
The Healthy and Diseased Retina Seen through Neuron–Glia Interactions

 , ,
, ,  , , , , , ,
, , , , , ,  , , ,
, , ,  ,
,  , , , and
, , , and 
{kind=link}
{kind=link}
Abstract
1. Introduction
2. The Organization of the Retina
3. Neurotransmitters
3.1. Glutamate
3.2. γ-Aminobutyric Acid (GABA)
3.3. Dopamine
3.4. The Endocannabinoid System
3.5. TRP Channels
3.6. Adenosine
3.7. Neuropeptides: PACAP
3.8. Nitric Oxide
4. Gliotransmitters
4.1. Nucleotide Receptors
4.2. Nucleotides and Retinal Cell Proliferation
4.3. Nucleotides and Retinal Cell Migration
4.4. Nucleotides and the Induction of Cell Death in the Retina
4.5. P2X7 Glial Receptors and Retinal Development
5. Antioxidants
5.1. Glutathione
5.2. Vitamin C
6. Reciprocal Interactions between Retinal Transmitters
6.1. Dopamine and Adenosine
6.2. Glutamate and Adenosine
6.3. Glutamate and Vitamin C
6.4. Glutamate and GABA
6.5. Dopamine and Glutamate
6.6. Endocannabinoid and Dopamine
6.7. Dopamine, Glutamate, and Vitamin C
6.8. Adenosine, Vitamin C, and Nitric Oxide
7. The Diseased Retina
7.1. Glaucoma
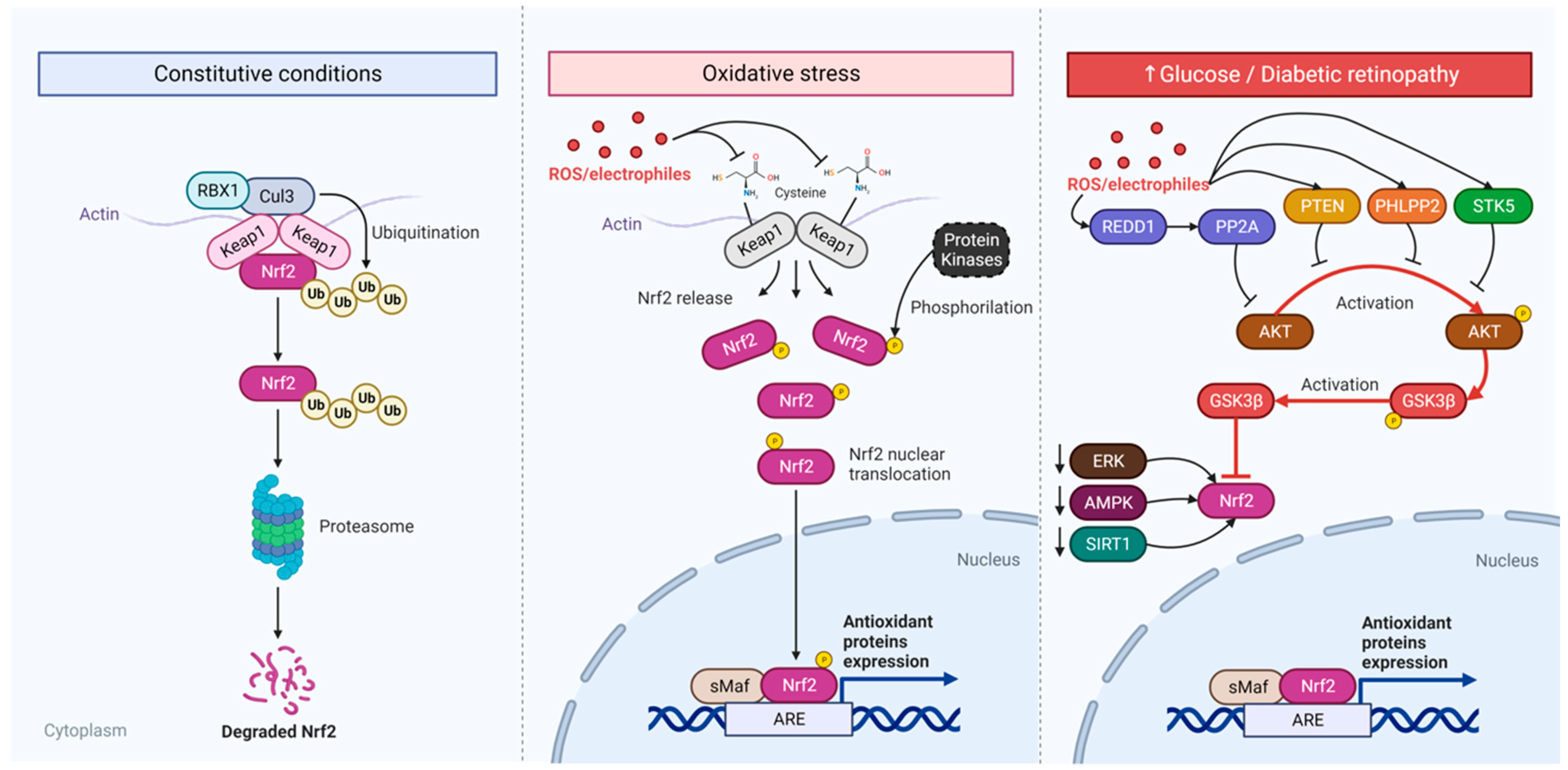
7.2. Diabetic Retina
8. Investigation of Innovative Therapeutic Strategies
8.1. Neuroprotection
8.2. Gene Therapy and the Future of Vision Recovery
8.3. Cell Reprogramming
9. Conclusions
10. Limitations of the Present Review
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
References
- Vergara, M.N.; Canto-Soler, M.V. Rediscovering the chick embryo as a model to study retinal development. Neural Dev. 2012, 7, 22. [Google Scholar] [CrossRef]
- Belecky-Adams, T.L.; Haynes, T.; Wilson, J.M.; Del Rio-Tsonis, K. Chapter 8-The Chick as a Model for Retina Development and Regeneration. In Animal Models in Eye Research; Tsonis, P.A., Ed.; Academic Press: London, UK, 2008; pp. 102–119. [Google Scholar] [CrossRef]
- Cebulla, C.M.; Zelinka, C.P.; Scott, M.A.; Lubow, M.; Bingham, A.; Rasiah, S.; Mahmoud, A.M.; Fischer, A.J. A chick model of retinal detachment: Cone rich and novel. PLoS ONE 2012, 7, e44257. [Google Scholar] [CrossRef]
- Al Sabaani, N. Exendin-4 inhibits high glucose-induced oxidative stress in retinal pigment epithelial cells by modulating the expression and activation of p(66)Shc. Cutan. Ocul. Toxicol. 2021, 40, 175–186. [Google Scholar] [CrossRef]
- Ventura, A.L.M.; De Mello, F.G.; De Melo Reis, R.A. Methods of dopamine research in retina cells. Methods Mol. Biol. 2013, 964, 25–42. [Google Scholar] [CrossRef]
- Tempone, M.H.; Freitas, H.R.; Schitine, C.S.; de Melo Reis, R.A. Visualizing Shifts on Neuron-Glia Circuit with the Calcium Imaging Technique. J. Vis. Exp. 2022, 8, e63338. [Google Scholar] [CrossRef]
- Arthur, P.; Muok, L.; Nathani, A.; Zeng, E.Z.; Sun, L.; Li, Y.; Singh, M. Bioengineering Human Pluripotent Stem Cell-Derived Retinal Organoids and Optic Vesicle-Containing Brain Organoids for Ocular Diseases. Cells 2022, 11, 3429. [Google Scholar] [CrossRef]
- Gardino, P.F. Neurochemical phenotype and birthdating of specific cell populations in the chick retina. An. Acad. Bras. Cienc. 2010, 82, 595–608. [Google Scholar] [CrossRef][Green Version]
- Yamagata, M.; Yan, W.; Sanes, J.R. A cell atlas of the chick retina based on single-cell transcriptomics. eLife 2021, 10, e63907. [Google Scholar] [CrossRef]
- Li, M.; Xu, N.; Bian, P.; Tian, X.; Wang, X.; Wang, Y.; Jia, X.; Heller, R.; Wang, M.; Wang, F.; et al. De Novo Assembly of 20 Chicken Genomes Reveals the Undetectable Phenomenon for Thousands of Core Genes on Microchromosomes and Subtelomeric Regions. Mol. Biol. Evol. 2023, 39, msac066. [Google Scholar] [CrossRef]
- Hoon, M.; Okawa, H.; Della Santina, L.; Wong, R.O.L. Functional architecture of the retina: Development and disease. Prog. Retin. Eye Res. 2014, 42, 44–84. [Google Scholar] [CrossRef] [PubMed]
- Reichenbach, A.; Bringmann, A. New functions of Müller cells. Glia 2013, 61, 651–678. [Google Scholar] [CrossRef]
- Karl, M.O.; Reh, T.A. Regenerative medicine for retinal diseases: Activating endogenous repair mechanisms. Trends Mol. Med. 2010, 16, 193–202. [Google Scholar] [CrossRef]
- Vecino, E.; Rodriguez, F.D.; Ruzafa, N.; Pereiro, X.; Sharma, S.C. Glia-neuron interactions in the mammalian retina. Prog. Retin. Eye Res. 2016, 51, 1–40. [Google Scholar] [CrossRef]
- Seifert, M.; Baden, T.; Osorio, D. The retinal basis of vision in chicken. In Seminars in Cell and Developmental Biology; Elsevier Ltd.: Amsterdam, The Netherlands, 2020. [Google Scholar] [CrossRef]
- Baden, T.; Osorio, D. The Retinal Basis of Vertebrate Color Vision. Annu. Rev. Vis. Sci. 2019, 5, 177–200. [Google Scholar] [CrossRef]
- Barnstable, C.J. Glutamate and GABA in retinal circuitry. Curr. Opin. Neurobiol. 1993, 3, 520–525. [Google Scholar] [CrossRef]
- Münch, T.A.; da Silveira, R.A.; Siegert, S.; Viney, T.J.; Awatramani, G.B.; Roska, B. Approach sensitivity in the retina processed by a multifunctional neural circuit. Nat. Neurosci. 2009, 12, 1308–1316. [Google Scholar] [CrossRef]
- Pourcho, R.G. Neurotransmitters in the retina. Curr. Eye Res. 1996, 15, 797–803. [Google Scholar] [CrossRef]
- Martins, R.A.; Pearson, R.A. Control of cell proliferation by neurotransmitters in the developing vertebrate retina. Brain Res. 2008, 1192, 37–60. [Google Scholar] [CrossRef]
- Ferreira, I.L.; Duarte, C.B.; Carvalho, A.P. Ca2+ influx through glutamate receptor-associated channels in retina cells correlates with neuronal cell death. Eur. J. Pharmacol. 1996, 302, 153–162. [Google Scholar] [CrossRef]
- Rodríguez Villanueva, J.; Martín Esteban, J.; Rodríguez Villanueva, L.J. Retinal Cell Protection in Ocular Excitotoxicity Diseases. Possible Alternatives Offered by Microparticulate Drug Delivery Systems and Future Prospects. Pharmaceutics 2020, 12, 94. [Google Scholar] [CrossRef]
- Carpi-Santos, R.; de Melo Reis, R.A.; Gomes, F.C.A.; Calaza, K.C. Contribution of Müller cells in the diabetic retinopathy development: Focus on Oxidative Stress and Inflammation. Antioxidants 2022, 11, 617. [Google Scholar] [CrossRef] [PubMed]
- Santos, A.E.; Carvalho, A.L.; Lopes, M.C.; Carvalho, A.P. Differential postreceptor signaling events triggered by excitotoxic stimulation of different ionotropic glutamate receptors in retinal neurons. J. Neurosci. Res. 2001, 66, 643–655. [Google Scholar] [CrossRef] [PubMed]
- Lambuk, L.; Jafri, A.J.A.; Iezhitsa, I.; Agarwal, R.; Bakar, N.S.; Agarwal, P.; Abdullah, A.; Ismail, N.M. Dose-dependent effects of NMDA on retinal and optic nerve morphology in rats. Int. J. Ophthalmol. 2019, 12, 746–753. [Google Scholar] [CrossRef] [PubMed]
- Rosenstein, R.E. New actors in optic neuritis pathogenesis: An Editorial for “Influence of retinal NMDA receptor activity during autoimmune optic neuritis” on page 693. J. Neurochem. 2020, 153, 671–673. [Google Scholar] [CrossRef] [PubMed]
- Ikonomidou, C.; Turski, L. Why did NMDA receptor antagonists fail clinical trials for stroke and traumatic brain injury? Lancet Neurol. 2002, 1, 383–386. [Google Scholar] [CrossRef] [PubMed]
- Traynelis, S.F.; Wollmuth, L.P.; McBain, C.J.; Menniti, F.S.; Vance, K.M.; Ogden, K.K.; Hansen, K.B.; Yuan, H.; Myers, S.J.; Dingledine, R. Glutamate receptor ion channels: Structure, regulation, and function. Pharmacol. Rev. 2010, 62, 405–496. [Google Scholar] [CrossRef] [PubMed]
- Pinto, M.C.X.; Kihara, A.H.; Goulart, V.A.M.; Tonelli, F.M.P.; Gomes, K.N.; Ulrich, H.; Resende, R.R. Calcium signaling and cell proliferation. Cell. Signal. 2015, 27, 2139–2149. [Google Scholar] [CrossRef]
- de Melo Reis, R.A.; Freitas, H.R.; de Mello, F.G. Cell Calcium Imaging as a Reliable Method to Study Neuron-Glial Circuits. Front. Neurosci. 2020, 14, 569361. [Google Scholar] [CrossRef]
- Dawson, T.M.; Dawson, V.L. Chapter Four-Nitric Oxide Signaling in Neurodegeneration and Cell Death. In Advances in Pharmacology; Pasternak, G.W., Coyle, J.T., Eds.; Academic Press: Cambridge, MA, USA, 2018; Volume 82, pp. 57–83. [Google Scholar]
- Marshall, J.; Wong, K.Y.; Rupasinghe, C.N.; Tiwari, R.; Zhao, X.; Berberoglu, E.D.; Sinkler, C.; Liu, J.; Lee, I.; Parang, K.; et al. Inhibition of N-Methyl-D-aspartate-induced Retinal Neuronal Death by Polyarginine Peptides Is Linked to the Attenuation of Stress-induced Hyperpolarization of the Inner Mitochondrial Membrane Potential. J. Biol. Chem. 2015, 290, 22030–22048. [Google Scholar] [CrossRef]
- Martel, M.A.; Ryan, T.J.; Bell, K.F.; Fowler, J.H.; McMahon, A.; Al-Mubarak, B.; Komiyama, N.H.; Horsburgh, K.; Kind, P.C.; Grant, S.G.; et al. The subtype of GluN2 C-terminal domain determines the response to excitotoxic insults. Neuron 2012, 74, 543–556. [Google Scholar] [CrossRef]
- Opere, C.A.; Heruye, S.; Njie-Mbye, Y.F.; Ohia, S.E.; Sharif, N.A. Regulation of Excitatory Amino Acid Transmission in the Retina: Studies on Neuroprotection. J. Ocul. Pharmacol. Ther. 2018, 34, 107–118. [Google Scholar] [CrossRef]
- Park, Y.H.; Broyles, H.V.; He, S.; McGrady, N.R.; Li, L.; Yorio, T. Involvement of AMPA Receptor and Its Flip and Flop Isoforms in Retinal Ganglion Cell Death Following Oxygen/Glucose Deprivation. Investig. Ophthalmol. Vis. Sci. 2016, 57, 508–526. [Google Scholar] [CrossRef]
- Cossenza, M.; Cadilhe, D.V.; Coutinho, R.N.; Paes-de-Carvalho, R. Inhibition of protein synthesis by activation of NMDA receptors in cultured retinal cells: A new mechanism for the regulation of nitric oxide production. J. Neurochem. 2006, 97, 1481–1493. [Google Scholar] [CrossRef]
- Gladulich, L.F.H.; Peixoto-Rodrigues, M.C.; Campello-Costa, P.; Paes-de-Carvalho, R.; Cossenza, M. NMDA-induced nitric oxide generation and CREB activation in central nervous system is dependent on eukaryotic elongation factor 2 kinase. Biochim. Biophys. Acta Mol. Cell Res. 2020, 1867, 118783. [Google Scholar] [CrossRef]
- Carlberg, U.; Nilsson, A.; Nygård, O. Functional properties of phosphorylated elongation factor 2. Eur. J. Biochem. 1990, 191, 639–645. [Google Scholar] [CrossRef]
- Nairn, A.C.; Matsushita, M.; Nastiuk, K.; Horiuchi, A.; Mitsui, K.; Shimizu, Y.; Palfrey, H.C. Elongation factor-2 phosphorylation and the regulation of protein synthesis by calcium. Prog. Mol. Subcell. Biol. 2001, 27, 91–129. [Google Scholar] [CrossRef]
- Price, N.T.; Redpath, N.T.; Severinov, K.V.; Campbell, D.G.; Russell, J.M.; Proud, C.G. Identification of the phosphorylation sites in elongation factor-2 from rabbit reticulocytes. FEBS Lett. 1991, 282, 253–258. [Google Scholar] [CrossRef]
- Rodnina, M.V.; Savelsbergh, A.; Wintermeyer, W. Dynamics of translation on the ribosome: Molecular mechanics of translocation. FEMS Microbiol. Rev. 1999, 23, 317–333. [Google Scholar] [CrossRef]
- Ryazanov, A.G.; Shestakova, E.A.; Natapov, P.G. Phosphorylation of elongation factor 2 by EF-2 kinase affects rate of translation. Nature 1988, 334, 170–173. [Google Scholar] [CrossRef]
- Scheetz, A.J.; Nairn, A.C.; Constantine-Paton, M. N-methyl-D-aspartate receptor activation and visual activity induce elongation factor-2 phosphorylation in amphibian tecta: A role for N-methyl-D-aspartate receptors in controlling protein synthesis. Proc. Natl. Acad. Sci. USA 1997, 94, 14770–14775. [Google Scholar] [CrossRef] [PubMed]
- Hsu, W.L.; Chung, H.W.; Wu, C.Y.; Wu, H.I.; Lee, Y.T.; Chen, E.C.; Fang, W.; Chang, Y.C. Glutamate Stimulates Local Protein Synthesis in the Axons of Rat Cortical Neurons by Activating α-Amino-3-hydroxy-5-methyl-4-isoxazolepropionic Acid (AMPA) Receptors and Metabotropic Glutamate Receptors. J. Biol. Chem. 2015, 290, 20748–20760. [Google Scholar] [CrossRef] [PubMed]
- Dieterich, D.C.; Hodas, J.J.; Gouzer, G.; Shadrin, I.Y.; Ngo, J.T.; Triller, A.; Tirrell, D.A.; Schuman, E.M. In situ visualization and dynamics of newly synthesized proteins in rat hippocampal neurons. Nat. Neurosci. 2010, 13, 897–905. [Google Scholar] [CrossRef] [PubMed]
- Scheetz, A.J.; Nairn, A.C.; Constantine-Paton, M. NMDA receptor-mediated control of protein synthesis at developing synapses. Nat. Neurosci. 2000, 3, 211–216. [Google Scholar] [CrossRef]
- Verpelli, C.; Piccoli, G.; Zibetti, C.; Zanchi, A.; Gardoni, F.; Huang, K.; Brambilla, D.; Di Luca, M.; Battaglioli, E.; Sala, C. Synaptic activity controls dendritic spine morphology by modulating eEF2-dependent BDNF synthesis. J. Neurosci. 2010, 30, 5830–5842. [Google Scholar] [CrossRef] [PubMed]
- Cossenza, M.; Socodato, R.; Mejía-García, T.A.; Domith, I.; Portugal, C.C.; Gladulich, L.F.H.; Duarte-Silva, A.T.; Khatri, L.; Antoine, S.; Hofmann, F.; et al. Protein synthesis inhibition promotes nitric oxide generation and activation of CGKII-dependent downstream signaling pathways in the retina. Biochim. Biophys. Acta Mol. Cell Res. 2020, 1867, 118732. [Google Scholar] [CrossRef] [PubMed]
- Numakawa, T.; Suzuki, S.; Kumamaru, E.; Adachi, N.; Richards, M.; Kunugi, H. BDNF function and intracellular signaling in neurons. Histol. Histopathol. 2010, 25, 237–258. [Google Scholar] [CrossRef]
- Schmid, R.S.; Graff, R.D.; Schaller, M.D.; Chen, S.; Schachner, M.; Hemperly, J.J.; Maness, P.F. NCAM stimulates the Ras-MAPK pathway and CREB phosphorylation in neuronal cells. J. Neurobiol. 1999, 38, 542–558. [Google Scholar] [CrossRef]
- Singh, L.; Bhatti, R. Signaling Pathways Involved in the Neuroprotective Effect of Osthole: Evidence and Mechanisms. Mol. Neurobiol. 2023, 1–19. [Google Scholar] [CrossRef]
- Luhmann, H.J.; Kirischuk, S.; Sinning, A.; Kilb, W. Early GABAergic circuitry in the cerebral cortex. Curr. Opin. Neurobiol. 2014, 26, 72–78. [Google Scholar] [CrossRef]
- Mosinger, J.L.; Yazulla, S.; Studholme, K.M. GABA-like immunoreactivity in the vertebrate retina: A species comparison. Exp. Eye Res. 1986, 42, 631–644. [Google Scholar] [CrossRef]
- Wu, C.; Sun, D. GABA receptors in brain development, function, and injury. Metab. Brain Dis. 2015, 30, 367–379. [Google Scholar] [CrossRef]
- Siucinska, E. Γ-Aminobutyric acid in adult brain: An update. Behav. Brain Res. 2019, 376, 112224. [Google Scholar] [CrossRef]
- Nuss, P. Anxiety disorders and GABA neurotransmission: A disturbance of modulation. Neuropsychiatr. Dis. Treat. 2015, 11, 165–175. [Google Scholar] [CrossRef] [PubMed]
- Wässle, H. Parallel processing in the mammalian retina. Nat. Rev. Neurosci. 2004, 5, 747–757. [Google Scholar] [CrossRef] [PubMed]
- De Sampaio Schitine, C.; Kubrusly, R.C.; De Melo Reis, R.A.; Yamasaki, E.N.; De Mello, M.C.; De Mello, F.G. GABA uptake by purified avian Müller glia cells in culture. Neurotox. Res. 2007, 12, 145–153. [Google Scholar] [CrossRef] [PubMed]
- Ferreira, D.D.; Stutz, B.; de Mello, F.G.; Reis, R.A.; Kubrusly, R.C. Caffeine potentiates the release of GABA mediated by NMDA receptor activation: Involvement of A1 adenosine receptors. Neuroscience 2014, 281, 208–215. [Google Scholar] [CrossRef] [PubMed]
- Frederick, J.M. The emergence of GABA-accumulating neurons during retinal histogenesis in the embryonic chick. Exp. Eye Res. 1987, 45, 933–945. [Google Scholar] [CrossRef]
- Hokoç, J.N.; Ventura, A.L.; Gardino, P.F.; De Mello, F.G. Developmental immunoreactivity for GABA and GAD in the avian retina: Possible alternative pathway for GABA synthesis. Brain Res. 1990, 532, 197–202. [Google Scholar] [CrossRef]
- Sun, H.; Crossland, W.J. Quantitative assessment of localization and colocalization of glutamate, aspartate, glycine, and GABA immunoreactivity in the chick retina. Anat. Rec. 2000, 260, 158–179. [Google Scholar] [CrossRef]
- Lee, S.E.; Lee, Y.; Lee, G.H. The regulation of glutamic acid decarboxylases in GABA neurotransmission in the brain. Arch. Pharm. Res. 2019, 42, 1031–1039. [Google Scholar] [CrossRef]
- Soghomonian, J.J.; Martin, D.L. Two isoforms of glutamate decarboxylase: Why? Trends Pharmacol. Sci. 1998, 19, 500–505. [Google Scholar] [CrossRef] [PubMed]
- Yamasaki, E.N.; Barbosa, V.D.; De Mello, F.G.; Hokoc, J.N. GABAergic system in the developing mammalian retina: Dual sources of GABA at early stages of postnatal development. Int. J. Dev. Neurosci. 1999, 17, 201–213. [Google Scholar] [CrossRef]
- Madsen, K.K.; White, H.S.; Schousboe, A. Neuronal and non-neuronal GABA transporters as targets for antiepileptic drugs. Pharmacol. Ther. 2010, 125, 394–401. [Google Scholar] [CrossRef]
- Pinal, C.S.; Tobin, A.J. Uniqueness and redundancy in GABA production. Perspect. Dev. Neurobiol. 1998, 5, 109–118. [Google Scholar]
- Wu, Z.; Guo, Z.; Gearing, M.; Chen, G. Tonic inhibition in dentate gyrus impairs long-term potentiation and memory in an Alzheimer’s [corrected] disease model. Nat. Commun. 2014, 5, 4159. [Google Scholar] [CrossRef] [PubMed]
- Kwak, H.; Koh, W.; Kim, S.; Song, K.; Shin, J.I.; Lee, J.M.; Lee, E.H.; Bae, J.Y.; Ha, G.E.; Oh, J.E.; et al. Astrocytes Control Sensory Acuity via Tonic Inhibition in the Thalamus. Neuron 2020, 108, 691–706. [Google Scholar] [CrossRef]
- Krantis, A. GABA in the Mammalian Enteric Nervous System. News Physiol. Sci. 2000, 15, 284–290. [Google Scholar] [CrossRef] [PubMed]
- De, A.; Dos, S.; Nora, H.; Yamasaki, E.; Gardino, P.; Mello, F. Regulation of glutamic acid decarboxylase of chick and rat retina cells by GABA and excitatory amino acids. An. Acad. Bras. Ciênc. 2000, 72, 438. [Google Scholar] [CrossRef]
- Sequerra, E.B.; Gardino, P.; Hedin-Pereira, C.; de Mello, F.G. Putrescine as an important source of GABA in the postnatal rat subventricular zone. Neuroscience 2007, 146, 489–493. [Google Scholar] [CrossRef]
- Kim, J.I.; Ganesan, S.; Luo, S.X.; Wu, Y.W.; Park, E.; Huang, E.J.; Chen, L.; Ding, J.B. Aldehyde dehydrogenase 1a1 mediates a GABA synthesis pathway in midbrain dopaminergic neurons. Science 2015, 350, 102–106. [Google Scholar] [CrossRef]
- Magri, C.; Giacopuzzi, E.; La Via, L.; Bonini, D.; Ravasio, V.; Elhussiny, M.E.A.; Orizio, F.; Gangemi, F.; Valsecchi, P.; Bresciani, R.; et al. A novel homozygous mutation in GAD1 gene described in a schizophrenic patient impairs activity and dimerization of GAD67 enzyme. Sci. Rep. 2018, 8, 15470. [Google Scholar] [CrossRef] [PubMed]
- Fletcher, E.L.; Phipps, J.A.; Ward, M.M.; Puthussery, T.; Wilkinson-Berka, J.L. Neuronal and glial cell abnormality as predictors of progression of diabetic retinopathy. Curr. Pharm. Des. 2007, 13, 2699–2712. [Google Scholar] [CrossRef] [PubMed]
- Malomouzh, A.; Ilyin, V.; Nikolsky, E. Components of the GABAergic signaling in the peripheral cholinergic synapses of vertebrates: A review. Amino Acids 2019, 51, 1093–1102. [Google Scholar] [CrossRef]
- Eskandari, S.; Willford, S.L.; Anderson, C.M. Revised Ion/Substrate Coupling Stoichiometry of GABA Transporters. Adv. Neurobiol. 2017, 16, 85–116. [Google Scholar] [CrossRef]
- Gether, U.; Andersen, P.H.; Larsson, O.M.; Schousboe, A. Neurotransmitter transporters: Molecular function of important drug targets. Trends Pharmacol. Sci. 2006, 27, 375–383. [Google Scholar] [CrossRef] [PubMed]
- Kubrusly, R.C.; da Cunha, M.C.; Reis, R.A.; Soares, H.; Ventura, A.L.; Kurtenbach, E.; de Mello, M.C.; de Mello, F.G. Expression of functional receptors and transmitter enzymes in cultured Muller cells. Brain Res. 2005, 1038, 141–149. [Google Scholar] [CrossRef] [PubMed]
- Scimemi, A. Structure, function, and plasticity of GABA transporters. Front. Cell Neurosci. 2014, 8, 161. [Google Scholar] [CrossRef] [PubMed]
- Calaza, K.C.; Gardino, P.F.; de Mello, F.G. Transporter mediated GABA release in the retina: Role of excitatory amino acids and dopamine. Neurochem. Int. 2006, 49, 769–777. [Google Scholar] [CrossRef]
- Schwartz, E.A. Transport-mediated synapses in the retina. Physiol. Rev. 2002, 82, 875–891. [Google Scholar] [CrossRef]
- Leviel, V. Dopamine release mediated by the dopamine transporter, facts and consequences. J. Neurochem. 2011, 118, 475–489. [Google Scholar] [CrossRef]
- Nicholls, D.; Attwell, D. The release and uptake of excitatory amino acids. Trends Pharmacol. Sci. 1990, 11, 462–468. [Google Scholar] [CrossRef]
- Roux, M.J.; Supplisson, S. Neuronal and glial glycine transporters have different stoichiometries. Neuron 2000, 25, 373–383. [Google Scholar] [CrossRef]
- Calaza Kda, C.; de Mello, M.C.; de Mello, F.G.; Gardino, P.F. Local differences in GABA release induced by excitatory amino acids during retina development: Selective activation of NMDA receptors by aspartate in the inner retina. Neurochem. Res. 2003, 28, 1475–1485. [Google Scholar] [CrossRef] [PubMed]
- Yazulla, S.; Kleinschmidt, J. Dopamine blocks carrier-mediated release of GABA from retinal horizontal cells. Brain Res. 1982, 233, 211–215. [Google Scholar] [CrossRef] [PubMed]
- Do Nascimento, J.L.; Kubrusly, R.C.; Reis, R.A.; De Mello, M.C.; De Mello, F.G. Atypical effect of dopamine in modulating the functional inhibition of NMDA receptors of cultured retina cells. Eur. J. Pharmacol. 1998, 343, 103–110. [Google Scholar] [CrossRef]
- Maggesissi, R.S.; Gardino, P.F.; Guimarães-Souza, E.M.; Paes-de-Carvalho, R.; Silva, R.B.; Calaza, K.C. Modulation of GABA release by nitric oxide in the chick retina: Different effects of nitric oxide depending on the cell population. Vision Res. 2009, 49, 2494–2502. [Google Scholar] [CrossRef] [PubMed]
- Ferreira, I.L.; Duarte, C.B.; Santos, P.F.; Carvalho, C.M.; Carvalho, A.P. Release of [3H]GABA evoked by glutamate receptor agonists in cultured chick retina cells: Effect of Ca2+. Brain Res. 1994, 664, 252–256. [Google Scholar] [CrossRef]
- Melone, M.; Ciappelloni, S.; Conti, F. Plasma membrane transporters GAT-1 and GAT-3 contribute to heterogeneity of GABAergic synapses in neocortex. Front. Neuroanat. 2014, 8, 72. [Google Scholar] [CrossRef]
- do Nascimento, J.L.; Ventura, A.L.; Paes de Carvalho, R. Veratridine- and glutamate-induced release of [3H]-GABA from cultured chick retina cells: Possible involvement of a GAT-1-like subtype of GABA transporter. Brain Res. 1998, 798, 217–222. [Google Scholar] [CrossRef]
- Borges-Martins, V.P.P.; Ferreira, D.D.P.; Souto, A.C.; Oliveira Neto, J.G.; Pereira-Figueiredo, D.; da Costa Calaza, K.; de Jesus Oliveira, K.; Manhaes, A.C.; de Melo Reis, R.A.; Kubrusly, R.C.C. Caffeine regulates GABA transport via A1R blockade and cAMP signaling. Neurochem. Int. 2019, 131, 104550. [Google Scholar] [CrossRef]
- Tapia, R.; Arias, C. Selective stimulation of neurotransmitter release from chick retina by kainic and glutamic acids. J. Neurochem. 1982, 39, 1169–1178. [Google Scholar] [CrossRef]
- Calaza, K.C.; de Mello, F.G.; Gardino, P.F. GABA release induced by aspartate-mediated activation of NMDA receptors is modulated by dopamine in a selective subpopulation of amacrine cells. J. Neurocytol. 2001, 30, 181–193. [Google Scholar] [CrossRef]
- Pohl-Guimarães, F.; Calaza Kda, C.; Yamasaki, E.N.; Kubrusly, R.C.; Reis, R.A. Ethanol increases GABA release in the embryonic avian retina. Int. J. Dev. Neurosci. 2010, 28, 189–194. [Google Scholar] [CrossRef]
- Cristóvão-Ferreira, S.; Vaz, S.H.; Ribeiro, J.A.; Sebastião, A.M. Adenosine A2A receptors enhance GABA transport into nerve terminals by restraining PKC inhibition of GAT-1. J. Neurochem. 2009, 109, 336–347. [Google Scholar] [CrossRef]
- Ehinger, B.; Florén, I. Quantitation of the uptake of indoleamines and dopamine in the rabbit retina. Exp. Eye Res. 1978, 26, 1–11. [Google Scholar] [CrossRef]
- Feldkaemper, M.; Schaeffel, F. An updated view on the role of dopamine in myopia. Exp. Eye Res. 2013, 114, 106–119. [Google Scholar] [CrossRef]
- Reis, R.A.; Ventura, A.L.; Kubrusly, R.C.; de Mello, M.C.; de Mello, F.G. Dopaminergic signaling in the developing retina. Brain Res. Rev. 2007, 54, 181–188. [Google Scholar] [CrossRef]
- Lankford, K.L.; DeMello, F.G.; Klein, W.L. D1-type dopamine receptors inhibit growth cone motility in cultured retina neurons: Evidence that neurotransmitters act as morphogenic growth regulators in the developing central nervous system. Proc. Natl. Acad. Sci. USA 1988, 85, 4567–4571. [Google Scholar] [CrossRef]
- Gardino, P.F.; dos Santos, R.M.; Hokoç, J.N. Histogenesis and topographical distribution of tyrosine hydroxylase immunoreactive amacrine cells in the developing chick retina. Brain Res. Dev. Brain Res. 1993, 72, 226–236. [Google Scholar] [CrossRef]
- Kubrusly, R.C.; Guimarães, M.Z.; Vieira, A.P.; Hokoç, J.N.; Casarini, D.E.; de Mello, M.C.; de Mello, F.G. L-DOPA supply to the neuro retina activates dopaminergic communication at the early stages of embryonic development. J. Neurochem. 2003, 86, 45–54. [Google Scholar] [CrossRef]
- Ming, M.; Li, X.; Fan, X.; Yang, D.; Li, L.; Chen, S.; Gu, Q.; Le, W. Retinal pigment epithelial cells secrete neurotrophic factors and synthesize dopamine: Possible contribution to therapeutic effects of RPE cell transplantation in Parkinson’s disease. J. Transl. Med. 2009, 7, 53. [Google Scholar] [CrossRef] [PubMed]
- de Mello, M.C.; Ventura, A.L.; Paes de Carvalho, R.; Klein, W.L.; de Mello, F.G. Regulation of dopamine- and adenosine-dependent adenylate cyclase systems of chicken embryo retina cells in culture. Proc. Natl. Acad. Sci. USA 1982, 79, 5708–5712. [Google Scholar] [CrossRef] [PubMed]
- Callier, S.; Snapyan, M.; Le Crom, S.; Prou, D.; Vincent, J.D.; Vernier, P. Evolution and cell biology of dopamine receptors in vertebrates. Biol. Cell 2003, 95, 489–502. [Google Scholar] [CrossRef]
- Soares, H.C.; de Melo Reis, R.A.; De Mello, F.G.; Ventura, A.L.; Kurtenbach, E. Differential expression of D(1A) and D(1B) dopamine receptor mRNAs in the developing avian retina. J. Neurochem. 2000, 75, 1071–1075. [Google Scholar] [CrossRef]
- de Mello, M.C.; Pinheiro, M.C.; de Mello, F.G. Transient expression of an atypical D1-like dopamine receptor system during avian retina differentiation. Braz. J. Med. Biol. Res. 1996, 29, 1035–1044. [Google Scholar] [PubMed]
- Kubrusly, R.C.; Ventura, A.L.; de Melo Reis, R.A.; Serra, G.C.; Yamasaki, E.N.; Gardino, P.F.; de Mello, M.C.; de Mello, F.G. Norepinephrine acts as D1-dopaminergic agonist in the embryonic avian retina: Late expression of beta1-adrenergic receptor shifts norepinephrine specificity in the adult tissue. Neurochem. Int. 2007, 50, 211–218. [Google Scholar] [CrossRef]
- Paes de Carvalho, R.; de Mello, F.G. Expression of A1 adenosine receptors modulating dopamine-dependent cyclic AMP accumulation in the chick embryo retina. J. Neurochem. 1985, 44, 845–851. [Google Scholar] [CrossRef] [PubMed]
- Guimarães, M.Z.; Hokoç, J.N.; Duvoisin, R.; Reis, R.A.; De Mello, F.G. Dopaminergic retinal cell differentiation in culture: Modulation by forskolin and dopamine. Eur. J. Neurosci. 2001, 13, 1931–1937. [Google Scholar] [CrossRef]
- Borba, J.C.; Henze, I.P.; Silveira, M.S.; Kubrusly, R.C.; Gardino, P.F.; de Mello, M.C.; Hokoc, J.N.; de Mello, F.G. Pituitary adenylate cyclase-activating polypeptide (PACAP) can act as determinant of the tyrosine hydroxylase phenotype of dopaminergic cells during retina development. Brain Res. Dev. Brain Res. 2005, 156, 193–201. [Google Scholar] [CrossRef]
- Katona, I.; Freund, T.F. Endocannabinoid signaling as a synaptic circuit breaker in neurological disease. Nat. Med. 2008, 14, 923–930. [Google Scholar] [CrossRef]
- Heifets, B.D.; Castillo, P.E. Endocannabinoid signaling and long-term synaptic plasticity. Annu. Rev. Physiol. 2009, 71, 283–306. [Google Scholar] [CrossRef] [PubMed]
- Bockmann, E.C.; Brito, R.; Madeira, L.F.; da Silva Sampaio, L.; de Melo Reis, R.A.; França, G.R.; Calaza, K.D.C. The Role of Cannabinoids in CNS Development: Focus on Proliferation and Cell Death. Cell Mol. Neurobiol. 2022, 43, 1469–1485. [Google Scholar] [CrossRef] [PubMed]
- Miranzadeh Mahabadi, H.; Bhatti, H.; Laprairie, R.B.; Taghibiglou, C. Cannabinoid receptors distribution in mouse cortical plasma membrane compartments. Mol. Brain 2021, 14, 89. [Google Scholar] [CrossRef]
- Fernández-Ruiz, J.J.; Berrendero, F.; Hernández, M.L.; Romero, J.; Ramos, J.A. Role of endocannabinoids in brain development. Life Sci. 1999, 65, 725–736. [Google Scholar] [CrossRef] [PubMed]
- da Silva Sampaio, L.; Kubrusly, R.C.C.; Colli, Y.P.; Trindade, P.P.; Ribeiro-Resende, V.T.; Einicker-Lamas, M.; Paes-de-Carvalho, R.; Gardino, P.F.; de Mello, F.G.; De Melo Reis, R.A. Cannabinoid Receptor Type 1 Expression in the Developing Avian Retina: Morphological and Functional Correlation With the Dopaminergic System. Front. Cell Neurosci. 2018, 12, 58. [Google Scholar] [CrossRef] [PubMed]
- Kubrusly, R.C.C.; Gunter, A.; Sampaio, L.; Martins, R.S.; Schitine, C.S.; Trindade, P.; Fernandes, A.; Borelli-Torres, R.; Miya-Coreixas, V.S.; Rego Costa, A.C.; et al. Neuro-glial cannabinoid receptors modulate signaling in the embryonic avian retina. Neurochem. Int. 2018, 112, 27–37. [Google Scholar] [CrossRef] [PubMed]
- Jo, A.O.; Noel, J.M.; Lakk, M.; Yarishkin, O.; Ryskamp, D.A.; Shibasaki, K.; McCall, M.A.; Križaj, D. Mouse retinal ganglion cell signalling is dynamically modulated through parallel anterograde activation of cannabinoid and vanilloid pathways. J. Physiol. 2017, 595, 6499–6516. [Google Scholar] [CrossRef]
- Straiker, A.; Sullivan, J.M. Cannabinoid receptor activation differentially modulates ion channels in photoreceptors of the tiger salamander. J. Neurophysiol. 2003, 89, 2647–2654. [Google Scholar] [CrossRef]
- Gallo Afflitto, G.; Aiello, F.; Scuteri, D.; Bagetta, G.; Nucci, C. CB(1)R, CB(2)R and TRPV1 expression and modulation in in vivo, animal glaucoma models: A systematic review. Biomed. Pharmacother. 2022, 150, 112981. [Google Scholar] [CrossRef]
- Cairns, E.A.; Baldridge, W.H.; Kelly, M.E. The Endocannabinoid System as a Therapeutic Target in Glaucoma. Neural Plast. 2016, 2016, 9364091. [Google Scholar] [CrossRef]
- Nucci, C.; Gasperi, V.; Tartaglione, R.; Cerulli, A.; Terrinoni, A.; Bari, M.; De Simone, C.; Agrò, A.F.; Morrone, L.A.; Corasaniti, M.T.; et al. Involvement of the endocannabinoid system in retinal damage after high intraocular pressure-induced ischemia in rats. Investig. Ophthalmol. Vis. Sci. 2007, 48, 2997–3004. [Google Scholar] [CrossRef] [PubMed]
- Rapino, C.; Tortolani, D.; Scipioni, L.; Maccarrone, M. Neuroprotection by (endo)Cannabinoids in Glaucoma and Retinal Neurodegenerative Diseases. Curr. Neuropharmacol. 2018, 16, 959–970. [Google Scholar] [CrossRef]
- Schlicker, E.; Timm, J.; Göthert, M. Cannabinoid receptor-mediated inhibition of dopamine release in the retina. Naunyn Schmiedebergs Arch. Pharmacol. 1996, 354, 791–795. [Google Scholar] [CrossRef]
- Buckley, N.E.; Hansson, S.; Harta, G.; Mezey, E. Expression of the CB1 and CB2 receptor messenger RNAs during embryonic development in the rat. Neuroscience 1998, 82, 1131–1149. [Google Scholar] [CrossRef]
- Diacou, R.; Nandigrami, P.; Fiser, A.; Liu, W.; Ashery-Padan, R.; Cvekl, A. Cell fate decisions, transcription factors and signaling during early retinal development. Prog. Retin. Eye Res. 2022, 91, 101093. [Google Scholar] [CrossRef]
- Schwitzer, T.; Schwan, R.; Angioi-Duprez, K.; Giersch, A.; Laprevote, V. The Endocannabinoid System in the Retina: From Physiology to Practical and Therapeutic Applications. Neural Plast. 2016, 2016, 2916732. [Google Scholar] [CrossRef]
- Straiker, A.; Stella, N.; Piomelli, D.; Mackie, K.; Karten, H.J.; Maguire, G. Cannabinoid CB1 receptors and ligands in vertebrate retina: Localization and function of an endogenous signaling system. Proc. Natl. Acad. Sci. USA 1999, 96, 14565–14570. [Google Scholar] [CrossRef]
- Matsuda, S.; Kanemitsu, N.; Nakamura, A.; Mimura, Y.; Ueda, N.; Kurahashi, Y.; Yamamoto, S. Metabolism of Anandamide, an Endogenous Cannabinoid Receptor Ligand, in Porcine Ocular Tissues. Exp. Eye Res. 1997, 64, 707–711. [Google Scholar] [CrossRef]
- Freitas, H.R.; Isaac, A.R.; Silva, T.M.; Diniz, G.O.F.; Dos Santos Dabdab, Y.; Bockmann, E.C.; Guimaraes, M.Z.P.; da Costa Calaza, K.; de Mello, F.G.; Ventura, A.L.M.; et al. Cannabinoids Induce Cell Death and Promote P2X7 Receptor Signaling in Retinal Glial Progenitors in Culture. Mol. Neurobiol. 2019, 56, 6472–6486. [Google Scholar] [CrossRef]
- Yates, C.F.; Huang, J.Y.; Protti, D.A. Tonic Endocannabinoid Levels Modulate Retinal Signaling. Int. J. Environ. Res. Public. Health 2022, 19, 12460. [Google Scholar] [CrossRef]
- Begbie, J.; Doherty, P.; Graham, A. Cannabinoid receptor, CB1, expression follows neuronal differentiation in the early chick embryo. J. Anat. 2004, 205, 213–218. [Google Scholar] [CrossRef] [PubMed]
- Leonelli, M.; Britto, L.R.G.; Chaves, G.P.; Torrão, A.S. Developmental expression of cannabinoid receptors in the chick retinotectal system. Dev. Brain Res. 2005, 156, 176–182. [Google Scholar] [CrossRef] [PubMed]
- Hu, S.S.; Arnold, A.; Hutchens, J.M.; Radicke, J.; Cravatt, B.F.; Wager-Miller, J.; Mackie, K.; Straiker, A. Architecture of cannabinoid signaling in mouse retina. J. Comp. Neurol. 2010, 518, 3848–3866. [Google Scholar] [CrossRef]
- Felder, C.C.; Glass, M. Cannabinoid receptors and their endogenous agonists. Annu. Rev. Pharmacol. Toxicol. 1998, 38, 179–200. [Google Scholar] [CrossRef] [PubMed]
- Warrier, A.; Wilson, M. Endocannabinoid signaling regulates spontaneous transmitter release from embryonic retinal amacrine cells. Vis. Neurosci. 2007, 24, 25–35. [Google Scholar] [CrossRef] [PubMed]
- Chaves, G.P.; Nogueira, T.C.A.; Britto, L.R.G.; Bordin, S.; Torrão, A.S. Retinal removal up-regulates cannabinoid CB1 receptors in the chick optic tectum. J. Neurosci. Res. 2008, 86, 1626–1634. [Google Scholar] [CrossRef]
- Araújo, D.S.M.; Miya-Coreixas, V.S.; Pandolfo, P.; Calaza, K.C. Cannabinoid receptors and TRPA1 on neuroprotection in a model of retinal ischemia. Exp. Eye Res. 2017, 154, 116–125. [Google Scholar] [CrossRef]
- Faria, R.X.; Freitas, H.R.; Reis, R.A.M. P2X7 receptor large pore signaling in avian Müller glial cells. J. Bioenerg. Biomembr. 2017, 49, 215–229. [Google Scholar] [CrossRef]
- Faria, R.X.; Reis, R.A.; Ferreira, L.G.; Cezar-de-Mello, P.F.; Moraes, M.O. P2X7R large pore is partially blocked by pore forming proteins antagonists in astrocytes. J. Bioenerg. Biomembr. 2016, 48, 309–324. [Google Scholar] [CrossRef]
- Zhao, Y.-F.; Tang, Y.; Illes, P. Astrocytic and Oligodendrocytic P2X7 Receptors Determine Neuronal Functions in the CNS. Front. Mol. Neurosci. 2021, 14, 641570. [Google Scholar] [CrossRef]
- Freitas, H.R.; Reis, R.A.M.; Ventura, A.L.M.; Franca, G.R. Interaction between cannabinoid and nucleotide systems as a new mechanism of signaling in retinal cell death. Neural Regen. Res. 2019, 14, 2093–2094. [Google Scholar] [CrossRef]
- De Melo Reis, R.A.; Schitine, C.S.; Köfalvi, A.; Grade, S.; Cortes, L.; Gardino, P.F.; Malva, J.O.; de Mello, F.G. Functional identification of cell phenotypes differentiating from mice retinal neurospheres using single cell calcium imaging. Cell Mol. Neurobiol. 2011, 31, 835–846. [Google Scholar] [CrossRef]
- Campbell, W.A.; Blum, S.; Reske, A.; Hoang, T.; Blackshaw, S.; Fischer, A.J. Cannabinoid signaling promotes the de-differentiation and proliferation of Müller glia-derived progenitor cells. Glia 2021, 69, 2503–2521. [Google Scholar] [CrossRef] [PubMed]
- Cosens, D.J.; Manning, A. Abnormal electroretinogram from a Drosophila mutant. Nature 1969, 224, 285–287. [Google Scholar] [CrossRef] [PubMed]
- Gees, M.; Owsianik, G.; Nilius, B.; Voets, T. TRP channels. Compr. Physiol. 2012, 2, 563–608. [Google Scholar] [CrossRef]
- Zhao, Y.; McVeigh, B.M.; Moiseenkova-Bell, V.Y. Structural Pharmacology of TRP Channels. J. Mol. Biol. 2021, 433, 166914. [Google Scholar] [CrossRef]
- Bisogno, T.; Delton-Vandenbroucke, I.; Milone, A.; Lagarde, M.; Di Marzo, V. Biosynthesis and inactivation of N-arachidonoylethanolamine (anandamide) and N-docosahexaenoylethanolamine in bovine retina. Arch. Biochem. Biophys. 1999, 370, 300–307. [Google Scholar] [CrossRef] [PubMed]
- Bazan, N.G. Metabolism of arachidonic acid in the retina and retinal pigment epithelium: Biological effects of oxygenated metabolites of arachidonic acid. Prog. Clin. Biol. Res. 1989, 312, 15–37. [Google Scholar]
- Sawamura, S.; Shirakawa, H.; Nakagawa, T.; Mori, Y.; Kaneko, S. Frontiers in Neuroscience TRP Channels in the Brain: What Are They There for? In Neurobiology of TRP Channels; Emir, T.L.R., Ed.; CRC Press/Taylor & Francis © 2018 by Taylor & Francis Group, LLC.: Boca Raton, FL, USA, 2017; pp. 295–322. [Google Scholar] [CrossRef]
- Thébault, S. Minireview: Insights into the role of TRP channels in the retinal circulation and function. Neurosci. Lett. 2021, 765, 136285. [Google Scholar] [CrossRef]
- Gilliam, J.C.; Wensel, T.G. TRP channel gene expression in the mouse retina. Vision. Res. 2011, 51, 2440–2452. [Google Scholar] [CrossRef]
- Rychkov, G.; Barritt, G.J. TRPC1 Ca(2+)-permeable channels in animal cells. Handb. Exp. Pharmacol. 2007, 179, 23–52. [Google Scholar] [CrossRef]
- Lakk, M.; Young, D.; Baumann, J.M.; Jo, A.O.; Hu, H.; Križaj, D. Polymodal TRPV1 and TRPV4 Sensors Colocalize but Do Not Functionally Interact in a Subpopulation of Mouse Retinal Ganglion Cells. Front. Cell Neurosci. 2018, 12, 353. [Google Scholar] [CrossRef] [PubMed]
- Molnar, T.; Barabas, P.; Birnbaumer, L.; Punzo, C.; Kefalov, V.; Križaj, D. Store-operated channels regulate intracellular calcium in mammalian rods. J. Physiol. 2012, 590, 3465–3481. [Google Scholar] [CrossRef] [PubMed]
- Tóth, A.; Czikora, A.; Pásztor, E.T.; Dienes, B.; Bai, P.; Csernoch, L.; Rutkai, I.; Csató, V.; Mányiné, I.S.; Pórszász, R.; et al. Vanilloid receptor-1 (TRPV1) expression and function in the vasculature of the rat. J. Histochem. Cytochem. 2014, 62, 129–144. [Google Scholar] [CrossRef] [PubMed]
- Crousillac, S.; LeRouge, M.; Rankin, M.; Gleason, E. Immunolocalization of TRPC channel subunits 1 and 4 in the chicken retina. Vis. Neurosci. 2003, 20, 453–463. [Google Scholar] [CrossRef] [PubMed]
- Da Silva, N.; Herron, C.E.; Stevens, K.; Jollimore, C.A.; Barnes, S.; Kelly, M.E. Metabotropic receptor-activated calcium increases and store-operated calcium influx in mouse Müller cells. Investig. Ophthalmol. Vis. Sci. 2008, 49, 3065–3073. [Google Scholar] [CrossRef] [PubMed]
- Witkovsky, P.; Gábriel, R.; Krizaj, D. Anatomical and neurochemical characterization of dopaminergic interplexiform processes in mouse and rat retinas. J. Comp. Neurol. 2008, 510, 158–174. [Google Scholar] [CrossRef] [PubMed]
- Maddox, J.W.; Khorsandi, N.; Gleason, E. TRPC5 is required for the NO-dependent increase in dendritic Ca(2+) and GABA release from chick retinal amacrine cells. J. Neurophysiol. 2018, 119, 262–273. [Google Scholar] [CrossRef]
- Morgans, C.W.; Zhang, J.; Jeffrey, B.G.; Nelson, S.M.; Burke, N.S.; Duvoisin, R.M.; Brown, R.L. TRPM1 is required for the depolarizing light response in retinal ON-bipolar cells. Proc. Natl. Acad. Sci. USA 2009, 106, 19174–19178. [Google Scholar] [CrossRef]
- Hasan, N.; Pangeni, G.; Cobb, C.A.; Ray, T.A.; Nettesheim, E.R.; Ertel, K.J.; Lipinski, D.M.; McCall, M.A.; Gregg, R.G. Presynaptic Expression of LRIT3 Transsynaptically Organizes the Postsynaptic Glutamate Signaling Complex Containing TRPM1. Cell Rep. 2019, 27, 3107–3116.e3103. [Google Scholar] [CrossRef]
- Anastassov, I.A.; Wang, W.; Dunn, F.A. Synaptogenesis and synaptic protein localization in the postnatal development of rod bipolar cell dendrites in mouse retina. J. Comp. Neurol. 2019, 527, 52–66. [Google Scholar] [CrossRef] [PubMed]
- Kozuka, T.; Chaya, T.; Tamalu, F.; Shimada, M.; Fujimaki-Aoba, K.; Kuwahara, R.; Watanabe, S.I.; Furukawa, T. The TRPM1 Channel Is Required for Development of the Rod ON Bipolar Cell-AII Amacrine Cell Pathway in the Retinal Circuit. J. Neurosci. 2017, 37, 9889–9900. [Google Scholar] [CrossRef]
- Takeuchi, H.; Horie, S.; Moritoh, S.; Matsushima, H.; Hori, T.; Kimori, Y.; Kitano, K.; Tsubo, Y.; Tachibana, M.; Koike, C. Different Activity Patterns in Retinal Ganglion Cells of TRPM1 and mGluR6 Knockout Mice. Biomed. Res. Int. 2018, 2018, 2963232. [Google Scholar] [CrossRef] [PubMed]
- Meléndez García, R.; Arredondo Zamarripa, D.; Arnold, E.; Ruiz-Herrera, X.; Noguez Imm, R.; Baeza Cruz, G.; Adán, N.; Binart, N.; Riesgo-Escovar, J.; Goffin, V.; et al. Prolactin protects retinal pigment epithelium by inhibiting sirtuin 2-dependent cell death. EBioMedicine 2016, 7, 35–49. [Google Scholar] [CrossRef] [PubMed]
- Malko, P.; Syed Mortadza, S.A.; McWilliam, J.; Jiang, L.H. TRPM2 Channel in Microglia as a New Player in Neuroinflammation Associated with a Spectrum of Central Nervous System Pathologies. Front. Pharmacol. 2019, 10, 239. [Google Scholar] [CrossRef] [PubMed]
- Webster, C.M.; Tworig, J.; Caval-Holme, F.; Morgans, C.W.; Feller, M.B. The Impact of Steroid Activation of TRPM3 on Spontaneous Activity in the Developing Retina. eNeuro 2020, 22, 7. [Google Scholar] [CrossRef]
- McGahon, M.K.; Fernández, J.A.; Dash, D.P.; McKee, J.; Simpson, D.A.; Zholos, A.V.; McGeown, J.G.; Curtis, T.M. TRPV2 Channels Contribute to Stretch-Activated Cation Currents and Myogenic Constriction in Retinal Arterioles. Investig. Ophthalmol. Vis. Sci. 2016, 57, 5637–5647. [Google Scholar] [CrossRef]
- Souza Monteiro de Araújo, D.; De Logu, F.; Adembri, C.; Rizzo, S.; Janal, M.N.; Landini, L.; Magi, A.; Mattei, G.; Cini, N.; Pandolfo, P.; et al. TRPA1 mediates damage of the retina induced by ischemia and reperfusion in mice. Cell Death Dis. 2020, 11, 633. [Google Scholar] [CrossRef]
- Davis, J.B.; Gray, J.; Gunthorpe, M.J.; Hatcher, J.P.; Davey, P.T.; Overend, P.; Harries, M.H.; Latcham, J.; Clapham, C.; Atkinson, K.; et al. Vanilloid receptor-1 is essential for inflammatory thermal hyperalgesia. Nature 2000, 405, 183–187. [Google Scholar] [CrossRef]
- Dhaka, A.; Uzzell, V.; Dubin, A.E.; Mathur, J.; Petrus, M.; Bandell, M.; Patapoutian, A. TRPV1 is activated by both acidic and basic pH. J. Neurosci. 2009, 29, 153–158. [Google Scholar] [CrossRef]
- Clapham, D.E. TRP channels as cellular sensors. Nature 2003, 426, 517–524. [Google Scholar] [CrossRef] [PubMed]
- Di Marzo, V. Endocannabinoids: Synthesis and degradation. Rev. Physiol. Biochem. Pharmacol. 2008, 160, 1–24. [Google Scholar] [CrossRef] [PubMed]
- Benítez-Angeles, M.; Morales-Lázaro, S.L.; Juárez-González, E.; Rosenbaum, T. TRPV1: Structure, Endogenous Agonists, and Mechanisms. Int. J. Mol. Sci. 2020, 21, 3421. [Google Scholar] [CrossRef]
- Jo, A.O.; Ryskamp, D.A.; Phuong, T.T.; Verkman, A.S.; Yarishkin, O.; MacAulay, N.; Križaj, D. TRPV4 and AQP4 Channels Synergistically Regulate Cell Volume and Calcium Homeostasis in Retinal Müller Glia. J. Neurosci. 2015, 35, 13525–13537. [Google Scholar] [CrossRef]
- Ryskamp, D.A.; Redmon, S.; Jo, A.O.; Križaj, D. TRPV1 and Endocannabinoids: Emerging Molecular Signals that Modulate Mammalian Vision. Cells 2014, 3, 914–938. [Google Scholar] [CrossRef] [PubMed]
- Sappington, R.M.; Calkins, D.J. Contribution of TRPV1 to microglia-derived IL-6 and NFkappaB translocation with elevated hydrostatic pressure. Investig. Ophthalmol. Vis. Sci. 2008, 49, 3004–3017. [Google Scholar] [CrossRef] [PubMed]
- Sappington, R.M.; Sidorova, T.; Long, D.J.; Calkins, D.J. TRPV1: Contribution to retinal ganglion cell apoptosis and increased intracellular Ca2+ with exposure to hydrostatic pressure. Investig. Ophthalmol. Vis. Sci. 2009, 50, 717–728. [Google Scholar] [CrossRef]
- Yazulla, S. Endocannabinoids in the retina: From marijuana to neuroprotection. Prog. Retin. Eye Res. 2008, 27, 501–526. [Google Scholar] [CrossRef]
- Leonelli, M.; Martins, D.O.; Kihara, A.H.; Britto, L.R. Ontogenetic expression of the vanilloid receptors TRPV1 and TRPV2 in the rat retina. Int. J. Dev. Neurosci. 2009, 27, 709–718. [Google Scholar] [CrossRef]
- Shen, Y.; Heimel, J.A.; Kamermans, M.; Peachey, N.S.; Gregg, R.G.; Nawy, S. A transient receptor potential-like channel mediates synaptic transmission in rod bipolar cells. J. Neurosci. 2009, 29, 6088–6093. [Google Scholar] [CrossRef]
- Glaser, S.T.; Deutsch, D.G.; Studholme, K.M.; Zimov, S.; Yazulla, S. Endocannabinoids in the intact retina: 3 H-anandamide uptake, fatty acid amide hydrolase immunoreactivity and hydrolysis of anandamide. Vis. Neurosci. 2005, 22, 693–705. [Google Scholar] [CrossRef] [PubMed]
- Bisogno, T.; Hanus, L.; De Petrocellis, L.; Tchilibon, S.; Ponde, D.E.; Brandi, I.; Moriello, A.S.; Davis, J.B.; Mechoulam, R.; Di Marzo, V. Molecular targets for cannabidiol and its synthetic analogues: Effect on vanilloid VR1 receptors and on the cellular uptake and enzymatic hydrolysis of anandamide. Br. J. Pharmacol. 2001, 134, 845–852. [Google Scholar] [CrossRef]
- Anand, U.; Jones, B.; Korchev, Y.; Bloom, S.R.; Pacchetti, B.; Anand, P.; Sodergren, M.H. CBD Effects on TRPV1 Signaling Pathways in Cultured DRG Neurons. J. Pain. Res. 2020, 13, 2269–2278. [Google Scholar] [CrossRef] [PubMed]
- de Almeida, D.L.; Devi, L.A. Diversity of molecular targets and signaling pathways for CBD. Pharmacol. Res. Perspect. 2020, 8, e00682. [Google Scholar] [CrossRef]
- Yazulla, S.; Studholme, K.M. Vanilloid receptor like 1 (VRL1) immunoreactivity in mammalian retina: Colocalization with somatostatin and purinergic P2X1 receptors. J. Comp. Neurol. 2004, 474, 407–418. [Google Scholar] [CrossRef]
- Thermos, K. Functional mapping of somatostatin receptors in the retina: A review. Vision. Res. 2003, 43, 1805–1815. [Google Scholar] [CrossRef] [PubMed]
- Snyder, S.H. Adenosine as a neuromodulator. Annu. Rev. Neurosci. 1985, 8, 103–124. [Google Scholar] [CrossRef]
- Fredholm, B.B.; Chen, J.F.; Cunha, R.A.; Svenningsson, P.; Vaugeois, J.M. Adenosine and brain function. Int. Rev. Neurobiol. 2005, 63, 191–270. [Google Scholar] [CrossRef]
- Blazynski, C.; Perez, M.T. Adenosine in vertebrate retina: Localization, receptor characterization, and function. Cell Mol. Neurobiol. 1991, 11, 463–484. [Google Scholar] [CrossRef]
- Shewan, D.; Dwivedy, A.; Anderson, R.; Holt, C.E. Age-related changes underlie switch in netrin-1 responsiveness as growth cones advance along visual pathway. Nat. Neurosci. 2002, 5, 955–962. [Google Scholar] [CrossRef]
- Zhang, M.; Budak, M.T.; Lu, W.; Khurana, T.S.; Zhang, X.; Laties, A.M.; Mitchell, C.H. Identification of the A3 adenosine receptor in rat retinal ganglion cells. Mol. Vis. 2006, 12, 937–948. [Google Scholar]
- Portugal, C.C.; da Encarnação, T.G.; Sagrillo, M.A.; Pereira, M.R.; Relvas, J.B.; Socodato, R.; Paes-de-Carvalho, R. Activation of adenosine A3 receptors regulates vitamin C transport and redox balance in neurons. Free Radic. Biol. Med. 2021, 163, 43–55. [Google Scholar] [CrossRef]
- Duarte-Silva, A.T.; Ximenes, L.G.R.; Guimarães-Souza, M.; Domith, I.; Paes-de-Carvalho, R. Chemical signaling in the developing avian retina: Focus on cyclic AMP and AKT-dependent pathways. Front. Cell Dev. Biol. 2022, 10, 1058925. [Google Scholar] [CrossRef] [PubMed]
- Paes de Carvalho, R. Development of A1 adenosine receptors in the chick embryo retina. J. Neurosci. Res. 1990, 25, 236–242. [Google Scholar] [CrossRef] [PubMed]
- Paes de Carvalho, R.; de Mello, F.G. Adenosine-elicited accumulation of adenosine 3′, 5′-cyclic monophosphate in the chick embryo retina. J. Neurochem. 1982, 38, 493–500. [Google Scholar] [CrossRef] [PubMed]
- de Carvalho, R.P.; Braas, K.M.; Adler, R.; Snyder, S.H. Developmental regulation of adenosine A1 receptors, uptake sites and endogenous adenosine in the chick retina. Brain Res. Dev. Brain Res. 1992, 70, 87–95. [Google Scholar] [CrossRef]
- dos Santos-Rodrigues, A.; Ferreira, J.M.; Paes-de-Carvalho, R. Differential adenosine uptake in mixed neuronal/glial or purified glial cultures of avian retinal cells: Modulation by adenosine metabolism and the ERK cascade. Biochem. Biophys. Res. Commun. 2011, 414, 175–180. [Google Scholar] [CrossRef] [PubMed]
- Pereira, M.R.; Hang, V.R.; Vardiero, E.; de Mello, F.G.; Paes-de-Carvalho, R. Modulation of A1 adenosine receptor expression by cell aggregation and long-term activation of A2a receptors in cultures of avian retinal cells: Involvement of the cyclic AMP/PKA pathway. J. Neurochem. 2010, 113, 661–673. [Google Scholar] [CrossRef]
- Paes-de-Carvalho, R.; Maia, G.A.; Ferreira, J.M. Adenosine regulates the survival of avian retinal neurons and photoreceptors in culture. Neurochem. Res. 2003, 28, 1583–1590. [Google Scholar] [CrossRef]
- Ferreira, J.M.; Paes-de-Carvalho, R. Long-term activation of adenosine A(2a) receptors blocks glutamate excitotoxicity in cultures of avian retinal neurons. Brain Res. 2001, 900, 169–176. [Google Scholar] [CrossRef]
- Socodato, R.; Brito, R.; Calaza, K.C.; Paes-de-Carvalho, R. Developmental regulation of neuronal survival by adenosine in the in vitro and in vivo avian retina depends on a shift of signaling pathways leading to CREB phosphorylation or dephosphorylation. J. Neurochem. 2011, 116, 227–239. [Google Scholar] [CrossRef] [PubMed]
- Paes de Carvalho, R.; Braas, K.M.; Snyder, S.H.; Adler, R. Analysis of adenosine immunoreactivity, uptake, and release in purified cultures of developing chick embryo retinal neurons and photoreceptors. J. Neurochem. 1990, 55, 1603–1611. [Google Scholar] [CrossRef]
- Paes-de-Carvalho, R.; Dias, B.V.; Martins, R.A.; Pereira, M.R.; Portugal, C.C.; Lanfredi, C. Activation of glutamate receptors promotes a calcium-dependent and transporter-mediated release of purines in cultured avian retinal cells: Possible involvement of calcium/calmodulin-dependent protein kinase II. Neurochem. Int. 2005, 46, 441–451. [Google Scholar] [CrossRef]
- Langer, I.; Jeandriens, J.; Couvineau, A.; Sanmukh, S.; Latek, D. Signal Transduction by VIP and PACAP Receptors. Biomedicines 2022, 10, 406. [Google Scholar] [CrossRef] [PubMed]
- Hirabayashi, T.; Nakamachi, T.; Shioda, S. Discovery of PACAP and its receptors in the brain. J. Headache Pain. 2018, 19, 28. [Google Scholar] [CrossRef]
- May, V.; Parsons, R.L. G Protein-Coupled Receptor Endosomal Signaling and Regulation of Neuronal Excitability and Stress Responses: Signaling Options and Lessons From the PAC1 Receptor. J. Cell Physiol. 2017, 232, 698–706. [Google Scholar] [CrossRef]
- Onali, P.; Olianas, M.C. PACAP is a potent and highly effective stimulator of adenylyl cyclase activity in the retinas of different mammalian species. Brain Res. 1994, 641, 132–134. [Google Scholar] [CrossRef]
- Denes, V.; Geck, P.; Mester, A.; Gabriel, R. Pituitary Adenylate Cyclase-Activating Polypeptide: 30 Years in Research Spotlight and 600 Million Years in Service. J. Clin. Med. 2019, 8, 1488. [Google Scholar] [CrossRef] [PubMed]
- Shioda, S.; Takenoya, F.; Wada, N.; Hirabayashi, T.; Seki, T.; Nakamachi, T. Pleiotropic and retinoprotective functions of PACAP. Anat. Sci. Int. 2016, 91, 313–324. [Google Scholar] [CrossRef]
- Njaine, B.; Martins, R.A.; Santiago, M.F.; Linden, R.; Silveira, M.S. Pituitary adenylyl cyclase-activating polypeptide controls the proliferation of retinal progenitor cells through downregulation of cyclin D1. Eur. J. Neurosci. 2010, 32, 311–321. [Google Scholar] [CrossRef]
- Njaine, B.; Rocha-Martins, M.; Vieira-Vieira, C.H.; De-Melo, L.D.; Linden, R.; Braas, K.; May, V.; Martins, R.A.; Silveira, M.S. Pleiotropic functions of pituitary adenylyl cyclase-activating polypeptide on retinal ontogenesis: Involvement of KLF4 in the control of progenitor cell proliferation. J. Mol. Neurosci. 2014, 54, 430–442. [Google Scholar] [CrossRef]
- Fleming, R.L.; Silveira, M.S.; Santos, L.E.; Henze, I.P.; Gardino, P.F.; de Mello, M.C.; de Mello, F.G. Pituitary adenylyl cyclase-activating polypeptide receptor re-sensitization induces plastic changes in the dopaminergic phenotype in the mature avian retina. J. Neurochem. 2013, 124, 621–631. [Google Scholar] [CrossRef] [PubMed]
- Silveira, M.S.; Costa, M.R.; Bozza, M.; Linden, R. Pituitary adenylyl cyclase-activating polypeptide prevents induced cell death in retinal tissue through activation of cyclic AMP-dependent protein kinase. J. Biol. Chem. 2002, 277, 16075–16080. [Google Scholar] [CrossRef]
- Denes, V.; Hideg, O.; Nyisztor, Z.; Lakk, M.; Godri, Z.; Berta, G.; Geck, P.; Gabriel, R. The Neuroprotective Peptide PACAP1-38 Contributes to Horizontal Cell Development in Postnatal Rat Retina. Investig. Ophthalmol. Vis. Sci. 2019, 60, 770–778. [Google Scholar] [CrossRef]
- Seki, T.; Itoh, H.; Nakamachi, T.; Endo, K.; Wada, Y.; Nakamura, K.; Shioda, S. Suppression of rat retinal ganglion cell death by PACAP following transient ischemia induced by high intraocular pressure. J. Mol. Neurosci. 2011, 43, 30–34. [Google Scholar] [CrossRef]
- Danyadi, B.; Szabadfi, K.; Reglodi, D.; Mihalik, A.; Danyadi, T.; Kovacs, Z.; Batai, I.; Tamas, A.; Kiss, P.; Toth, G.; et al. PACAP application improves functional outcome of chronic retinal ischemic injury in rats-evidence from electroretinographic measurements. J. Mol. Neurosci. 2014, 54, 293–299. [Google Scholar] [CrossRef]
- Kvarik, T.; Mammel, B.; Reglodi, D.; Kovacs, K.; Werling, D.; Bede, B.; Vaczy, A.; Fabian, E.; Toth, G.; Kiss, P.; et al. PACAP Is Protective in a Rat Model of Retinopathy of Prematurity. J. Mol. Neurosci. 2016, 60, 179–185. [Google Scholar] [CrossRef] [PubMed]
- Kvarik, T.; Reglodi, D.; Werling, D.; Vaczy, A.; Kovari, P.; Szabo, E.; Kovacs, K.; Hashimoto, H.; Ertl, T.; Gyarmati, J.; et al. The Protective Effects of Endogenous PACAP in Oxygen-Induced Retinopathy. J. Mol. Neurosci. 2021, 71, 2546–2557. [Google Scholar] [CrossRef]
- Patko, E.; Szabo, E.; Vaczy, A.; Molitor, D.; Tari, E.; Li, L.; Csutak, A.; Toth, G.; Reglodi, D.; Atlasz, T. Protective Effects of Pituitary Adenylate-Cyclase-Activating Polypeptide on Retinal Vasculature and Molecular Responses in a Rat Model of Moderate Glaucoma. Int. J. Mol. Sci. 2023, 24, 13256. [Google Scholar] [CrossRef] [PubMed]
- Atlasz, T.; Szabadfi, K.; Kiss, P.; Marton, Z.; Griecs, M.; Hamza, L.; Gaal, V.; Biro, Z.; Tamas, A.; Hild, G.; et al. Effects of PACAP in UV-A radiation-induced retinal degeneration models in rats. J. Mol. Neurosci. 2011, 43, 51–57. [Google Scholar] [CrossRef] [PubMed]
- Gábriel, R.; Pöstyéni, E.; Dénes, V. Neuroprotective Potential of Pituitary Adenylate Cyclase Activating Polypeptide in Retinal Degenerations of Metabolic Origin. Front. Neurosci. 2019, 13, 1031. [Google Scholar] [CrossRef]
- Wang, T.; Li, Y.; Guo, M.; Dong, X.; Liao, M.; Du, M.; Wang, X.; Yin, H.; Yan, H. Exosome-Mediated Delivery of the Neuroprotective Peptide PACAP38 Promotes Retinal Ganglion Cell Survival and Axon Regeneration in Rats With Traumatic Optic Neuropathy. Front. Cell Dev. Biol. 2021, 9, 659783. [Google Scholar] [CrossRef]
- Van, C.; Condro, M.C.; Ko, H.H.; Hoang, A.Q.; Zhu, R.; Lov, K.; Ricaflanca, P.T.; Diep, A.L.; Nguyen, N.N.M.; Lipshutz, G.S.; et al. Targeted deletion of PAC1 receptors in retinal neurons enhances neuron loss and axonopathy in a model of multiple sclerosis and optic neuritis. Neurobiol. Dis. 2021, 160, 105524. [Google Scholar] [CrossRef]
- Goldstein, I.M.; Ostwald, P.; Roth, S. Nitric oxide: A review of its role in retinal function and disease. Vision. Res. 1996, 36, 2979–2994. [Google Scholar] [CrossRef] [PubMed]
- Toda, N.; Nakanishi-Toda, M. Nitric oxide: Ocular blood flow, glaucoma, and diabetic retinopathy. Prog. Retin. Eye Res. 2007, 26, 205–238. [Google Scholar] [CrossRef] [PubMed]
- Cossenza, M.; Socodato, R.; Portugal, C.C.; Domith, I.C.; Gladulich, L.F.; Encarnação, T.G.; Calaza, K.C.; Mendonça, H.R.; Campello-Costa, P.; Paes-de-Carvalho, R. Nitric oxide in the nervous system: Biochemical, developmental, and neurobiological aspects. Vitam. Horm. 2014, 96, 79–125. [Google Scholar] [CrossRef]
- Cossenza, M.; Paes de Carvalho, R. L-arginine uptake and release by cultured avian retinal cells: Differential cellular localization in relation to nitric oxide synthase. J. Neurochem. 2000, 74, 1885–1894. [Google Scholar] [CrossRef] [PubMed]
- Do, K.Q.; Grima, G.; Benz, B.; Salt, T.E. Glial-neuronal transfer of arginine and S-nitrosothiols in nitric oxide transmission. Ann. N. Y. Acad. Sci. 2002, 962, 81–92. [Google Scholar] [CrossRef]
- Grima, G.; Benz, B.; Do, K.Q. Glutamate-induced release of the nitric oxide precursor, arginine, from glial cells. Eur. J. Neurosci. 1997, 9, 2248–2258. [Google Scholar] [CrossRef]
- Grima, G.; Benz, B.; Do, K.Q. Glial-derived arginine, the nitric oxide precursor, protects neurons from NMDA-induced excitotoxicity. Eur. J. Neurosci. 2001, 14, 1762–1770. [Google Scholar] [CrossRef]
- Bredt, D.S.; Ferris, C.D.; Snyder, S.H. Nitric oxide synthase regulatory sites. Phosphorylation by cyclic AMP-dependent protein kinase, protein kinase C, and calcium/calmodulin protein kinase; identification of flavin and calmodulin binding sites. J. Biol. Chem. 1992, 267, 10976–10981. [Google Scholar] [CrossRef] [PubMed]
- Lamas, S.; Marsden, P.A.; Li, G.K.; Tempst, P.; Michel, T. Endothelial nitric oxide synthase: Molecular cloning and characterization of a distinct constitutive enzyme isoform. Proc. Natl. Acad. Sci. USA 1992, 89, 6348–6352. [Google Scholar] [CrossRef]
- Cho, H.J.; Xie, Q.W.; Calaycay, J.; Mumford, R.A.; Swiderek, K.M.; Lee, T.D.; Nathan, C. Calmodulin is a subunit of nitric oxide synthase from macrophages. J. Exp. Med. 1992, 176, 599–604. [Google Scholar] [CrossRef] [PubMed]
- Garthwaite, J. Nitric oxide signalling in the nervous system. Semin. Neurosci. 1993, 5, 171–180. [Google Scholar] [CrossRef]
- Garthwaite, J.; Boulton, C.L. Nitric oxide signaling in the central nervous system. Annu. Rev. Physiol. 1995, 57, 683–706. [Google Scholar] [CrossRef] [PubMed]
- Garthwaite, J.; Charles, S.L.; Chess-Williams, R. Endothelium-derived relaxing factor release on activation of NMDA receptors suggests role as intercellular messenger in the brain. Nature 1988, 336, 385–388. [Google Scholar] [CrossRef]
- Garthwaite, J.; Garthwaite, G. Cellular origins of cyclic GMP responses to excitatory amino acid receptor agonists in rat cerebellum in vitro. J. Neurochem. 1987, 48, 29–39. [Google Scholar] [CrossRef]
- Brenman, J.E.; Bredt, D.S. Synaptic signaling by nitric oxide. Curr. Opin. Neurobiol. 1997, 7, 374–378. [Google Scholar] [CrossRef]
- Brenman, J.E.; Chao, D.S.; Gee, S.H.; McGee, A.W.; Craven, S.E.; Santillano, D.R.; Wu, Z.; Huang, F.; Xia, H.; Peters, M.F.; et al. Interaction of nitric oxide synthase with the postsynaptic density protein PSD-95 and alpha1-syntrophin mediated by PDZ domains. Cell 1996, 84, 757–767. [Google Scholar] [CrossRef]
- Dawson, T.M.; Bredt, D.S.; Fotuhi, M.; Hwang, P.M.; Snyder, S.H. Nitric oxide synthase and neuronal NADPH diaphorase are identical in brain and peripheral tissues. Proc. Natl. Acad. Sci. USA 1991, 88, 7797–7801. [Google Scholar] [CrossRef]
- Hope, B.T.; Michael, G.J.; Knigge, K.M.; Vincent, S.R. Neuronal NADPH Diaphorase is a Nitric Oxide Synthase. Proc. Natl. Acad. Sci. USA 1991, 88, 2811–2814. [Google Scholar] [CrossRef]
- Kurenny, D.E.; Moroz, L.L.; Turner, R.W.; Sharkey, K.A.; Barnes, S. Modulation of ion channels in rod photoreceptors by nitric oxide. Neuron 1994, 13, 315–324. [Google Scholar] [CrossRef]
- Yamamoto, R.; Bredt, D.S.; Snyder, S.H.; Stone, R.A. The localization of nitric oxide synthase in the rat eye and related cranial ganglia. Neuroscience 1993, 54, 189–200. [Google Scholar] [CrossRef]
- Vielma, A.H.; Retamal, M.A.; Schmachtenberg, O. Nitric oxide signaling in the retina: What have we learned in two decades? Brain Res. 2012, 1430, 112–125. [Google Scholar] [CrossRef] [PubMed]
- Andrade da Costa, B.L.; Hokoç, J.N. Coexistence of GAD-65 and GAD-67 with tyrosine hydroxylase and nitric oxide synthase in amacrine and interplexiform cells of the primate, Cebus apella. Vis. Neurosci. 2003, 20, 153–163. [Google Scholar] [CrossRef] [PubMed]
- Vardi, N.; Auerbach, P. Specific cell types in cat retina express different forms of glutamic acid decarboxylase. J. Comp. Neurol. 1995, 351, 374–384. [Google Scholar] [CrossRef]
- Socodato, R.; Brito, R.; Portugal, C.C.; de Oliveira, N.A.; Calaza, K.C.; Paes-de-Carvalho, R. The nitric oxide-cGKII system relays death and survival signals during embryonic retinal development via AKT-induced CREB1 activation. Cell Death Differ. 2014, 21, 915–928. [Google Scholar] [CrossRef] [PubMed]
- Pang, J.J.; Gao, F.; Wu, S.M. Light responses and morphology of bNOS-immunoreactive neurons in the mouse retina. J. Comp. Neurol. 2010, 518, 2456–2474. [Google Scholar] [CrossRef]
- Blom, J.; Giove, T.; Deshpande, M.; Eldred, W.D. Characterization of nitric oxide signaling pathways in the mouse retina. J. Comp. Neurol. 2012, 520, 4204–4217. [Google Scholar] [CrossRef]
- Tekmen-Clark, M.; Gleason, E. Nitric oxide production and the expression of two nitric oxide synthases in the avian retina. Vis. Neurosci. 2013, 30, 91–103. [Google Scholar] [CrossRef]
- Djamgoz, M.B.; Sekaran, S.; Angotzi, A.R.; Haamedi, S.; Vallerga, S.; Hirano, J.; Yamada, M. Light-adaptive role of nitric oxide in the outer retina of lower vertebrates: A brief review. Philos. Trans. R. Soc. Lond. B Biol. Sci. 2000, 355, 1199–1203. [Google Scholar] [CrossRef] [PubMed]
- Giove, T.J.; Deshpande, M.M.; Eldred, W.D. Identification of alternate transcripts of neuronal nitric oxide synthase in the mouse retina. J. Neurosci. Res. 2009, 87, 3134–3142. [Google Scholar] [CrossRef] [PubMed]
- Shi, Q.; Teves, M.M.; Lillywhite, A.; Pagtalunan, E.B.; Stell, W.K. Light adaptation in the chick retina: Dopamine, nitric oxide, and gap-junction coupling modulate spatiotemporal contrast sensitivity. Exp. Eye Res. 2020, 195, 108026. [Google Scholar] [CrossRef] [PubMed]
- Sato, M.; Ohtsuka, T.; Stell, W.K. Endogenous nitric oxide enhances the light-response of cones during light-adaptation in the rat retina. Vision. Res. 2011, 51, 131–137. [Google Scholar] [CrossRef][Green Version]
- DeVries, S.H.; Schwartz, E.A. Modulation of an electrical synapse between solitary pairs of catfish horizontal cells by dopamine and second messengers. J. Physiol. 1989, 414, 351–375. [Google Scholar] [CrossRef] [PubMed]
- Mills, S.L.; Massey, S.C. Differential properties of two gap junctional pathways made by AII amacrine cells. Nature 1995, 377, 734–737. [Google Scholar] [CrossRef]
- Ding, J.D.; Weinberg, R.J. Distribution of soluble guanylyl cyclase in rat retina. J. Comp. Neurol. 2007, 500, 734–745. [Google Scholar] [CrossRef] [PubMed]
- Hirooka, K.; Kourennyi, D.E.; Barnes, S. Calcium channel activation facilitated by nitric oxide in retinal ganglion cells. J. Neurophysiol. 2000, 83, 198–206. [Google Scholar] [CrossRef]
- Wexler, E.M.; Stanton, P.K.; Nawy, S. Nitric oxide depresses GABAA receptor function via coactivation of cGMP-dependent kinase and phosphodiesterase. J. Neurosci. 1998, 18, 2342–2349. [Google Scholar] [CrossRef]
- McMahon, D.G.; Ponomareva, L.V. Nitric oxide and cGMP modulate retinal glutamate receptors. J. Neurophysiol. 1996, 76, 2307–2315. [Google Scholar] [CrossRef]
- McMahon, D.G.; Schmidt, K.F. Horizontal cell glutamate receptor modulation by NO: Mechanisms and functional implications for the first visual synapse. Vis. Neurosci. 1999, 16, 425–433. [Google Scholar] [CrossRef]
- Ientile, R.; Pedale, S.; Picciurro, V.; Macaione, V.; Fabiano, C.; Macaione, S. Nitric oxide mediates NMDA-evoked [3H]GABA release from chick retina cells. FEBS Lett. 1997, 417, 345–348. [Google Scholar] [CrossRef]
- Ientile, R.; Picciurro, V.; Pedale, S.; Nucci, C.; Malecka, B.; Nisticò, G.; Macaione, S. Nitric oxide enhances amino acid release from immature chick embryo retina. Neurosci. Lett. 1996, 219, 79–82. [Google Scholar] [CrossRef]
- Yu, D.; Eldred, W.D. Nitric oxide stimulates gamma-aminobutyric acid release and inhibits glycine release in retina. J. Comp. Neurol. 2005, 483, 278–291. [Google Scholar] [CrossRef] [PubMed]
- Tsukaguchi, H.; Tokui, T.; Mackenzie, B.; Berger, U.V.; Chen, X.Z.; Wang, Y.; Brubaker, R.F.; Hediger, M.A. A family of mammalian Na+-dependent L-ascorbic acid transporters. Nature 1999, 399, 70–75. [Google Scholar] [CrossRef]
- Portugal, C.C.; da Encarnação, T.G.; Socodato, R.; Moreira, S.R.; Brudzewsky, D.; Ambrósio, A.F.; Paes-de-Carvalho, R. Nitric oxide modulates sodium vitamin C transporter 2 (SVCT-2) protein expression via protein kinase G (PKG) and nuclear factor-κB (NF-κB). J. Biol. Chem. 2012, 287, 3860–3872. [Google Scholar] [CrossRef] [PubMed]
- Portugal, C.C.; Miya, V.S.; Calaza Kda, C.; Santos, R.A.; Paes-de-Carvalho, R. Glutamate receptors modulate sodium-dependent and calcium-independent vitamin C bidirectional transport in cultured avian retinal cells. J. Neurochem. 2009, 108, 507–520. [Google Scholar] [CrossRef] [PubMed]
- Socodato, R.E.; Magalhaes, C.R.; Paes-de-Carvalho, R. Glutamate and nitric oxide modulate ERK and CREB phosphorylation in the avian retina: Evidence for direct signaling from neurons to Muller glial cells. J. Neurochem. 2009, 108, 417–429. [Google Scholar] [CrossRef]
- Moriyama, S.; Hiasa, M. Expression of Vesicular Nucleotide Transporter in the Mouse Retina. Biol. Pharm. Bull. 2016, 39, 564–569. [Google Scholar] [CrossRef][Green Version]
- Xia, J.; Lim, J.C.; Lu, W.; Beckel, J.M.; Macarak, E.J.; Laties, A.M.; Mitchell, C.H. Neurons respond directly to mechanical deformation with pannexin-mediated ATP release and autostimulation of P2X7 receptors. J. Physiol. 2012, 590, 2285–2304. [Google Scholar] [CrossRef]
- Mitchell, C.H. Release of ATP by a human retinal pigment epithelial cell line: Potential for autocrine stimulation through subretinal space. J. Physiol. 2001, 534, 193–202. [Google Scholar] [CrossRef] [PubMed]
- Santos, P.F.; Caramelo, O.L.; Carvalho, A.P.; Duarte, C.B. Characterization of ATP release from cultures enriched in cholinergic amacrine-like neurons. J. Neurobiol. 1999, 41, 340–348. [Google Scholar] [CrossRef]
- Newman, E.A. Calcium increases in retinal glial cells evoked by light-induced neuronal activity. J. Neurosci. 2005, 25, 5502–5510. [Google Scholar] [CrossRef] [PubMed]
- Uckermann, O.; Wolf, A.; Kutzera, F.; Kalisch, F.; Beck-Sickinger, A.G.; Wiedemann, P.; Reichenbach, A.; Bringmann, A. Glutamate release by neurons evokes a purinergic inhibitory mechanism of osmotic glial cell swelling in the rat retina: Activation by neuropeptide Y. J. Neurosci. Res. 2006, 83, 538–550. [Google Scholar] [CrossRef] [PubMed]
- Loiola, E.C.; Ventura, A.L. Release of ATP from avian Müller glia cells in culture. Neurochem. Int. 2011, 58, 414–422. [Google Scholar] [CrossRef]
- Ventura, A.L.M.; Dos Santos-Rodrigues, A.; Mitchell, C.H.; Faillace, M.P. Purinergic signaling in the retina: From development to disease. Brain Res. Bull. 2019, 151, 92–108. [Google Scholar] [CrossRef] [PubMed]
- de Almeida-Pereira, L.; Magalhães, C.F.; Repossi, M.G.; Thorstenberg, M.L.P.; Sholl-Franco, A.; Coutinho-Silva, R.; Ventura, A.L.M.; Fragel-Madeira, L. Adenine Nucleotides Control Proliferation In Vivo of Rat Retinal Progenitors by P2Y(1) Receptor. Mol. Neurobiol. 2017, 54, 5142–5155. [Google Scholar] [CrossRef]
- Jacques, F.J.; Silva, T.M.; da Silva, F.E.; Ornelas, I.M.; Ventura, A.L.M. Nucleotide P2Y13-stimulated phosphorylation of CREB is required for ADP-induced proliferation of late developing retinal glial progenitors in culture. Cell Signal 2017, 35, 95–106. [Google Scholar] [CrossRef]
- Sugioka, M.; Fukuda, Y.; Yamashita, M. Ca2+ responses to ATP via purinoceptors in the early embryonic chick retina. J. Physiol. 1996, 493 Pt 3, 855–863. [Google Scholar] [CrossRef]
- Pearson, R.; Catsicas, M.; Becker, D.; Mobbs, P. Purinergic and muscarinic modulation of the cell cycle and calcium signaling in the chick retinal ventricular zone. J. Neurosci. 2002, 22, 7569–7579. [Google Scholar] [CrossRef]
- Pearson, R.A.; Catsicas, M.; Becker, D.L.; Bayley, P.; Lüneborg, N.L.; Mobbs, P. Ca(2+) signalling and gap junction coupling within and between pigment epithelium and neural retina in the developing chick. Eur. J. Neurosci. 2004, 19, 2435–2445. [Google Scholar] [CrossRef]
- Sanches, G.; de Alencar, L.S.; Ventura, A.L. ATP induces proliferation of retinal cells in culture via activation of PKC and extracellular signal-regulated kinase cascade. Int. J. Dev. Neurosci. 2002, 20, 21–27. [Google Scholar] [CrossRef] [PubMed]
- França, G.R.; Freitas, R.C.; Ventura, A.L. ATP-induced proliferation of developing retinal cells: Regulation by factors released from postmitotic cells in culture. Int. J. Dev. Neurosci. 2007, 25, 283–291. [Google Scholar] [CrossRef]
- Sholl-Franco, A.; Fragel-Madeira, L.; Macama Ada, C.; Linden, R.; Ventura, A.L. ATP controls cell cycle and induces proliferation in the mouse developing retina. Int. J. Dev. Neurosci. 2010, 28, 63–73. [Google Scholar] [CrossRef]
- Nunes, P.H.; Calaza Kda, C.; Albuquerque, L.M.; Fragel-Madeira, L.; Sholl-Franco, A.; Ventura, A.L. Signal transduction pathways associated with ATP-induced proliferation of cell progenitors in the intact embryonic retina. Int. J. Dev. Neurosci. 2007, 25, 499–508. [Google Scholar] [CrossRef] [PubMed]
- Sugioka, M.; Zhou, W.L.; Hofmann, H.D.; Yamashita, M. Ca2+ mobilization and capacitative Ca2+ entry regulate DNA synthesis in cultured chick retinal neuroepithelial cells. Int. J. Dev. Neurosci. 1999, 17, 163–172. [Google Scholar] [CrossRef]
- Sugioka, M.; Zhou, W.L.; Hofmann, H.D.; Yamashita, M. Involvement of P2 purinoceptors in the regulation of DNA synthesis in the neural retina of chick embryo. Int. J. Dev. Neurosci. 1999, 17, 135–144. [Google Scholar] [CrossRef] [PubMed]
- Yamashita, M. From neuroepithelial cells to neurons: Changes in the physiological properties of neuroepithelial stem cells. Arch. Biochem. Biophys. 2013, 534, 64–70. [Google Scholar] [CrossRef]
- Ornelas, I.M.; Ventura, A.L. Involvement of the PI3K/AKT pathway in ATP-induced proliferation of developing retinal cells in culture. Int. J. Dev. Neurosci. 2010, 28, 503–511. [Google Scholar] [CrossRef]
- Moll, V.; Weick, M.; Milenkovic, I.; Kodal, H.; Reichenbach, A.; Bringmann, A. P2Y receptor-mediated stimulation of Müller glial DNA synthesis. Investig. Ophthalmol. Vis. Sci. 2002, 43, 766–773. [Google Scholar]
- Milenkovic, I.; Weick, M.; Wiedemann, P.; Reichenbach, A.; Bringmann, A. P2Y receptor-mediated stimulation of Müller glial cell DNA synthesis: Dependence on EGF and PDGF receptor transactivation. Investig. Ophthalmol. Vis. Sci. 2003, 44, 1211–1220. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Ornelas, I.M.; Silva, T.M.; Fragel-Madeira, L.; Ventura, A.L. Inhibition of PI3K/Akt pathway impairs G2/M transition of cell cycle in late developing progenitors of the avian embryo retina. PLoS ONE 2013, 8, e53517. [Google Scholar] [CrossRef] [PubMed]
- Massé, K.; Bhamra, S.; Eason, R.; Dale, N.; Jones, E.A. Purine-mediated signalling triggers eye development. Nature 2007, 449, 1058–1062. [Google Scholar] [CrossRef]
- Gampe, K.; Haverkamp, S.; Robson, S.C.; Gachet, C.; Hüser, L.; Acker-Palmer, A.; Zimmermann, H. NTPDase2 and the P2Y1 receptor are not required for mammalian eye formation. Purinergic Signal 2015, 11, 155–160. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Lewis, G.P.; Chapin, E.A.; Luna, G.; Linberg, K.A.; Fisher, S.K. The fate of Müller’s glia following experimental retinal detachment: Nuclear migration, cell division, and subretinal glial scar formation. Mol. Vis. 2010, 16, 1361–1372. [Google Scholar] [PubMed]
- Reichenbach, A.; Bringmann, A. Role of Purines in Müller Glia. J. Ocul. Pharmacol. Ther. 2016, 32, 518–533. [Google Scholar] [CrossRef] [PubMed]
- Silva, T.M.; França, G.R.; Ornelas, I.M.; Loiola, E.C.; Ulrich, H.; Ventura, A.L. Involvement of nucleotides in glial growth following scratch injury in avian retinal cell monolayer cultures. Purinergic Signal 2015, 11, 183–201. [Google Scholar] [CrossRef]
- Resta, V.; Novelli, E.; Vozzi, G.; Scarpa, C.; Caleo, M.; Ahluwalia, A.; Solini, A.; Santini, E.; Parisi, V.; Di Virgilio, F.; et al. Acute retinal ganglion cell injury caused by intraocular pressure spikes is mediated by endogenous extracellular ATP. Eur. J. Neurosci. 2007, 25, 2741–2754. [Google Scholar] [CrossRef]
- Anccasi, R.M.; Ornelas, I.M.; Cossenza, M.; Persechini, P.M.; Ventura, A.L. ATP induces the death of developing avian retinal neurons in culture via activation of P2X7 and glutamate receptors. Purinergic Signal 2013, 9, 15–29. [Google Scholar] [CrossRef]
- Zhang, X.; Zhang, M.; Laties, A.M.; Mitchell, C.H. Stimulation of P2X7 receptors elevates Ca2+ and kills retinal ganglion cells. Investig. Ophthalmol. Vis. Sci. 2005, 46, 2183–2191. [Google Scholar] [CrossRef]
- Hu, H.; Lu, W.; Zhang, M.; Zhang, X.; Argall, A.J.; Patel, S.; Lee, G.E.; Kim, Y.C.; Jacobson, K.A.; Laties, A.M.; et al. Stimulation of the P2X7 receptor kills rat retinal ganglion cells in vivo. Exp. Eye Res. 2010, 91, 425–432. [Google Scholar] [CrossRef] [PubMed]
- Sugiyama, T.; Oku, H.; Shibata, M.; Fukuhara, M.; Yoshida, H.; Ikeda, T. Involvement of P2X7 receptors in the hypoxia-induced death of rat retinal neurons. Investig. Ophthalmol. Vis. Sci. 2010, 51, 3236–3243. [Google Scholar] [CrossRef][Green Version]
- Niyadurupola, N.; Sidaway, P.; Ma, N.; Rhodes, J.D.; Broadway, D.C.; Sanderson, J. P2X7 receptor activation mediates retinal ganglion cell death in a human retina model of ischemic neurodegeneration. Investig. Ophthalmol. Vis. Sci. 2013, 54, 2163–2170. [Google Scholar] [CrossRef]
- Campagno, K.E.; Lu, W.; Jassim, A.H.; Albalawi, F.; Cenaj, A.; Tso, H.Y.; Clark, S.P.; Sripinun, P.; Gómez, N.M.; Mitchell, C.H. Rapid morphologic changes to microglial cells and upregulation of mixed microglial activation state markers induced by P2X7 receptor stimulation and increased intraocular pressure. J. Neuroinflamm. 2021, 18, 217. [Google Scholar] [CrossRef]
- Hu, X.; Zhao, G.L.; Xu, M.X.; Zhou, H.; Li, F.; Miao, Y.; Lei, B.; Yang, X.L.; Wang, Z. Interplay between Müller cells and microglia aggravates retinal inflammatory response in experimental glaucoma. J. Neuroinflamm. 2021, 18, 303. [Google Scholar] [CrossRef] [PubMed]
- Kakurai, K.; Sugiyama, T.; Kurimoto, T.; Oku, H.; Ikeda, T. Involvement of P2X(7) receptors in retinal ganglion cell death after optic nerve crush injury in rats. Neurosci. Lett. 2013, 534, 237–241. [Google Scholar] [CrossRef] [PubMed]
- Xue, B.; Xie, Y.; Xue, Y.; Hu, N.; Zhang, G.; Guan, H.; Ji, M. Involvement of P2X(7) receptors in retinal ganglion cell apoptosis induced by activated Müller cells. Exp. Eye Res. 2016, 153, 42–50. [Google Scholar] [CrossRef]
- Franke, H.; Klimke, K.; Brinckmann, U.; Grosche, J.; Francke, M.; Sperlagh, B.; Reichenbach, A.; Liebert, U.G.; Illes, P. P2X(7) receptor-mRNA and -protein in the mouse retina; changes during retinal degeneration in BALBCrds mice. Neurochem. Int. 2005, 47, 235–242. [Google Scholar] [CrossRef]
- Puthussery, T.; Fletcher, E. Extracellular ATP induces retinal photoreceptor apoptosis through activation of purinoceptors in rodents. J. Comp. Neurol. 2009, 513, 430–440. [Google Scholar] [CrossRef]
- Notomi, S.; Hisatomi, T.; Kanemaru, T.; Takeda, A.; Ikeda, Y.; Enaida, H.; Kroemer, G.; Ishibashi, T. Critical involvement of extracellular ATP acting on P2RX7 purinergic receptors in photoreceptor cell death. Am. J. Pathol. 2011, 179, 2798–2809. [Google Scholar] [CrossRef]
- Cao, M.; Huang, X.; Zou, J.; Peng, Y.; Wang, Y.; Zheng, X.; Tang, L.; Zhang, L. Attenuation of Microglial Activation and Pyroptosis by Inhibition of P2X7 Pathway Promotes Photoreceptor Survival in Experimental Retinal Detachment. Investig. Ophthalmol. Vis. Sci. 2023, 64, 34. [Google Scholar] [CrossRef]
- Rice, M.E.; Russo-Menna, I. Differential compartmentalization of brain ascorbate and glutathione between neurons and glia. Neuroscience 1998, 82, 1213–1223. [Google Scholar] [CrossRef] [PubMed]
- Raj Rai, S.; Bhattacharyya, C.; Sarkar, A.; Chakraborty, S.; Sircar, E.; Dutta, S.; Sengupta, R. Glutathione: Role in Oxidative/Nitrosative Stress, Antioxidant Defense, and Treatments. ChemistrySelect 2021, 6, 4566–4590. [Google Scholar] [CrossRef]
- Gu, F.; Chauhan, V.; Chauhan, A. Glutathione redox imbalance in brain disorders. Curr. Opin. Clin. Nutr. Metab. Care 2015, 18, 89–95. [Google Scholar] [CrossRef] [PubMed]
- Bjørklund, G.; Tinkov, A.A.; Hosnedlová, B.; Kizek, R.; Ajsuvakova, O.P.; Chirumbolo, S.; Skalnaya, M.G.; Peana, M.; Dadar, M.; El-Ansary, A.; et al. The role of glutathione redox imbalance in autism spectrum disorder: A review. Free Radic. Biol. Med. 2020, 160, 149–162. [Google Scholar] [CrossRef] [PubMed]
- Freitas, H.R.; Reis, R.A. Glutathione induces GABA release through P2X7R activation on Muller glia. Neurogenesis 2017, 4, e1283188. [Google Scholar] [CrossRef]
- Freitas, H.R.; Ferraz, G.; Ferreira, G.C.; Ribeiro-Resende, V.T.; Chiarini, L.B.; do Nascimento, J.L.; Matos Oliveira, K.R.; Pereira Tde, L.; Ferreira, L.G.; Kubrusly, R.C.; et al. Glutathione-Induced Calcium Shifts in Chick Retinal Glial Cells. PLoS ONE 2016, 11, e0153677. [Google Scholar] [CrossRef] [PubMed]
- Pow, D.V.; Crook, D.K. Immunocytochemical evidence for the presence of high levels of reduced glutathione in radial glial cells and horizontal cells in the rabbit retina. Neurosci. Lett. 1995, 193, 25–28. [Google Scholar] [CrossRef]
- Schütte, M.; Werner, P. Redistribution of glutathione in the ischemic rat retina. Neurosci. Lett. 1998, 246, 53–56. [Google Scholar] [CrossRef]
- Castagné, V.; Clarke, P.G.H. Inhibition of glutathione synthesis can enhance cycloheximide-induced protection of developing neurons against axotomy. Dev. Brain Res. 1997, 102, 285–290. [Google Scholar] [CrossRef]
- Castagné, V.; Clarke, P.G.H. Cooperation between glutathione depletion and protein synthesis inhibition against naturally occurring neuronal death. Neuroscience 1998, 86, 895–902. [Google Scholar] [CrossRef] [PubMed]
- Corpe, C.P.; Tu, H.; Eck, P.; Wang, J.; Faulhaber-Walter, R.; Schnermann, J.; Margolis, S.; Padayatty, S.; Sun, H.; Wang, Y.; et al. Vitamin C transporter Slc23a1 links renal reabsorption, vitamin C tissue accumulation, and perinatal survival in mice. J. Clin. Investig. 2010, 120, 1069–1083. [Google Scholar] [CrossRef]
- Ferrada, L.; Magdalena, R.; Barahona, M.J.; Ramírez, E.; Sanzana, C.; Gutiérrez, J.; Nualart, F. Two Distinct Faces of Vitamin C: AA vs. DHA. Antioxidants 2021, 10, 215. [Google Scholar] [CrossRef]
- Padayatty, S.J.; Levine, M. Vitamin C: The known and the unknown and Goldilocks. Oral. Dis. 2016, 22, 463–493. [Google Scholar] [CrossRef] [PubMed]
- Diliberto, E.J., Jr.; Allen, P.L. Semidehydroascorbate as a product of the enzymic conversion of dopamine to norepinephrine. Coupling of semidehydroascorbate reductase to dopamine-beta-hydroxylase. Mol. Pharmacol. 1980, 17, 421–426. [Google Scholar] [PubMed]
- Qiu, S.; Li, L.; Weeber, E.J.; May, J.M. Ascorbate transport by primary cultured neurons and its role in neuronal function and protection against excitotoxicity. J. Neurosci. Res. 2007, 85, 1046–1056. [Google Scholar] [CrossRef]
- Eldridge, C.F.; Bunge, M.B.; Bunge, R.P.; Wood, P.M. Differentiation of axon-related Schwann cells in vitro. I. Ascorbic acid regulates basal lamina assembly and myelin formation. J. Cell Biol. 1987, 105, 1023–1034. [Google Scholar] [CrossRef]
- Covarrubias-Pinto, A.; Acuña, A.I.; Beltrán, F.A.; Torres-Díaz, L.; Castro, M.A. Old Things New View: Ascorbic Acid Protects the Brain in Neurodegenerative Disorders. Int. J. Mol. Sci. 2015, 16, 28194–28217. [Google Scholar] [CrossRef]
- Kocot, J.; Luchowska-Kocot, D.; Kiełczykowska, M.; Musik, I.; Kurzepa, J. Does Vitamin C Influence Neurodegenerative Diseases and Psychiatric Disorders? Nutrients 2017, 9, 659. [Google Scholar] [CrossRef]
- Moretti, M.; Fraga, D.B.; Rodrigues, A.L.S. Ascorbic Acid to Manage Psychiatric Disorders. CNS Drugs 2017, 31, 571–583. [Google Scholar] [CrossRef]
- Renner, O.; Burkard, M.; Michels, H.; Vollbracht, C.; Sinnberg, T.; Venturelli, S. Parenteral high-dose ascorbate—A possible approach for the treatment of glioblastoma (Review). Int. J. Oncol. 2021, 58, 1–17. [Google Scholar] [CrossRef]
- De Mello, F.G. The ontogeny of dopamine-dependent increase of adenosine 3’,5’-cyclic monophosphate in the chick retina. J. Neurochem. 1978, 31, 1049–1053. [Google Scholar] [CrossRef] [PubMed]
- Majumdar, S. Role of glutamate in the development of visual pathways. Front. Ophthalmol. 2023, 3, 1147769. [Google Scholar] [CrossRef]
- Domith, I.; Socodato, R.; Portugal, C.C.; Munis, A.F.; Duarte-Silva, A.T.; Paes-de-Carvalho, R. Vitamin C modulates glutamate transport and NMDA receptor function in the retina. J. Neurochem. 2018, 144, 408–420. [Google Scholar] [CrossRef] [PubMed]
- Telegina, D.V.; Antonenko, A.K.; Fursova, A.Z.; Kolosova, N.G. The glutamate/GABA system in the retina of male rats: Effects of aging, neurodegeneration, and supplementation with melatonin and antioxidant SkQ1. Biogerontology 2022, 23, 571–585. [Google Scholar] [CrossRef]
- do Nascimento, J.L.; de Mello, F.G. Induced release of gamma-aminobutyric acid by a carrier-mediated, high-affinity uptake of L-glutamate in cultured chick retina cells. J. Neurochem. 1985, 45, 1820–1827. [Google Scholar] [CrossRef]
- Schitine, C.S.; de Mello, F.G.; Reis, R.A. Neurochemical plasticity of Müller cells after retinal injury: Overexpression of GAT-3 may potentiate excitotoxicity. Neural Regen. Res. 2015, 10, 1376–1378. [Google Scholar] [CrossRef]
- de Almeida, O.M.; Gardino, P.F.; Loureiro dos Santos, N.E.; Yamasaki, E.N.; de Mello, M.C.; Hokoç, J.N.; de Mello, F.G. Opposite roles of GABA and excitatory amino acids on the control of GAD expression in cultured retina cells. Brain Res. 2002, 925, 89–99. [Google Scholar] [CrossRef]
- Socodato, R.; Santiago, F.N.; Portugal, C.C.; Domith, I.; Encarnação, T.G.; Loiola, E.C.; Ventura, A.L.; Cossenza, M.; Relvas, J.B.; Castro, N.G.; et al. Dopamine promotes NMDA receptor hypofunction in the retina through D(1) receptor-mediated Csk activation, Src inhibition and decrease of GluN2B phosphorylation. Sci. Rep. 2017, 7, 40912. [Google Scholar] [CrossRef]
- Lowry, W.E.; Huang, J.; Ma, Y.C.; Ali, S.; Wang, D.; Williams, D.M.; Okada, M.; Cole, P.A.; Huang, X.Y. Csk, a critical link of g protein signals to actin cytoskeletal reorganization. Dev. Cell 2002, 2, 733–744. [Google Scholar] [CrossRef]
- Salter, M.W.; Kalia, L.V. Src kinases: A hub for NMDA receptor regulation. Nat. Rev. Neurosci. 2004, 5, 317–328. [Google Scholar] [CrossRef] [PubMed]
- Batty, N.J.; Fenrich, K.K.; Fouad, K. The role of cAMP and its downstream targets in neurite growth in the adult nervous system. Neurosci. Lett. 2017, 652, 56–63. [Google Scholar] [CrossRef]
- Lankford, K.; De Mello, F.G.; Klein, W.L. A transient embryonic dopamine receptor inhibits growth cone motility and neurite outgrowth in a subset of avian retina neurons. Neurosci. Lett. 1987, 75, 169–174. [Google Scholar] [CrossRef]
- da Encarnação, T.G.; Portugal, C.C.; Nogueira, C.E.; Santiago, F.N.; Socodato, R.; Paes-de-Carvalho, R. Dopamine Promotes Ascorbate Release from Retinal Neurons: Role of D(1) Receptors and the Exchange Protein Directly Activated by cAMP type 2 (EPAC2). Mol. Neurobiol. 2018, 55, 7858–7871. [Google Scholar] [CrossRef] [PubMed]
- Portugal, C.C.; da Encarnacao, T.G.; Domith, I.; Dos Santos Rodrigues, A.; de Oliveira, N.A.; Socodato, R.; Paes-de-Carvalho, R. Dopamine-Induced Ascorbate Release From Retinal Neurons Involves Glutamate Release, Activation of AMPA/Kainate Receptors and Downstream Signaling Pathways. Front. Neurosci. 2019, 13, 453. [Google Scholar] [CrossRef]
- Paes-De-Carvalho, R. Adenosine as a signaling molecule in the retina: Biochemical and developmental aspects. An. Acad. Bras. Cienc. 2002, 74, 437–451. [Google Scholar] [CrossRef]
- Garthwaite, J. Glutamate, nitric oxide and cell-cell signalling in the nervous system. Trends Neurosci. 1991, 14, 60–67. [Google Scholar] [CrossRef] [PubMed]
- Sohocki, M.M.; Daiger, S.P.; Bowne, S.J.; Rodriquez, J.A.; Northrup, H.; Heckenlively, J.R.; Birch, D.G.; Mintz-Hittner, H.; Ruiz, R.S.; Lewis, R.A.; et al. Prevalence of mutations causing retinitis pigmentosa and other inherited retinopathies. Hum. Mutat. 2001, 17, 42–51. [Google Scholar] [CrossRef]
- Kumaran, N.; Michaelides, M.; Smith, A.J.; Ali, R.R.; Bainbridge, J.W.B. Retinal gene therapy. Br. Med. Bull. 2018, 126, 13–25. [Google Scholar] [CrossRef]
- Carss, K.J.; Arno, G.; Erwood, M.; Stephens, J.; Sanchis-Juan, A.; Hull, S.; Megy, K.; Grozeva, D.; Dewhurst, E.; Malka, S.; et al. Comprehensive Rare Variant Analysis via Whole-Genome Sequencing to Determine the Molecular Pathology of Inherited Retinal Disease. Am. J. Hum. Genet. 2017, 100, 75–90. [Google Scholar] [CrossRef]
- Wong, W.L.; Su, X.; Li, X.; Cheung, C.M.; Klein, R.; Cheng, C.Y.; Wong, T.Y. Global prevalence of age-related macular degeneration and disease burden projection for 2020 and 2040: A systematic review and meta-analysis. Lancet Glob. Health 2014, 2, e106–e116. [Google Scholar] [CrossRef]
- Fleckenstein, M.; Keenan, T.D.L.; Guymer, R.H.; Chakravarthy, U.; Schmitz-Valckenberg, S.; Klaver, C.C.; Wong, W.T.; Chew, E.Y. Age-related macular degeneration. Nat. Rev. Dis. Primers 2021, 7, 31. [Google Scholar] [CrossRef] [PubMed]
- Saaddine, J.B.; Honeycutt, A.A.; Narayan, K.M.; Zhang, X.; Klein, R.; Boyle, J.P. Projection of diabetic retinopathy and other major eye diseases among people with diabetes mellitus: United States, 2005–2050. Arch. Ophthalmol. 2008, 126, 1740–1747. [Google Scholar] [CrossRef]
- Lucchesi, M.; Marracci, S.; Amato, R.; Filippi, L.; Cammalleri, M.; Dal Monte, M. Neurosensory Alterations in Retinopathy of Prematurity: A Window to Neurological Impairments Associated to Preterm Birth. Biomedicines 2022, 10, 1603. [Google Scholar] [CrossRef]
- Quigley, H.A. Understanding Glaucomatous Optic Neuropathy: The Synergy between Clinical Observation and Investigation. Annu. Rev. Vis. Sci. 2016, 2, 235–254. [Google Scholar] [CrossRef] [PubMed]
- Amerasinghe, N.; Zhang, J.; Thalamuthu, A.; He, M.; Vithana, E.N.; Viswanathan, A.; Wong, T.Y.; Foster, P.J.; Aung, T. The heritability and sibling risk of angle closure in Asians. Ophthalmology 2011, 118, 480–485. [Google Scholar] [CrossRef]
- Nickells, R.W. Apoptosis of retinal ganglion cells in glaucoma: An update of the molecular pathways involved in cell death. Surv. Ophthalmol. 1999, 43 (Suppl. S1), S151–S161. [Google Scholar] [CrossRef] [PubMed]
- Tham, Y.C.; Li, X.; Wong, T.Y.; Quigley, H.A.; Aung, T.; Cheng, C.Y. Global prevalence of glaucoma and projections of glaucoma burden through 2040: A systematic review and meta-analysis. Ophthalmology 2014, 121, 2081–2090. [Google Scholar] [CrossRef]
- Bravo Filho, V.T.; Ventura, R.U.; Brandt, C.T.; Sarteschi, C.; Ventura, M.C. Visual impairment impact on the quality of life of the elderly population that uses the public health care system from the western countryside of Pernambuco State, Brazil. Arq. Bras. Oftalmol. 2012, 75, 161–165. [Google Scholar] [CrossRef]
- Allison, K.; Patel, D.; Alabi, O. Epidemiology of Glaucoma: The Past, Present, and Predictions for the Future. Cureus 2020, 12, e11686. [Google Scholar] [CrossRef]
- Lusthaus, J.; Goldberg, I. Current management of glaucoma. Med. J. Aust. 2019, 210, 180–187. [Google Scholar] [CrossRef]
- Lo, J.; Mehta, K.; Dhillon, A.; Huang, Y.K.; Luo, Z.; Nam, M.H.; Al Diri, I.; Chang, K.C. Therapeutic strategies for glaucoma and optic neuropathies. Mol. Aspects Med. 2023, 94, 101219. [Google Scholar] [CrossRef] [PubMed]
- Killer, H.E.; Pircher, A. Normal tension glaucoma: Review of current understanding and mechanisms of the pathogenesis. Eye 2018, 32, 924–930. [Google Scholar] [CrossRef] [PubMed]
- Heng, L.Z.; Comyn, O.; Peto, T.; Tadros, C.; Ng, E.; Sivaprasad, S.; Hykin, P.G. Diabetic retinopathy: Pathogenesis, clinical grading, management and future developments. Diabet. Med. 2013, 30, 640–650. [Google Scholar] [CrossRef]
- Lechner, J.; O’Leary, O.E.; Stitt, A.W. The pathology associated with diabetic retinopathy. Vision. Res. 2017, 139, 7–14. [Google Scholar] [CrossRef] [PubMed]
- Wang, W.; Lo, A.C.Y. Diabetic Retinopathy: Pathophysiology and Treatments. Int. J. Mol. Sci. 2018, 19, 1816. [Google Scholar] [CrossRef]
- Zheng, Y.; He, M.; Congdon, N. The worldwide epidemic of diabetic retinopathy. Indian J. Ophthalmol. 2012, 60, 428–431. [Google Scholar] [CrossRef] [PubMed]
- Ting, D.S.; Cheung, G.C.; Wong, T.Y. Diabetic retinopathy: Global prevalence, major risk factors, screening practices and public health challenges: A review. Clin. Exp. Ophthalmol. 2016, 44, 260–277. [Google Scholar] [CrossRef]
- Leasher, J.L.; Bourne, R.R.; Flaxman, S.R.; Jonas, J.B.; Keeffe, J.; Naidoo, K.; Pesudovs, K.; Price, H.; White, R.A.; Wong, T.Y.; et al. Global Estimates on the Number of People Blind or Visually Impaired by Diabetic Retinopathy: A Meta-analysis From 1990 to 2010. Diabetes Care 2016, 39, 1643–1649. [Google Scholar] [CrossRef]
- Barber, A.J.; Baccouche, B. Neurodegeneration in diabetic retinopathy: Potential for novel therapies. Vision Res. 2017, 139, 82–92. [Google Scholar] [CrossRef]
- Seki, M.; Tanaka, T.; Nawa, H.; Usui, T.; Fukuchi, T.; Ikeda, K.; Abe, H.; Takei, N. Involvement of brain-derived neurotrophic factor in early retinal neuropathy of streptozotocin-induced diabetes in rats: Therapeutic potential of brain-derived neurotrophic factor for dopaminergic amacrine cells. Diabetes 2004, 53, 2412–2419. [Google Scholar] [CrossRef] [PubMed]
- Gastinger, M.J.; Singh, R.S.; Barber, A.J. Loss of cholinergic and dopaminergic amacrine cells in streptozotocin-diabetic rat and Ins2Akita-diabetic mouse retinas. Investig. Ophthalmol. Vis. Sci. 2006, 47, 3143–3150. [Google Scholar] [CrossRef] [PubMed]
- Miya-Coreixas, V.S.; Maggesissi Santos, R.; Carpi Santos, R.; Gardino, P.F.; Calaza, K. Regulation of GABA content by glucose in the chick retina. Exp. Eye Res. 2013, 115, 206–215. [Google Scholar] [CrossRef]
- Carpi-Santos, R.; Ferreira, M.J.; Pereira Netto, A.D.; Giestal-de-Araujo, E.; Ventura, A.L.M.; Cossenza, M.; Calaza, K.C. Early changes in system [Formula: See text] and glutathione in the retina of diabetic rats. Exp. Eye Res. 2016, 146, 35–42. [Google Scholar] [CrossRef] [PubMed]
- Carpi-Santos, R.; Calaza, K.C. Alterations in System x(c)(-) Expression in the Retina of Type 1 Diabetic Rats and the Role of Nrf2. Mol. Neurobiol. 2018, 55, 7941–7948. [Google Scholar] [CrossRef]
- Wong, T.Y.; Cheung, C.M.; Larsen, M.; Sharma, S.; Simó, R. Diabetic retinopathy. Nat. Rev. Dis. Primers 2016, 2, 16012. [Google Scholar] [CrossRef]
- Barber, A.J.; Lieth, E.; Khin, S.A.; Antonetti, D.A.; Buchanan, A.G.; Gardner, T.W. Neural apoptosis in the retina during experimental and human diabetes. Early onset and effect of insulin. J. Clin. Investig. 1998, 102, 783–791. [Google Scholar] [CrossRef]
- Mendonca, H.R.; Carpi-Santos, R.; da Costa Calaza, K.; Blanco Martinez, A.M. Neuroinflammation and oxidative stress act in concert to promote neurodegeneration in the diabetic retina and optic nerve: Galectin-3 participation. Neural Regen. Res. 2020, 15, 625–635. [Google Scholar] [CrossRef]
- Carpineto, P.; Toto, L.; Aloia, R.; Ciciarelli, V.; Borrelli, E.; Vitacolonna, E.; Di Nicola, M.; Di Antonio, L.; Mastropasqua, R. Neuroretinal alterations in the early stages of diabetic retinopathy in patients with type 2 diabetes mellitus. Eye 2016, 30, 673–679. [Google Scholar] [CrossRef]
- Zhao, X.; Wang, J.; Li, P.; Tang, L.; Bai, Y. Casein Kinase 2-Interacting Protein-1 Alleviates High Glucose-Reduced Autophagy, Oxidative Stress, and Apoptosis in Retinal Pigment Epithelial Cells via Activating the p62/KEAP1/NRF2 Signaling Pathway. J. Ophthalmol. 2021, 2021, 6694050. [Google Scholar] [CrossRef]
- Lopes de Faria, J.M.; Duarte, D.A.; Simó, R.; García-Ramirez, M.; Dátilo, M.N.; Pasqualetto, F.C.; Lopes de Faria, J.B. δ Opioid Receptor Agonism Preserves the Retinal Pigmented Epithelial Cell Tight Junctions and Ameliorates the Retinopathy in Experimental Diabetes. Investig. Ophthalmol. Vis. Sci. 2019, 60, 3842–3853. [Google Scholar] [CrossRef]
- Feng, L.; Liang, L.; Zhang, S.; Yang, J.; Yue, Y.; Zhang, X. HMGB1 downregulation in retinal pigment epithelial cells protects against diabetic retinopathy through the autophagy-lysosome pathway. Autophagy 2022, 18, 320–339. [Google Scholar] [CrossRef] [PubMed]
- Janani, R.; Anitha, R.E.; Perumal, M.K.; Divya, P.; Baskaran, V. Astaxanthin mediated regulation of VEGF through HIF1α and XBP1 signaling pathway: An insight from ARPE-19 cell and streptozotocin mediated diabetic rat model. Exp. Eye Res. 2021, 206, 108555. [Google Scholar] [CrossRef]
- Gao, L.M.; Fu, S.; Liu, F.; Wu, H.B.; Li, W.J. Astragalus Polysaccharide Regulates miR-182/Bcl-2 Axis to Relieve Metabolic Memory through Suppressing Mitochondrial Damage-Mediated Apoptosis in Retinal Pigment Epithelial Cells. Pharmacology 2021, 106, 520–533. [Google Scholar] [CrossRef]
- Giacco, F.; Brownlee, M. Oxidative stress and diabetic complications. Circ. Res. 2010, 107, 1058–1070. [Google Scholar] [CrossRef]
- Kowluru, R.A.; Mishra, M. Oxidative stress, mitochondrial damage and diabetic retinopathy. Biochim. Biophys. Acta 2015, 1852, 2474–2483. [Google Scholar] [CrossRef] [PubMed]
- Rodríguez, M.L.; Pérez, S.; Mena-Mollá, S.; Desco, M.C.; Ortega, Á.L. Oxidative Stress and Microvascular Alterations in Diabetic Retinopathy: Future Therapies. Oxid. Med. Cell Longev. 2019, 2019, 4940825. [Google Scholar] [CrossRef] [PubMed]
- Sun, F.; Sun, Y.; Zhu, J.; Wang, X.; Ji, C.; Zhang, J.; Chen, S.; Yu, Y.; Xu, W.; Qian, H. Mesenchymal stem cells-derived small extracellular vesicles alleviate diabetic retinopathy by delivering NEDD4. Stem Cell Res. Ther. 2022, 13, 293. [Google Scholar] [CrossRef]
- Tang, X.; Li, X.; Zhang, D.; Han, W. Astragaloside-IV alleviates high glucose-induced ferroptosis in retinal pigment epithelial cells by disrupting the expression of miR-138-5p/Sirt1/Nrf2. Bioengineered 2022, 13, 8240–8254. [Google Scholar] [CrossRef]
- Li, R.; Ye, Z.; Yang, W.; Xu, Y.J.; Tan, C.P.; Liu, Y. Blueberry Anthocyanins from Commercial Products: Structure Identification and Potential for Diabetic Retinopathy Amelioration. Molecules 2022, 27, 7475. [Google Scholar] [CrossRef]
- D’Agata, V.; D’Amico, A.G.; Maugeri, G.; Bucolo, C.; Rossi, S.; Giunta, S. Carnosol attenuates high glucose damage in human retinal endothelial cells through regulation of ERK/Nrf2/HO-1 pathway. J. Asian Nat. Prod. Res. 2023, 25, 783–795. [Google Scholar] [CrossRef]
- Albert-Garay, J.S.; Riesgo-Escovar, J.R.; Salceda, R. High glucose concentrations induce oxidative stress by inhibiting Nrf2 expression in rat Müller retinal cells in vitro. Sci. Rep. 2022, 12, 1261. [Google Scholar] [CrossRef]
- Liu, X.; Liu, Y.; Chen, L.; Zhang, Z.; Cui, L.; Wei, T. Loss of pleckstrin homology domain and leucine-rich repeat protein phosphatase 2 has protective effects on high glucose-injured retinal ganglion cells via the effect on the Akt-GSK-3β-Nrf2 pathway. Inflamm. Res. 2023, 72, 373–385. [Google Scholar] [CrossRef] [PubMed]
- Fang, J.; Bai, W.; Yang, L. Astaxanthin inhibits oxidative stress and apoptosis in diabetic retinopathy. Acta Histochem. 2023, 125, 152069. [Google Scholar] [CrossRef]
- Yang, X.; Li, D. Tricin attenuates diabetic retinopathy by inhibiting oxidative stress and angiogenesis through regulating Sestrin2/Nrf2 signaling. Hum. Exp. Toxicol. 2023, 42, 9603271231171642. [Google Scholar] [CrossRef] [PubMed]
- Bannai, S.; Tateishi, N. Role of membrane transport in metabolism and function of glutathione in mammals. J. Membr. Biol. 1986, 89, 1–8. [Google Scholar] [CrossRef] [PubMed]
- Zhou, Z.; Li, H.; Bai, S.; Xu, Z.; Jiao, Y. Loss of serine/threonine protein kinase 25 in retinal ganglion cells ameliorates high glucose-elicited damage through regulation of the AKT-GSK-3β/Nrf2 pathway. Biochem. Biophys. Res. Commun. 2022, 600, 87–93. [Google Scholar] [CrossRef] [PubMed]
- Hayes, J.D.; Chowdhry, S.; Dinkova-Kostova, A.T.; Sutherland, C. Dual regulation of transcription factor Nrf2 by Keap1 and by the combined actions of β-TrCP and GSK-3. Biochem. Soc. Trans. 2015, 43, 611–620. [Google Scholar] [CrossRef]
- Rojo, A.I.; Sagarra, M.R.; Cuadrado, A. GSK-3beta down-regulates the transcription factor Nrf2 after oxidant damage: Relevance to exposure of neuronal cells to oxidative stress. J. Neurochem. 2008, 105, 192–202. [Google Scholar] [CrossRef]
- Rojo, A.I.; Rada, P.; Egea, J.; Rosa, A.O.; López, M.G.; Cuadrado, A. Functional interference between glycogen synthase kinase-3 beta and the transcription factor Nrf2 in protection against kainate-induced hippocampal cell death. Mol. Cell Neurosci. 2008, 39, 125–132. [Google Scholar] [CrossRef]
- Giacco, F.; Du, X.; Carratú, A.; Gerfen, G.J.; D’Apolito, M.; Giardino, I.; Rasola, A.; Marin, O.; Divakaruni, A.S.; Murphy, A.N.; et al. GLP-1 Cleavage Product Reverses Persistent ROS Generation After Transient Hyperglycemia by Disrupting an ROS-Generating Feedback Loop. Diabetes 2015, 64, 3273–3284. [Google Scholar] [CrossRef] [PubMed]
- Miller, W.P.; Toro, A.L.; Barber, A.J.; Dennis, M.D. REDD1 Activates a ROS-Generating Feedback Loop in the Retina of Diabetic Mice. Investig. Ophthalmol. Vis. Sci. 2019, 60, 2369–2379. [Google Scholar] [CrossRef]
- Miller, W.P.; Sunilkumar, S.; Giordano, J.F.; Toro, A.L.; Barber, A.J.; Dennis, M.D. The stress response protein REDD1 promotes diabetes-induced oxidative stress in the retina by Keap1-independent Nrf2 degradation. J. Biol. Chem. 2020, 295, 7350–7361. [Google Scholar] [CrossRef] [PubMed]
- Schrufer, T.L.; Antonetti, D.A.; Sonenberg, N.; Kimball, S.R.; Gardner, T.W.; Jefferson, L.S. Ablation of 4E-BP1/2 prevents hyperglycemia-mediated induction of VEGF expression in the rodent retina and in Muller cells in culture. Diabetes 2010, 59, 2107–2116. [Google Scholar] [CrossRef]
- Dennis, M.D.; Kimball, S.R.; Fort, P.E.; Jefferson, L.S. Regulated in development and DNA damage 1 is necessary for hyperglycemia-induced vascular endothelial growth factor expression in the retina of diabetic rodents. J. Biol. Chem. 2015, 290, 3865–3874. [Google Scholar] [CrossRef] [PubMed]
- Hao, Y.; Gao, X. Diosgenin protects retinal pigment epithelial cells from inflammatory damage and oxidative stress induced by high glucose by activating AMPK/Nrf2/HO-1 pathway. Immun. Inflamm. Dis. 2022, 10, e698. [Google Scholar] [CrossRef]
- Barouch, F.C.; Miyamoto, K.; Allport, J.R.; Fujita, K.; Bursell, S.E.; Aiello, L.P.; Luscinskas, F.W.; Adamis, A.P. Integrin-mediated neutrophil adhesion and retinal leukostasis in diabetes. Investig. Ophthalmol. Vis. Sci. 2000, 41, 1153–1158. [Google Scholar]
- Joussen, A.M.; Poulaki, V.; Le, M.L.; Koizumi, K.; Esser, C.; Janicki, H.; Schraermeyer, U.; Kociok, N.; Fauser, S.; Kirchhof, B.; et al. A central role for inflammation in the pathogenesis of diabetic retinopathy. FASEB J. 2004, 18, 1450–1452. [Google Scholar] [CrossRef]
- Kasza, M.; Meleg, J.; Vardai, J.; Nagy, B., Jr.; Szalai, E.; Damjanovich, J.; Csutak, A.; Ujhelyi, B.; Nagy, V. Plasma E-selectin levels can play a role in the development of diabetic retinopathy. Graefes Arch. Clin. Exp. Ophthalmol. 2017, 255, 25–30. [Google Scholar] [CrossRef]
- Miyamoto, K.; Khosrof, S.; Bursell, S.E.; Rohan, R.; Murata, T.; Clermont, A.C.; Aiello, L.P.; Ogura, Y.; Adamis, A.P. Prevention of leukostasis and vascular leakage in streptozotocin-induced diabetic retinopathy via intercellular adhesion molecule-1 inhibition. Proc. Natl. Acad. Sci. USA 1999, 96, 10836–10841. [Google Scholar] [CrossRef]
- Schröder, S.; Palinski, W.; Schmid-Schönbein, G.W. Activated monocytes and granulocytes, capillary nonperfusion, and neovascularization in diabetic retinopathy. Am. J. Pathol. 1991, 139, 81–100. [Google Scholar] [PubMed]
- Boss, J.D.; Singh, P.K.; Pandya, H.K.; Tosi, J.; Kim, C.; Tewari, A.; Juzych, M.S.; Abrams, G.W.; Kumar, A. Assessment of Neurotrophins and Inflammatory Mediators in Vitreous of Patients With Diabetic Retinopathy. Investig. Ophthalmol. Vis. Sci. 2017, 58, 5594–5603. [Google Scholar] [CrossRef]
- Koleva-Georgieva, D.N.; Sivkova, N.P.; Terzieva, D. Serum inflammatory cytokines IL-1beta, IL-6, TNF-alpha and VEGF have influence on the development of diabetic retinopathy. Folia Med. 2011, 53, 44–50. [Google Scholar] [CrossRef]
- Rangasamy, S.; McGuire, P.G.; Franco Nitta, C.; Monickaraj, F.; Oruganti, S.R.; Das, A. Chemokine mediated monocyte trafficking into the retina: Role of inflammation in alteration of the blood-retinal barrier in diabetic retinopathy. PLoS ONE 2014, 9, e108508. [Google Scholar] [CrossRef] [PubMed]
- Suzuki, Y.; Nakazawa, M.; Suzuki, K.; Yamazaki, H.; Miyagawa, Y. Expression profiles of cytokines and chemokines in vitreous fluid in diabetic retinopathy and central retinal vein occlusion. Jpn. J. Ophthalmol. 2011, 55, 256–263. [Google Scholar] [CrossRef]
- Abcouwer, S.F. Müller Cell-Microglia Cross Talk Drives Neuroinflammation in Diabetic Retinopathy. Diabetes 2017, 66, 261–263. [Google Scholar] [CrossRef]
- Sorrentino, F.S.; Allkabes, M.; Salsini, G.; Bonifazzi, C.; Perri, P. The importance of glial cells in the homeostasis of the retinal microenvironment and their pivotal role in the course of diabetic retinopathy. Life Sci. 2016, 162, 54–59. [Google Scholar] [CrossRef]
- Xu, Z.; Li, S.; Li, K.; Wang, X.; Li, X.; An, M.; Yu, X.; Long, X.; Zhong, R.; Liu, Q.; et al. Urolithin A ameliorates diabetic retinopathy via activation of the Nrf2/HO-1 pathway. Endocr. J. 2022, 69, 971–982. [Google Scholar] [CrossRef]
- Mansour, S.E.; Browning, D.J.; Wong, K.; Flynn, H.W., Jr.; Bhavsar, A.R. The Evolving Treatment of Diabetic Retinopathy. Clin. Ophthalmol. 2020, 14, 653–678. [Google Scholar] [CrossRef]
- Jakus, V.; Rietbrock, N. Advanced glycation end-products and the progress of diabetic vascular complications. Physiol. Res. 2004, 53, 131–142. [Google Scholar] [CrossRef]
- Haritoglou, C.; Gerss, J.; Sauerland, C.; Kampik, A.; Ulbig, M.W. Effect of calcium dobesilate on occurrence of diabetic macular oedema (CALDIRET study): Randomised, double-blind, placebo-controlled, multicentre trial. Lancet 2009, 373, 1364–1371. [Google Scholar] [CrossRef]
- Mayer-Davis, E.J.; Bell, R.A.; Reboussin, B.A.; Rushing, J.; Marshall, J.A.; Hamman, R.F. Antioxidant nutrient intake and diabetic retinopathy: The San Luis Valley Diabetes Study. Ophthalmology 1998, 105, 2264–2270. [Google Scholar] [CrossRef]
- Millen, A.E.; Klein, R.; Folsom, A.R.; Stevens, J.; Palta, M.; Mares, J.A. Relation between intake of vitamins C and E and risk of diabetic retinopathy in the Atherosclerosis Risk in Communities Study. Am. J. Clin. Nutr. 2004, 79, 865–873. [Google Scholar] [CrossRef] [PubMed]
- Yang, J.; Hua, Z.; Zheng, Z.; Ma, X.; Zhu, L.; Li, Y. Acteoside inhibits high glucose-induced oxidative stress injury in RPE cells and the outer retina through the Keap1/Nrf2/ARE pathway. Exp. Eye Res. 2023, 232, 109496. [Google Scholar] [CrossRef] [PubMed]
- Li, S.; Lu, S.; Wang, L.; Liu, S.; Zhang, L.; Du, J.; Wu, Z.; Huang, X. Effects of amygdalin on ferroptosis and oxidative stress in diabetic retinopathy progression via the NRF2/ARE signaling pathway. Exp. Eye Res. 2023, 234, 109569. [Google Scholar] [CrossRef]
- Blaustein, M.P.; Hamlyn, J.M. Ouabain, endogenous ouabain and ouabain-like factors: The Na(+) pump/ouabain receptor, its linkage to NCX, and its myriad functions. Cell Calcium 2020, 86, 102159. [Google Scholar] [CrossRef] [PubMed]
- Blanco, G. Na,K-ATPase subunit heterogeneity as a mechanism for tissue-specific ion regulation. Semin. Nephrol. 2005, 25, 292–303. [Google Scholar] [CrossRef]
- Wetzel, R.K.; Arystarkhova, E.; Sweadner, K.J. Cellular and subcellular specification of Na,K-ATPase alpha and beta isoforms in the postnatal development of mouse retina. J. Neurosci. 1999, 19, 9878–9889. [Google Scholar] [CrossRef]
- Maturana-Teixeira, S.; Braga, L.E.; Carpi Santos, R.; Calaza Kda, C.; Giestal-de-Araujo, E.; Leão-Ferreira, L.R. The (Na(+)/K(+))-ATPase activity in the developing rat retina: The role of insulin-like growth factor-I (IGF-I). Cell Mol. Neurobiol. 2015, 35, 243–254. [Google Scholar] [CrossRef]
- Demontis, G.C.; Ratto, G.M.; Bisti, S.; Cervetto, L. Effect of blocking the Na+/K+ ATPase on Ca2+ extrusion and light adaptation in mammalian retinal rods. Biophys. J. 1995, 69, 439–450. [Google Scholar] [CrossRef]
- Namekata, K.; Harada, C.; Kohyama, K.; Matsumoto, Y.; Harada, T. Interleukin-1 stimulates glutamate uptake in glial cells by accelerating membrane trafficking of Na+/K+-ATPase via actin depolymerization. Mol. Cell Biol. 2008, 28, 3273–3280. [Google Scholar] [CrossRef]
- Country, M.W. Retinal metabolism: A comparative look at energetics in the retina. Brain Res. 2017, 1672, 50–57. [Google Scholar] [CrossRef]
- Nagaoka, K.; Kurauchi, Y.; Asano, D.; Morita, A.; Sakamoto, K.; Nakahara, T. Pharmacological inhibition of Na(+)/K(+)-ATPase induces neurovascular degeneration and glial cell alteration in the rat retina. Exp. Eye Res. 2022, 220, 109107. [Google Scholar] [CrossRef] [PubMed]
- McGinn, T.E.; Galicia, C.A.; Leoni, D.C.; Partington, N.; Mitchell, D.M.; Stenkamp, D.L. Rewiring the Regenerated Zebrafish Retina: Reemergence of Bipolar Neurons and Cone-Bipolar Circuitry Following an Inner Retinal Lesion. Front. Cell Dev. Biol. 2019, 7, 95. [Google Scholar] [CrossRef] [PubMed]
- Barrett, L.M.; Mitchell, D.M.; Meighan, P.C.; Varnum, M.D.; Stenkamp, D.L. Dynamic functional and structural remodeling during retinal regeneration in zebrafish. Front. Mol. Neurosci. 2022, 15, 1070509. [Google Scholar] [CrossRef] [PubMed]
- Corrêa Gde, R.; Cunha, K.C.; dos Santos, A.A.; de Araujo, E.G. The trophic effect of ouabain on retinal ganglion cell is mediated by EGF receptor and PKC delta activation. Neurochem. Res. 2010, 35, 1343–1352. [Google Scholar] [CrossRef]
- Sarkies, N. Traumatic optic neuropathy. Eye 2004, 18, 1122–1125. [Google Scholar] [CrossRef]
- Almasieh, M.; Wilson, A.M.; Morquette, B.; Cueva Vargas, J.L.; Di Polo, A. The molecular basis of retinal ganglion cell death in glaucoma. Prog. Retin. Eye Res. 2012, 31, 152–181. [Google Scholar] [CrossRef]
- Tribble, J.R.; Hui, F.; Quintero, H.; El Hajji, S.; Bell, K.; Di Polo, A.; Williams, P.A. Neuroprotection in glaucoma: Mechanisms beyond intraocular pressure lowering. Mol. Aspects Med. 2023, 92, 101193. [Google Scholar] [CrossRef]
- Fry, L.E.; Fahy, E.; Chrysostomou, V.; Hui, F.; Tang, J.; van Wijngaarden, P.; Petrou, S.; Crowston, J.G. The coma in glaucoma: Retinal ganglion cell dysfunction and recovery. Prog. Retin. Eye Res. 2018, 65, 77–92. [Google Scholar] [CrossRef]
- Berkelaar, M.; Clarke, D.B.; Wang, Y.C.; Bray, G.M.; Aguayo, A.J. Axotomy results in delayed death and apoptosis of retinal ganglion cells in adult rats. J. Neurosci. 1994, 14, 4368–4374. [Google Scholar] [CrossRef] [PubMed]
- de Araujo, E.G.; Linden, R. Trophic factors produced by retinal cells increase the survival of retinal ganglion cells in vitro. Eur. J. Neurosci. 1993, 5, 1181–1188. [Google Scholar] [CrossRef] [PubMed]
- Yin, Y.; Benowitz, L.I. In Vitro and In Vivo Methods for Studying Retinal Ganglion Cell Survival and Optic Nerve Regeneration. Methods Mol. Biol. 2018, 1695, 187–205. [Google Scholar] [CrossRef] [PubMed]
- Kügler, S.; Straten, G.; Kreppel, F.; Isenmann, S.; Liston, P.; Bähr, M. The X-linked inhibitor of apoptosis (XIAP) prevents cell death in axotomized CNS neurons in vivo. Cell Death Differ. 2000, 7, 815–824. [Google Scholar] [CrossRef] [PubMed]
- Isenmann, S.; Kretz, A.; Cellerino, A. Molecular determinants of retinal ganglion cell development, survival, and regeneration. Prog. Retin. Eye Res. 2003, 22, 483–543. [Google Scholar] [CrossRef] [PubMed]
- Kroeger, H.; Chiang, W.C.; Felden, J.; Nguyen, A.; Lin, J.H. ER stress and unfolded protein response in ocular health and disease. FEBS J. 2019, 286, 399–412. [Google Scholar] [CrossRef] [PubMed]
- Fudalej, E.; Justyniarska, M.; Kasarełło, K.; Dziedziak, J.; Szaflik, J.P.; Cudnoch-Jędrzejewska, A. Neuroprotective Factors of the Retina and Their Role in Promoting Survival of Retinal Ganglion Cells: A Review. Ophthalmic Res. 2021, 64, 345–355. [Google Scholar] [CrossRef]
- de Rezende Corrêa, G.; Araujo dos Santos, A.; Frederico Leite Fontes, C.; Giestal de Araujo, E. Ouabain induces an increase of retinal ganglion cell survival in vitro: The involvement of protein kinase C. Brain Res. 2005, 1049, 89–94. [Google Scholar] [CrossRef]
- Mázala-de-Oliveira, T.; de Figueiredo, C.S.; de Rezende Corrêa, G.; da Silva, M.S.; Miranda, R.L.; de Azevedo, M.A.; Cossenza, M.; Dos Santos, A.A.; Giestal-de-Araujo, E. Ouabain-Na(+)/K(+)-ATPase Signaling Regulates Retinal Neuroinflammation and ROS Production Preventing Neuronal Death by an Autophagy-Dependent Mechanism Following Optic Nerve Axotomy In Vitro. Neurochem. Res. 2022, 47, 723–738. [Google Scholar] [CrossRef]
- Salles von-Held-Ventura, J.; Mázala-de-Oliveira, T.; Cândida da Rocha Oliveira, A.; Granja, M.G.; Gonçalves-de-Albuquerque, C.F.; Castro-Faria-Neto, H.C.; Giestal-de-Araujo, E. The trophic effect of ouabain on retinal ganglion cells is mediated by IL-1β and TNF-α. Biochem. Biophys. Res. Commun. 2016, 478, 378–384. [Google Scholar] [CrossRef]
- Ail, D.; Ren, D.; Brazhnikova, E.; Nouvel-Jaillard, C.; Bertin, S.; Mirashrafi, S.B.; Fisson, S.; Dalkara, D. Systemic and local immune responses to intraocular AAV vector administration in non-human primates. Mol. Ther. Methods Clin. Dev. 2022, 24, 306–316. [Google Scholar] [CrossRef]
- Bennett, J.; Maguire, A.M. Lessons Learned from the Development of the First FDA-Approved Gene Therapy Drug, Voretigene Neparvovec-rzyl. Cold Spring Harb. Perspect. Med. 2023, 13, a041307. [Google Scholar] [CrossRef] [PubMed]
- Haider, N.B.; Ikeda, A.; Naggert, J.K.; Nishina, P.M. Genetic modifiers of vision and hearing. Hum. Mol. Genet. 2002, 11, 1195–1206. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Dipple, K.M.; McCabe, E.R. Modifier genes convert “simple” Mendelian disorders to complex traits. Mol. Genet. Metab. 2000, 71, 43–50. [Google Scholar] [CrossRef]
- Toms, M.; Ward, N.; Moosajee, M. Nuclear Receptor Subfamily 2 Group E Member 3 (NR2E3): Role in Retinal Development and Disease. Genes 2023, 14, 1325. [Google Scholar] [CrossRef] [PubMed]
- Maeder, M.L.; Stefanidakis, M.; Wilson, C.J.; Baral, R.; Barrera, L.A.; Bounoutas, G.S.; Bumcrot, D.; Chao, H.; Ciulla, D.M.; DaSilva, J.A.; et al. Development of a gene-editing approach to restore vision loss in Leber congenital amaurosis type 10. Nat. Med. 2019, 25, 229–233. [Google Scholar] [CrossRef] [PubMed]
- Nirenberg, S.; Pandarinath, C. Retinal prosthetic strategy with the capacity to restore normal vision. Proc. Natl. Acad. Sci. USA 2012, 109, 15012–15017. [Google Scholar] [CrossRef]
- Batabyal, S.; Gajjeraman, S.; Pradhan, S.; Bhattacharya, S.; Wright, W.; Mohanty, S. Sensitization of ON-bipolar cells with ambient light activatable multi-characteristic opsin rescues vision in mice. Gene Ther. 2021, 28, 162–176. [Google Scholar] [CrossRef]
- Sahel, J.A.; Boulanger-Scemama, E.; Pagot, C.; Arleo, A.; Galluppi, F.; Martel, J.N.; Esposti, S.D.; Delaux, A.; de Saint Aubert, J.B.; de Montleau, C.; et al. Partial recovery of visual function in a blind patient after optogenetic therapy. Nat. Med. 2021, 27, 1223–1229. [Google Scholar] [CrossRef]
- Miltner, A.M.; La Torre, A. Retinal Ganglion Cell Replacement: Current Status and Challenges Ahead. Dev. Dyn. 2019, 248, 118–128. [Google Scholar] [CrossRef]
- Lahne, M.; Nagashima, M.; Hyde, D.R.; Hitchcock, P.F. Reprogramming Müller Glia to Regenerate Retinal Neurons. Annu. Rev. Vis. Sci. 2020, 6, 171–193. [Google Scholar] [CrossRef] [PubMed]
- Goldman, D. Muller glial cell reprogramming and retina regeneration. Nat. Rev. Neurosci. 2014, 15, 431–442. [Google Scholar] [CrossRef] [PubMed]
- Blackshaw, S. Why Has the Ability to Regenerate Following CNS Injury Been Repeatedly Lost over the Course of Evolution? Front. Neurosci. 2022, 16, 831062. [Google Scholar] [CrossRef]
- Hoang, T.; Wang, J.; Boyd, P.; Wang, F.; Santiago, C.; Jiang, L.; Yoo, S.; Lahne, M.; Todd, L.J.; Jia, M.; et al. Gene regulatory networks controlling vertebrate retinal regeneration. Science 2020, 370, eabb8598. [Google Scholar] [CrossRef]
- Pollak, J.; Wilken, M.S.; Ueki, Y.; Cox, K.E.; Sullivan, J.M.; Taylor, R.J.; Levine, E.M.; Reh, T.A. ASCL1 reprograms mouse Muller glia into neurogenic retinal progenitors. Development 2013, 140, 2619–2631. [Google Scholar] [CrossRef] [PubMed]
- Ueki, Y.; Wilken, M.S.; Cox, K.E.; Chipman, L.; Jorstad, N.; Sternhagen, K.; Simic, M.; Ullom, K.; Nakafuku, M.; Reh, T.A. Transgenic expression of the proneural transcription factor Ascl1 in Muller glia stimulates retinal regeneration in young mice. Proc. Natl. Acad. Sci. USA 2015, 112, 13717–13722. [Google Scholar] [CrossRef]
- Jorstad, N.L.; Wilken, M.S.; Grimes, W.N.; Wohl, S.G.; VandenBosch, L.S.; Yoshimatsu, T.; Wong, R.O.; Rieke, F.; Reh, T.A. Stimulation of functional neuronal regeneration from Muller glia in adult mice. Nature 2017, 548, 103–107. [Google Scholar] [CrossRef]
- Todd, L.; Hooper, M.J.; Haugan, A.K.; Finkbeiner, C.; Jorstad, N.; Radulovich, N.; Wong, C.K.; Donaldson, P.C.; Jenkins, W.; Chen, Q.; et al. Efficient stimulation of retinal regeneration from Muller glia in adult mice using combinations of proneural bHLH transcription factors. Cell Rep. 2021, 37, 109857. [Google Scholar] [CrossRef]
- Todd, L.; Jenkins, W.; Finkbeiner, C.; Hooper, M.J.; Donaldson, P.C.; Pavlou, M.; Wohlschlegel, J.; Ingram, N.; Rieke, F.; Reh, T.A.; et al. Reprogramming Müller glia to regenerate ganglion-like cells in adult mouse retina with developmental transcription factors. Sci. Adv. 2022, 8, eabq7219. [Google Scholar] [CrossRef]
- Xiao, D.; Jin, K.; Qiu, S.; Lei, Q.; Huang, W.; Chen, H.; Su, J.; Xu, Q.; Xu, Z.; Gou, B.; et al. In vivo Regeneration of Ganglion Cells for Vision Restoration in Mammalian Retinas. Front. Cell Dev. Biol. 2021, 9, 755544. [Google Scholar] [CrossRef]
- Zhou, H.; Su, J.; Hu, X.; Zhou, C.; Li, H.; Chen, Z.; Xiao, Q.; Wang, B.; Wu, W.; Sun, Y.; et al. Glia-to-Neuron Conversion by CRISPR-CasRx Alleviates Symptoms of Neurological Disease in Mice. Cell 2020, 181, 590–603. [Google Scholar] [CrossRef] [PubMed]
- Yao, K.; Qiu, S.; Wang, Y.V.; Park, S.J.H.; Mohns, E.J.; Mehta, B.; Liu, X.; Chang, B.; Zenisek, D.; Crair, M.C.; et al. Restoration of vision after de novo genesis of rod photoreceptors in mammalian retinas. Nature 2018, 560, 484–488. [Google Scholar] [CrossRef] [PubMed]
- Le, N.; Appel, H.; Pannullo, N.; Hoang, T.; Blackshaw, S. Ectopic insert-dependent neuronal expression of GFAP promoter-driven AAV constructs in adult mouse retina. Front. Cell Dev. Biol. 2022, 10, 914386. [Google Scholar] [CrossRef] [PubMed]
- Wang, L.L.; Zhang, C.L. Therapeutic Potential of PTBP1 Inhibition, If Any, Is Not Attributed to Glia-to-Neuron Conversion. Annu. Rev. Neurosci. 2023, 46, 1–15. [Google Scholar] [CrossRef] [PubMed]
- Xie, Y.; Zhou, J.; Chen, B. Critical examination of Ptbp1-mediated glia-to-neuron conversion in the mouse retina. Cell Rep. 2022, 39, 110960. [Google Scholar] [CrossRef]
- Blackshaw, S.; Sanes, J.R. Turning lead into gold: Reprogramming retinal cells to cure blindness. J. Clin. Investig. 2021, 131, e146134. [Google Scholar] [CrossRef]
- Ling, J.P.; Bygrave, A.M.; Santiago, C.P.; Carmen-Orozco, R.P.; Trinh, V.T.; Yu, M.; Li, Y.; Liu, Y.; Bowden, K.D.; Duncan, L.H.; et al. Cell-specific regulation of gene expression using splicing-dependent frameshifting. Nat. Commun. 2022, 13, 5773. [Google Scholar] [CrossRef]
- Gao, Y.; Fang, K.; Yan, Z.; Zhang, H.; Geng, G.; Wu, W.; Xu, D.; Zhang, H.; Zhong, N.; Wang, Q.; et al. Develop an efficient and specific AAV-based labeling system for Muller glia in mice. Sci. Rep. 2022, 12, 22410. [Google Scholar] [CrossRef]
- Tresenrider, A.; Hooper, M.; Todd, L.; Kierney, F.; Blasdel, N.; Trapnell, C.; Reh, T.A. A multiplexed, single-cell sequencing screen identifies compounds that increase neurogenic reprogramming of murine Muller glia. bioRxiv 2023. [Google Scholar] [CrossRef]
- Oliveira-Valenca, V.M.; Bosco, A.; Vetter, M.L.; Silveira, M.S. On the Generation and Regeneration of Retinal Ganglion Cells. Front. Cell Dev. Biol. 2020, 8, 581136. [Google Scholar] [CrossRef]
- Soucy, J.R.; Aguzzi, E.A.; Cho, J.; Gilhooley, M.J.; Keuthan, C.; Luo, Z.; Monavarfeshani, A.; Saleem, M.A.; Wang, X.W.; Wohlschlegel, J.; et al. Retinal ganglion cell repopulation for vision restoration in optic neuropathy: A roadmap from the RReSTORe Consortium. Mol. Neurodegener. 2023, 18, 64. [Google Scholar] [CrossRef] [PubMed]


Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Tempone, M.H.; Borges-Martins, V.P.; César, F.; Alexandrino-Mattos, D.P.; de Figueiredo, C.S.; Raony, Í.; dos Santos, A.A.; Duarte-Silva, A.T.; Dias, M.S.; Freitas, H.R.; et al. The Healthy and Diseased Retina Seen through Neuron–Glia Interactions. Int. J. Mol. Sci. 2024, 25, 1120. https://doi.org/10.3390/ijms25021120
Tempone MH, Borges-Martins VP, César F, Alexandrino-Mattos DP, de Figueiredo CS, Raony Í, dos Santos AA, Duarte-Silva AT, Dias MS, Freitas HR, et al. The Healthy and Diseased Retina Seen through Neuron–Glia Interactions. International Journal of Molecular Sciences. 2024; 25(2):1120. https://doi.org/10.3390/ijms25021120
Chicago/Turabian StyleTempone, Matheus H., Vladimir P. Borges-Martins, Felipe César, Dio Pablo Alexandrino-Mattos, Camila S. de Figueiredo, Ícaro Raony, Aline Araujo dos Santos, Aline Teixeira Duarte-Silva, Mariana Santana Dias, Hércules Rezende Freitas, and et al. 2024. "The Healthy and Diseased Retina Seen through Neuron–Glia Interactions" International Journal of Molecular Sciences 25, no. 2: 1120. https://doi.org/10.3390/ijms25021120
APA StyleTempone, M. H., Borges-Martins, V. P., César, F., Alexandrino-Mattos, D. P., de Figueiredo, C. S., Raony, Í., dos Santos, A. A., Duarte-Silva, A. T., Dias, M. S., Freitas, H. R., de Araújo, E. G., Ribeiro-Resende, V. T., Cossenza, M., P. Silva, H., P. de Carvalho, R., Ventura, A. L. M., Calaza, K. C., Silveira, M. S., Kubrusly, R. C. C., & de Melo Reis, R. A. (2024). The Healthy and Diseased Retina Seen through Neuron–Glia Interactions. International Journal of Molecular Sciences, 25(2), 1120. https://doi.org/10.3390/ijms25021120