]




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Sample | Clean Bases | Mapped Reads | Mapping Rate (%) | Average Depth (X) | Bisulfite Conversion Rate (%) |
|---|---|---|---|---|---|
| DY5O | 248,189,440 | 213,850,619 | 86.16 | 29.58 | 99.14 |
| DY5CMS | 301,409,704 | 262,053,058 | 86.94 | 36.00 | 99.33 |
| Background | Sample | Number of Methylated Cytosine | Mean Methylation Level (%) |
|---|---|---|---|
| C | DY5CMS | 53,612,497 | 27.32 |
| DY5O | 51,332,141 | 27.18 | |
| CG | DY5CMS | 14,155,733 | 80.43 |
| DY5O | 14,101,006 | 79.96 | |
| CHG | DY5CMS | 12,223,221 | 58.72 |
| DY5O | 12,149,595 | 58.69 | |
| CHH | DY5CMS | 27,233,543 | 12.42 |
| DY5O | 25,081,540 | 12.28 |
| Target Area | Sample | DNA Total Methylation Level (%) | mCG (%) | mCHG (%) | mCHH (%) |
|---|---|---|---|---|---|
| Chr7: 1,633,239–1,633,426 | CMS | 12 | 57.94 | 4.5 | 1 |
| O | 13 | 62.05 | 36.46 | 20.45 | |
| Chr3: 51,195,686–51,195,932 | CMS | 14 | 85.08 | 37.21 | 1.98 |
| O | 10 | 69.42 | 0 | 2.02 | |
| Chr1: 7,437,126–7,437,376 | CMS | 9 | 16.23 | 66.57 | 6.7 |
| O | 5 | 3.05 | 6.27 | 6.23 |
| Gene Name | Location | Methylation Status | Gene Annotation |
|---|---|---|---|
| BVRB_3g061680 | gene | Hypo | MADS-box transcription factor family protein |
| BVRB_6g133580 | gene, prompter | Hypo | WRKY transcription factor family protein |
| BVRB_2g032900 | promoter | Hyper | AP2/ERF and B3 domain-containing transcription factor family protein |
| BVRB_2g047050 | gene | Hyper | NADH dehydrogenase |
| BVRB_6g137940 | gene | Hyper | galacturonosyltransferase |
| BVRB_003480 | gene | Hypo | UDP-Glycosyltransferase superfamily protein |
| BVRB_004020 | gene, prompter | Hyper | pyruvate dehydrogenase E1 component subunit beta-3 |
| BVRB_9g208010 | gene | Hyper | ABC transporter A family protein |
| BVRB_4g073090 | gene | Hypo | ABC transporter G family protein |
| BVRB_2g036570 | gene | Hypo | PPR superfamily protein |
| BVRB_3g049380 | gene | Hypo | ACO family protein |
| BVRB_7g172330 | promoter | Hypo | TDR transcription factor family protein |
| Primer | Primer Sequence | Location | Length (bp) |
|---|---|---|---|
| Primer1 | F:TATTATTTTAAGGGATGGATGTTTTG | Chr7 | 228 |
| R:ACTCCAAATTCACAACTATAACCTTCT | 1,633,303–1,633,462 | ||
| Primer2 | F:TGGGTTGATTTAGGTTTATTTGAAT | Chr5 | 249 |
| R:ACAAAAAATACATACCCCCTCAAT | 51,195,686–51,195,932 | ||
| Primer3 | F:TGGGGTTAATAATTGTTAGTTTTAT | Chr1 | 249 |
| R:TCACCACCCCATCTAAAATATATAC | 7,437,126–7,437,376 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Weng, J.; Wang, H.; Cheng, D.; Liu, T.; Zeng, D.; Dai, C.; Luo, C. The Effects of DNA Methylation on Cytoplasmic Male Sterility in Sugar Beet. Int. J. Mol. Sci. 2024, 25, 1118. https://doi.org/10.3390/ijms25021118
Weng J, Wang H, Cheng D, Liu T, Zeng D, Dai C, Luo C. The Effects of DNA Methylation on Cytoplasmic Male Sterility in Sugar Beet. International Journal of Molecular Sciences. 2024; 25(2):1118. https://doi.org/10.3390/ijms25021118
Chicago/Turabian StyleWeng, Jiamin, Hui Wang, Dayou Cheng, Tianjiao Liu, Deyong Zeng, Cuihong Dai, and Chengfei Luo. 2024. "The Effects of DNA Methylation on Cytoplasmic Male Sterility in Sugar Beet" International Journal of Molecular Sciences 25, no. 2: 1118. https://doi.org/10.3390/ijms25021118
APA StyleWeng, J., Wang, H., Cheng, D., Liu, T., Zeng, D., Dai, C., & Luo, C. (2024). The Effects of DNA Methylation on Cytoplasmic Male Sterility in Sugar Beet. International Journal of Molecular Sciences, 25(2), 1118. https://doi.org/10.3390/ijms25021118