
Navigating the Cytokine Seas: Targeting Cytokine Signaling Pathways in Cancer Therapy



Abstract
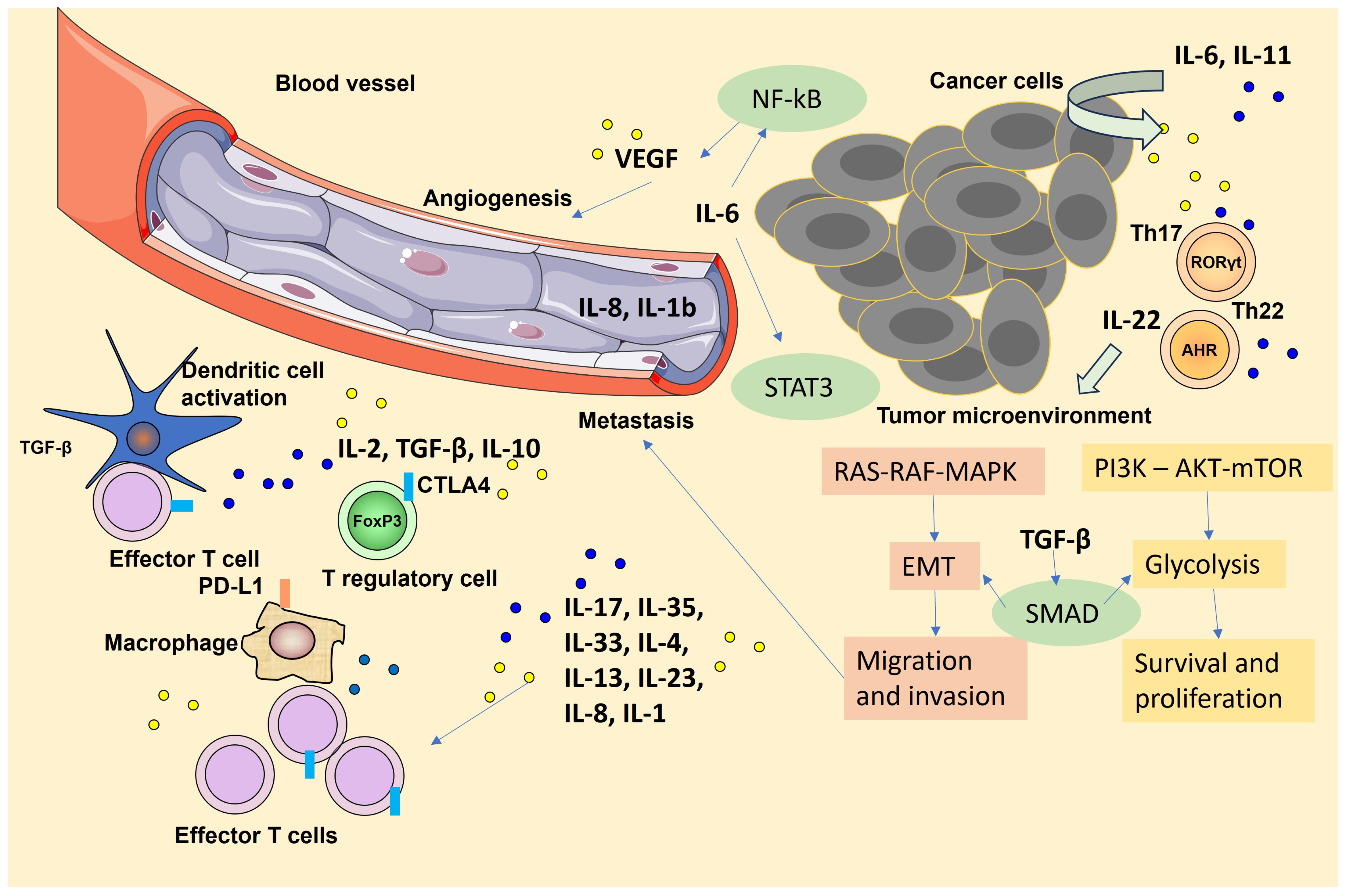
1. Introduction
2. Cytokines Induced NFκB Signaling Pathway That Promotes Tumor Growth
3. Cytokine-Induced STAT Signaling Pathway Promoting Tumor Invasion and Metastasis
4. TGF-β Signaling Pathway
5. Cytokine-Induced Signaling Pathway Related to Epithelial-Mesenchymal Transition and Tumor Metastasis
6. Targeting Cytokine Signaling in Cancer Therapy: Objectives
7. Recent Advancements in Cytokine-Based Cancer Treatments: Clinical Trials Data
8. Future Directions and Challenges in Emerging Cytokine Targets and Novel Therapies
9. Conclusions
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Nicholson, L.B. The immune system. Essays Biochem. 2016, 60, 275–301. [Google Scholar] [CrossRef] [PubMed]
- Chang, M.; Jin, W.; Sun, S.C. Peli1 facilitates TRIF-dependent Toll-like receptor signaling and proinflammatory cytokine production. Nat. Immunol. 2009, 10, 1089–1095. [Google Scholar] [CrossRef] [PubMed]
- Moraga, I.; Spangler, J.; Mendoza, J.L.; Garcia, K.C. Multifarious determinants of cytokine receptor signaling specificity. Adv. Immunol. 2014, 121, 1–39. [Google Scholar] [PubMed]
- Haan, C.; Kreis, S.; Margue, C.; Behrmann, I. Jaks and cytokine receptors-An intimate relationship. Biochem. Pharmacol. 2006, 72, 1538–1546. [Google Scholar] [CrossRef] [PubMed]
- Kalliolias, G.D.; Ivashkiv, L.B. TNF biology, pathogenic mechanisms and emerging therapeutic strategies. Nat. Rev. Rheumatol. 2016, 12, 49–62. [Google Scholar] [CrossRef]
- Dunn, G.P.; Koebel, C.M.; Schreiber, R.D. Interferons, immunity and cancer immunoediting. Nat. Rev. Immunol. 2006, 6, 836–848. [Google Scholar] [CrossRef]
- Grivennikov, S.I.; Greten, F.R.; Karin, M. Immunity, inflammation, and cancer. Cell 2010, 140, 883–899. [Google Scholar] [CrossRef]
- Crusz, S.M.; Balkwill, F.R. Inflammation and cancer: Advances and new agents. Nat. Rev. Clin. Oncol. 2015, 12, 584–596. [Google Scholar] [CrossRef]
- Masetti, R.; Zama, D.; Urbini, M.; Astolfi, A.; Libri, V.; Vendemini, F.; Morello, W.; Rondelli, R.; Prete, A.; Pession, A. Impact of inflammatory cytokine gene polymorphisms on developing acute graft-versus-host disease in children undergoing allogeneic hematopoietic stem cell transplantation. J. Immunol. Res. 2015, 2015, 248264. [Google Scholar] [CrossRef]
- Hanahan, D.; Weinberg, R.A. Hallmarks of cancer: The next generation. Cell 2011, 144, 646–674. [Google Scholar] [CrossRef]
- Lippitz, B.E. Cytokine patterns in patients with cancer: A systematic review. Lancet Oncol. 2013, 14, e218–e228. [Google Scholar] [CrossRef] [PubMed]
- Park, M.H.; Hong, J.T. Roles of NF-κB in cancer and inflammatory diseases and their therapeutic approaches. Cells 2016, 5, 15. [Google Scholar] [CrossRef] [PubMed]
- Amani, H.; Ajami, M.; Maleki, S.N.; Pazoki-Toroudi, H.; Daglia, M.; Sokeng, A.J.T.; Di Lorenzo, A.; Nabavi, S.F.; Devi, K.P.; Nabavi, S.M. Targeting signal transducers and activators of ranscription (STAT) in human cancer by dietary polyphenolic antioxidants. Biochimie 2017, 142, 63–79. [Google Scholar] [CrossRef] [PubMed]
- Atsaves, V.; Leventaki, V.; Rassidakis, G.Z.; Claret, F.X. AP-1 transcription factors as regulators of immune responses in cancer. Cancers 2019, 11, 1037. [Google Scholar] [CrossRef] [PubMed]
- Balkwill, F.; Mantovani, A. Inflammation and cancer: Back to Virchow? Lancet 2001, 357, 539–545. [Google Scholar] [CrossRef] [PubMed]
- Aggarwal, B.B.; Vijayalekshmi, R.V.; Sung, B. Targeting inflammatory pathways for prevention and therapy of cancer: Short-term friend, long-term foe. Clin. Cancer Res. 2009, 15, 425–430. [Google Scholar] [CrossRef] [PubMed]
- Stanilov, N.; Miteva, L.; Mintchev, N.; Stanilova, S. High expression of Foxp3, IL-23p19 and survivin mRNA in colorectal carcinoma. Int. J. Color. Dis. 2009, 24, 151–157. [Google Scholar] [CrossRef]
- Stanilov, N.; Karakolev, I.; Deliysky, T.; Jovchev, J.; Stanilova, S. Association of insulin-like growth factor-I receptor polymorphism with colorectal cancer development. Mol. Biol. Rep. 2014, 41, 8099–8106. [Google Scholar] [CrossRef]
- Israelsson, P.; Dehlin, E.; Nagaev, I.; Lundin, E.; Ottander, U.; Mincheva-Nilsson, L. Cytokine mRNA and protein expression by cell cultures of epithelial ovarian cancer-Methodological considerations on the choice of analytical method for cytokine analyses. Am. J. Reprod. Immunol. 2020, 84, e13249. [Google Scholar] [CrossRef]
- Dinarello, C.A. Overview of the IL-1 family in innate inflammation and acquired immunity. Immunol. Rev. 2018, 281, 8–27. [Google Scholar] [CrossRef]
- Fields, J.K.; Günther, S.; Sundberg, E.J. Structural Basis of IL-1 Family Cytokine Signaling. Front. Immunol. 2019, 10, 1412. [Google Scholar] [CrossRef] [PubMed]
- Kim, J.S.; Kim, E.J.; Kim, H.S.; Kurie, J.M.; Ahn, Y.H. MKK4 activates non-canonical NFκB signaling by promoting NFκB2-p100 processing. Biochem. Biophys. Res. Commun. 2017, 491, 337–342. [Google Scholar] [CrossRef] [PubMed]
- Sun, S.C. The noncanonical NF-κB pathway. Immunol. Rev. 2012, 246, 125–140. [Google Scholar] [CrossRef] [PubMed]
- Baud, V.; Karin, M. Is NF-κB a good target for cancer therapy? Hopes and pitfalls. Nat. Rev. Drug Discov. 2009, 8, 33–40. [Google Scholar] [CrossRef]
- Ben-Neriah, Y.; Karin, M. Inflammation meets cancer with NF-κB as the matchmaker. Nat. Immunol. 2011, 12, 715–723. [Google Scholar] [CrossRef] [PubMed]
- Wang, W.; Nag, S.A.; Zhang, R. Targeting the NFκB Signaling Pathways for Breast Cancer Prevention and Therapy. Curr. Med. Chem. 2015, 22, 264–289. [Google Scholar] [CrossRef] [PubMed]
- Stratton, M.R.; Campbell, P.J.; Futreal, P.A. The cancer genome. Nature 2009, 458, 719–724. [Google Scholar] [CrossRef]
- Xia, L.; Tan, S.; Zhou, Y.; Lin, J.; Wang, H.; Oyang, L.; Tian, Y.; Liu, L.; Su, M.; Wang, H.; et al. Role of the NFκB-signaling pathway in cancer. Onco. Targets. Ther. 2018, 11, 2063–2073. [Google Scholar] [CrossRef]
- Wu, C.Y.; Ke, Y.; Zeng, Y.F.; Zhang, Y.W.; Yu, H.J. Anticancer activity of Astragalus polysaccharide in human non-small cell lung cancer cells. Cancer Cell Int. 2017, 17, 115. [Google Scholar] [CrossRef]
- Vaisitti, T.; Gaudino, F.; Ouk, S.; Moscvin, M.; Vitale, N.; Serra, S.; Deaglio, S. Targeting metabolism and survival in chronic lymphocytic leukemia and Richter syndrome cells by a novel NF-κB inhibitor. Haematologica 2017, 102, 1878–1889. [Google Scholar] [CrossRef]
- Barbosa, M.L.; da Conceicao, R.A.; Fraga, A.G.; Guerra Manssour Fraga, A.; Dias Camarinha, B.; Cristina de Carvalho Silva, G.; Gilcler Ferreira Lima, A. NF-κB signaling pathway inhibitors as anticancer drug candidates. Anticancer. Agents. Med. Chem. 2017, 17, 483–490. [Google Scholar] [CrossRef]
- Dinarello, C.A.; van der Meer, J.W. Treating inflammation by blocking interleukin-1 in humans. Semin. Immunol. 2013, 25, 469–484. [Google Scholar] [CrossRef]
- Lust, J.A.; Lacy, M.Q.; Zeldenrust, S.R.; Witzig, T.E.; Moon-Tasson, L.L.; Dinarello, C.A.; Donovan, K.A. Reduction in C-reactive protein indicates successful targeting of the IL-1/IL-6 axis resulting in improved survival in early stage multiple myeloma. Am. J. Hematol. 2016, 91, 571–574. [Google Scholar] [CrossRef] [PubMed]
- Ron, A.N.; Voronov, E. Immunotherapeutic approaches of IL-1 neutralization in the tumor microenvironment. J. Leukoc. Biol. 2017, 102, 293–306. [Google Scholar]
- Rodig, S.J.; Meraz, M.A.; White, J.M.; Lampe, P.A.; Riley, J.K.; Arthur, C.D.; Schreiber, R.D. Disruption of the Jak1 gene demonstrates obligatory and nonredundant roles of the Jaks in cytokine-induced biologic responses. Cell 1998, 93, 373–383. [Google Scholar] [CrossRef]
- Hu, X.; Li, J.; Fu, M.; Zhao, X.; Wang, W. The JAK/STAT signaling pathway: From bench to clinic. Sig. Transduct. Target. Ther. 2021, 6, 402. [Google Scholar] [CrossRef]
- Darnell, J.E., Jr. STATs and gene regulation. Science 1997, 277, 1630–1635. [Google Scholar] [CrossRef]
- Levy, D.E.; Darnell, J.E., Jr. Stats: Transcriptional control and biological impact. Nat. Rev. Mol. Cell Biol. 2002, 3, 651–662. [Google Scholar] [CrossRef]
- Siveen, K.S.; Sikka, S.; Surana, R.; Dai, X.; Zhang, J.; Kumar, A.P.; Tan, B.K.; Sethi, G.; Bishayee, A. Targeting the STAT3 signaling pathway in cancer: Role of synthetic and natural inhibitors. Biochim. Biophys. Acta 2014, 1845, 136–154. [Google Scholar] [CrossRef] [PubMed]
- Avalle, L.; Camporeale, A.; Camperi, A.; Poli, V. STAT3 in cancer: A double edged sword. Cytokine 2017, 98, 42–50. [Google Scholar] [CrossRef] [PubMed]
- Shao, F.; Pang, X.; Baeg, G.H. Targeting the JAK/STAT signaling pathway for breast cancer. Curr. Med. Hem. 2021, 28, 5137–5151. [Google Scholar] [CrossRef]
- Grivennikov, S.; Karin, E.; Terzic, J.; Mucida, D.; Yu, G.Y.; Vallabhapurapu, S.; Scheller, J.; Rose-John, S.; Cheroutre, H.; Eckmann, L.; et al. IL-6 and STAT3 are required for survival of intestinal epithelial cells and development of colitis-associated cancer. Cancer Cell 2009, 15, 103–113. [Google Scholar] [CrossRef] [PubMed]
- Jarnicki, A.; Putoczki, T.; Ernst, M. Stat3: Linking inflammation to epithelial cancer-more than a “gut” feeling? Cell Div. 2010, 5, 1–15. [Google Scholar] [CrossRef]
- Bromberg, J. Signal transducers and activators of transcription as regulators of growth, apoptosis and breast development. Breast Cancer Res. 2000, 2, 86–90. [Google Scholar] [CrossRef] [PubMed]
- Reed, J.C. Mechanisms of apoptosis avoidance in cancer. Curr. Opin. Oncol. 1999, 11, 68–75. [Google Scholar] [CrossRef]
- Zhao, S.H.; Zhao, F.; Zheng, J.Y.; Gao, L.F.; Zhao, X.J.; Cui, M.H. Knockdown of stat3 ex-pression by RNAi inhibits in vitro growth of human ovarian cancer. Radiol. Oncol. 2011, 45, 196–203. [Google Scholar] [CrossRef]
- DeMichele, A.; Gray, R.; Horn, M.; Chen, J.; Aplenc, R.; Vaughan, W.P.; Tallman, M.S. Host genetic variants in the interleukin-6 promoter predict poor outcome in patients with estrogen receptor-positive, node-positive breast cancer. Cancer Res. 2009, 69, 4184–4191. [Google Scholar] [CrossRef]
- Johnson, D.E.; O’Keefe, R.A.; Grandis, J.R. Targeting the IL-6/JAK/STAT3 signalling axis in cancer. Nat. Rev. Clin. Oncol. 2018, 15, 234–248. [Google Scholar] [CrossRef]
- Niu, G.; Wright, K.L.; Huang, M.; Song, L.; Haura, E.; Turkson, J.; Zhang, S.; Wang, T.; Sinibaldi, D.; Coppola, D.; et al. Constitutive STAT-3 activity up-regulates VEGF expression and tumor angiogenesis. Oncogene 2002, 21, 2000–2008. [Google Scholar] [CrossRef]
- Dechow, T.N.; Pedranzini, L.; Leitch, A.; Leslie, K.; Gerald, W.L.; Linkov, I.; Bromberg, J.F. Requirement of matrix metalloproteinase-9 for the transformation of human mammary epithelial cells by STAT-3-C. Proc. Natl. Acad. Sci. USA 2004, 101, 10602–10607. [Google Scholar] [CrossRef] [PubMed]
- Sun, R.; Liu, Z.; Qiu, B.; Chen, T.; Li, Z.; Zhang, X.; Xu, Y.; Zhang, Z. Annexin10 promotes extrahepatic cholangiocarcinoma metastasis by facilitating EMT via PLA2G4A/PGE2/STAT3 pathway. EBioMedicine 2019, 47, 142–155. [Google Scholar] [CrossRef] [PubMed]
- Ma, Y.; Zhao, S.; Shen, S.; Fang, S.; Ye, Z.; Shi, Z.; Hong, A. A novel recombinant slow-release TNF α-derived peptide effectively inhibits tumor growth and angiogensis. Sci. Rep. 2015, 5, 13595. [Google Scholar] [CrossRef] [PubMed]
- Lopes, J.E.; Fisher, J.L.; Flick, H.L.; Wang, C.; Sun, L.; Ernstoff, M.S.; Losey, H.C. ALKS 4230: A novel engineered IL-2 fusion protein with an improved cellular selectivity profile for cancer immunotherapy. J. Immunother. Cancer 2020, 8, 673. [Google Scholar] [CrossRef] [PubMed]
- Murer, P.; Neri, D. Antibody-cytokine fusion proteins: A novel class of biopharmaceuticals for the therapy of cancer and of chronic inflammation. N. Biotechnol. 2019, 52, 42–53. [Google Scholar] [CrossRef] [PubMed]
- Kampan, N.C.; Xiang, S.D.; McNally, O.M.; Stephens, A.N.; Quinn, M.A.; Plebanski, M. Immunotherapeutic interleukin-6 or interleukin-6 receptor blockade in cancer: Challenges and opportunities. Curr. Med. Chem. 2018, 25, 4785–4806. [Google Scholar] [CrossRef] [PubMed]
- Rodriguez-Barrueco, R.; Yu, J.; Saucedo-Cuevas, L.P.; Olivan, M.; Llobet-Navas, D.; Putcha, P.; Silva, J.M. Inhibition of the autocrine IL-6-JAK2-STAT3-calprotectin axis as targeted therapy for HR-/HER2+ breast cancers. Genes Dev. 2015, 29, 1631–1648. [Google Scholar] [CrossRef]
- Xue, C.; Yao, Q.; Gu, X.; Shi, Q.; Yuan, X.; Chu, Q.; Li, L. Evolving cognition of the JAK-STAT signaling pathway: Autoimmune disorders and cancer. Sig. Transduct. Target. Ther. 2023, 8, 204. [Google Scholar] [CrossRef]
- Chikuma, S.; Kanamori, M.; Mise-Omata, S.; Yoshimura, A. Suppressors of cytokine signaling: Potential im-mune checkpoint molecules for cancer immunotherapy. Cancer Sci. 2017, 108, 574–580. [Google Scholar] [CrossRef]
- Gu, Y.; Mohammad, I.S.; Liu, Z. Overview of the STAT-3 signaling pathway in cancer and the development of specific inhibitors (Review). Oncol. Lett. 2020, 19, 2585–2594. [Google Scholar] [CrossRef]
- Yang, J.; Wang, L.; Guan, X.; Qin, J.J. Inhibiting STAT3 signaling pathway by natural products for cancer prevention and therapy: In vitro and in vivo activity and mechanisms of action. Pharm. Res. 2022, 182, 106357. [Google Scholar] [CrossRef]
- Todaro, G.J.; De Larco, J.E.; Fryling, C.; Johnson, P.A.; Sporn, M.B. Transforming Growth Factors (TGFs): Properties and Possible Mechanisms of Action. J. Supramol. Struct. Cel. Biochem. 1981, 15, 287–301. [Google Scholar] [CrossRef] [PubMed]
- Morikawa, M.; Derynck, R.; Miyazono, K. TGF-β and the TGF-β Family: Context-Dependent Roles in Cell and Tissue Physiology. Cold Spring Harb. Perspect. Biol. 2016, 8, a021873. [Google Scholar] [CrossRef] [PubMed]
- Poniatowski, Ł.A.; Wojdasiewicz, P.; Gasik, R.; Szukiewicz, D. Transforming growth factor Beta family: Insight into the role of growth factors in regulation of fracture healing biology and potential clinical applications. Mediat. Inflamm. 2015, 2015, 137823. [Google Scholar] [CrossRef] [PubMed]
- Wendt, M.K.; Tian, M.; Schiemann, W.P. Deconstructing the mechanisms and consequences of TGF-beta-induced EMT during cancer progression. Cell Tissue Res. 2012, 347, 85–101. [Google Scholar] [CrossRef]
- Zhang, Y.; Alexander, P.B.; Wang, X.F. TGF-β Family Signaling in the Control of Cell Proliferation and Survival. Cold Spring Harb. Perspect. Biol. 2017, 9, a022145. [Google Scholar] [CrossRef]
- Santibañez, J.F.; Quintanilla, M.; Bernabeu, C. TGF-β/TGF-β Receptor System and its Role in Physiological and Pathological Conditions. Clin. Sci. 2011, 121, 233–251. [Google Scholar] [CrossRef] [PubMed]
- Huang, T.; David, L.; Mendoza, V.; Yang, Y.; Villarreal, M.; De, K.; Sun, L.; Fang, X.; López-Casillas, F.; Wrana, J.L.; et al. TGF-β signalling is mediated by two autonomously functioning TβRI:TβRII pairs. EMBO J. 2011, 30, 1263–1276. [Google Scholar] [CrossRef]
- Heldin, C.H.; Moustakas, A. Signaling Receptors for TGF-β Family Members. Cold Spring Harb. Perspect. Biol. 2016, 8, a022053. [Google Scholar] [CrossRef]
- Wrana, J.L.; Attisano, L. The Smad Pathway. Cytokine Growth Factor. Rev. 2000, 11, 5–13. [Google Scholar] [CrossRef]
- Xie, F.; Zhang, Z.; Van Dam, H.; Zhang, L.; Zhou, F. Regulation of TGF-β Superfamily Signaling by SMAD Mono-Ubiquitination. Cells 2014, 3, 981–993. [Google Scholar] [CrossRef]
- Macias, M.J.; Martin-Malpartida, P.; Massagué, J. Structural Determinants of Smad Function in TGF-β Signaling. Trends Biochem. Sci. 2015, 40, 296–308. [Google Scholar] [CrossRef] [PubMed]
- Ahmed, S.; Bradshaw, A.D.; Gera, S.; Dewan, M.Z.; Xu, R. The TGF-β/Smad4 Signaling Pathway in Pancreatic Carcinogenesis and its Clinical Significance. J. Clin. Med. 2017, 6, 5. [Google Scholar] [CrossRef] [PubMed]
- Massague, J. TGF beta in cancer. Cell 2008, 134, 215–230. [Google Scholar] [CrossRef] [PubMed]
- Vander Ark, A.; Cao, J.; Li, X. TGF-β receptors: In and beyond TGF-β signaling. Cell. Signal. 2018, 52, 112–120. [Google Scholar] [CrossRef] [PubMed]
- Principe, D.R.; Doll, J.A.; Bauer, J.; Jung, B.; Munshi, H.G.; Bartholin, L.; Grippo, P.J. TGF-beta: Duality of function between tumor prevention and carcinogenesis. J. Natl. Cancer Inst. 2014, 106, djt369. [Google Scholar] [CrossRef]
- Mukherjee, P.; Winter, S.L.; Alexandrow, M.G. Cell Cycle Arrest by Transforming Growth Factor Beta1 Near G1/S Is Mediated by Acute Abrogation of Prereplication Complex Activation Involving an Rb-MCM Interaction. Mol. Cel. Biol. 2010, 30, 845–856. [Google Scholar] [CrossRef]
- Samanta, D.; Datta, P.K. Alterations in the Smad Pathway in Human Cancers. Front. Biosci. 2012, 17, 1281–1293. [Google Scholar] [CrossRef]
- Vidakovic, A.; Rueda, O.M.; Vervoort, S.J.; Sati Batra, A.; Goldgraben, M.A.; Uribe-Lewis, S.; Caldas, C. Context-Specific Effects of TGF-Β/smad3 in Cancer Are Modulated by the Epigenome. Cell Rep. 2015, 13, 2480–2490. [Google Scholar] [CrossRef]
- Monteleone, I.; Pallone, F.; Monteleone, G. Th17-related cytokines: New players in the control of chronic intestinal inflammation. BMC Med. 2011, 9, 122. [Google Scholar] [CrossRef]
- Marie, J.C.; Letterio, J.J.; Gavin, M.; Rudensky, A.Y. TGF-beta1 maintains suppressor function and Foxp3 expression in CD4+CD25+ regulatory T cells. J. Exp. Med. 2005, 201, 1061–1067. [Google Scholar] [CrossRef]
- Stanilova, S.; Stanilov, N.; Julianov, A.; Manolova, I.; Miteva, L. Transforming growth factor-β1 gene promoter-509C/T polymorphism in association with expression affects colorectal cancer development and depends on gender. PLoS ONE 2018, 13, e0201775. [Google Scholar] [CrossRef] [PubMed]
- Melzer, C.; von der Ohe, J.; Otterbein, H.; Ungefroren, H.; Hass, R. Changes in uPA, PAI-1, and TGF-β Production during Breast Cancer Cell Interaction with Human Mesenchymal Stroma/Stem-like Cells (MSC). Int. J. Mol. Sci. 2019, 20, 2630. [Google Scholar] [CrossRef] [PubMed]
- Baba, A.B.; Rah, B.; Bhat, G.R.; Mushtaq, I.; Parveen, S.; Hassan, R.; Hameed Zargar, M.; Afroze, D. Transforming Growth Factor-Beta (TGF-β) Signaling in Cancer-A Betrayal Within. Front. Pharmacol. 2022, 13, 791272. [Google Scholar] [CrossRef] [PubMed]
- Tsubakihara, Y.; Moustakas, A. Epithelial-Mesenchymal Transition and Metastasis under the Control of Transforming Growth Factor β. Int. J. Mol. Sci. 2018, 19, 3672. [Google Scholar] [CrossRef] [PubMed]
- Betts, G.; Jones, E.; Junaid, S.; El-Shanawany, T.; Scurr, M.; Mizen, P.; Kumar, M.; Jones, S.; Rees, B.; Williams, G.; et al. Suppression of tumour-specific CD4+ T cells by regulatory T cells is associated with progression of human colorectal cancer. Gut 2012, 61, 1163–1171. [Google Scholar] [CrossRef] [PubMed]
- Miteva, L.D.; Stanilov, N.S.; Cirovski, G.M.; Stanilova, S.A. Upregulation of Treg-related genes in addition with IL6 showed the significant role for the distant metastasis in colorectal cancer. Cancer Microenviron. 2017, 10, 69–76. [Google Scholar] [CrossRef] [PubMed]
- Morris, J.C.; Tan, A.R.; Olencki, T.E.; Shapiro, G.I.; Dezube, B.J.; Reiss, M.; Lawrence, D.P. Phase I Study of GC1008 (Fresolimumab): A Human Anti-transforming Growth Factor-Beta (TGFβ) Monoclonal Antibody in Patients with Advanced Malignant Melanoma or Renal Cell Carcinoma. PLoS ONE 2014, 9, e90353. [Google Scholar] [CrossRef]
- Zhong, Z.; Carroll, K.D.; Policarpio, D.; Osborn, C.; Gregory, M.; Bassi, R.; Wu, Y. Anti-transforming Growth Factor Beta Receptor II Antibody Has Therapeutic Efficacy against Primary Tumor Growth and Metastasis through Multieffects on Cancer, Stroma, and Immune Cells. Clin. Cancer Res. 2010, 16, 1191–1205. [Google Scholar] [CrossRef]
- Yap, T.A.; Lakhani, N.J.; Araujo, D.V.; Rodon Ahnert, J.; Chandana, S.R.; Sharma, M.; Siu, L.L. AVID200, First-In-Class TGF-Beta 1 and 3 Selective and Potent Inhibitor: Safety and Biomarker Results of a Phase I Monotherapy Dose-Escalation Study in Patients with Advanced Solid Tumors. J. Clin. Oncol. 2020, 38 (Suppl. 15), 3587. [Google Scholar] [CrossRef]
- Oettle, H.; Hilbig, A.; Seufferlein, T.; Tsianakas, A.; Luger, T.; Schmid, R.M.; Wichert, G.; Endlicher, E.; Garbe, C.; Kaehler, K.K.; et al. Phase I/II Study with Trabedersen (AP 12009) Monotherapy for the Treatment of Patients with Advanced Pancreatic Cancer, Malignant Melanoma, and Colorectal Carcinoma. Jco 2011, 29, 2513. [Google Scholar] [CrossRef]
- Shi, X.; Yang, J.; Deng, S.; Xu, H.; Wu, D.; Zeng, Q.; Wang, S.; Hu, T.; Wu, F.; Zhou, H. TGF-β signaling in the tumor metabolic microenvironment and targeted therapies. J. Hematol. Oncol. 2022, 15, 135. [Google Scholar] [CrossRef] [PubMed]
- Velikova, T.V.; Miteva, L.; Stanilov, N.; Spassova, Z.; Stanilova, S.A. Interleukin-6 compared to the other Th17/Treg related cytokines in inflammatory bowel disease and colorectal cancer. World J. Gastroenterol. 2020, 26, 1912–1925. [Google Scholar] [CrossRef]
- Dufour, A.M.; Alvarez, M.; Russo, B.; Chizzolini, C. Interleukin-6 and Type-I Collagen Production by Systemic Sclerosis Fibroblasts Are Differentially Regulated by interleukin-17A in the Presence of Transforming Growth Factor-Beta 1. Front. Immunol. 2018, 9, 1865. [Google Scholar] [CrossRef] [PubMed]
- Ribatti, D.; Tamma, R.; Annese, T. Epithelial-Mesenchymal Transition in Cancer: A Historical Overview. Transl. Oncol. 2020, 13, 100773. [Google Scholar] [CrossRef] [PubMed]
- Jing, Y.; Han, Z.; Zhang, S.; Liu, Y.; Wei, L. Epithelial-Mesenchymal Transition in tumor microenvironment. Cell Biosci. 2011, 1, 29. [Google Scholar] [CrossRef] [PubMed]
- Ray, I.; Michael, A.; Meira, L.B.; Ellis, P.E. The Role of Cytokines in Epithelial-Mesenchymal Transition in Gynaecological Cancers: A Systematic Review. Cells 2023, 12, 416. [Google Scholar] [CrossRef] [PubMed]
- Zuo, X.; Jiao, J.; Wang, J.; Ning, Y.J.; Liu, N.; Qin, B.W. Mechanism by which Human Papillomavirus 16 Infection Interferes with Progression and Immune Evasion of Cervical Cancer by Acting on Th9 Cytokines. Indian J. Pharm. Sci. 2022, 84, 40–47. [Google Scholar] [CrossRef]
- Zhang, J.; Zhu, D.; Lv, Q.; Yi, Y.; Li, F.; Zhang, W. The key role of astrocyte elevated gene-1 in CCR6-induced EMT in cervical cancer. Tumour. Biol. 2015, 36, 9763–9767. [Google Scholar] [CrossRef]
- Chen, H.-Y.; Chiang, Y.-F.; Huang, J.-S.; Huang, T.-C.; Shih, Y.-H.; Wang, K.-L.; Ali, M.; Hong, Y.-H.; Shieh, T.-M.; Hsia, S.-M. Isoliquiritigenin Reverses Epithelial-Mesenchymal Transition Through Modulation of the TGF-β/Smad Signaling Pathway in Endometrial Cancer. Cancers 2021, 13, 1236. [Google Scholar] [CrossRef]
- Wang, L.; Zhuang, T.; Li, F.; Wei, W. Fluorene-9-bisphenol inhibits epithelial-mesenchymal transition of human endometrial cancer Ishikawa cells by repressing TGF-β signaling pathway. Environ. Sci. Pollut. Res. Int. 2019, 26, 27407–27413. [Google Scholar] [CrossRef]
- Buyuk, B.; Jin, S.; Ye, K. Epithelial-to-Mesenchymal Transition Signaling Pathways Responsible for Breast Cancer Metastasis. Cell. Mol. Bioeng. 2021, 15, 1–13. [Google Scholar] [CrossRef] [PubMed]
- Martins-Lima, C.; Chianese, U.; Benedetti, R.; Altucci, L.; Jerónimo, C.; Correia, M.P. Tumor microenvironment and epithelial-mesenchymal transition in bladder cancer: Cytokines in the game? Front. Mol. Biosci. 2023, 9, 1070383. [Google Scholar] [CrossRef] [PubMed]
- Berraondo, P.; Sanmamed, M.F.; Ochoa, M.C.; Etxeberria, I.; Aznar, M.A.; Pérez-Gracia, J.L.; Melero, I. Cytokines in clinical cancer immunotherapy. Br. J. Cancer 2019, 120, 6–15. [Google Scholar] [CrossRef]
- Conlon, K.C.; Miljkovic, M.D.; Waldmann, T.A. Cytokines in the Treatment of Cancer. J. Interferon Cytokine Res. Off. J. Int. Soc. Interferon Cytokine Res. 2019, 39, 6–21. [Google Scholar] [CrossRef] [PubMed]
- Chmielewski, M.; Abken, H. TRUCKs: The fourth generation of CARs. Expert Opin. Biol. Ther. 2015, 15, 1145–1154. [Google Scholar] [CrossRef]
- Rallis, K.S.; Corrigan, A.E.; Dadah, H.; George, A.M.; Keshwara, S.M.; Sideris, M.; Szabados, B. Cytokine-based Cancer Immunotherapy: Challenges and Opportunities for IL-10. Anticancer. Res. 2021, 41, 3247–3252. [Google Scholar] [CrossRef] [PubMed]
- Chen, D.S.; Mellman, I. Elements of cancer immunity and the cancer-immune set point. Nature 2017, 541, 321–330. [Google Scholar] [CrossRef]
- Heimberger, A.B.; Tripathi, S.; Platanias, L.C. Targeting Cytokines and Their Pathways for the Treatment of Cancer. Cancers 2023, 15, 5224. [Google Scholar] [CrossRef]
- Song, Y.; Liu, Y.; Teo, H.Y.; Liu, H. Targeting Cytokine Signals to Enhance γδT Cell-Based Cancer Immunotherapy. Front. Immunol. 2022, 13, 914839. [Google Scholar] [CrossRef]
- Bonati, L.; Tang, L. Cytokine engineering for targeted cancer immunotherapy. Curr. Opin. Chem. Biol. 2021, 62, 43–52. [Google Scholar] [CrossRef]
- Fu, Y.; Tang, R.; Zhao, X. Engineering cytokines for cancer immunotherapy: A systematic review. Front. Immunol. 2023, 14, 1218082. [Google Scholar] [CrossRef] [PubMed]
- Atallah-Yunes, S.A.; Robertson, M.J. Cytokine Based Immunotherapy for Cancer and Lymphoma: Biology, Challenges and Future Perspectives. Front. Immunol. 2022, 13, 872010. [Google Scholar] [CrossRef]
- Soler, M.F.; Abaurrea, A.; Azcoaga, P.; Araujo, A.M.; Caffarel, M.M. New perspectives in cancer immunotherapy: Targeting IL-6 cytokine family. J. Immunother. Cancer 2023, 11, e007530. [Google Scholar] [CrossRef]
- Guo, Y.; Xu, F.; Lu, T.; Duan, Z.; Zhang, Z. Interleukin-6 signaling pathway in targeted therapy for cancer. Cancer Treat. Rev. 2012, 38, 904–910. [Google Scholar] [CrossRef]

{kind=link}
| Target Cytokine/ Cytokine Receptor | Intervention, Combinations | Cancer Type | Mechanisms of Immunotherapy | Clinical Trials |
|---|---|---|---|---|
| TNF-α | Nivolumab + ipilimumab + certolizumab or infliximab | Advanced melanoma | Blockade of activation-immune cell death of tumor-infiltrating lymphocytes | NCT03293784 |
| TGF-β | Galunisertib + nivolumab; Galunisertib + durvalumab; Fresolimumab + radiotherapy; M7824 (dual block of TGF-β and PD-L1) | Advanced Hepatocellular Carcinoma, Metastatic Pancreatic Cancer, Early Stage Non-small Cell Lung Cancer, Previously Treated Advanced Adenocarcinoma of the Pancreas | Inflammation of immune-excluded tumours | NCT02423343 NCT02734160 NCT02581787 NCT03451773 |
| LIF | AZD0171, durvalumab, gemcitabine, nab-paclitaxel; AZD0171, durvalumab, oleclumab, monalizumab, MEDI5752, dato-DXd, pemetrexed, carboplatin, cisplatin, paclitaxel | Locally advanced or metastatic solid tumors NSCLC | Blocking LIF/LIFR to inhibit tumor growth and DNA damage responses | NCT04999969 NCT05061550 |
| CSF-1 | Cabiralizumab + nivolumab; Pexidartinib + durvalumab; Pexidartinib + durvalumab or tremelimumab; Pexidartinib + pembrolizumab | Non-small Cell Lung Cancer or Renal Cell Carcinoma, Metastatic/Advanced Pancreatic or Colorectal Cancers, Advanced Solid Tumors, Advanced Melanoma and Other Solid Tumors | Suppression of tumor-associated myeloid cell | NCT03502330 NCT02777710 NCT02718911 NCT02452424 |
| IL-2 | Nivolumab; pembrolzumab, ipilimumab, enoblituzumab; TASO-001; NKTR-214 + atezolizumab; NKTR-214 + nivolumab; NKTR-214 + nivolumab + ipilimumab; Cergutuzumab amunaleukin + atezolizumab; RO6874281 + trastuzumab or cetuximab; RO6874281 + atezolizumab; RO6874281 + atezolizumab + bevacizumab | Melanoma, RCC, ovarian cancer, solid tumors | Expansion of NK and T lymphocytes | NCT03991130 NCT02964078 NCTO4562129 NCT04630769 NCT04862767 NCT03138889 NCT02983045 NCT03282344 NCT03435640 NCT02350673 NCT02627274 NCT03386721 NCT03063762 |
| IL-6R | ERY974, tocilizumab, atezolizumab, bevacizumab, Ipilimumab, nivolumab, tiragolumab, conventional surgery, radiation, TPST-1120, RO7247669, Sarilumab, relatlimab, carboplatin, gemcitabine, linagliptin, ipatasertib, sacituzumab govitecan, evolocumab, etrumadenant, enfortumab vedotin, niraparib, Hu5F9-G4, cisplatin, gemcitabine, Obinutuzumab, cibisatamab, Capecitabine, SGN-LIV1A, gemcitabine + carboplatin or eribulin, selicrelumab, nab-Paclitaxel, cobimetinib, RO6958688, docetaxel, CPI-444, pemetrexed | HCC; Advanced Melanoma; NSCLC; Urothelial Carcinoma, Bladder Cancer; Recurrent Glioblastoma, Diffuse Astrocytoma; Advanced Liver Cancers; Pancreatic Cancer; Colorectal Cancer; Prostate Cancer; Head and Neck Squamous Cell Carcinoma; Triple Negative Breast Cancer. | Blocking IL-6 receptor to suppress cancer invasiveness and metastasizing | NCT05022927 NCT04940299 NCT04729959 NCT04691817 NCT04524871 NCT04258150 NCT03999749 NCT03869190 NCT03866239 NCT03821246 NCT03708224 NCT03424005 NCT03337698 NCT05428007 |
| IL-6 | Siltuximab, spartalizumab | Metastatic pancreatic adenocarcinoma | Blocking IL-6 receptor to suppress cancer invasiveness and metastasizing | NCT04191421 |
| IL-8 | mAb anti-IL-8 + nivolumab | Advanced Cancers | Suppression of tumor-associated myeloid cell | NCT03400332 |
| IL-10 | Pegilodecakin + FOLFOX | Metastatic Pancreatic Cancer | Blockade of activation-immune cell death of tumor-infiltrating lymphocytes | NCT02923921 |
| IL-12 | DC/tumor vaccine; cetuximab; pembrolizumab; Transduced TILs; Electroporated plasmid; Electroporated plasmid + pembrolizumab | Glioma, head and neck cancer, solid tumors | Promotion of NK cells and Th1 CD4+ and CD8+ lymphocytes | NCT04388033 NCT01468896 NCT03030378 NCT01236573 NCT01579318 NCT00323206 NCT01502293 NCT02345330 NCT02493361 NCT03132675 |
| IL-15 | Mogamulizumab, nivolumab, ipilimumab, avelumab; ALT-803; IL-15 + T or NK cells; IL15 + alemtuzumab; IL15 + rituximab | CTCL, solid tumors, PTCL | Expansion of NK and T lymphocytes | NCT04185220 NCT03388632 NCT03905135 NCT02989844 NCT01875601 NCT02465957 NCT01385423 NCT1369888 NCT02689453 NCT02384954 |
| IL-27 | SRF388, pembrolizumab SRF388, atezolizumab, bevacizumab, placebo | Advanced ccRCC or HCC, or anti-PD(L)1 relapsed/refractory advanced NSCLC HCC | IL-27 possesses direct antitumor activity and indirectly suppresses tumor development by influencing the tumor microenvironment | NCT04374877 NCT05359861 |
| CCL2, CCL3, CCL5 | CCR2/CCR5 inhibitor + nivolumab or chemotherapy | Advanced Solid Tumors | Suppression of tumor-associated myeloid cell | NCT03184870 |
| VEGF | Bevacizumab + atezolizumab | untreated metastatic renal cell carcinoma | Suppression of tumor-associated myeloid cell | NCT01984242 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Stanilov, N.; Velikova, T.; Stanilova, S. Navigating the Cytokine Seas: Targeting Cytokine Signaling Pathways in Cancer Therapy. Int. J. Mol. Sci. 2024, 25, 1009. https://doi.org/10.3390/ijms25021009
Stanilov N, Velikova T, Stanilova S. Navigating the Cytokine Seas: Targeting Cytokine Signaling Pathways in Cancer Therapy. International Journal of Molecular Sciences. 2024; 25(2):1009. https://doi.org/10.3390/ijms25021009
Chicago/Turabian StyleStanilov, Noyko, Tsvetelina Velikova, and Spaska Stanilova. 2024. "Navigating the Cytokine Seas: Targeting Cytokine Signaling Pathways in Cancer Therapy" International Journal of Molecular Sciences 25, no. 2: 1009. https://doi.org/10.3390/ijms25021009
APA StyleStanilov, N., Velikova, T., & Stanilova, S. (2024). Navigating the Cytokine Seas: Targeting Cytokine Signaling Pathways in Cancer Therapy. International Journal of Molecular Sciences, 25(2), 1009. https://doi.org/10.3390/ijms25021009