
SCN1A Channels a Wide Range of Epileptic Phenotypes: Report of Novel and Known Variants with Variable Presentations

 , , , and
, , , and
Abstract
1. Introduction
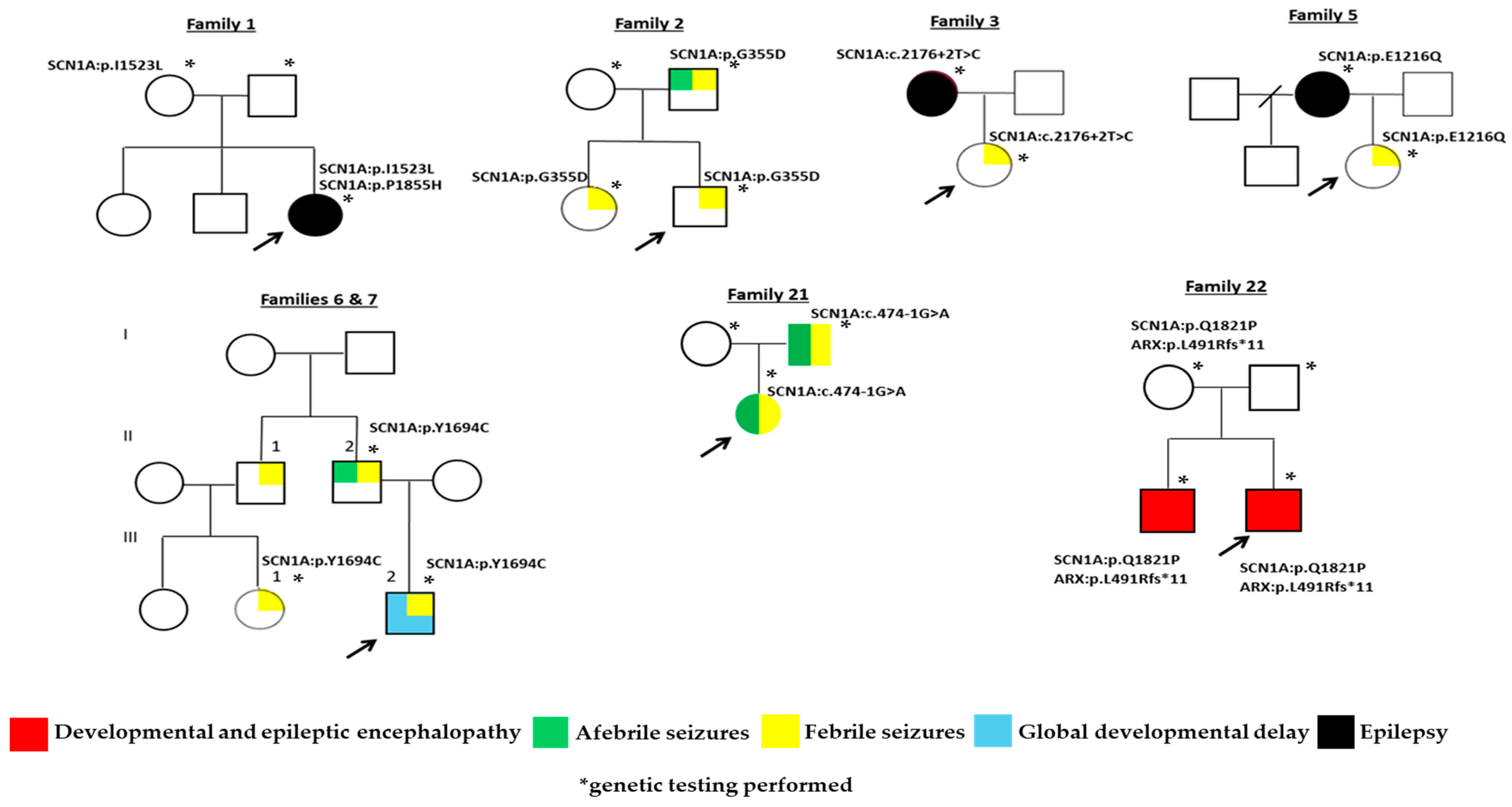
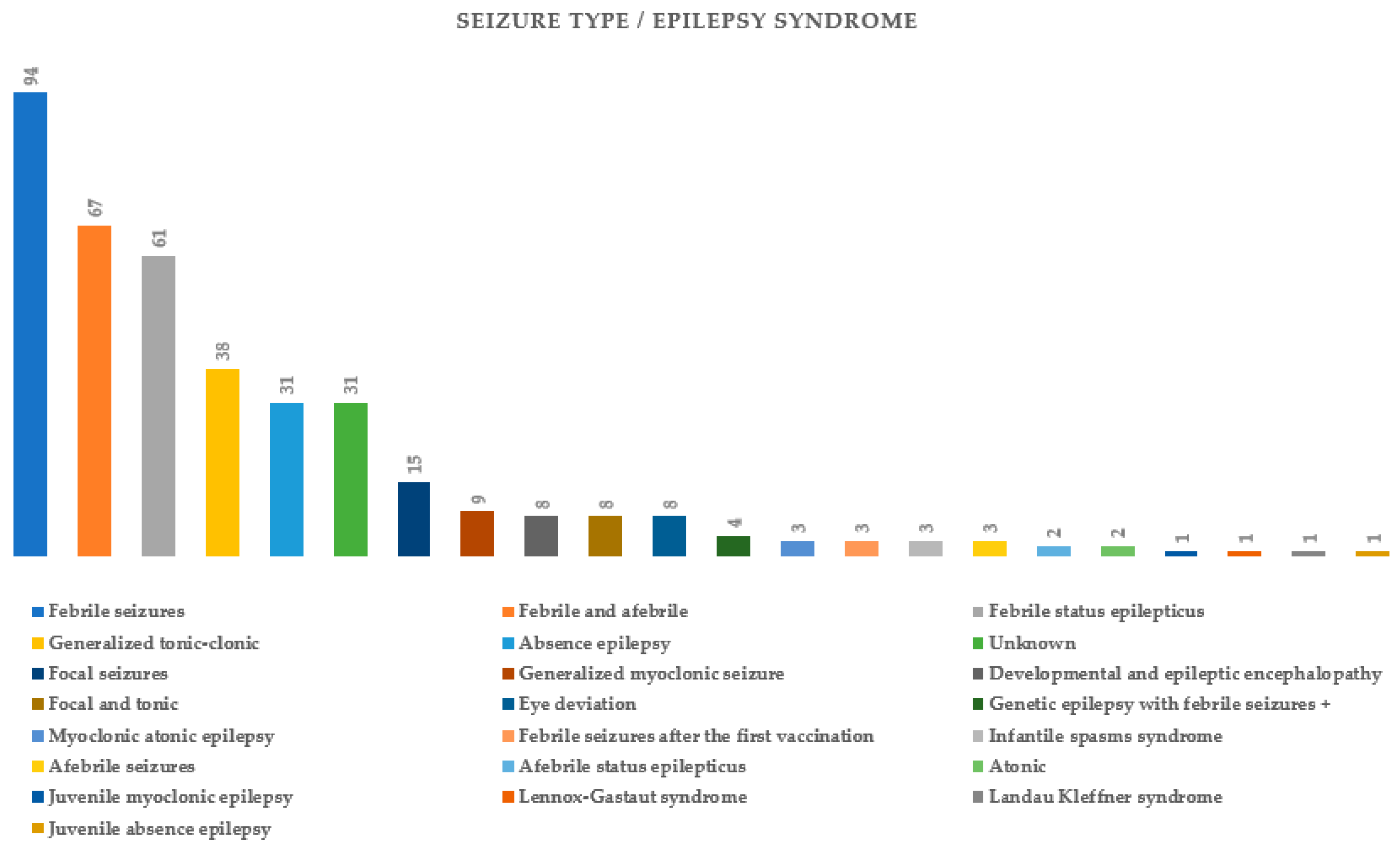
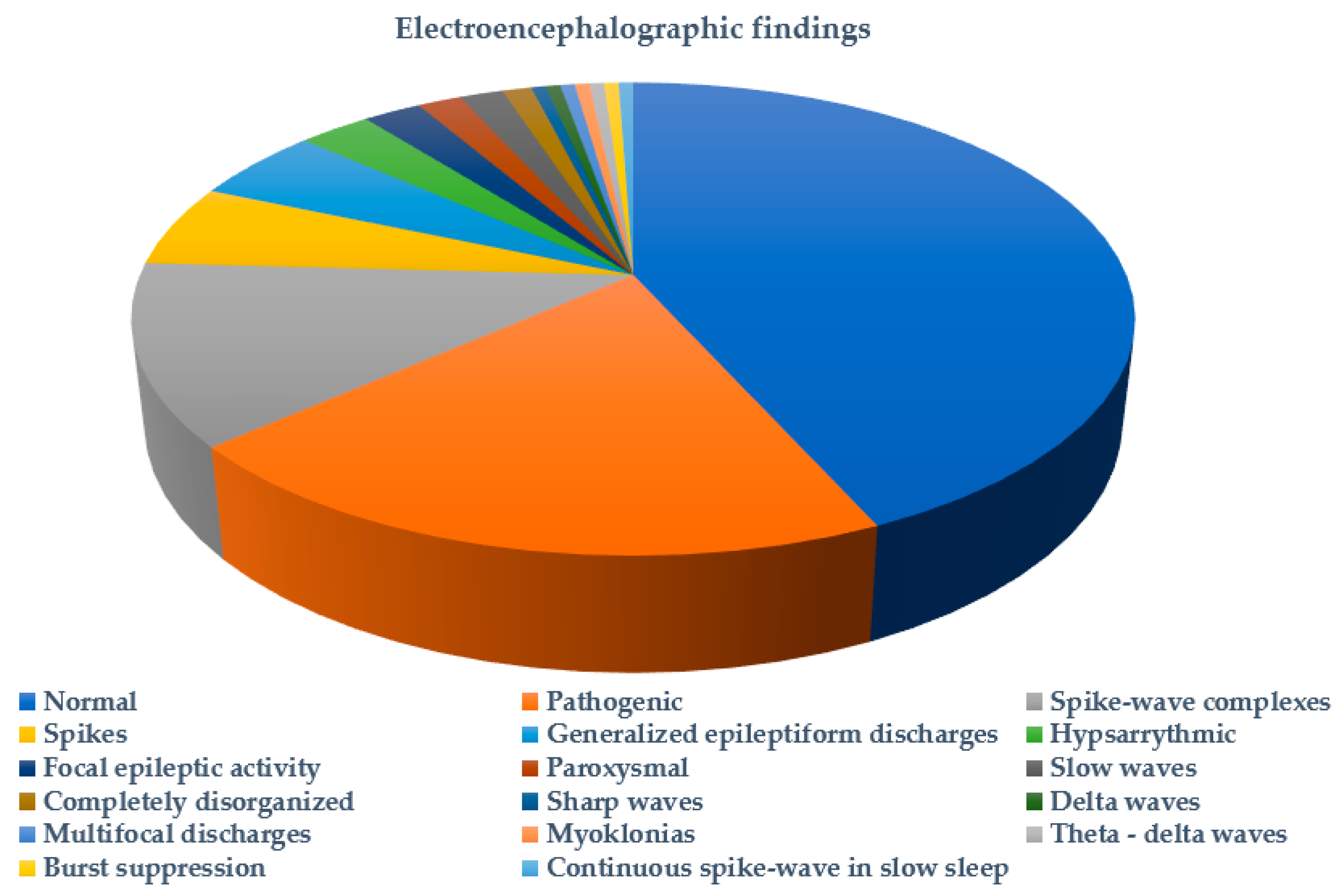
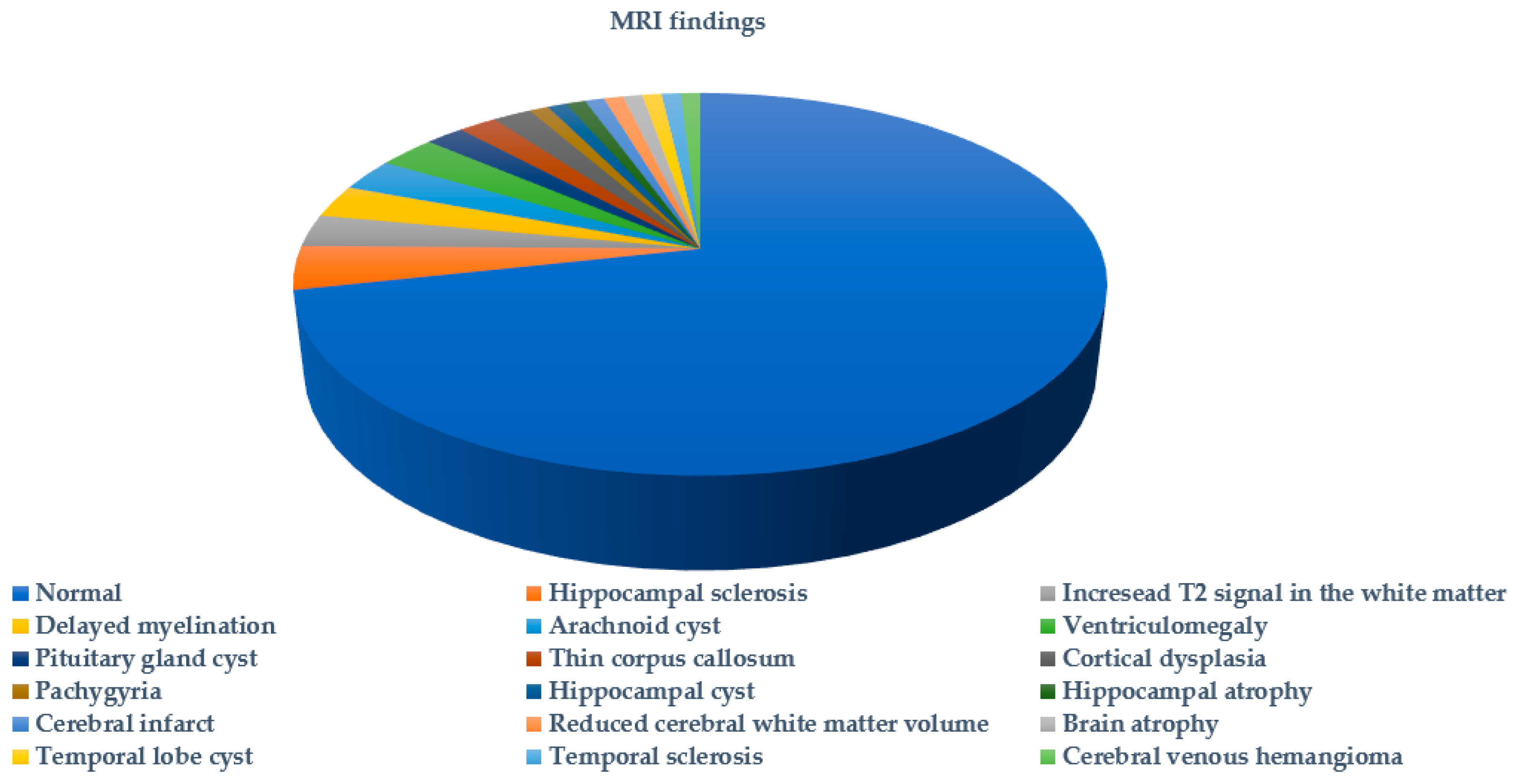
2. Results
3. Discussion
4. Materials and Methods
4.1. Patients
4.2. Methods
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Fisher, R.S.; Cross, J.H.; D’Souza, C.; French, J.A.; Haut, S.R.; Higurashi, N.; Hirsch, E.; Jansen, F.E.; Lagae, L.; Moshé, S.L.; et al. Instruction manual for the ILAE 2017 operational classification of seizure types. Epilepsia 2017, 58, 531–542. [Google Scholar] [CrossRef] [PubMed]
- Scheffer, I.E.; Berkovic, S.; Capovilla, G.; Connolly, M.B.; French, J.; Guilhoto, L.; Hirsch, E.; Jain, S.; Mathern, G.W.; Moshé, S.L.; et al. ILAE classification of the epilepsies: Position paper of the ILAE Commission for Classification and Terminology. Epilepsia 2017, 58, 512–521. [Google Scholar] [CrossRef] [PubMed]
- Hong, R.; Zheng, T.; Marra, V.; Yang, D.; Liu, J.K. Multi-scale modelling of the epileptic brain: Advantages of computational therapy exploration. J. Neural Eng. 2024, 21, 021002. [Google Scholar] [CrossRef] [PubMed]
- Gokben, S.; Onay, H.; Yilmaz, S.; Atik, T.; Serdaroglu, G.; Tekin, H.; Ozkinay, F. Targeted next generation sequencing: The diagnostic value in early-onset epileptic encephalopathy. Acta Neurol. Belg. 2017, 117, 131–138. [Google Scholar] [CrossRef] [PubMed]
- Mei, D.; Parrini, E.; Marini, C.; Guerrini, R. The Impact of Next-Generation Sequencing on the Diagnosis and Treatment of Epilepsy in Paediatric Patients. Mol. Diagn. Ther. 2017, 21, 357–373. [Google Scholar] [CrossRef]
- Staley, K. Molecular mechanisms of epilepsy. Nat. Neurosci. 2015, 18, 367–372. [Google Scholar] [CrossRef]
- Stafstrom, C.E. Epilepsy: A review of selected clinical syndromes and advances in basic science. J. Cereb. Blood Flow Metab. 2006, 26, 983–1004. [Google Scholar] [CrossRef]
- Perucca, E.; French, J.; Bialer, M. Development of new antiepileptic drugs: Challenges, incentives, and recent advances. Lancet Neurol. 2007, 6, 793–804. [Google Scholar] [CrossRef]
- Brunklaus, A.; Lal, D. Sodium channel epilepsies and neurodevelopmental disorders: From disease mechanisms to clinical application. Dev. Med. Child Neurol. 2020, 62, 784–792. [Google Scholar] [CrossRef]
- Stafstrom, C.E. Severe epilepsy syndromes of early childhood: The link between genetics and pathophysiology with a focus on SCN1A mutations. J. Child Neurol. 2009, 24, 15s–23s. [Google Scholar] [CrossRef]
- Chen, C.; Fang, F.; Wang, X.; Lv, J.; Wang, X.; Jin, H. Phenotypic and Genotypic Characteristics of SCN1A Associated Seizure Diseases. Front. Mol. Neurosci. 2022, 15, 821012. [Google Scholar] [CrossRef] [PubMed]
- Meng, H.; Xu, H.Q.; Yu, L.; Lin, G.W.; He, N.; Su, T.; Shi, Y.W.; Li, B.; Wang, J.; Liu, X.R.; et al. The SCN1A mutation database: Updating information and analysis of the relationships among genotype, functional alteration, and phenotype. Hum. Mutat. 2015, 36, 573–580. [Google Scholar] [CrossRef] [PubMed]
- Dravet, C. The core Dravet syndrome phenotype. Epilepsia 2011, 52 (Suppl. 2), 3–9. [Google Scholar] [CrossRef]
- Scheffer, I.E.; Nabbout, R. SCN1A-related phenotypes: Epilepsy and beyond. Epilepsia 2019, 60 (Suppl. 3), S17–S24. [Google Scholar] [CrossRef] [PubMed]
- Steel, D.; Symonds, J.D.; Zuberi, S.M.; Brunklaus, A. Dravet syndrome and its mimics: Beyond SCN1A. Epilepsia 2017, 58, 1807–1816. [Google Scholar] [CrossRef] [PubMed]
- Mei, D.; Cetica, V.; Marini, C.; Guerrini, R. Dravet syndrome as part of the clinical and genetic spectrum of sodium channel epilepsies and encephalopathies. Epilepsia 2019, 60 (Suppl. 3), S2–S7. [Google Scholar] [CrossRef] [PubMed]
- Goldberg-Stern, H.; Aharoni, S.; Afawi, Z.; Bennett, O.; Appenzeller, S.; Pendziwiat, M.; Kuhlenbäumer, G.; Basel-Vanagaite, L.; Shuper, A.; Korczyn, A.D.; et al. Broad phenotypic heterogeneity due to a novel SCN1A mutation in a family with genetic epilepsy with febrile seizures plus. J. Child Neurol. 2014, 29, 221–226. [Google Scholar] [CrossRef] [PubMed]
- Jaber, D.; Gitiaux, C.; Blesson, S.; Marguet, F.; Buard, D.; Varela Salgado, M.; Kaminska, A.; Saada, J.; Fallet-Bianco, C.; Martinovic, J.; et al. De novo mutations of SCN1A are responsible for arthrogryposis broadening the SCN1A-related phenotypes. J. Med. Genet. 2021, 58, 737–742. [Google Scholar] [CrossRef]
- Scheffer, I.E.; Liao, J. Deciphering the concepts behind “Epileptic encephalopathy” and “Developmental and epileptic encephalopathy”. Eur. J. Paediatr. Neurol. EJPN Off. J. Eur. Paediatr. Neurol. Soc. 2020, 24, 11–14. [Google Scholar] [CrossRef]
- Zhou, P.; He, N.; Zhang, J.W.; Lin, Z.J.; Wang, J.; Yan, L.M.; Meng, H.; Tang, B.; Li, B.M.; Liu, X.R.; et al. Novel mutations and phenotypes of epilepsy-associated genes in epileptic encephalopathies. Genes Brain Behav. 2018, 17, e12456. [Google Scholar] [CrossRef]
- Wheless, J.W.; Fulton, S.P.; Mudigoudar, B.D. Dravet Syndrome: A Review of Current Management. Pediatr. Neurol. 2020, 107, 28–40. [Google Scholar] [CrossRef] [PubMed]
- Veltra, D.; Tilemis, F.N.; Marinakis, N.M.; Svingou, M.; Mitrakos, A.; Kosma, K.; Tsoutsou, I.; Makrythanasis, P.; Theodorou, V.; Katsalouli, M.; et al. Combined exome analysis and exome depth assessment achieve a high diagnostic yield in an epilepsy case series, revealing significant genomic heterogeneity and novel mechanisms. Expert Rev. Mol. Diagn. 2023, 23, 85–103. [Google Scholar] [CrossRef]
- Brunklaus, A.; Ellis, R.; Stewart, H.; Aylett, S.; Reavey, E.; Jefferson, R.; Jain, R.; Chakraborty, S.; Jayawant, S.; Zuberi, S.M. Homozygous mutations in the SCN1A gene associated with genetic epilepsy with febrile seizures plus and Dravet syndrome in 2 families. Eur. J. Paediatr. Neurol. EJPN Off. J. Eur. Paediatr. Neurol. Soc. 2015, 19, 484–488. [Google Scholar] [CrossRef] [PubMed]
- Marco Hernández, A.V.; Tomás Vila, M.; Caro Llopis, A.; Monfort, S.; Martinez, F. Case Report: Novel Homozygous Likely Pathogenic SCN1A Variant with Autosomal Recessive Inheritance and Review of the Literature. Front. Neurol. 2021, 12, 784892. [Google Scholar] [CrossRef]
- Martin, M.S.; Dutt, K.; Papale, L.A.; Dubé, C.M.; Dutton, S.B.; de Haan, G.; Shankar, A.; Tufik, S.; Meisler, M.H.; Baram, T.Z.; et al. Altered function of the SCN1A voltage-gated sodium channel leads to gamma-aminobutyric acid-ergic (GABAergic) interneuron abnormalities. J. Biol. Chem. 2010, 285, 9823–9834. [Google Scholar] [CrossRef]
- Stephenson, J.D.; Laskowski, R.A.; Nightingale, A.; Hurles, M.E.; Thornton, J.M. VarMap: A web tool for mapping genomic coordinates to protein sequence and structure and retrieving protein structural annotations. Bioinformatics 2019, 35, 4854–4856. [Google Scholar] [CrossRef] [PubMed]
- Zuberi, S.M.; Brunklaus, A.; Birch, R.; Reavey, E.; Duncan, J.; Forbes, G.H. Genotype-phenotype associations in SCN1A-related epilepsies. Neurology 2011, 76, 594–600. [Google Scholar] [CrossRef]
- Catarino, C.B.; Liu, J.Y.; Liagkouras, I.; Gibbons, V.S.; Labrum, R.W.; Ellis, R.; Woodward, C.; Davis, M.B.; Smith, S.J.; Cross, J.H.; et al. Dravet syndrome as epileptic encephalopathy: Evidence from long-term course and neuropathology. Brain J. Neurol. 2011, 134, 2982–3010. [Google Scholar] [CrossRef] [PubMed]
- Kato, M.; Dobyns, W.B. X-linked lissencephaly with abnormal genitalia as a tangential migration disorder causing intractable epilepsy: Proposal for a new term, “interneuronopathy”. J. Child Neurol. 2005, 20, 392–397. [Google Scholar] [CrossRef]
- Posey, J.E.; Harel, T.; Liu, P.; Rosenfeld, J.A.; James, R.A.; Coban Akdemir, Z.H.; Walkiewicz, M.; Bi, W.; Xiao, R.; Ding, Y.; et al. Resolution of Disease Phenotypes Resulting from Multilocus Genomic Variation. N. Engl. J. Med. 2017, 376, 21–31. [Google Scholar] [CrossRef]
- Martins Custodio, H.; Clayton, L.M.; Bellampalli, R.; Pagni, S.; Silvennoinen, K.; Caswell, R.; Brunklaus, A.; Guerrini, R.; Koeleman, B.P.C.; Lemke, J.R.; et al. Widespread genomic influences on phenotype in Dravet syndrome, a ‘monogenic’ condition. Brain J. Neurol. 2023, 146, 3885–3897. [Google Scholar] [CrossRef] [PubMed]
- Richards, S.; Aziz, N.; Bale, S.; Bick, D.; Das, S.; Gastier-Foster, J.; Grody, W.W.; Hegde, M.; Lyon, E.; Spector, E.; et al. Standards and guidelines for the interpretation of sequence variants: A joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet. Med. 2015, 17, 405–424. [Google Scholar] [CrossRef] [PubMed]
- Verbeek, N.E.; van der Maas, N.A.; Jansen, F.E.; van Kempen, M.J.; Lindhout, D.; Brilstra, E.H. Prevalence of SCN1A-related dravet syndrome among children reported with seizures following vaccination: A population-based ten-year cohort study. PLoS ONE 2013, 8, e65758. [Google Scholar] [CrossRef]
- Yang, X.; Liu, A.; Xu, X.; Yang, X.; Zeng, Q.; Ye, A.Y.; Yu, Z.; Wang, S.; Huang, A.Y.; Wu, X.; et al. Genomic mosaicism in paternal sperm and multiple parental tissues in a Dravet syndrome cohort. Sci. Rep. 2017, 7, 15677. [Google Scholar] [CrossRef]
- Xu, X.; Yang, X.; Wu, Q.; Liu, A.; Yang, X.; Ye, A.Y.; Huang, A.Y.; Li, J.; Wang, M.; Yu, Z.; et al. Amplicon Resequencing Identified Parental Mosaicism for Approximately 10% of “de novo” SCN1A Mutations in Children with Dravet Syndrome. Hum. Mutat. 2015, 36, 861–872. [Google Scholar] [CrossRef]
- Djémié, T.; Weckhuysen, S.; von Spiczak, S.; Carvill, G.L.; Jaehn, J.; Anttonen, A.K.; Brilstra, E.; Caglayan, H.S.; de Kovel, C.G.; Depienne, C.; et al. Pitfalls in genetic testing: The story of missed SCN1A mutations. Mol. Genet. Genom. Med. 2016, 4, 457–464. [Google Scholar] [CrossRef]
- de Lange, I.M.; Koudijs, M.J.; van ‘t Slot, R.; Sonsma, A.C.M.; Mulder, F.; Carbo, E.C.; van Kempen, M.J.A.; Nijman, I.J.; Ernst, R.F.; Savelberg, S.M.C.; et al. Assessment of parental mosaicism in SCN1A-related epilepsy by single-molecule molecular inversion probes and next-generation sequencing. J. Med. Genet. 2019, 56, 75–80. [Google Scholar] [CrossRef] [PubMed]
- Zucca, C.; Redaelli, F.; Epifanio, R.; Zanotta, N.; Romeo, A.; Lodi, M.; Veggiotti, P.; Airoldi, G.; Panzeri, C.; Romaniello, R.; et al. Cryptogenic epileptic syndromes related to SCN1A: Twelve novel mutations identified. Arch. Neurol. 2008, 65, 489–494. [Google Scholar] [CrossRef]
- Cetica, V.; Chiari, S.; Mei, D.; Parrini, E.; Grisotto, L.; Marini, C.; Pucatti, D.; Ferrari, A.; Sicca, F.; Specchio, N.; et al. Clinical and genetic factors predicting Dravet syndrome in infants with SCN1A mutations. Neurology 2017, 88, 1037–1044. [Google Scholar] [CrossRef]
- Lindy, A.S.; Stosser, M.B.; Butler, E.; Downtain-Pickersgill, C.; Shanmugham, A.; Retterer, K.; Brandt, T.; Richard, G.; McKnight, D.A. Diagnostic outcomes for genetic testing of 70 genes in 8565 patients with epilepsy and neurodevelopmental disorders. Epilepsia 2018, 59, 1062–1071. [Google Scholar] [CrossRef]
- Xu, X.; Zhang, Y.; Sun, H.; Liu, X.; Yang, X.; Xiong, H.; Jiang, Y.; Bao, X.; Wang, S.; Yang, Z.; et al. Early clinical features and diagnosis of Dravet syndrome in 138 Chinese patients with SCN1A mutations. Brain Dev. 2014, 36, 676–681. [Google Scholar] [CrossRef] [PubMed]
- Desmet, F.O.; Hamroun, D.; Lalande, M.; Collod-Beroud, G.; Claustres, M.; Beroud, C. Human Splicing Finder: An online bioinformatics tool to predict splicing signals. Nucleic Acids Res. 2009, 37, e67. [Google Scholar] [CrossRef] [PubMed]
- Lemke, J.R.; Riesch, E.; Scheurenbrand, T.; Schubach, M.; Wilhelm, C.; Steiner, I.; Hansen, J.; Courage, C.; Gallati, S.; Bürki, S.; et al. Targeted next generation sequencing as a diagnostic tool in epileptic disorders. Epilepsia 2012, 53, 1387–1398. [Google Scholar] [CrossRef]
- Brunklaus, A.; Leu, C.; Gramm, M.; Pérez-Palma, E.; Iqbal, S.; Lal, D. Time to move beyond genetics towards biomedical data-driven translational genomic research in severe paediatric epilepsies. Eur. J. Paediatr. Neurol. EJPN Off. J. Eur. Paediatr. Neurol. Soc. 2020, 24, 35–39. [Google Scholar] [CrossRef] [PubMed]
- Schuster, J.; Lu, X.; Dang, Y.; Klar, J.; Wenz, A.; Dahl, N.; Chen, X. Epigenetic insights into GABAergic development in Dravet Syndrome iPSC and therapeutic implications. bioRxiv 2023. [Google Scholar] [CrossRef]
- Kearney, J.A. Genetic modifiers of neurological disease. Curr. Opin. Genet. Dev. 2011, 21, 349–353. [Google Scholar] [CrossRef]
- Brunklaus, A.; Schorge, S.; Smith, A.D.; Ghanty, I.; Stewart, K.; Gardiner, S.; Du, J.; Pérez-Palma, E.; Symonds, J.D.; Collier, A.C.; et al. SCN1A variants from bench to bedside-improved clinical prediction from functional characterization. Hum. Mutat. 2020, 41, 363–374. [Google Scholar] [CrossRef] [PubMed]
- Brunklaus, A.; Pérez-Palma, E.; Ghanty, I.; Xinge, J.; Brilstra, E.; Ceulemans, B.; Chemaly, N.; de Lange, I.; Depienne, C.; Guerrini, R.; et al. Development and Validation of a Prediction Model for Early Diagnosis of SCN1A-Related Epilepsies. Neurology 2022, 98, e1163–e1174. [Google Scholar] [CrossRef]
- Yamagata, T.; Raveau, M.; Kobayashi, K.; Miyamoto, H.; Tatsukawa, T.; Ogiwara, I.; Itohara, S.; Hensch, T.K.; Yamakawa, K. CRISPR/dCas9-based Scn1a gene activation in inhibitory neurons ameliorates epileptic and behavioral phenotypes of Dravet syndrome model mice. Neurobiol. Dis. 2020, 141, 104954. [Google Scholar] [CrossRef]
- Colasante, G.; Lignani, G.; Brusco, S.; Di Berardino, C.; Carpenter, J.; Giannelli, S.; Valassina, N.; Bido, S.; Ricci, R.; Castoldi, V.; et al. dCas9-Based Scn1a Gene Activation Restores Inhibitory Interneuron Excitability and Attenuates Seizures in Dravet Syndrome Mice. Mol. Ther. 2020, 28, 235–253. [Google Scholar] [CrossRef]
- Tanenhaus, A.; Stowe, T.; Young, A.; McLaughlin, J.; Aeran, R.; Lin, I.W.; Li, J.; Hosur, R.; Chen, M.; Leedy, J.; et al. Cell-Selective Adeno-Associated Virus-Mediated SCN1A Gene Regulation Therapy Rescues Mortality and Seizure Phenotypes in a Dravet Syndrome Mouse Model and Is Well Tolerated in Nonhuman Primates. Hum. Gene Ther. 2022, 33, 579–597. [Google Scholar] [CrossRef] [PubMed]
- Zeng, B.; Zhang, H.; Lu, Q.; Fu, Q.; Yan, Y.; Lu, W.; Ma, P.; Feng, C.; Qin, J.; Luo, L.; et al. Identification of five novel SCN1A variants. Front. Behav. Neurosci. 2023, 17, 1272748. [Google Scholar] [CrossRef] [PubMed]
- Tilemis, F.N.; Marinakis, N.M.; Veltra, D.; Svingou, M.; Kekou, K.; Mitrakos, A.; Tzetis, M.; Kosma, K.; Makrythanasis, P.; Traeger-Synodinos, J.; et al. Germline CNV Detection through Whole-Exome Sequencing (WES) Data Analysis Enhances Resolution of Rare Genetic Diseases. Genes 2023, 14, 1490. [Google Scholar] [CrossRef]
- Riggs, E.R.; Andersen, E.F.; Cherry, A.M.; Kantarci, S.; Kearney, H.; Patel, A.; Raca, G.; Ritter, D.I.; South, S.T.; Thorland, E.C.; et al. Technical standards for the interpretation and reporting of constitutional copy-number variants: A joint consensus recommendation of the American College of Medical Genetics and Genomics (ACMG) and the Clinical Genome Resource (ClinGen). Genet. Med. Off. J. Am. Coll. Med. Genet. 2020, 22, 245–257. [Google Scholar] [CrossRef] [PubMed]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Variant (Reference Transcript GRCh37: NM_001165963.4) | Population Frequency | GnomAD | VarMap | SIFT | Revel | Location in the Channel | Expected Impact on Protein Function |
|---|---|---|---|---|---|---|---|
| c.4567A>Τ:p.I1523L | - | - | Likely deleterious (0.39) | 0.001 | 0.777 | DIII–DIV loop | Unknown |
| c.5564C>A:p.P1855H | - | - | Likely deleterious (0.94) | 0 | 0.939 | C-terminal | Unknown |
| c.2176+2T>C | - | - | N.A. | N.A. | N.A. | DII, S4 | LOF |
| c.3646G>C:p.E1216Q | - | - | Likely deleterious (0.59) | 0 | 0.909 | DIII, S1 | Unknown |
| c.4331C>A:p.S1444Y | - | - | Likely deleterious (1.02) | 0 | 0.894 | DIII, loop S5–S6 | Unknown |
| c.1130_1131delGAinsAC:p.R377H | - | - | N.A. | N.A. | N.A. | DI, loop S5–S6 | Unknown |
| c.1574_1580delCTGAGGA:p.E526Afs*16 | - | - | N.A. | N.A. | N.A. | Loop DI–DII | LOF |
| c.4620A>G:p.R1540R | - | - | N.A. | N.A. | N.A. | DIV, S1 | Unknown |
| c.5462A>C:p.Q1821P | - | - | Likely deleterious (0.39) | 0.001 | 0.893 | C-terminal | Unknown |
| P | G | Age of Diagnosis | Variant (Reference Transcript GRCh37: NM_001165963.4) | ACMG/ ACMG and ClinGen Classification (for CNVs) | Inheritance | Clinical Presentation | EEG | MRI | Treatment |
|---|---|---|---|---|---|---|---|---|---|
| 1 | F | 1.5 y | c.4567A>Τ p.I1523L | Likely pathogenic (PM1, PM2, PP2, PP3) | Maternal | Seizure onset at 1 y. | Normal. | Increased Τ2 and FLAIR signals in the subcortical white matter in the frontal lobes. | LEV, VPA. |
| c.5564C>A p.P1855H | Pathogenic (PS2, PM1, PM2, PM5, PP2, PP3) | De novo | |||||||
| 2 | M | 7 m | c.1064G>A p.G355D | Pathogenic (PM1, ΡΜ2, ΡΡ2, PP3, PP5) | Paternal | FS at 7 m. | N.A. Diagnosis after first seizure. | N.A. | N.A. |
| 3 | F | 11 m | c.2176+2T>C | Pathogenic (PVS1, PP3, PM2) | Maternal |
| Normal. | N.A. | N.A. |
| 4 | M | 8 y | c.5536_5539delAAAC p.K1846Sfs*10 | Pathogenic (PVS1, PP3, PP5, PM2) | N.A. |
| Normal. | Normal. | LEV, VPA, RIS. |
| 5 | F | 3 y | c.3646G>C p.E1216Q | Pathogenic (PΜ1, PM2, PP2, PP3, PM5) | Maternal |
| N.A. | Normal. | VPA. |
| 6 | M | 1.5 y | c.5081A>G p.Y1694C | Likely pathogenic (PM1, PM2, PP2, PP3, PP5) | Paternal |
| N.A. Diagnosis after first seizure. | N.A. | VPA. |
| 7 | F | 9 m | c.5081A>G p.Y1694C | Likely pathogenic (PM1, PM2, PP2, PP3, PP5) | Paternal | FS onset at 9 m. | N.A. Diagnosis after first seizure. | N.A. | N.A. |
| 8 | F | 1 y | c.429_430delGT p.F144Yfs*5 | Pathogenic (PVS1, PM1, PP3, PP5) | N.A. |
| Normal. | Normal. | LEV, VPA. |
| 9 | M | 6 y | c.1060G>C p.A354P | Pathogenic (PS2, PΜ1, PM2, PP2, PP3) | De novo | DEE onset at 6 m. | Pathogenic. | N.A. | LEV, VPA, CLB. |
| 10 | M | 3 y | c.4331C>A p.S1444Y | Pathogenic (PS2, PM2, PP2, PP3, PP5) | De novo |
| Normal. | Normal. | N.A. |
| 11 | M | 4 y | c.4933C>T p.R1645X | Pathogenic (PVS1, PM2, PP3, PP5) | N.A. |
| N.A. | Normal. | VPA. |
| 12 | F | 3.5 y | c.2593C>T p.R865X | Pathogenic (PS2, PVS1, PP3, PP5, PM2) | De novo |
| Myoclonus and photosensitivity. | Normal. | PB, VPA, LTG, CLB. |
| 13 | F | 3 y | c.5388T>A p.S1796R | Pathogenic (PS2, PM1, PM2, PP2, PP3, PP5) | De novo | Complex FS and AFS onset: 2 y. | N.A. | N.A. | N.A. |
| 14 | F | 2 y | c.1130_1131delGAinsAC p.R377H | Pathogenic (PM1, PM2, PM5, PP2, PP3) | N.A. | Multiple FS episodes. Diagnosis during Influenza B infection. | N.A. | N.A. | N.A. |
| 15 | F | 18 y | c.1574_1580delCTGAGGA p.E526Afs*16 | Pathogenic (PS2, PVS1, PM2, PP3, PP5) | De novo |
| Initially pathogenic and then with photosensitivity. Now normal. | Venous hemangioma of the pituitary gland. | Now without treatment. Previously seizure aggravation was reported with the use of CBZ. |
| 16 | F | 5 y | chr2:166,848,222–167,005,693del size: 157.5 kb contains the whole SCN1A gene. | Pathogenic (1) (2B, 2C, 4L) | N.A. |
| Normal. | Normal. | LEV, VPA. Seizure aggravation was reported with the use of OXC. |
| 17 | M | 2.5 y | c.3762T>A p.Y1254X | Pathogenic (PS2, PVS1, PM2, PP3, PP5) | De novo |
| Nonspecific findings. | Normal. | LEV, VPA, TPM. |
| 18 | M | 2 y | c.1702C>T p.R568X | Pathogenic (PS2, PVS1, PM2, PP3, PP5) | De novo |
| Pathogenic. | Atrophy of the left temporal lobe with progressive atrophy and gliosis/sclerosis of the associated hippocampus. | LEV, VPA, LCM. |
| 19 | F | 2 y | c.4620A>G p.R1540R | VUS (PS2, PM2) | De novo |
| Delta waves. | Normal. | LEV. |
| 20 ‡ | M | 31 y | c.2244G>A p.W748X | Pathogenic (PVS1, PM2, PP5) | De novo |
| N.A. | N.A. | N.A. |
| 21 ‡ | F | 10 m | c.474-1G>A | Pathogenic (PVS1, PP5, PM2) | Paternal |
| N.A. | N.A. | LEV, VPA, CLB. |
| 22 ‡,^ | M | 6 m | c.5462A>C p.Q1821P | Likely pathogenic (PM2, PP2, PP3) | Maternal |
| Pathogenic. | Hypoplasia of corpus callosum. | PB, LEV. |
| ARX gene NM_139058.3: c.1472delT: p.L491Rfs*11 | Pathogenic (PVS1, PM2, PP3) | ||||||||
| 23 ‡,^ | M | 13 y | c.3985C>Τ p.R1329X | Pathogenic (PVS1, PM2, PP3, PP5) | N.A. |
| N.A. | Normal. | LEV, VPA. |
| 24 ‡‡,^ | M | 1.5 y | Chr2:166,892,579–167,005,693del (includes exons 1–16 of SCN1A gene) | Likely pathogenic (0.9–1A, 2B, 2C, 3A) | De novo |
| Normal. | Normal. | LEV, PHT. |
| GLI3 gene NM_000168.6:c.793delG:p.A265Pfs*45 | Pathogenic (PVS1, PM2, PP3) | ||||||||
| 25 | F | 1 y | c.1076A>G p.N359S | Pathogenic (PS2, PM1, PM2, PM5, PP3, PP5) | De novo |
| Normal. | N.A. | LEV, VPA. |
| Febrile seizures | 19.53% (67/343) |
| Epilepsy | 17.20% (59/343) |
| ASD | 0.87% (3/343) |
| Intellectual development disorder | 0.87% (3/343) |
| Psychiatric disorders | 0.58% (2/343) |
| Encephalitis | 0.29% (1/343) |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Veltra, D.; Theodorou, V.; Katsalouli, M.; Vorgia, P.; Niotakis, G.; Tsaprouni, T.; Pons, R.; Kosma, K.; Kampouraki, A.; Tsoutsou, I.; et al. SCN1A Channels a Wide Range of Epileptic Phenotypes: Report of Novel and Known Variants with Variable Presentations. Int. J. Mol. Sci. 2024, 25, 5644. https://doi.org/10.3390/ijms25115644
Veltra D, Theodorou V, Katsalouli M, Vorgia P, Niotakis G, Tsaprouni T, Pons R, Kosma K, Kampouraki A, Tsoutsou I, et al. SCN1A Channels a Wide Range of Epileptic Phenotypes: Report of Novel and Known Variants with Variable Presentations. International Journal of Molecular Sciences. 2024; 25(11):5644. https://doi.org/10.3390/ijms25115644
Chicago/Turabian StyleVeltra, Danai, Virginia Theodorou, Marina Katsalouli, Pelagia Vorgia, Georgios Niotakis, Triantafyllia Tsaprouni, Roser Pons, Konstantina Kosma, Afroditi Kampouraki, Irene Tsoutsou, and et al. 2024. "SCN1A Channels a Wide Range of Epileptic Phenotypes: Report of Novel and Known Variants with Variable Presentations" International Journal of Molecular Sciences 25, no. 11: 5644. https://doi.org/10.3390/ijms25115644
APA StyleVeltra, D., Theodorou, V., Katsalouli, M., Vorgia, P., Niotakis, G., Tsaprouni, T., Pons, R., Kosma, K., Kampouraki, A., Tsoutsou, I., Makrythanasis, P., Kekou, K., Traeger-Synodinos, J., & Sofocleous, C. (2024). SCN1A Channels a Wide Range of Epileptic Phenotypes: Report of Novel and Known Variants with Variable Presentations. International Journal of Molecular Sciences, 25(11), 5644. https://doi.org/10.3390/ijms25115644