
From Gut to Brain: Uncovering Potential Serum Biomarkers Connecting Inflammatory Bowel Diseases to Neurodegenerative Diseases

 , , ,
, , , 
{kind=link}
{kind=link}
Abstract
1. Introduction
1.1. Inflammatory Bowel Diseases
- There is an overactive immune response in the gut, leading to inflammation and damage to the intestinal tissue. This immune response involves various immune cells, including T cells, B cells, and macrophages.
- The imbalance of gut bacteria is present in IBDs, leading to inflammation and immune activation.
- Genetic factors have been associated with an increased risk of developing IBDs. These genetic factors can affect the immune system, barrier functions of the gut, and the body’s response to inflammation.
- Environmental factors, such as diet, smoking, and infections, can play a role in triggering or exacerbating IBDs. These factors can interact with the gut microbiota and the immune system, contributing to inflammation.
1.2. Gut–Brain Axis
1.3. Neurodegenerative Diseases
1.4. Gut–Brain Axis, Intestinal Microbiome, and Neurodegenerative Diseases
2. Aim
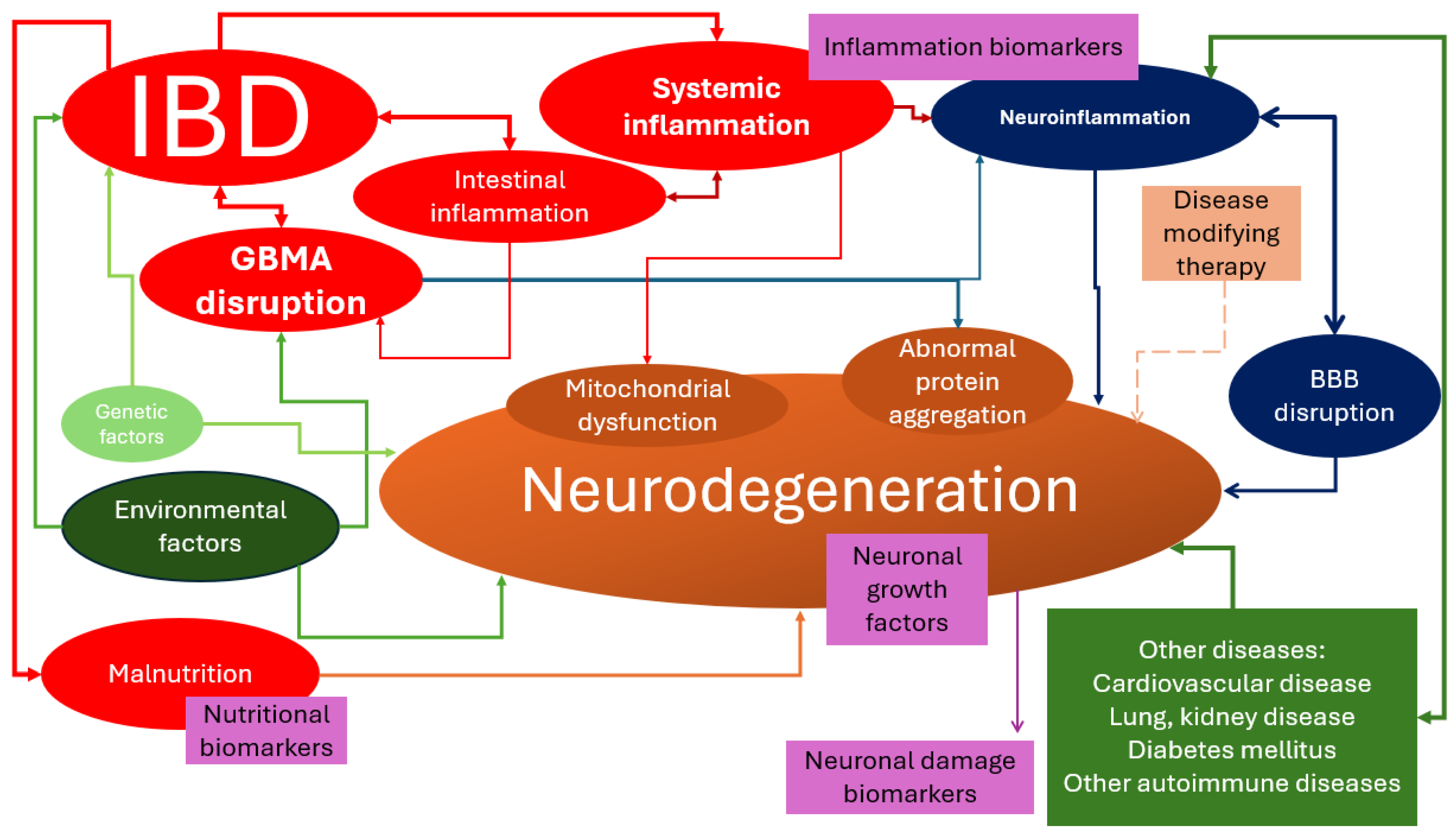
3. Is Neurodegeneration an Extraintestinal Manifestation of IBDs?
- Shared genetic loci were identified between IBDs and neurodegenerative diseases. For example, the LRRK2 gene has been implicated in both PD and CD [47].
- Chronic inflammation is a hallmark of both IBDs and neurodegenerative diseases. Inflammatory mediators and immune cells can cross the BBB and contribute to neuroinflammation, neuronal damage, and neurodegeneration [43,47,48,49]. Increased oxidative stress, resulting from an imbalance between reactive oxygen species (ROS) production and antioxidant defense mechanisms, is implicated in the pathogenesis of both IBDs and neurodegenerative diseases [43]. The integrity of the BBB might be compromised in IBDs, allowing the entry of inflammatory mediators and immune cells into the CNS [43]. Some of the inflammatory blood biomarkers associated with neurodegeneration include cytokines (such as IL-6, IL-8, and TNF-α), chemokines (such as MCP-1), growth factors (such as VEGF), and immune-related proteins (such as ICAM-1 and VCAM-1). These biomarkers have been found to be altered in the serum of patients with AD and MCI [50,51,52]. Elevated levels of C-reactive protein (CRP) have been associated with an increased risk of AD and PD [53,54]. Increased levels of IL-6 have been observed in AD, PD, and other neurodegenerative diseases [54]. TNF-alpha has been implicated in the pathogenesis of AD, PD, and other neurodegenerative diseases [54]. Markers of microglial activation, such as CD68 and HLA-DR, have been associated with neurodegenerative diseases [55]. Elevated levels of IL-1β have been found in the brains of individuals with AD and PD [54].
- Abnormal protein aggregation is a characteristic feature of many neurodegenerative diseases, such as the accumulation of alpha-synuclein in PD, tau and amyloid-beta in AD, or TDP-43 in frontotemporal lobe degeneration [43,56,57]. It has been suggested that similar protein aggregation processes may occur in the gut of IBD patients, contributing to neurodegenerative pathology [58]. For example, abnormal aggregation of alpha-synuclein has been detected in the enteric neurons of some patients with UC [59].
- Environmental factors, including gut microbiota composition, diet, and exposure to toxins, may influence the development and progression of both IBDs and neurodegenerative diseases. These factors can interact with genetic susceptibility to modulate disease risk [47]. Nutritional deficits that have been studied in relation to the development of neurodegenerative diseases include deficiencies of essential nutrients such as long-chain polyunsaturated fatty acids, vitamins (such as vitamin E), and mineral elements [60]. Malnutrition and low body mass index (BMI) have also been associated with a higher risk of dementia and mortality [61]. Additionally, chronic over-nutrition and metabolic disorders, such as diabetes, hypertension, dyslipidemia, and atherosclerosis, have been linked to neurodegenerative diseases [62]. Deficiencies of vitamins such as B1, B12, and vitamin D have also been associated with an increased risk of neurodegenerative diseases [63,64,65].
- Mitochondrial dysfunction, characterized by impaired energy production and increased production of ROS, has been observed in both IBDs and neurodegenerative diseases. The dysfunction of mitochondria can lead to neuronal cell death and contribute to the progression of neurodegeneration [43].
- The disruption of the neurotrophic factors, which play an important role in the survival, growth, and maintenance of neurons, might exist in both IBDs and neurodegenerative diseases, leading to neuronal dysfunction and degeneration [43].
- GBA disruption, especially of the intestinal barrier functions, and immune responses in IBDs can lead to systemic inflammation and neuroinflammation, which are associated with neurodegenerative diseases [47,48]. The disruption of the gut–brain axis can affect various brain structures and functions. The insular cortex is involved in processing and integrating sensory information from the gut, such as pain and visceral sensations, and it might be affected by GBA disruption. The cingulate cortex plays a role in regulating emotions and pain perception. Dysfunction in the GBA can influence the functioning of the cingulate cortex, leading to emotional and pain-related disturbances. The hypothalamus is a brain structure involved in regulating various physiological processes, including appetite, metabolism, stress response, and sleep. The disruption of the GBA can also affect hypothalamic function [66,67]. Dysfunction in the GBA has been associated with cognitive impairments, such as difficulties in memory, attention, and executive function. It might also lead to mood disorders, such as anxiety or depression [68]. An increase in neuroinflammation has also been observed in GBA disruption, which might also lead to neurodegeneration [66]. Due to GBA’s effect on the production and regulation of neurotransmitters, such as serotonin and dopamine, its disruption might imbalance these neurotransmitters [68].
- Adverse effects of therapies such as high-dose corticosteroids have been associated with cognitive impairment, emotional disturbance, and behavioral changes in children and adolescents [69]. However, the use of anti-TNF-α inhibitors is associated with a lower risk of developing AD and PD in patients with IBDs as they may reduce inflammation and modulate the gut microbiome [42,70].
4. Nutritional Deficiency Biomarkers
4.1. Vitamin D3
4.2. Vitamin B12
4.3. Vitamin B9
4.4. Homocysteine
4.5. Vitamin B6
4.6. Vitamin B1
5. Neuronal Damage Biomarkers
5.1. Neuron-Specific Enolase
5.2. Neurofilament Light Chain
5.3. S100 Proteins
6. Neurotrophic Factors
6.1. Brain-Derived Neurotrophic Factor
6.2. Glial Cell-Derived Neurotrophic Factor
7. Inflammatory Biomarkers
7.1. C-Reactive Protein
7.2. Serum Amyloid
7.3. IL-6
7.4. LP2-Associated Phospholipase
7.5. Prostaglandin E2
7.6. IL-1β
7.7. TNF-α
7.8. Paraoxonases
8. Conclusions and Future Insights
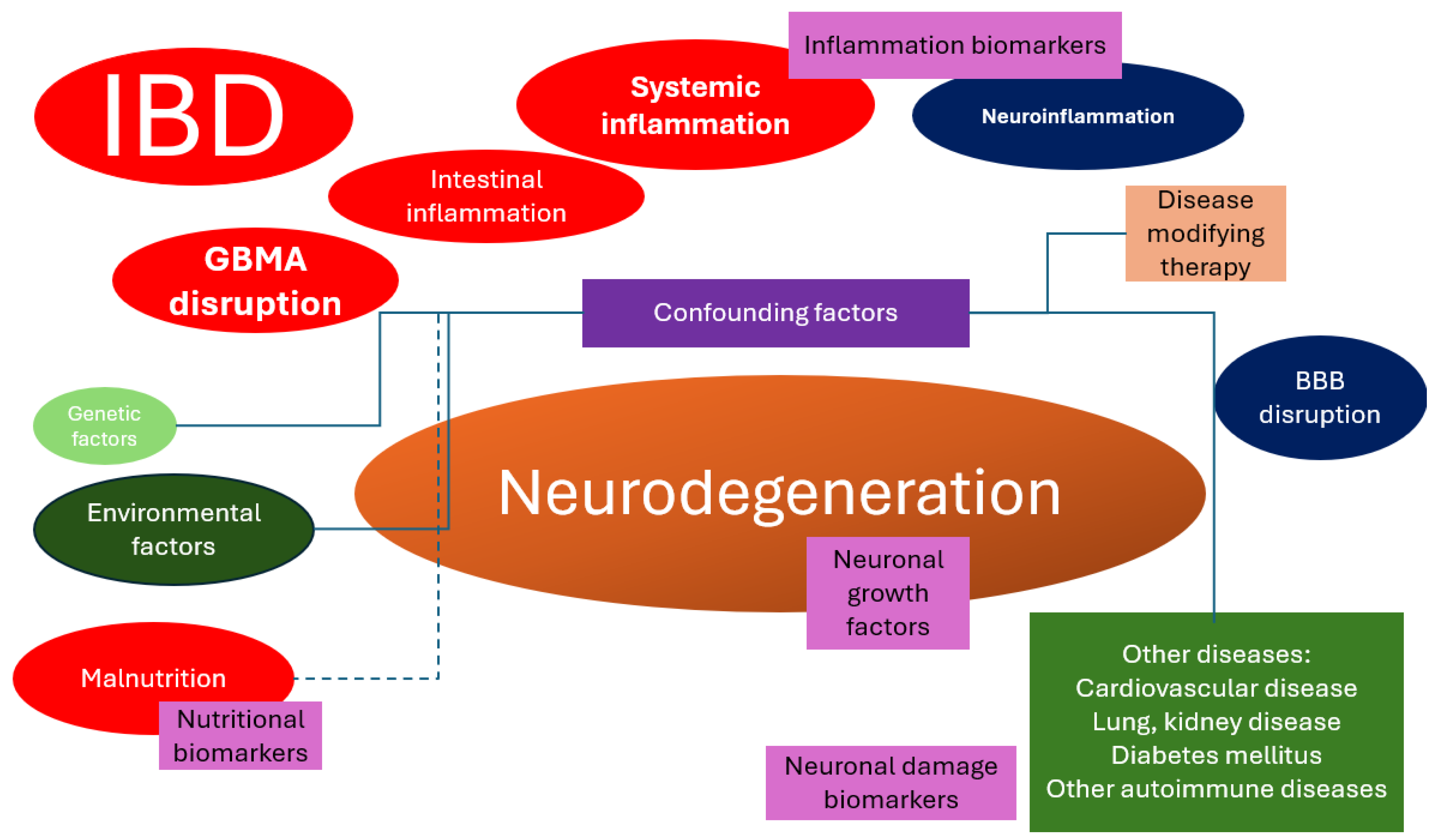
- Exploring the common pathways: (1) changes in the gut microbiome and metabolites that are secreted by the gut microbiome, which bypass the intestinal barrier, reach the bloodstream, and finally cross over the BBB and (2) systemic inflammation and how it affects the cerebral circulation. The important aspect of this is to identify whether the structural changes in the brain are the effects of IBDs or the cumulative effects of inflammation and comorbidities.
- Inflammatory pathway: research regarding correlations between the inflammatory biomarker levels and the development of neurodegenerative diseases.
- Therapeutic interventions regarding the effects of probiotics and whether they could prevent the development of neurodegenerative diseases. Interventional studies focusing on dietary modifications or supplementation to increase vitamin levels could provide valuable insights. However, patients with malabsorption issues may require parenteral supplementation. Additionally, strategies such as probiotics to enhance vitamin production in the gut microbiome could be explored.
- Neuroimaging: the serum levels of biomarkers should be correlated to the degree of cerebral atrophy or metabolism, but this requires prospective studies. This hypothesis, which is very expensive to explore, should be tested soon.
- Lifestyle factors associated with IBDs: different diet regimens could potentially influence the gut microbiome, thus influencing the development of neurodegenerative diseases. Different diets might also change the serum levels of vitamins, which have been linked to neurodegeneration.
- Genetic risk: patients diagnosed with IBDs should be questioned for the family history of neurodegenerative diseases to be included in prospective observational studies to test the hypothesis of whether there are common genetic factors between IBDs and neurodegenerative diseases.
- Medication effects: observational prospective studies must be conducted regarding the development of MCI and neurodegenerative diseases in patients with IBDs using different disease-modifying therapies.
Author Contributions
Funding
Conflicts of Interest
Abbreviations
References
- Maaser, C.; Sturm, A.; Vavricka, S.R.; Kucharzik, T.; Fiorino, G.; Annese, V.; Calabrese, E.; Baumgart, D.C.; Bettenworth, D.; Borralho Nunes, P.; et al. ECCO-ESGAR Guideline for Diagnostic Assessment in IBD Part 1: Initial diagnosis, monitoring of known IBD, detection of complications. J. Crohns Colitis. 2019, 13, 144–164. [Google Scholar] [CrossRef] [PubMed]
- Wang, R.; Li, Z.; Liu, S.; Zhang, D. Global, regional and national burden of inflammatory bowel disease in 204 countries and territories from 1990 to 2019: A systematic analysis based on the Global Burden of Disease Study 2019. BMJ Open 2023, 13, e065186. [Google Scholar] [CrossRef] [PubMed]
- Lee, S.H.; Kwon, J.E.; Cho, M.-L. Immunological pathogenesis of inflammatory bowel disease. Intest. Res. 2018, 16, 26–42. [Google Scholar] [CrossRef] [PubMed]
- Kim, D.H.; Cheon, J.H. Pathogenesis of Inflammatory Bowel Disease and Recent Advances in Biologic Therapies. Immune Netw. 2017, 17, 25–40. [Google Scholar] [CrossRef] [PubMed]
- Viscido, A.; Aratari, A.; Maccioni, F.; Signore, A.; Caprilli, R. Inflammatory bowel diseases: Clinical update of practical guidelines. Nucl. Med. Commun. 2005, 26, 649–655. [Google Scholar] [CrossRef] [PubMed]
- Bonaz, B.L.; Bernstein, C.N. Brain-gut interactions in inflammatory bowel disease. Gastroenterology 2013, 144, 36–49. [Google Scholar] [CrossRef]
- Gîlcă-Blanariu, G.E.; Șchiopu, C.G.; Ștefănescu, G.; Mihai, C.; Diaconescu, S.; Afrăsânie, V.A.; Lupu, V.V.; Lupu, A.; Boloș, A.; Ștefănescu, C. The Intertwining Roads between Psychological Distress and Gut Microbiota in Inflammatory Bowel Disease. Microorganisms 2023, 11, 2268. [Google Scholar] [CrossRef]
- Abautret-Daly, Á.; Dempsey, E.; Parra-Blanco, A.; Medina, C.; Harkin, A. Gut-brain actions underlying comorbid anxiety and depression associated with inflammatory bowel disease. Acta Neuropsychiatr. 2018, 30, 275–296. [Google Scholar] [CrossRef]
- Stolzer, I.; Scherer, E.; Süß, P.; Rothhammer, V.; Winner, B.; Neurath, M.F.; Günther, C. Impact of Microbiome–Brain Communication on Neuroinflammation and Neurodegeneration. Int. J. Mol. Sci. 2023, 24, 14925. [Google Scholar] [CrossRef]
- Banfi, D.; Moro, E.; Bosi, A.; Bistoletti, M.; Cerantola, S.; Crema, F.; Maggi, F.; Giron, M.C.; Giaroni, C.; Baj, A. Impact of Microbial Metabolites on Microbiota-Gut-Brain Axis in Inflammatory Bowel Disease. Int. J. Mol. Sci. 2021, 22, 1623. [Google Scholar] [CrossRef]
- Kovacs, G.G. Concepts and classification of neurodegenerative diseases. Handb. Clin. Neurol. 2017, 145, 301–307. [Google Scholar]
- Levin, M.C.; Douglas, J.N.; Meyers, L.; Lee, S.; Shin, Y.; Gardner, L.A. Neurodegeneration in multiple sclerosis involves multiple pathogenic mechanisms. Degener. Neurol. Neuromuscul. Dis. 2014, 4, 49–63. [Google Scholar] [CrossRef] [PubMed]
- Gao, H.-M.; Hong, J.-S. Why neurodegenerative diseases are progressive: Uncontrolled inflammation drives disease progression. Trends Immunol. 2008, 29, 357–365. [Google Scholar] [CrossRef] [PubMed]
- Cullinane, P.W.; Wrigley, S.; Bezerra Parmera, J.; Valerio, F.; Millner, T.O.; Shaw, K.; De Pablo-Fernandez, E.; Warner, T.T.; Jaunmuktane, Z. Pathology of neurodegenerative disease for the general neurologist. Pract. Neurol. 2023. [Google Scholar] [CrossRef] [PubMed]
- Choonara, Y.E.; Pillay, V.; Du Toit, L.C.; Modi, G.; Naidoo, D.; Ndesendo, V.M.K.; Sibambo, S.R. Trends in the molecular pathogenesis and clinical therapeutics of common neurodegenerative disorders. Int. J. Mol. Sci. 2009, 10, 2510–2557. [Google Scholar] [CrossRef]
- Taipa, R.; Pinho, J.; Melo-Pires, M. Clinico-pathological correlations of the most common neurodegenerative dementias. Front. Neurol. 2012, 3, 68. [Google Scholar] [CrossRef]
- Attems, J.; Jellinger, K. Neuropathological correlates of cerebral multimorbidity. Curr. Alzheimer Res. 2013, 10, 569–577. [Google Scholar] [CrossRef]
- Giannakopoulos, P.; Gold, G.; von Gunten, A.; Hof, P.R.; Bouras, C. Pathological substrates of cognitive decline in Alzheimer’s disease. Front. Neurol. Neurosci. 2009, 24, 20–29. [Google Scholar]
- Fereshtehnejad, S.-M.; Vosoughi, K.; Heydarpour, P.; Sepanlou, S.G.; Farzadfar, F.; Tehrani-Banihashemi, A.; Malekzadeh, R.; Sahraian, M.A.; Vollset, S.E.; Naghavi, M.; et al. Burden of neurodegenerative diseases in the Eastern Mediterranean Region, 1990–2016: Findings from the Global Burden of Disease Study 2016. Eur. J. Neurol. 2019, 26, 1252–1265. [Google Scholar] [CrossRef]
- Lopes, M.A.; Bottino, C.M.C. Prevalence of dementia in several regions of the world: Analysis of epidemiologic studies from 1994 to 2000. Arq. Neuropsiquiatr. 2002, 60, 61–69. [Google Scholar] [CrossRef]
- Gauthier, S.; Reisberg, B.; Zaudig, M.; Petersen, R.C.; Ritchie, K.; Broich, K.; Belleville, S.; Brodaty, H.; Bennett, D.; Chertkow, H.; et al. Mild cognitive impairment. Lancet 2006, 367, 1262–1270. [Google Scholar] [CrossRef] [PubMed]
- Overton, M.; Pihlsgård, M.; Elmståhl, S. Prevalence and Incidence of Mild Cognitive Impairment across Subtypes, Age, and Sex. Dement. Geriatr. Cogn. Disord. 2019, 47, 219–232. [Google Scholar] [CrossRef] [PubMed]
- Nie, H.; Xu, Y.; Liu, B.; Zhang, Y.; Lei, T.; Hui, X.; Zhang, L.; Wu, Y. The prevalence of mild cognitive impairment about elderly population in China: A meta-analysis. Int. J. Geriatr. Psychiatry. 2011, 26, 558–563. [Google Scholar] [CrossRef] [PubMed]
- Hu, C.; Yu, D.; Sun, X.; Zhang, M.; Wang, L.; Qin, H. The prevalence and progression of mild cognitive impairment among clinic and community populations: A systematic review and meta-analysis. Int. Psychogeriatr. 2017, 29, 1595–1608. [Google Scholar] [CrossRef] [PubMed]
- Shen, X.N.; Lu, Y.; Tan, C.T.Y.; Liu, L.Y.; Yu, J.T.; Feng, L.; Larbi, A. Identification of inflammatory and vascular markers associated with mild cognitive impairment. Aging 2019, 11, 2403–2419. [Google Scholar] [CrossRef] [PubMed]
- Lauriola, M.; D’Onofrio, G.; Ciccone, F.; Germano, C.; Cascavilla, L.; Paris, F.; Greco, A. Relationship of Homocysteine Plasma Levels with Mild Cognitive Impairment, Alzheimer’s Disease, Vascular Dementia, Psychobehavioral, and Functional Complications. J. Alzheimer’s Dis. 2021, 82, 235–248. [Google Scholar] [CrossRef]
- Zhou, X.; Wang, Q.; An, P.; Du, Y.; Zhao, J.; Song, A.; Huang, G. Relationship between folate, vitamin B12, homocysteine, transaminase and mild cognitive impairment in China: A case-control study. Int. J. Food Sci. Nutr. 2020, 71, 315–324. [Google Scholar] [CrossRef]
- Chen, Y.X.; Liang, N.; Li, X.L.; Yang, S.H.; Wang, Y.P.; Shi, N.N. Diagnosis and Treatment for Mild Cognitive Impairment: A Systematic Review of Clinical Practice Guidelines and Consensus Statements. Front. Neurol. 2021, 12, 719849. [Google Scholar] [CrossRef] [PubMed]
- Ticinesi, A.; Tana, C.; Nouvenne, A.; Prati, B.; Lauretani, F.; Meschi, T. Gut microbiota, cognitive frailty and dementia in older individuals: A systematic review. Clin. Interv. Aging 2018, 13, 1497–1511. [Google Scholar] [CrossRef]
- Kandpal, M.; Indari, O.; Baral, B.; Jakhmola, S.; Tiwari, D.; Bhandari, V.; Pandey, R.K.; Bala, K.; Sonawane, A.; Jha, H.C. Dysbiosis of Gut Microbiota from the Perspective of the Gut-Brain Axis: Role in the Provocation of Neurological Disorders. Metabolites 2022, 12, 1064. [Google Scholar] [CrossRef]
- Ghezzi, L.; Cantoni, C.; Rotondo, E.; Galimberti, D. The Gut Microbiome-Brain Crosstalk in Neurodegenerative Diseases. Biomedicines 2022, 10, 1486. [Google Scholar] [CrossRef] [PubMed]
- D’Antongiovanni, V.; Pellegrini, C.; Antonioli, L.; Ippolito, C.; Segnani, C.; Benvenuti, L.; D’Amati, A.; Errede, M.; Virgintino, D.; Fornai, M.; et al. Enteric Glia and Brain Astroglia: Complex Communication in Health and Disease along the Gut-Brain Axis. Neurosci. Rev. J. Bringing Neurobiol. Neurol. Psychiatry 2023, 10738584231163460. [Google Scholar] [CrossRef] [PubMed]
- Franco, C.D.; Sagar, R.S.; Bokhari, S.F.H. From Microbes to Memories: Challenges and Future Perspectives Regarding the Gut-Brain Axis for Improved Cognitive Health in Alzheimer’s. Cureus 2024, 16, e52795. [Google Scholar] [CrossRef] [PubMed]
- Zheng, Y.; Bonfili, L.; Wei, T.; Eleuteri, A.M. Understanding the Gut-Brain Axis and Its Therapeutic Implications for Neurodegenerative Disorders. Nutrients 2023, 15, 4631. [Google Scholar] [CrossRef] [PubMed]
- Loh, J.S.; Mak, W.Q.; Tan, L.K.S.; Ng, C.X.; Chan, H.H.; Yeow, S.H.; Foo, J.B.; Ong, Y.S.; How, C.W.; Khaw, K.Y. Microbiota-gut-brain axis and its therapeutic applications in neurodegenerative diseases. Signal Transduct. Target. Ther. 2024, 9, 37. [Google Scholar]
- Lyon, L. “All disease begins in the gut”: Was Hippocrates right? Brain 2018, 141, e20. [Google Scholar] [CrossRef] [PubMed]
- Giraldo, G. Counting to 100: A First Look at Cuba’s National Centenarian Study. MEDICC Rev. 2009, 11, 17–19. [Google Scholar]
- Kinsella, K.G. Changes in life expectancy 1900–1990. Am. J. Clin. Nutr. 1992, 55 (Suppl. S6), 1196S–1202S. [Google Scholar] [CrossRef] [PubMed]
- Caselli, G.; Egidi, V. A new insight into morbidity and mortality transition in Italy. Genus 1991, 47, 1–29. [Google Scholar]
- Kim, G.H.; Lee, Y.C.; Kim, T.J.; Kim, E.R.; Hong, S.N.; Chang, D.K.; Kim, Y.H. Risk of Neurodegenerative Diseases in Patients with Inflammatory Bowel Disease: A Nationwide Population-based Cohort Study. J. Crohns Colitis. 2022, 16, 436–443. [Google Scholar] [CrossRef]
- Zeng, R.; Wang, J.; Zheng, C.; Jiang, R.; Tong, S.; Wu, H.; Zhuo, Z.; Yang, Q.; Leung, F.W.; Sha, W.; et al. Lack of Causal Associations of Inflammatory Bowel Disease with Parkinson’s Disease and Other Neurodegenerative Disorders. Mov. Disord. 2023, 38, 1082–1088. [Google Scholar] [CrossRef]
- Zamani, M.; Ebrahimtabar, F.; Alizadeh-Tabari, S.; Kasner, S.E.; Elkind, M.S.V.; Ananthakrishnan, A.N.; Choden, T.; Rubin, D.T.; Malekzadeh, R. Risk of Common Neurological Disorders in Adult Patients with Inflammatory Bowel Disease: A Systematic Review and Meta-analysis. Inflamm. Bowel Dis. 2024. [Google Scholar] [CrossRef]
- Hey, G.E.; Vedam-Mai, V.; Beke, M.; Amaris, M.; Ramirez-Zamora, A. The Interface between Inflammatory Bowel Disease, Neuroinflammation, and Neurological Disorders. Semin. Neurol. 2023, 43, 572–582. [Google Scholar] [CrossRef]
- Wang, D.; Zhang, X.; Du, H. Inflammatory bowel disease: A potential pathogenic factor of Alzheimer’s disease. Prog. Neuropsychopharmacol. Biol. Psychiatry 2022, 119, 110610. [Google Scholar] [CrossRef] [PubMed]
- Knuesel, T.; Mohajeri, M.H. The Role of the Gut Microbiota in the Development and Progression of Major Depressive and Bipolar Disorder. Nutrients 2021, 14, 37. [Google Scholar] [CrossRef]
- Marchesi, V.T. An alternative interpretation of the amyloid Aβ hypothesis with regard to the pathogenesis of Alzheimer’s disease. Proc. Natl. Acad. Sci. USA 2005, 102, 9093–9098. [Google Scholar] [CrossRef]
- Lee, H.-S.; Lobbestael, E.; Vermeire, S.; Sabino, J.; Cleynen, I. Inflammatory bowel disease and Parkinson’s disease: Common pathophysiological links. Gut 2021, 70, 408–417. [Google Scholar] [CrossRef] [PubMed]
- El-Hakim, Y.; Bake, S.; Mani, K.K.; Sohrabji, F. Impact of intestinal disorders on central and peripheral nervous system diseases. Neurobiol. Dis. 2022, 165, 105627. [Google Scholar] [CrossRef] [PubMed]
- Zong, J.; Yang, Y.; Wang, H.; Zhang, H.; Yang, X.; Yang, X. The two-directional prospective association between inflammatory bowel disease and neurodegenerative disorders: A systematic review and meta-analysis based on longitudinal studies. Front. Immunol. 2024, 15, 1325908. [Google Scholar] [CrossRef]
- Abbatecola, A.M.; Giuliani, A.; Biscetti, L.; Scisciola, L.; Battista, P.; Barbieri, M.; Sabbatinelli, J.; Olivieri, F. Circulating biomarkers of inflammaging and Alzheimer’s disease to track age-related trajectories of dementia: Can we develop a clinically relevant composite combination? Ageing Res. Rev. 2024, 96, 102257. [Google Scholar] [CrossRef]
- Delaby, C.; Gabelle, A.; Blum, D.; Schraen-Maschke, S.; Moulinier, A.; Boulanghien, J.; Séverac, D.; Buée, L.; Rème, T.; Lehmann, S. Central Nervous System and Peripheral Inflammatory Processes in Alzheimer’s Disease: Biomarker Profiling Approach. Front. Neurol. 2015, 6, 181. [Google Scholar] [CrossRef] [PubMed]
- Gambino, C.M.; Sasso, B.L.; Bivona, G.; Agnello, L.; Ciaccio, M. Aging and Neuroinflammatory Disorders: New Biomarkers and Therapeutic Targets. Curr. Pharm. Des. 2019, 25, 4168–4174. [Google Scholar] [CrossRef] [PubMed]
- Andican, G.; Konukoglu, D.; Bozluolcay, M.; Bayülkem, K.; Firtiına, S.; Burcak, G. Plasma oxidative and inflammatory markers in patients with idiopathic Parkinson’s disease. Acta Neurol. Belg. 2012, 112, 155–159. [Google Scholar] [CrossRef] [PubMed]
- Amin, J.; Erskine, D.; Donaghy, P.C.; Surendranathan, A.; Swann, P.; Kunicki, A.P.; Boche, D.; Holmes, C.; McKeith, I.G.; O’Brien, J.T.; et al. Inflammation in dementia with Lewy bodies. Neurobiol. Dis. 2022, 168, 105698. [Google Scholar] [CrossRef] [PubMed]
- Lee, S.Y.H.; Yates, N.J.; Tye, S.J. Inflammatory Mechanisms in Parkinson’s Disease: From Pathogenesis to Targeted Therapies. Neuroscientist 2022, 28, 485–506. [Google Scholar] [CrossRef] [PubMed]
- Takalo, M.; Salminen, A.; Soininen, H.; Hiltunen, M.; Haapasalo, A. Protein aggregation and degradation mechanisms in neurodegenerative diseases. Am. J. Neurodegener. Dis. 2013, 2, 1–14. [Google Scholar]
- Moda, F.; Ciullini, A.; Dellarole, I.L.; Lombardo, A.; Campanella, N.; Bufano, G.; Cazzaniga, F.A.; Giaccone, G. Secondary Protein Aggregates in Neurodegenerative Diseases: Almost the Rule Rather than the Exception. Front. Biosci. 2023, 28, 255. [Google Scholar] [CrossRef] [PubMed]
- Prigent, A.; Chapelet, G.; De Guilhem de Lataillade, A.; Oullier, T.; Durieu, E.; Bourreille, A.; Duchalais, E.; Hardonnière, K.; Neunlist, M.; Noble, W.; et al. Tau accumulates in Crohn’s disease gut. FASEB J. 2020, 34, 9285–9296. [Google Scholar] [CrossRef] [PubMed]
- Gibo, N.; Hamaguchi, T.; Miki, Y.; Yamamura, T.; Nakaguro, M.; Ito, M.; Nakamuara, M.; Kawashima, H.; Hirayama, M.; Hirooka, Y.; et al. Examination of Abnormal Alpha-synuclein Aggregates in the Enteric Neural Plexus in Patients with Ulcerative Colitis. J. Gastrointestin Liver Dis. 2022, 31, 290–300. [Google Scholar] [CrossRef]
- Businaro, R.; Vauzour, D.; Sarris, J.; Münch, G.; Gyengesi, E.; Brogelli, L.; Zuzarte, P. Therapeutic Opportunities for Food Supplements in Neurodegenerative Disease and Depression. Front. Nutr. 2021, 8, 669846. [Google Scholar] [CrossRef]
- Bianchi, V.E.; Herrera, P.F.; Laura, R. Effect of nutrition on neurodegenerative diseases. A systematic review. Nutr. Neurosci. 2021, 24, 810–834. [Google Scholar] [CrossRef] [PubMed]
- Pandareesh, M.D.; Kandikattu, H.K.; Razack, S.; Amruta, N.; Choudhari, R.; Vikram, A.; Doddapattar, P. Nutrition and Nutraceuticals in Neuroinflammatory and Brain Metabolic Stress: Implications for Neurodegenerative Disorders. CNS Neurol. Disord. Drug Targets 2018, 17, 680–688. [Google Scholar] [CrossRef]
- Liu, D.; Ke, Z.; Luo, J. Thiamine Deficiency and Neurodegeneration: The Interplay Among Oxidative Stress, Endoplasmic Reticulum Stress, and Autophagy. Mol. Neurobiol. 2017, 54, 5440–5448. [Google Scholar] [CrossRef]
- Luthra, N.S.; Marcus, A.H.; Hills, N.K.; Christine, C.W. Vitamin B12 measurements across neurodegenerative disorders. J. Clin. Mov. Disord. 2020, 7, 3–6. [Google Scholar] [CrossRef]
- Kumar, R.R.; Singh, L.; Thakur, A.; Singh, S.; Kumar, B. Role of Vitamins in Neurodegenerative Diseases: A Review. CNS Neurol. Disord. Drug Targets 2022, 21, 766–773. [Google Scholar] [CrossRef]
- Sundman, M.H.; Chen, N.-K.; Subbian, V.; Chou, Y.-H. The bidirectional gut-brain-microbiota axis as a potential nexus between traumatic brain injury, inflammation, and disease. Brain Behav. Immun. 2017, 66, 31–44. [Google Scholar] [CrossRef] [PubMed]
- Kharrazian, D. Traumatic Brain Injury and the Effect on the Brain-Gut Axis. Altern. Ther. Health Med. 2015, 21 (Suppl. S3), 28–32. [Google Scholar] [PubMed]
- Gupta, S.; Dinesh, S.; Sharma, S. Bridging the Mind and Gut: Uncovering the Intricacies of Neurotransmitters, Neuropeptides, and their Influence on Neuropsychiatric Disorders. Cent. Nerv. Syst. Agents Med. Chem. 2024, 24, 2–21. [Google Scholar] [CrossRef]
- Mrakotsky, C.; Forbes, P.W.; Bernstein, J.H.; Grand, R.J.; Bousvaros, A.; Szigethy, E.; Waber, D.P. Acute cognitive and behavioral effects of systemic corticosteroids in children treated for inflammatory bowel disease. J. Int. Neuropsychol. Soc. 2013, 19, 96–109. [Google Scholar] [CrossRef]
- Aggarwal, M.; Alkhayyat, M.; Abou Saleh, M.; Sarmini, M.T.; Singh, A.; Garg, R.; Garg, P.; Mansoor, E.; Padival, R.; Cohen, B.L. Alzheimer Disease Occurs More Frequently In Patients With Inflammatory Bowel Disease: Insight From a Nationwide Study. J. Clin. Gastroenterol. 2023, 57, 501–507. [Google Scholar] [CrossRef]
- Hébuterne, X.; Filippi, J.; Al-Jaouni, R.; Schneider, S. Nutritional consequences and nutrition therapy in Crohn’s disease. Gastroenterol. Clin. Biol. 2009, 33 (Suppl. S3), S235–S244. [Google Scholar] [CrossRef] [PubMed]
- Mijac, D.D.; Janković, G.L.J.; Jorga, J.; Krstić, M.N. Nutritional status in patients with active inflammatory bowel disease: Prevalence of malnutrition and methods for routine nutritional assessment. Eur. J. Intern. Med. 2010, 21, 315–319. [Google Scholar] [CrossRef] [PubMed]
- Jabłońska, B.; Mrowiec, S. Nutritional Status and Its Detection in Patients with Inflammatory Bowel Diseases. Nutrients 2023, 15, 1991. [Google Scholar] [CrossRef]
- Viganò, C.; Palermo, A.; Mulinacci, G.; Pirola, L.; Losco, A.; Meucci, G.; Saibeni, S.; Pastorelli, L.; Amato, A.; Gatti, M.; et al. Prevalence of Disease-Related Malnutrition and Micronutrients Deficit in Patients with Inflammatory Bowel Disease: A Multicentric Cross-Sectional Study by the GSMII (Inflammatory Bowel Disease Study Group). Inflamm. Bowel Dis. 2023, izad146. [Google Scholar] [CrossRef]
- Vagianos, K.; Bector, S.; McConnell, J.; Bernstein, C.N. Nutrition assessment of patients with inflammatory bowel disease. JPEN J. Parenter. Enter. Nutr. 2007, 31, 311–319. [Google Scholar] [CrossRef] [PubMed]
- Hwang, C.; Ross, V.; Mahadevan, U. Micronutrient deficiencies in inflammatory bowel disease: From A to zinc. Inflamm. Bowel Dis. 2012, 18, 1961–1981. [Google Scholar] [CrossRef] [PubMed]
- Del Pinto, R.; Ferri, C.; Cominelli, F. Vitamin D Axis in Inflammatory Bowel Diseases: Role, Current Uses and Future Perspectives. Int. J. Mol. Sci. 2017, 18, 2360. [Google Scholar] [CrossRef] [PubMed]
- Fakhoury, H.M.A.; Kvietys, P.R.; AlKattan, W.; Al Anouti, F.; Elahi, M.A.; Karras, S.N.; Grant, W.B. Vitamin D and intestinal homeostasis: Barrier, microbiota, and immune modulation. J. Steroid Biochem. Mol. Biol. 2020, 200, 105663. [Google Scholar] [CrossRef]
- Bartosik-Psujek, H.; Tabarkiewicz, J.; Pocinska, K.; Stelmasiak, Z.; Rolinski, J. Immunomodulatory effects of vitamin D on monocyte-derived dendritic cells in multiple sclerosis. Mult. Scler. 2010, 16, 1513–1516. [Google Scholar] [CrossRef]
- Jiao, K.-P.; Li, S.-M.; Lv, W.-Y.; Jv, M.-L.; He, H.-Y. Vitamin D3 repressed astrocyte activation following lipopolysaccharide stimulation in vitro and in neonatal rats. Neuroreport 2017, 28, 492–497. [Google Scholar] [CrossRef]
- Correale, J.; Ysrraelit, M.C.; Gaitán, M.I. Gender differences in 1,25 dihydroxyvitamin D3 immunomodulatory effects in multiple sclerosis patients and healthy subjects. J. Immunol. 2010, 185, 4948–4958. [Google Scholar] [CrossRef] [PubMed]
- Janjusevic, M.; Gagno, G.; Fluca, A.L.; Padoan, L.; Beltrami, A.P.; Sinagra, G.; Moretti, R.; Aleksova, A. The peculiar role of vitamin D in the pathophysiology of cardiovascular and neurodegenerative diseases. Life Sci. 2022, 289, 120193. [Google Scholar] [CrossRef] [PubMed]
- Wang, W.; Li, Y.; Meng, X. Vitamin D and neurodegenerative diseases. Heliyon 2023, 9, e12877. [Google Scholar] [CrossRef]
- Kasatkina, L.A.; Tarasenko, A.S.; Krupko, O.O.; Kuchmerovska, T.M.; Lisakovska, O.O.; Trikash, I.O. Vitamin D deficiency induces the excitation/inhibition brain imbalance and the proinflammatory shift. Int. J. Biochem. Cell Biol. 2020, 119, 105665. [Google Scholar] [CrossRef]
- Skv, M.; Abraham, S.M.; Eshwari, O.; Golla, K.; Jhelum, P.; Maity, S.; Komal, P. Tremendous Fidelity of Vitamin D3 in Age-related Neurological Disorders. Mol. Neurobiol. 2024, 1–28. [Google Scholar] [CrossRef]
- Boontanrart, M.; Hall, S.D.; Spanier, J.A.; Hayes, C.E.; Olson, J.K. Vitamin D3 alters microglia immune activation by an IL-10 dependent SOCS3 mechanism. J. Neuroimmunol. 2016, 292, 126–136. [Google Scholar] [CrossRef] [PubMed]
- Uberti, F.; Trotta, F.; Cavalli, R.; Galla, R.; Caldera, F.; Ferrari, S.; Mulè, S.; Brovero, A.; Molinari, C.; Pagliaro, P.; et al. Enhancing Vitamin D3 Efficacy: Insights from Complexation with Cyclodextrin Nanosponges and Its Impact on Gut-Brain Axes in Physiology and IBS Syndrome. Int. J. Mol. Sci. 2024, 25, 2189. [Google Scholar] [CrossRef] [PubMed]
- Lai, R.-H.; Hsu, Y.-Y.; Shie, F.-S.; Huang, C.-C.; Chen, M.-H.; Juang, J.-L. Non-genomic rewiring of vitamin D receptor to p53 as a key to Alzheimer’s disease. Aging Cell 2021, 20, e13509. [Google Scholar] [CrossRef] [PubMed]
- Caviezel, D.; Maissen, S.; Niess, J.H.; Kiss, C.; Hruz, P. High Prevalence of Vitamin D Deficiency among Patients with Inflammatory Bowel Disease. Inflamm. Intest. Dis. 2018, 2, 200–210. [Google Scholar] [CrossRef]
- Frigstad, S.O.; Høivik, M.; Jahnsen, J.; Dahl, S.R.; Cvancarova, M.; Grimstad, T.; Berset, I.P.; Huppertz-Hauss, G.; Hovde, Ø.; Torp, R.; et al. Vitamin D deficiency in inflammatory bowel disease: Prevalence and predictors in a Norwegian outpatient population. Scand. J. Gastroenterol. 2017, 52, 100–106. [Google Scholar] [CrossRef]
- Zhao, J.; Wang, Y.; Gu, Q.; Du, Z.; Chen, W. The association between serum vitamin D and inflammatory bowel disease. Medicine 2019, 98, e15233. [Google Scholar] [CrossRef] [PubMed]
- Briani, C.; Dalla Torre, C.; Citton, V.; Manara, R.; Pompanin, S.; Binotto, G.; Adami, F. Cobalamin deficiency: Clinical picture and radiological findings. Nutrients 2013, 5, 4521–4539. [Google Scholar] [CrossRef] [PubMed]
- Tamura, J.; Kubota, K.; Murakami, H.; Sawamura, M.; Matsushima, T.; Tamura, T.; Saitoh, T.; Kurabayshi, H.; Naruse, T. Immunomodulation by vitamin B12: Augmentation of CD8+ T lymphocytes and natural killer (NK) cell activity in vitamin B12-deficient patients by methyl-B12 treatment. Clin. Exp. Immunol. 1999, 116, 28–32. [Google Scholar] [CrossRef] [PubMed]
- Calderón-Ospina, C.A.; Nava-Mesa, M.O. B Vitamins in the nervous system: Current knowledge of the biochemical modes of action and synergies of thiamine, pyridoxine, and cobalamin. CNS Neurosci. Ther. 2020, 26, 5–13. [Google Scholar] [CrossRef] [PubMed]
- Weir, D.G.; Scott, J.M. The biochemical basis of the neuropathy in cobalamin deficiency. Baillieres Clin. Haematol. 1995, 8, 479–497. [Google Scholar] [CrossRef] [PubMed]
- Miller, A.; Korem, M.; Almog, R.; Galboiz, Y. Vitamin B12, demyelination, remyelination and repair in multiple sclerosis. J. Neurol. Sci. 2005, 233, 93–97. [Google Scholar] [CrossRef]
- Serin, H.M.; Arslan, E.A. Neurological symptoms of vitamin B12 deficiency: Analysis of pediatric patients. Acta Clin. Croat. 2019, 58, 295–302. [Google Scholar] [CrossRef] [PubMed]
- Bermejo, F.; Algaba, A.; Guerra, I.; Chaparro, M.; De-La-Poza, G.; Valer, P.; Piqueras, B.; Bermejo, A.; García-Alonso, J.; Pérez, M.J.; et al. Should we monitor vitamin B12 and folate levels in Crohn’s disease patients? Scand. J. Gastroenterol. 2013, 48, 1272–1277. [Google Scholar] [CrossRef] [PubMed]
- Shaw, S.; Jayatilleke, E.; Meyers, S.; Colman, N.; Herzlich, B.; Herbert, V. The ileum is the major site of absorption of vitamin B12 analogues. Am. J. Gastroenterol. 1989, 84, 22–26. [Google Scholar]
- Filipsson, S.; Hultén, L.; Lindstedt, G. Malabsorption of fat and vitamin B12 before and after intestinal resection for Crohn’s disease. Scand. J. Gastroenterol. 1978, 13, 529–536. [Google Scholar] [CrossRef]
- Lambert, D.; Benhayoun, S.; Adjalla, C.; Gelot, M.A.; Renkes, P.; Felden, F.; Gérard, P.; Belleville, F.; Gaucher, P.; Guéant, J.-L.; et al. Crohn’s disease and vitamin B12 metabolism. Dig. Dis. Sci. 1996, 41, 1417–1422. [Google Scholar] [CrossRef] [PubMed]
- Moore, E.; Mander, A.; Ames, D.; Carne, R.; Sanders, K.; Watters, D. Cognitive impairment and vitamin B12: A review. Int. Psychogeriatr. 2012, 24, 541–556. [Google Scholar] [CrossRef] [PubMed]
- Wu, Y.; Zhao, Z.; Yang, N.; Xin, C.; Li, Z.; Xu, J.; Ma, B.; Lim, K.-L.; Li, L.; Wu, Q.; et al. Vitamin B12 Ameliorates the Pathological Phenotypes of Multiple Parkinson’s Disease Models by Alleviating Oxidative Stress. Antioxidants 2023, 12, 153. [Google Scholar] [CrossRef]
- Zhou, L.; Bai, X.; Huang, J.; Tan, Y.; Yang, Q. Vitamin B12 supplementation improves cognitive function in middle aged and elderly patients with cognitive impairment. Nutr. Hosp. 2023, 40, 724–731. [Google Scholar] [PubMed]
- Eastley, R.; Wilcock, G.K.; Bucks, R.S. Vitamin B12 deficiency in dementia and cognitive impairment: The effects of treatment on neuropsychological function. Int. J. Geriatr. Psychiatry 2000, 15, 226–233. [Google Scholar] [CrossRef]
- Gröber, U.; Kisters, K.; Schmidt, J. Neuroenhancement with vitamin B12-underestimated neurological significance. Nutrients 2013, 5, 5031–5045. [Google Scholar] [CrossRef] [PubMed]
- Robinson, N.; Grabowski, P.; Rehman, I. Alzheimer’s disease pathogenesis: Is there a role for folate? Mech. Ageing Dev. 2018, 174, 86–94. [Google Scholar] [CrossRef] [PubMed]
- Vyas, S.P.; Goswami, R. Calcitriol and Retinoic acid antagonize each other to suppress the production of IL-9 by Th9 cells. J. Nutr. Biochem. 2021, 96, 108788. [Google Scholar] [CrossRef] [PubMed]
- Cui, G.; Li, S.; Ye, H.; Yang, Y.; Huang, Q.; Chu, Y.; Shi, Z.; Zhang, X. Are neurodegenerative diseases associated with an increased risk of inflammatory bowel disease? A two-sample Mendelian randomization study. Front. Immunol. 2022, 13, 956005. [Google Scholar] [CrossRef]
- Xu, P.; Pang, D.; Zhou, J.; Li, S.; Chen, D.; Yu, B. Behavioral changes and brain epigenetic alterations induced by maternal deficiencies of B vitamins in a mouse model. Psychopharmacology 2021, 238, 1213–1222. [Google Scholar] [CrossRef]
- Saibeni, S.; Cattaneo, M.; Vecchi, M.; Zighetti, M.L.; Lecchi, A.; Lombardi, R.; Meucci, G.; Spina, L.; De Franchis, R. Low vitamin B(6) plasma levels, a risk factor for thrombosis, in inflammatory bowel disease: Role of inflammation and correlation with acute phase reactants. Am. J. Gastroenterol. 2003, 98, 112–117. [Google Scholar] [CrossRef] [PubMed]
- Pan, Y.; Liu, Y.; Guo, H.; Jabir, M.S.; Liu, X.; Cui, W.; Li, D. Associations between Folate and Vitamin B12 Levels and Inflammatory Bowel Disease: A Meta-Analysis. Nutrients 2017, 9, 382. [Google Scholar] [CrossRef]
- Levin, J.; Bötzel, K.; Giese, A.; Vogeser, M.; Lorenzl, S. Elevated levels of methylmalonate and homocysteine in Parkinson’s disease, progressive supranuclear palsy and amyotrophic lateral sclerosis. Dement. Geriatr. Cogn. Disord. 2010, 29, 553–559. [Google Scholar] [CrossRef] [PubMed]
- Hoffmann, J.; Busse, S.; von Hoff, F.; Borucki, K.; Frodl, T.; Busse, M. Association Between Homocysteine and Vitamin Levels in Demented Patients. J. Alzheimers Dis. 2021, 81, 1781–1792. [Google Scholar] [CrossRef] [PubMed]
- Wang, M.; Liu, Z.; Sun, W.; Yuan, Y.; Jiao, B.; Zhang, X.; Shen, L.; Jiang, H.; Xia, K.; Tang, B.; et al. Association Between Vitamins and Amyotrophic Lateral Sclerosis: A Center-Based Survey in Mainland China. Front. Neurol. 2020, 11, 488. [Google Scholar] [CrossRef] [PubMed]
- Guilland, J.-C.; Favier, A.; Potier de Courcy, G.; Galan, P.; Hercberg, S. Hyperhomocysteinemia: An independent risk factor or a simple marker of vascular disease? 1. Basic data. Pathol. Biol. 2003, 51, 101–110. [Google Scholar] [CrossRef]
- Kim, H.J.; Kim, M.K.; Kim, J.U.; Ha, H.Y.; Choi, B.Y. Major determinants of serum homocysteine concentrations in a Korean population. J. Korean Med. Sci. 2010, 25, 509–516. [Google Scholar] [CrossRef] [PubMed]
- Clarke, R.; Armitage, J. Vitamin supplements and cardiovascular risk: Review of the randomized trials of homocysteine-lowering vitamin supplements. Semin. Thromb. Hemost. 2000, 26, 341–348. [Google Scholar] [CrossRef] [PubMed]
- Dawson, H.; Collins, G.; Pyle, R.; Deep-Dixit, V.; Taub, D.D. The immunoregulatory effects of homocysteine and its intermediates on T-lymphocyte function. Mech. Ageing Dev. 2004, 125, 107–110. [Google Scholar] [CrossRef]
- Ientile, R.; Curro’, M.; Ferlazzo, N.; Condello, S.; Caccamo, D.; Pisani, F. Homocysteine, vitamin determinants and neurological diseases. Front. Biosci. 2010, 2, 359–372. [Google Scholar] [CrossRef]
- Rozycka, A.; Jagodzinski, P.P.; Kozubski, W.; Lianeri, M.; Dorszewska, J. Homocysteine Level and Mechanisms of Injury in Parkinson’s Disease as Related to MTHFR, MTR, and MTHFD1 Genes Polymorphisms and L-Dopa Treatment. Curr. Genomics. 2013, 14, 534–542. [Google Scholar] [CrossRef] [PubMed]
- Rajagopalan, P.; Hua, X.; Toga, A.W.; Jack, C.R.J.; Weiner, M.W.; Thompson, P.M. Homocysteine effects on brain volumes mapped in 732 elderly individuals. Neuroreport 2011, 22, 391–395. [Google Scholar] [CrossRef] [PubMed]
- Hooshmand, B.; Polvikoski, T.; Kivipelto, M.; Tanskanen, M.; Myllykangas, L.; Erkinjuntti, T.; Mäkelä, M.; Oinas, M.; Paetau, A.; Scheltens, P.; et al. Plasma homocysteine, Alzheimer and cerebrovascular pathology: A population-based autopsy study. Brain 2013, 136 Pt 9, 2707–2716. [Google Scholar] [CrossRef]
- Shea, T.B.; Lyons-Weiler, J.; Rogers, E. Homocysteine, folate deprivation and Alzheimer neuropathology. J. Alzheimers Dis. 2002, 4, 261–267. [Google Scholar] [CrossRef] [PubMed]
- Danese, S.; Sgambato, A.; Papa, A.; Scaldaferri, F.; Pola, R.; Sans, M.; Lovecchio, M.; Gasbarrini, G.; Cittadini, A.; Gasbarrini, A. Homocysteine triggers mucosal microvascular activation in inflammatory bowel disease. Am. J. Gastroenterol. 2005, 100, 886–895. [Google Scholar] [CrossRef]
- Morgenstern, I.; Raijmakers, M.T.M.; Peters, W.H.M.; Hoensch, H.; Kirch, W. Homocysteine, cysteine, and glutathione in human colonic mucosa: Elevated levels of homocysteine in patients with inflammatory bowel disease. Dig. Dis. Sci. 2003, 48, 2083–2090. [Google Scholar] [CrossRef]
- Koutroubakis, I.E.; Dilaveraki, E.; Vlachonikolis, I.G.; Vardas, E.; Vrentzos, G.; Ganotakis, E.; Mouzas, I.; Gravanis, A.; Emmanouel, D.; Kouroumalis, E. Hyperhomocysteinemia in Greek patients with inflammatory bowel disease. Dig. Dis. Sci. 2000, 45, 2347–2351. [Google Scholar] [CrossRef]
- Roblin, X.; Germain, E.; Phelip, J.M.; Ducros, V.; Pofelski, J.; Heluwaert, F.; Oltean, P.; Faucheron, J.L.; Bonaz, B. Factors associated with hyperhomocysteinemia in inflammatory bowel disease: Prospective study in 81 patients. La Rev. Med. Interne. 2006, 27, 106–110. [Google Scholar] [CrossRef]
- Owczarek, D.; Cibor, D.; Sałapa, K.; Jurczyszyn, A.; Mach, T. Homocysteine in patients with inflammatory bowel diseases. Przegl Lek. 2014, 71, 189–192. [Google Scholar]
- Romagnuolo, J.; Fedorak, R.N.; Dias, V.C.; Bamforth, F.; Teltscher, M. Hyperhomocysteinemia and inflammatory bowel disease: Prevalence and predictors in a cross-sectional study. Am. J. Gastroenterol. 2001, 96, 2143–2149. [Google Scholar] [CrossRef]
- Robinson, K.; Arheart, K.; Refsum, H.; Brattström, L.; Boers, G.; Ueland, P.; Rubba, P.; Palma-Reis, R.; Meleady, R.; Daly, L.; et al. Low circulating folate and vitamin B6 concentrations: Risk factors for stroke, peripheral vascular disease, and coronary artery disease. European COMAC Group. Circulation 1998, 97, 437–443. [Google Scholar] [CrossRef] [PubMed]
- Ma, F.; Wu, T.; Zhao, J.; Ji, L.; Song, A.; Zhang, M.; Huang, G. Plasma Homocysteine and Serum Folate and Vitamin B12 Levels in Mild Cognitive Impairment and Alzheimer’s Disease: A Case-Control Study. Nutrients 2017, 7, 725. [Google Scholar] [CrossRef] [PubMed]
- Shetty, A.; Konuri, A.; Bhat, N.; Moorkhot, S.; Raveendran, A.; Kumar, S.E.P.; Surendran, S. Effects of maternal vitamin deficiency on the microstructure of the maternal hippocampus and behavior in offspring. J. Taibah Univ. Med. Sci. 2023, 18, 1108–1114. [Google Scholar] [CrossRef] [PubMed]
- Ebadi, M. Regulation and function of pyridoxal phosphate in CNS. Neurochem. Int. 1981, 3, 181–205. [Google Scholar] [CrossRef] [PubMed]
- Feng, Z.; Hua, J.; Guo, F.; Liu, Z.; Zhao, Y.; Wu, W. A retrospective analysis of vitamin B6 deficiency and associated changes of gut microbes in Crohn’s disease. Eur. J. Clin. Nutr. 2023, 77, 1034–1043. [Google Scholar] [CrossRef] [PubMed]
- Clarke, R. Lowering blood homocysteine with folic acid-based supplements: Meta-analysis of randomised trials. Indian. Heart J. 2000, 52 (Suppl. S7), S59–S64. [Google Scholar] [PubMed]
- Modica, J.S.; Déry, C.; Canissario, R.; Logigian, E.; Bonno, D.; Stanton, M.; Dupré, N.; McDermott, M.P.; Bouchard, M.; Lang, A.E.; et al. A systematic review of the potential consequences of abnormal serum levels of vitamin B6 in people living with Parkinson’s disease. J. Neurol. Sci. 2023, 450, 120690. [Google Scholar] [CrossRef] [PubMed]
- Ko, J.W.; Jeon, S.; Kwon, Y.H. Dietary vitamin B6 restriction aggravates neurodegeneration in mice fed a high-fat diet. Life Sci. 2022, 309, 121041. [Google Scholar] [CrossRef]
- Castelnau, P.A.; Garrett, R.S.; Palinski, W.; Witztum, J.L.; Campbell, I.L.; Powell, H.C. Abnormal iron deposition associated with lipid peroxidation in transgenic mice expressing interleukin-6 in the brain. J. Neuropathol. Exp. Neurol. 1998, 57, 268–282. [Google Scholar] [CrossRef]
- Polegato, B.F.; Pereira, A.G.; Azevedo, P.S.; Costa, N.A.; Zornoff, L.A.M.; Paiva, S.A.R.; Minicucci, M.F. Role of Thiamin in Health and Disease. Nutr. Clin. Pr. Off. Publ. Am. Soc. Parenter. Enter. Nutr. 2019, 34, 558–564. [Google Scholar] [CrossRef]
- Bubko, I.; Gruber, B.M.; Anuszewska, E.L. The role of thiamine in neurodegenerative diseases. Postepy Hig. Med. Dosw. 2015, 69, 1096–1106. [Google Scholar]
- Vavricka, S.R.; Rogler, G. Intestinal absorption and vitamin levels: Is a new focus needed? Dig. Dis. 2012, 30 (Suppl. S3), 73–80. [Google Scholar] [CrossRef] [PubMed]
- Mańkowska-Wierzbicka, D.; Michalak, S.; Karczewski, J.; Dobrowolska, A.; Wierzbicka, A.; Stelmach-Mardas, M. Erythrocyte transketolase deficiency in patients suffering from Crohn’s disease. Eur. Rev. Med. Pharmacol. Sci. 2019, 23, 8501–8505. [Google Scholar] [PubMed]
- Zhang, Q.; Yang, G.; Li, W.; Fan, Z.; Sun, A.; Luo, J.; Ke, Z.-J. Thiamine deficiency increases β-secretase activity and accumulation of β-amyloid peptides. Neurobiol. Aging. 2011, 32, 42–53. [Google Scholar] [CrossRef] [PubMed]
- Gibson, G.E.; Zhang, H. Interactions of oxidative stress with thiamine homeostasis promote neurodegeneration. Neurochem. Int. 2002, 40, 493–504. [Google Scholar] [CrossRef]
- Bettendorff, L. Synthetic Thioesters of Thiamine: Promising Tools for Slowing Progression of Neurodegenerative Diseases. Int. J. Mol. Sci. 2023, 24, 11296. [Google Scholar] [CrossRef] [PubMed]
- Yuan, A.; Nixon, R.A. Neurofilament Proteins as Biomarkers to Monitor Neurological Diseases and the Efficacy of Therapies. Front. Neurosci. 2021, 15, 689938. [Google Scholar] [CrossRef]
- Khalil, M.; Teunissen, C.E.; Otto, M.; Piehl, F.; Sormani, M.P.; Gattringer, T.; Barro, C.; Kappos, L.; Comabella, M.; Fazekas, F.; et al. Neurofilaments as biomarkers in neurological disorders. Nat. Rev. Neurol. 2018, 14, 577–589. [Google Scholar] [CrossRef]
- Barro, C.; Chitnis, T.; Weiner, H.L. Blood neurofilament light: A critical review of its application to neurologic disease. Ann. Clin. Transl. Neurol. 2020, 7, 2508–2523. [Google Scholar] [CrossRef]
- Arslan, B.; Zetterberg, H. Neurofilament light chain as neuronal injury marker-what is needed to facilitate implementation in clinical laboratory practice? Clin. Chem. Lab. Med. 2023, 61, 1140–1149. [Google Scholar] [CrossRef]
- Gaetani, L.; Blennow, K.; Calabresi, P.; Di Filippo, M.; Parnetti, L.; Zetterberg, H. Neurofilament light chain as a biomarker in neurological disorders. J. Neurol. Neurosurg. Psychiatry. 2019, 90, 870–881. [Google Scholar] [CrossRef] [PubMed]
- Altaf, M.A.; Sood, M.R. The nervous system and gastrointestinal function. Dev. Disabil. Res. Rev. 2008, 14, 87–95. [Google Scholar] [CrossRef] [PubMed]
- Van Oudenhove, L.; Demyttenaere, K.; Tack, J.; Aziz, Q. Central nervous system involvement in functional gastrointestinal disorders. Best. Pr. Res. Clin. Gastroenterol. 2004, 18, 663–680. [Google Scholar] [CrossRef] [PubMed]
- Lomax, A.E.; Linden, D.R.; Mawe, G.M.; Sharkey, K.A. Effects of gastrointestinal inflammation on enteroendocrine cells and enteric neural reflex circuits. Auton. Neurosci. 2006, 126–127, 250–257. [Google Scholar] [CrossRef] [PubMed]
- Gross, K.J.; Pothoulakis, C. Role of neuropeptides in inflammatory bowel disease. Inflamm. Bowel Dis. 2007, 13, 918–932. [Google Scholar] [CrossRef]
- Holland, A.M.; Bon-Frauches, A.C.; Keszthelyi, D.; Melotte, V.; Boesmans, W. The enteric nervous system in gastrointestinal disease etiology. Cell Mol. Life Sci. 2021, 78, 4713–4733. [Google Scholar] [CrossRef]
- Li, Z.; Wang, X.; Wang, X.; Yi, X.; Wong, Y.K.; Wu, J.; Xie, F.; Hu, D.; Wang, Q.; Wang, J.; et al. Research progress on the role of extracellular vesicles in neurodegenerative diseases. Transl. Neurodegener. 2023, 12, 43. [Google Scholar] [CrossRef] [PubMed]
- Takeuchi, T. Pathogenic and protective roles of extracellular vesicles in neurodegenerative diseases. J. Biochem. 2021, 169, 181–186. [Google Scholar] [CrossRef]
- Whitehead, M.C.; Marangos, P.J.; Connolly, S.M.; Morest, D.K. Synapse formation is related to the onset of neuron-specific enolase immunoreactivity in the avian auditory and vestibular systems. Dev. Neurosci. 1982, 5, 298–307. [Google Scholar] [CrossRef]
- Schmechel, D.E.; Brightman, M.W.; Marangos, P.J. Neurons switch from non-neuronal enolase to neuron-specific enolase during differentiation. Brain Res. 1980, 190, 195–214. [Google Scholar] [CrossRef]
- Inoue, S.; Takahashi, H.; Kaneko, K. The fluctuations of neuron-specific enolase (NSE) levels of cerebrospinal fluid during bacterial meningitis: The relationship between the fluctuations of NSE levels and neurological complications or outcome. Acta Paediatr. Jpn. Overseas Ed. 1994, 36, 485–488. [Google Scholar] [CrossRef] [PubMed]
- Namjoshi, S.; Benson, H.A.E. Cyclic peptides as potential therapeutic agents for skin disorders. Biopolymers 2010, 94, 673–680. [Google Scholar] [CrossRef] [PubMed]
- Palumbo, B.; Siepi, D.; Sabalich, I.; Tranfaglia, C.; Parnetti, L. Cerebrospinal fluid neuron-specific enolase: A further marker of Alzheimer’s disease? Funct. Neurol. 2008, 23, 93–96. [Google Scholar] [PubMed]
- Tsukahara, A.; Hosokawa, T.; Nishioka, D.; Kotani, T.; Ishida, S.; Takeuchi, T.; Kimura, F.; Arawaka, S. Neuron-specific enolase level is a useful biomarker for distinguishing amyotrophic lateral sclerosis from cervical spondylotic myelopathy. Sci. Rep. 2021, 11, 22827. [Google Scholar] [CrossRef] [PubMed]
- Schmidt, F.M.; Mergl, R.; Stach, B.; Jahn, I.; Gertz, H.-J.; Schönknecht, P. Elevated levels of cerebrospinal fluid neuron-specific enolase (NSE) in Alzheimer’s disease. Neurosci. Lett. 2014, 570, 81–85. [Google Scholar] [CrossRef]
- Blennow, K.; Wallin, A.; Ekman, R. Neuron specific enolase in cerebrospinal fluid: A biochemical marker for neuronal degeneration in dementia disorders? J. Neural Transm. Park. Dis. Dement. Sect. 1994, 8, 183–191. [Google Scholar] [CrossRef]
- Busikova-Malenovska, P.; Danis, D.; Bencat, M.; Galfiova, P.; Kopani, M.; Labajova, V.; El Hassoun, O.; Porubsky, J.; Galatova, J. Neuron-specific enolase in the intestinal wall in Crohn´s disease. Bratisl. Lek. Listy. 2014, 115, 381–382. [Google Scholar] [CrossRef]
- Haque, A.; Polcyn, R.; Matzelle, D.; Banik, N.L. New Insights into the Role of Neuron-Specific Enolase in Neuro-Inflammation, Neurodegeneration, and Neuroprotection. Brain Sci. 2018, 8, 33. [Google Scholar] [CrossRef] [PubMed]
- Villanacci, V.; Bassotti, G.; Nascimbeni, R.; Antonelli, E.; Cadei, M.; Fisogni, S.; Salerni, B.; Geboes, K. Enteric nervous system abnormalities in inflammatory bowel diseases. Neurogastroenterol. Motil. 2008, 20, 1009–1016. [Google Scholar] [CrossRef]
- Bagnato, S.; Andriolo, M.; Boccagni, C.; Lucca, L.F.; De Tanti, A.; Pistarini, C.; Barone, T.; Galardi, G. Reduced Neuron-Specific Enolase Levels in Chronic Severe Traumatic Brain Injury. J. Neurotrauma 2020, 37, 423–427. [Google Scholar] [CrossRef]
- Heizmann, C.W.; Fritz, G.; Schäfer, B.W. S100 proteins: Structure, functions and pathology. Front. Biosci. 2002, 7, d1356-68. [Google Scholar]
- Kiss, B.; Ecsédi, P.; Simon, M.; Nyitray, L. Isolation and Characterization of S100 Protein-Protein Complexes. Methods Mol. Biol. 2019, 1929, 325–338. [Google Scholar]
- Cirillo, C.; Sarnelli, G.; Esposito, G.; Turco, F.; Steardo, L.; Cuomo, R. S100B protein in the gut: The evidence for enteroglial-sustained intestinal inflammation. World J. Gastroenterol. 2011, 17, 1261–1266. [Google Scholar] [CrossRef]
- Di Liddo, R.; Piccione, M.; Schrenk, S.; Dal Magro, C.; Cosma, C.; Padoan, A.; Contran, N.; Scapellato, M.L.; Pagetta, A.; Spica, V.R.; et al. S100B as a new fecal biomarker of inflammatory bowel diseases. Eur. Rev. Med. Pharmacol. Sci. 2020, 24, 323–332. [Google Scholar] [PubMed]
- Hellweg, R.; Jockers-Scherübl, M. Neurotrophic factors in memory disorders. Life Sci. 1994, 55, 2165–2169. [Google Scholar] [CrossRef]
- Meyer, M.; Rasmussen, J.Z. Neuronal growth factors--neurotrophins. Ugeskr Laeger 1999, 161, 2063–2070. [Google Scholar] [PubMed]
- Apfel, S.C.; Kessler, J.A. Neurotrophic factors in the therapy of peripheral neuropathy. Baillieres Clin. Neurol. 1995, 4, 593–606. [Google Scholar] [PubMed]
- Azman, K.F.; Zakaria, R. Recent Advances on the Role of Brain-Derived Neurotrophic Factor (BDNF) in Neurodegenerative Diseases. Int. J. Mol. Sci. 2022, 23, 6827. [Google Scholar] [CrossRef]
- Zuccato, C.; Cattaneo, E. Brain-derived neurotrophic factor in neurodegenerative diseases. Nat. Rev. Neurol. 2009, 5, 311–322. [Google Scholar] [CrossRef]
- Ibrahim, A.M.; Chauhan, L.; Bhardwaj, A.; Sharma, A.; Fayaz, F.; Kumar, B.; Alhashmi, M.; AlHajri, N.; Alam, S.; Pottoo, F.H. Brain-Derived Neurotropic Factor in Neurodegenerative Disorders. Biomedicines 2022, 10, 1143. [Google Scholar] [CrossRef]
- Konturek, T.J.; Martinez, C.; Niesler, B.; van der Voort, I.; Mönnikes, H.; Stengel, A.; Goebel-Stengel, M. The Role of Brain-Derived Neurotrophic Factor in Irritable Bowel Syndrome. Front. Psychiatry 2020, 11, 531385. [Google Scholar] [CrossRef] [PubMed]
- Yu, Y.-B.; Zuo, X.-L.; Zhao, Q.-J.; Chen, F.-X.; Yang, J.; Dong, Y.-Y.; Wang, P.; Li, Y.-Q. Brain-derived neurotrophic factor contributes to abdominal pain in irritable bowel syndrome. Gut 2012, 61, 685–694. [Google Scholar] [CrossRef] [PubMed]
- Sochal, M.; Ditmer, M.; Binienda, A.; Gabryelska, A.; Białasiewicz, P.; Talar-Wojnarowska, R.; Fichna, J.; Małecka-Wojciesko, E. Relation between Selected Sleep Parameters, Depression, Anti-Tumor Necrosis Factor Therapy, and the Brain-Derived Neurotrophic Factor Pathway in Inflammatory Bowel Disease. Metabolites 2023, 13, 450. [Google Scholar] [CrossRef] [PubMed]
- Liu, G.X.; Yang, Y.X.; Yan, J.; Zhang, T.; Zou, Y.P.; Huang, X.L.; Gan, H.T. Glial-derived neurotrophic factor reduces inflammation and improves delayed colonic transit in rat models of dextran sulfate sodium-induced colitis. Int. Immunopharmacol. 2014, 19, 145–152. [Google Scholar] [CrossRef] [PubMed]
- Chen, H.; Han, T.; Gao, L.; Zhang, D. The Involvement of Glial Cell-Derived Neurotrophic Factor in Inflammatory Bowel Disease. J. Interf. Cytokine Res. 2022, 42, 1–7. [Google Scholar] [CrossRef] [PubMed]
- Kearon, J.E.; Kocherry, S.C.; Zoumboulakis, D.; Rivera, D.; Lourenssen, S.R.; Blennerhassett, M.G. GDNF requires HIF-1α and RET activation for suppression of programmed cell death of enteric neurons by metabolic challenge. Mol. Cell Neurosci. 2021, 115, 103655. [Google Scholar] [CrossRef] [PubMed]
- Dragoni, G.; Innocenti, T.; Galli, A. Biomarkers of Inflammation in Inflammatory Bowel Disease: How Long before Abandoning Single-Marker Approaches? Dig. Dis. 2021, 39, 190–203. [Google Scholar] [CrossRef] [PubMed]
- Stavely, R.; Abalo, R.; Nurgali, K. Targeting Enteric Neurons and Plexitis for the Management of Inflammatory Bowel Disease. Curr. Drug Targets 2020, 21, 1428–1439. [Google Scholar] [CrossRef] [PubMed]
- Boggia, R.; Turrini, F.; Roggeri, A.; Olivero, G.; Cisani, F.; Bonfiglio, T.; Summa, M.; Grilli, M.; Caviglioli, G.; Alfei, S.; et al. Neuroinflammation in Aged Brain: Impact of the Oral Administration of Ellagic Acid Microdispersion. Int. J. Mol. Sci. 2020, 21, 3631. [Google Scholar] [CrossRef]
- Bajetto, A.; Bonavia, R.; Barbero, S.; Florio, T.; Schettini, G. Chemokines and their receptors in the central nervous system. Front. Neuroendocrinol. 2001, 22, 147–184. [Google Scholar] [CrossRef]
- Bene, L.; Füst, G.; Fekete, B.; Kovács, A.; Horváth, L.; Prohászka, Z.; Miklós, K.; Pálos, G.; Daha, M.; Farkas, H.; et al. High normal serum levels of C3 and C1 inhibitor, two acute-phase proteins belonging to the complement system, occur more frequently in patients with Crohn’s disease than ulcerative colitis. Dig. Dis. Sci. 2003, 48, 1186–1192. [Google Scholar] [CrossRef]
- Mavropoulou, E.; Mechie, N.-C.; Knoop, R.; Petzold, G.; Ellenrieder, V.; Kunsch, S.; Pilavakis, Y.; Amanzada, A. Association of serum interleukin-6 and soluble interleukin-2-receptor levels with disease activity status in patients with inflammatory bowel disease: A prospective observational study. PLoS ONE 2020, 15, e0233811. [Google Scholar] [CrossRef]
- Wilson, R.G.; Stevens, B.W.; Guo, A.Y.; Russell, C.N.; Thornton, A.; Cohen, M.A.; Sturgeon, H.C.; Giallourakis, C.; Khalili, H.; Nguyen, D.D.; et al. High C-Reactive Protein Is Associated with Poor Sleep Quality Independent of Nocturnal Symptoms in Patients with Inflammatory Bowel Disease. Dig. Dis. Sci. 2015, 60, 2136–2143. [Google Scholar] [CrossRef] [PubMed]
- Zheng, F.; Xie, W. High-sensitivity C-reactive protein and cognitive decline: The English Longitudinal Study of Ageing. Psychol. Med. 2018, 48, 1381–1389. [Google Scholar] [CrossRef] [PubMed]
- Komulainen, P.; Lakka, T.A.; Kivipelto, M.; Hassinen, M.; Penttilä, I.M.; Helkala, E.-L.; Gylling, H.; Nissinen, A.; Rauramaa, R. Serum high sensitivity C-reactive protein and cognitive function in elderly women. Age Ageing 2007, 36, 443–448. [Google Scholar] [CrossRef] [PubMed]
- Lewis, N.A.; Knight, J.E. Longitudinal associations between C-reactive protein and cognitive performance in normative cognitive ageing and dementia. Age Ageing 2021, 50, 2199–2205. [Google Scholar] [CrossRef]
- Dlugaj, M.; Gerwig, M.; Wege, N.; Siegrist, J.; Mann, K.; Bröcker-Preuss, M.; Dragano, N.; Moebus, S.; Jöckel, K.-H.; Bokhof, B.; et al. Elevated levels of high-sensitivity C-reactive protein are associated with mild cognitive impairment and its subtypes: Results of a population-based case-control study. J. Alzheimers Dis. 2012, 28, 503–514. [Google Scholar] [CrossRef] [PubMed]
- Jensen, L.E.; Whitehead, A.S. Regulation of serum amyloid A protein expression during the acute-phase response. Biochem. J. 1998, 334 Pt 3, 489–503. [Google Scholar] [CrossRef]
- Yokote, H.; Yagi, Y.; Watanabe, Y.; Amino, T.; Kamata, T.; Mizusawa, H. Serum amyloid A level is increased in neuromyelitis optica and atypical multiple sclerosis with smaller T2 lesion volume in brain MRI. J. Neuroimmunol. 2013, 259, 92–95. [Google Scholar] [CrossRef]
- Kawarabayashi, T.; Shoji, M.; Sato, M.; Sasaki, A.; Ho, L.; Eckman, C.B.; Prada, C.M.; Younkin, S.G.; Kobayashi, T.; Tada, N.; et al. Accumulation of beta-amyloid fibrils in pancreas of transgenic mice. Neurobiol. Aging 1996, 17, 215–222. [Google Scholar] [CrossRef]
- Shimizu, T.; Fukuda, H.; Murayama, S.; Izumiyama, N.; Shirasawa, T. Isoaspartate formation at position 23 of amyloid beta peptide enhanced fibril formation and deposited onto senile plaques and vascular amyloids in Alzheimer’s disease. J. Neurosci. Res. 2002, 70, 451–461. [Google Scholar] [CrossRef] [PubMed]
- Emmerling, M.R.; Spiegel, K.; Watson, M.D. Inhibiting the formation of classical C3-convertase on the Alzheimer’s beta-amyloid peptide. Immunopharmacology 1997, 38, 101–109. [Google Scholar] [CrossRef] [PubMed]
- Yarur, A.J.; Quintero, M.A.; Jain, A.; Czul, F.; Barkin, J.S.; Abreu, M.T. Serum Amyloid A as a Surrogate Marker for Mucosal and Histologic Inflammation in Patients with Crohn’s Disease. Inflamm. Bowel Dis. 2017, 23, 158–164. [Google Scholar] [CrossRef] [PubMed]
- Crouch, P.J.; Harding, S.-M.E.; White, A.R.; Camakaris, J.; Bush, A.I.; Masters, C.L. Mechanisms of A beta mediated neurodegeneration in Alzheimer’s disease. Int. J. Biochem. Cell Biol. 2008, 40, 181–198. [Google Scholar] [CrossRef] [PubMed]
- Shim, K.H.; Kang, M.J.; Youn, Y.C.; An, S.S.A.; Kim, S. Alpha-synuclein: A pathological factor with Aβ and tau and biomarker in Alzheimer’s disease. Alzheimers Res. Ther. 2022, 14, 201. [Google Scholar] [CrossRef] [PubMed]
- Quintanilla, R.A.; Orellana, D.I.; González-Billault, C.; Maccioni, R.B. Interleukin-6 induces Alzheimer-type phosphorylation of tau protein by deregulating the cdk5/p35 pathway. Exp. Cell Res. 2004, 295, 245–257. [Google Scholar] [CrossRef]
- Shan, C.; Zhang, C.; Zhang, C. The Role of IL-6 in Neurodegenerative Disorders. Neurochem. Res. 2024, 49, 834–846. [Google Scholar] [CrossRef] [PubMed]
- Economos, A.; Wright, C.B.; Moon, Y.P.; Rundek, T.; Rabbani, L.; Paik, M.C.; Sacco, R.L.; Elkind, M.S. Interleukin 6 plasma concentration associates with cognitive decline: The northern Manhattan study. Neuroepidemiology 2013, 40, 253–259. [Google Scholar] [CrossRef] [PubMed]
- Hosokawa, T.; Kusugami, K.; Ina, K.; Ando, T.; Shinoda, M.; Imada, A.; Ohsuga, M.; Sakai, T.; Matsuura, T.; Ito, K.; et al. Interleukin-6 and soluble interleukin-6 receptor in the colonic mucosa of inflammatory bowel disease. J. Gastroenterol. Hepatol. 1999, 14, 987–996. [Google Scholar] [CrossRef]
- Roberts, R.O.; Geda, Y.E.; Knopman, D.S.; Boeve, B.F.; Christianson, T.J.H.; Pankratz, V.S.; Kullo, I.J.; Tangalos, E.G.; Ivnik, R.J.; Petersen, R.C. Association of C-reactive protein with mild cognitive impairment. Alzheimers Dement. 2009, 5, 398–405. [Google Scholar] [CrossRef]
- Leach, C.A.; Hickey, D.M.; Ife, R.J.; Macphee, C.H.; Smith, S.A.; Tew, D.G. Lipoprotein-associated PLA2 inhibition—a novel, non-lipid lowering strategy for atherosclerosis therapy. Farmaco 2001, 56, 45–50. [Google Scholar] [CrossRef] [PubMed]
- Ikonomidis, I.; Michalakeas, C.A.; Lekakis, J.; Parissis, J.; Anastasiou-Nana, M. The role of lipoprotein-associated phospholipase A2 (Lp-PLA₂) in cardiovascular disease. Rev. Recent. Clin. Trials 2011, 6, 108–113. [Google Scholar] [CrossRef]
- Zalewski, A.; Macphee, C.; Nelson, J.J. Lipoprotein-associated phospholipase A2: A potential therapeutic target for atherosclerosis. Curr. Drug Targets Cardiovasc. Haematol. Disord. 2005, 5, 527–532. [Google Scholar] [CrossRef] [PubMed]
- Dohi, T.; Miyauchi, K.; Ohkawa, R.; Nakamura, K.; Thuboi, S.; Ogita, M.; Miyazaki, T.; Nishino, A.; Yokoyama, K.; Kurata, T.; et al. Higher lipoprotein-associated phospholipase A2 levels are associated with coronary atherosclerosis documented by coronary angiography. Ann. Clin. Biochem. 2012, 49 Pt 6, 527–533. [Google Scholar] [CrossRef]
- Charniot, J.C.; Khani-Bittar, R.; Albertini, J.P.; Giral, P.; Cherfils, C.; Cosson, C.; Guillerm, E.; Leprince, P.; Gandjbakhch, I.; Bonnefont-Rousselot, D. Interpretation of lipoprotein-associated phospholipase A2 levels is influenced by cardiac disease, comorbidities, extension of atherosclerosis and treatments. Int. J. Cardiol. 2013, 168, 132–138. [Google Scholar] [CrossRef]
- Haapamäki, M.M.; Grönroos, J.M.; Nurmi, H.; Söderlund, K.; Peuravuori, H.; Alanen, K.; Nevalainen, T.J. Elevated group II phospholipase A2 mass concentration in serum and colonic mucosa in Crohn’s disease. Clin. Chem. Lab. Med. 1998, 36, 751–755. [Google Scholar] [CrossRef] [PubMed]
- Andreadou, E.G.; Katsipis, G.; Tsolaki, M.; Pantazaki, A.A. Involvement and relationship of bacterial lipopolysaccharides and cyclooxygenases levels in Alzheimer’s Disease and Mild Cognitive Impairment patients. J. Neuroimmunol. 2021, 357, 577561. [Google Scholar] [CrossRef]
- Mutsaers, H.A.M.; Nørregaard, R. Prostaglandin E2 receptors as therapeutic targets in renal fibrosis. Kidney Res. Clin. Pract. 2022, 41, 4–13. [Google Scholar] [CrossRef]
- Wang, L.; Wu, Y.; Jia, Z.; Yu, J.; Huang, S. Roles of EP Receptors in the Regulation of Fluid Balance and Blood Pressure. Front. Endocrinol. 2022, 13, 875425. [Google Scholar] [CrossRef]
- Chen, S.-T.; Ji, S.; Guo, M.-N.; Chen, L.-H. Research advances of prostaglandin E(2) receptor 1 (EP1). Sheng Li Xue Bao 2024, 76, 105–118. [Google Scholar]
- Stock, J.L.; Shinjo, K.; Burkhardt, J.; Roach, M.; Taniguchi, K.; Ishikawa, T.; Kim, H.S.; Flannery, P.J.; Coffman, T.M.; McNeish, J.D.; et al. The prostaglandin E2 EP1 receptor mediates pain perception and regulates blood pressure. J. Clin. Investig. 2001, 107, 325–331. [Google Scholar] [CrossRef] [PubMed]
- Gomez, I.; Foudi, N.; Longrois, D.; Norel, X. The role of prostaglandin E2 in human vascular inflammation. Prostaglandins Leukot. Essent. Fat. Acids 2013, 89, 55–63. [Google Scholar] [CrossRef]
- Le Loupp, A.-G.; Bach-Ngohou, K.; Bourreille, A.; Boudin, H.; Rolli-Derkinderen, M.; Denis, M.G.; Neunlist, M.; Masson, D. Activation of the prostaglandin D2 metabolic pathway in Crohn’s disease: Involvement of the enteric nervous system. BMC Gastroenterol. 2015, 15, 112. [Google Scholar] [CrossRef]
- Zhang, J.; Rivest, S. Anti-inflammatory effects of prostaglandin E2 in the central nervous system in response to brain injury and circulating lipopolysaccharide. J. Neurochem. 2001, 76, 855–864. [Google Scholar] [CrossRef] [PubMed]
- Figueiredo-Pereira, M.E.; Rockwell, P.; Schmidt-Glenewinkel, T.; Serrano, P. Neuroinflammation and J2 prostaglandins: Linking impairment of the ubiquitin-proteasome pathway and mitochondria to neurodegeneration. Front. Mol. Neurosci. 2014, 7, 104. [Google Scholar] [CrossRef]
- Wei, L.-L.; Shen, Y.-D.; Zhang, Y.-C.; Hu, X.-Y.; Lu, P.-L.; Wang, L.; Chen, W. Roles of the prostaglandin E2 receptors EP subtypes in Alzheimer’s disease. Neurosci. Bull. 2010, 26, 77–84. [Google Scholar] [CrossRef] [PubMed]
- Minghetti, L. Cyclooxygenase-2 (COX-2) in inflammatory and degenerative brain diseases. J. Neuropathol. Exp. Neurol. 2004, 63, 901–910. [Google Scholar] [CrossRef]
- Cominelli, F.; Nast, C.C.; Clark, B.D.; Schindler, R.; Lierena, R.; Eysselein, V.E.; Thompson, R.C.; Dinarello, C.A. Interleukin 1 (IL-1) gene expression, synthesis, and effect of specific IL-1 receptor blockade in rabbit immune complex colitis. J. Clin. Investig. 1990, 86, 972–980. [Google Scholar] [CrossRef]
- Li, L.; Liu, Z.; Yang, X.; Yan, H.; Bao, S.; Fei, J. Bioluminescence imaging for IL-1β expression in experimental colitis. J. Inflamm. 2013, 10, 16. [Google Scholar] [CrossRef]
- Griffin, W.S.T.; Mrak, R.E. Interleukin-1 in the genesis and progression of and risk for development of neuronal degeneration in Alzheimer’s disease. J. Leukoc. Biol. 2002, 72, 233–238. [Google Scholar] [CrossRef]
- Berrill, J.W.; Gallacher, J.; Hood, K.; Green, J.T.; Matthews, S.B.; Campbell, A.K.; Smith, A.; Berrill, J.W.; Gallacher, J.; Hood, K.; et al. An observational study of cognitive function in patients with irritable bowel syndrome and inflammatory bowel disease. Neurogastroenterol. Motil. 2013, 25, 918-e704. [Google Scholar] [CrossRef]
- Huang, Z.-B.; Sheng, G.-Q. Interleukin-1β with learning and memory. Neurosci. Bull. 2010, 26, 455–468. [Google Scholar] [CrossRef] [PubMed]
- Silva, L.C.R.; Ortigosa, L.C.M.; Benard, G. Anti-TNF-α agents in the treatment of immune-mediated inflammatory diseases: Mechanisms of action and pitfalls. Immunotherapy 2010, 2, 817–833. [Google Scholar] [CrossRef] [PubMed]
- Gray, M.A.; Chao, C.-Y.; Staudacher, H.M.; Kolosky, N.A.; Talley, N.J.; Holtmann, G. Anti-TNFα therapy in IBD alters brain activity reflecting visceral sensory function and cognitive-affective biases. PLoS ONE 2018, 13, e0193542. [Google Scholar] [CrossRef] [PubMed]
- Sriram, K.; O’Callaghan, J.P. Divergent roles for tumor necrosis factor-alpha in the brain. J. Neuroimmune Pharmacol. 2007, 2, 140–153. [Google Scholar] [CrossRef] [PubMed]
- Fischer, R.; Maier, O. Interrelation of oxidative stress and inflammation in neurodegenerative disease: Role of TNF. Oxid. Med. Cell Longev. 2015, 2015, 610813. [Google Scholar] [CrossRef]
- Olmos, G.; Lladó, J. Tumor necrosis factor alpha: A link between neuroinflammation and excitotoxicity. Mediat. Inflamm. 2014, 2014, 861231. [Google Scholar] [CrossRef]
- Venters, H.D.; Dantzer, R.; Kelley, K.W. Tumor necrosis factor-alpha induces neuronal death by silencing survival signals generated by the type I insulin-like growth factor receptor. Ann. N. Y. Acad. Sci. 2000, 917, 210–220. [Google Scholar] [CrossRef]
- Aloe, L.; Fiore, M.; Probert, L.; Turrini, P.; Tirassa, P. Overexpression of tumour necrosis factor alpha in the brain of transgenic mice differentially alters nerve growth factor levels and choline acetyltransferase activity. Cytokine 1999, 11, 45–54. [Google Scholar] [CrossRef]
- Marín, M.; Moya, C.; Máñez, S. Mutual Influences between Nitric Oxide and Paraoxonase 1. Antioxidants 2019, 8, 619. [Google Scholar] [CrossRef]
- Voronin, M.V.; Lisitsyna, T.A.; Durnev, A.D.; Nasonov, E.L. Paraoxonase: Biological activity and clinical implications. Vestn Ross Akad. meditsinskikh Nauk. 2008, 9, 45–51. [Google Scholar]
- Li, Y.R.; Zhu, H.; Kauffman, M.; Danelisen, I.; Misra, H.P.; Ke, Y.; Jia, Z. Paraoxonases function as unique protectors against cardiovascular diseases and diabetes: Updated experimental and clinical data. Exp. Biol. Med. 2014, 239, 899–906. [Google Scholar] [CrossRef] [PubMed]
- Meisinger, C.; Freuer, D.; Bub, A.; Linseisen, J. Association between inflammatory markers and serum paraoxonase and arylesterase activities in the general population: A cross-sectional study. Lipids Health Dis. 2021, 20, 81. [Google Scholar] [CrossRef] [PubMed]
- Précourt, L.-P.; Marcil, V.; Ntimbane, T.; Taha, R.; Lavoie, J.-C.; Delvin, E.; Seidman, E.G.; Beaulieu, J.-F.; Levy, E. Antioxidative properties of paraoxonase 2 in intestinal epithelial cells. Am. J. Physiol. Gastrointest. Liver Physiol. 2012, 303, G623–G634. [Google Scholar] [CrossRef]
- Rothem, L.; Hartman, C.; Dahan, A.; Lachter, J.; Eliakim, R.; Shamir, R. Paraoxonases are associated with intestinal inflammatory diseases and intracellularly localized to the endoplasmic reticulum. Free Radic. Biol. Med. 2007, 43, 730–739. [Google Scholar] [CrossRef]
- Mouzaoui, S.; Djerdjouri, B.; Makhezer, N.; Kroviarski, Y.; El-Benna, J.; Dang, P.M.-C. Tumor necrosis factor-α-induced colitis increases NADPH oxidase 1 expression, oxidative stress, and neutrophil recruitment in the colon: Preventive effect of apocynin. Mediat. Inflamm. 2014, 2014, 312484. [Google Scholar] [CrossRef] [PubMed]
- Yamashita, J.; Iwamura, C.; Ito, T.; Narita, M.; Hara, Y.; Sasaki, T.; Masuda, D.; Takahashi, M.; Tsuchiya, M.; Hada, K.; et al. Paraoxonase-1 suppresses experimental colitis via the inhibition of IFN-γ production from CD4 T cells. J. Immunol. 2013, 191, 949–960. [Google Scholar] [CrossRef] [PubMed]
- Zhang, L.; Dong, W.; Ma, Y.; Bai, L.; Zhang, X.; Sun, C.; Li, J.; Zhang, L. Pon1 Deficiency Promotes Trem2 Pathway-Mediated Microglial Phagocytosis and Inhibits Pro-inflammatory Cytokines Release In Vitro and In Vivo. Mol. Neurobiol. 2022, 59, 4612–4629. [Google Scholar] [CrossRef] [PubMed]
- Jamwal, S.; Blackburn, J.K.; Elsworth, J.D. Expression of PON2 isoforms varies among brain regions in male and female African green monkeys. Free Radic. Biol. Med. 2022, 178, 215–218. [Google Scholar] [CrossRef]
- Parween, F.; Gupta, R.D. Insights into the role of paraoxonase 2 in human pathophysiology. J. Biosci. 2021, 46. [Google Scholar] [CrossRef]
- Androutsopoulos, V.P.; Kanavouras, K.; Tsatsakis, A.M. Role of paraoxonase 1 (PON1) in organophosphate metabolism: Implications in neurodegenerative diseases. Toxicol. Appl. Pharmacol. 2011, 256, 418–424. [Google Scholar] [CrossRef] [PubMed]
- Menini, T.; Gugliucci, A. Paraoxonase 1 in neurological disorders. Redox Rep. 2014, 19, 49–58. [Google Scholar] [CrossRef] [PubMed]
- Miner, S.E.; Evrovski, J.; Cole, D.E. Clinical chemistry and molecular biology of homocysteine metabolism: An update. Clin. Biochem. 1997, 30, 189–201. [Google Scholar] [CrossRef] [PubMed]
- Bresnick, A.R. S100 proteins as therapeutic targets. Biophys. Rev. 2018, 10, 1617–1629. [Google Scholar] [CrossRef]
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Sarb, O.-F.; Sarb, A.-D.; Iacobescu, M.; Vlad, I.-M.; Milaciu, M.-V.; Ciurmarnean, L.; Vacaras, V.; Tantau, A.-I. From Gut to Brain: Uncovering Potential Serum Biomarkers Connecting Inflammatory Bowel Diseases to Neurodegenerative Diseases. Int. J. Mol. Sci. 2024, 25, 5676. https://doi.org/10.3390/ijms25115676
Sarb O-F, Sarb A-D, Iacobescu M, Vlad I-M, Milaciu M-V, Ciurmarnean L, Vacaras V, Tantau A-I. From Gut to Brain: Uncovering Potential Serum Biomarkers Connecting Inflammatory Bowel Diseases to Neurodegenerative Diseases. International Journal of Molecular Sciences. 2024; 25(11):5676. https://doi.org/10.3390/ijms25115676
Chicago/Turabian StyleSarb, Oliviu-Florentiu, Adriana-Daniela Sarb, Maria Iacobescu, Irina-Maria Vlad, Mircea-Vasile Milaciu, Lorena Ciurmarnean, Vitalie Vacaras, and Alina-Ioana Tantau. 2024. "From Gut to Brain: Uncovering Potential Serum Biomarkers Connecting Inflammatory Bowel Diseases to Neurodegenerative Diseases" International Journal of Molecular Sciences 25, no. 11: 5676. https://doi.org/10.3390/ijms25115676
APA StyleSarb, O.-F., Sarb, A.-D., Iacobescu, M., Vlad, I.-M., Milaciu, M.-V., Ciurmarnean, L., Vacaras, V., & Tantau, A.-I. (2024). From Gut to Brain: Uncovering Potential Serum Biomarkers Connecting Inflammatory Bowel Diseases to Neurodegenerative Diseases. International Journal of Molecular Sciences, 25(11), 5676. https://doi.org/10.3390/ijms25115676