
Immunological Aspects of Cancer Cell Metabolism


Abstract
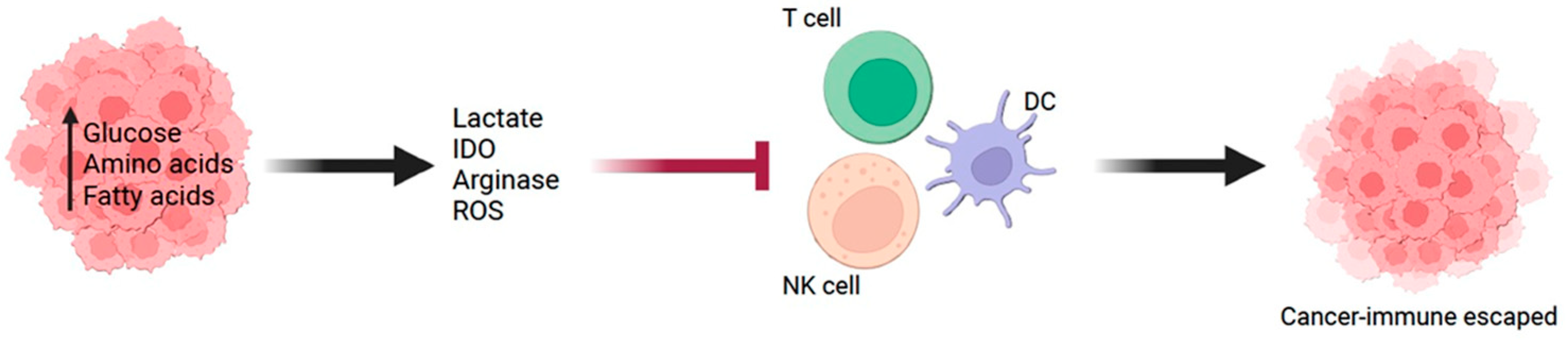
1. Background
2. Immunological Components of Tumor Microenvironment
3. Metabolic Changes of Cancer Cells
4. Metabolic Molecules in Cancer Cells Associated with Immunosuppressive TME
5. Metabolic Status in Cancer Cells Associated with Antitumor Immune Cell Function
6. Antioxidant Metabolism Associated with Cancer Cell Immune Escape
7. Targeting Cancer Cell Metabolism to Improve Immunotherapy Response
8. Future Directions
9. Concluding Remarks
Author Contributions
Funding
Conflicts of Interest
References
- Hanahan, D. Hallmarks of Cancer: New Dimensions. Cancer Discov. 2022, 12, 31–46. [Google Scholar] [CrossRef]
- Dunn, G.P.; Bruce, A.T.; Ikeda, H.; Old, L.J.; Schreiber, R.D. Cancer immunoediting: From immunosurveillance to tumor escape. Nat. Immunol. 2002, 3, 991–998. [Google Scholar] [CrossRef]
- Beatty, G.L.; Gladney, W.L. Immune escape mechanisms as a guide for cancer immunotherapy. Clin. Cancer Res. 2015, 21, 687–692. [Google Scholar] [CrossRef]
- Hanahan, D.; Coussens, L.M. Accessories to the crime: Functions of cells recruited to the tumor microenvironment. Cancer Cell 2012, 21, 309–322. [Google Scholar] [CrossRef]
- Quail, D.F.; Joyce, J.A. Microenvironmental regulation of tumor progression and metastasis. Nat. Med. 2013, 19, 1423–1437. [Google Scholar] [CrossRef]
- Chang, C.H.; Qiu, J.; O’Sullivan, D.; Buck, M.D.; Noguchi, T.; Curtis, J.D.; Chen, Q.; Gindin, M.; Gubin, M.M.; van der Windt, G.J.; et al. Metabolic Competition in the Tumor Microenvironment Is a Driver of Cancer Progression. Cell 2015, 162, 1229–1241. [Google Scholar] [CrossRef]
- Warburg, O.; Wind, F.; Negelein, E. The Metabolism of Tumors in the Body. J. Gen. Physiol. 1927, 8, 519–530. [Google Scholar] [CrossRef]
- Yu, M.; Yongzhi, H.; Chen, S.; Luo, X.; Lin, Y.; Zhou, Y.; Jin, H.; Hou, B.; Deng, Y.; Tu, L.; et al. The prognostic value of GLUT1 in cancers: A systematic review and meta-analysis. Oncotarget 2017, 8, 43356–43367. [Google Scholar] [CrossRef]
- Wu, L.; Jin, Y.; Zhao, X.; Tang, K.; Zhao, Y.; Tong, L.; Yu, X.; Xiong, K.; Luo, C.; Zhu, J.; et al. Tumor aerobic glycolysis confers immune evasion through modulating sensitivity to T cell-mediated bystander killing via TNF-alpha. Cell Metab. 2023, 35, 1580–1596.e1589. [Google Scholar] [CrossRef]
- Li, R.; Mei, S.; Ding, Q.; Wang, Q.; Yu, L.; Zi, F. A pan-cancer analysis of the role of hexokinase II (HK2) in human tumors. Sci. Rep. 2022, 12, 18807. [Google Scholar] [CrossRef]
- Zhang, J.; Pavlova, N.N.; Thompson, C.B. Cancer cell metabolism: The essential role of the nonessential amino acid, glutamine. EMBO J. 2017, 36, 1302–1315. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.; Zhao, T.; Li, Z.; Wang, L.; Yuan, S.; Sun, L. The role of ASCT2 in cancer: A review. Eur. J. Pharmacol. 2018, 837, 81–87. [Google Scholar] [CrossRef] [PubMed]
- Wen, Y.A.; Xing, X.; Harris, J.W.; Zaytseva, Y.Y.; Mitov, M.I.; Napier, D.L.; Weiss, H.L.; Mark Evers, B.; Gao, T. Adipocytes activate mitochondrial fatty acid oxidation and autophagy to promote tumor growth in colon cancer. Cell Death Dis. 2017, 8, e2593. [Google Scholar] [CrossRef] [PubMed]
- Yao, C.H.; Fowle-Grider, R.; Mahieu, N.G.; Liu, G.Y.; Chen, Y.J.; Wang, R.; Singh, M.; Potter, G.S.; Gross, R.W.; Schaefer, J.; et al. Exogenous Fatty Acids Are the Preferred Source of Membrane Lipids in Proliferating Fibroblasts. Cell Chem. Biol. 2016, 23, 483–493. [Google Scholar] [CrossRef] [PubMed]
- Mashima, T.; Seimiya, H.; Tsuruo, T. De novo fatty-acid synthesis and related pathways as molecular targets for cancer therapy. Br. J. Cancer 2009, 100, 1369–1372. [Google Scholar] [CrossRef] [PubMed]
- Santos, C.R.; Schulze, A. Lipid metabolism in cancer. FEBS J. 2012, 279, 2610–2623. [Google Scholar] [CrossRef] [PubMed]
- Rohrig, F.; Schulze, A. The multifaceted roles of fatty acid synthesis in cancer. Nat. Rev. Cancer 2016, 16, 732–749. [Google Scholar] [CrossRef]
- Xu, S.; Chaudhary, O.; Rodriguez-Morales, P.; Sun, X.; Chen, D.; Zappasodi, R.; Xu, Z.; Pinto, A.F.M.; Williams, A.; Schulze, I.; et al. Uptake of oxidized lipids by the scavenger receptor CD36 promotes lipid peroxidation and dysfunction in CD8(+) T cells in tumors. Immunity 2021, 54, 1561–1577.E7. [Google Scholar] [CrossRef]
- Tsai, C.H.; Chuang, Y.M.; Li, X.; Yu, Y.R.; Tzeng, S.F.; Teoh, S.T.; Lindblad, K.E.; Di Matteo, M.; Cheng, W.C.; Hsueh, P.C.; et al. Immunoediting instructs tumor metabolic reprogramming to support immune evasion. Cell Metab. 2023, 35, 118–133.e117. [Google Scholar] [CrossRef]
- Kaplan, D.H.; Shankaran, V.; Dighe, A.S.; Stockert, E.; Aguet, M.; Old, L.J.; Schreiber, R.D. Demonstration of an interferon gamma-dependent tumor surveillance system in immunocompetent mice. Proc. Natl. Acad. Sci. USA 1998, 95, 7556–7561. [Google Scholar] [CrossRef]
- Shankaran, V.; Ikeda, H.; Bruce, A.T.; White, J.M.; Swanson, P.E.; Old, L.J.; Schreiber, R.D. IFNgamma and lymphocytes prevent primary tumour development and shape tumour immunogenicity. Nature 2001, 410, 1107–1111. [Google Scholar] [CrossRef] [PubMed]
- Street, S.E.; Cretney, E.; Smyth, M.J. Perforin and interferon-gamma activities independently control tumor initiation, growth, and metastasis. Blood 2001, 97, 192–197. [Google Scholar] [CrossRef] [PubMed]
- Young, H.A.; Hardy, K.J. Role of interferon-gamma in immune cell regulation. J. Leukoc. Biol. 1995, 58, 373–381. [Google Scholar] [CrossRef] [PubMed]
- Garcia-Diaz, A.; Shin, D.S.; Moreno, B.H.; Saco, J.; Escuin-Ordinas, H.; Rodriguez, G.A.; Zaretsky, J.M.; Sun, L.; Hugo, W.; Wang, X.; et al. Interferon Receptor Signaling Pathways Regulating PD-L1 and PD-L2 Expression. Cell Rep. 2017, 19, 1189–1201. [Google Scholar] [CrossRef] [PubMed]
- Garcia-Gutierrez, L.; Delgado, M.D.; Leon, J. MYC Oncogene Contributions to Release of Cell Cycle Brakes. Genes 2019, 10, 244. [Google Scholar] [CrossRef] [PubMed]
- Dong, Y.; Tu, R.; Liu, H.; Qing, G. Regulation of cancer cell metabolism: Oncogenic MYC in the driver’s seat. Signal Transduct. Target. Ther. 2020, 5, 124. [Google Scholar] [CrossRef]
- Fischer, K.; Hoffmann, P.; Voelkl, S.; Meidenbauer, N.; Ammer, J.; Edinger, M.; Gottfried, E.; Schwarz, S.; Rothe, G.; Hoves, S.; et al. Inhibitory effect of tumor cell-derived lactic acid on human T cells. Blood 2007, 109, 3812–3819. [Google Scholar] [CrossRef]
- Bohn, T.; Rapp, S.; Luther, N.; Klein, M.; Bruehl, T.J.; Kojima, N.; Aranda Lopez, P.; Hahlbrock, J.; Muth, S.; Endo, S.; et al. Tumor immunoevasion via acidosis-dependent induction of regulatory tumor-associated macrophages. Nat. Immunol. 2018, 19, 1319–1329. [Google Scholar] [CrossRef] [PubMed]
- Mu, X.; Shi, W.; Xu, Y.; Xu, C.; Zhao, T.; Geng, B.; Yang, J.; Pan, J.; Hu, S.; Zhang, C.; et al. Tumor-derived lactate induces M2 macrophage polarization via the activation of the ERK/STAT3 signaling pathway in breast cancer. Cell Cycle 2018, 17, 428–438. [Google Scholar] [CrossRef]
- Yang, X.; Lu, Y.; Hang, J.; Zhang, J.; Zhang, T.; Huo, Y.; Liu, J.; Lai, S.; Luo, D.; Wang, L.; et al. Lactate-Modulated Immunosuppression of Myeloid-Derived Suppressor Cells Contributes to the Radioresistance of Pancreatic Cancer. Cancer Immunol. Res. 2020, 8, 1440–1451. [Google Scholar] [CrossRef]
- Prendergast, G.C.; Smith, C.; Thomas, S.; Mandik-Nayak, L.; Laury-Kleintop, L.; Metz, R.; Muller, A.J. Indoleamine 2,3-dioxygenase pathways of pathogenic inflammation and immune escape in cancer. Cancer Immunol. Immunother. 2014, 63, 721–735. [Google Scholar] [CrossRef] [PubMed]
- Song, X.; Si, Q.; Qi, R.; Liu, W.; Li, M.; Guo, M.; Wei, L.; Yao, Z. Indoleamine 2,3-Dioxygenase 1: A Promising Therapeutic Target in Malignant Tumor. Front. Immunol. 2021, 12, 800630. [Google Scholar] [CrossRef] [PubMed]
- Wainwright, D.A.; Balyasnikova, I.V.; Chang, A.L.; Ahmed, A.U.; Moon, K.S.; Auffinger, B.; Tobias, A.L.; Han, Y.; Lesniak, M.S. IDO expression in brain tumors increases the recruitment of regulatory T cells and negatively impacts survival. Clin. Cancer Res. 2012, 18, 6110–6121. [Google Scholar] [CrossRef] [PubMed]
- Holmgaard, R.B.; Zamarin, D.; Li, Y.; Gasmi, B.; Munn, D.H.; Allison, J.P.; Merghoub, T.; Wolchok, J.D. Tumor-Expressed IDO Recruits and Activates MDSCs in a Treg-Dependent Manner. Cell Rep. 2015, 13, 412–424. [Google Scholar] [CrossRef] [PubMed]
- Caldwell, R.B.; Toque, H.A.; Narayanan, S.P.; Caldwell, R.W. Arginase: An old enzyme with new tricks. Trends Pharmacol. Sci. 2015, 36, 395–405. [Google Scholar] [CrossRef] [PubMed]
- Czystowska-Kuzmicz, M.; Sosnowska, A.; Nowis, D.; Ramji, K.; Szajnik, M.; Chlebowska-Tuz, J.; Wolinska, E.; Gaj, P.; Grazul, M.; Pilch, Z.; et al. Small extracellular vesicles containing arginase-1 suppress T-cell responses and promote tumor growth in ovarian carcinoma. Nat. Commun. 2019, 10, 3000. [Google Scholar] [CrossRef] [PubMed]
- Lowe, M.M.; Boothby, I.; Clancy, S.; Ahn, R.S.; Liao, W.; Nguyen, D.N.; Schumann, K.; Marson, A.; Mahuron, K.M.; Kingsbury, G.A.; et al. Regulatory T cells use arginase 2 to enhance their metabolic fitness in tissues. JCI Insight 2019, 4, e129756. [Google Scholar] [CrossRef] [PubMed]
- Mao, F.Y.; Zhao, Y.L.; Lv, Y.P.; Teng, Y.S.; Kong, H.; Liu, Y.G.; Wu, X.L.; Hao, C.J.; Chen, W.; Duan, M.B.; et al. CD45(+)CD33(low)CD11b(dim) myeloid-derived suppressor cells suppress CD8(+) T cell activity via the IL-6/IL-8-arginase I axis in human gastric cancer. Cell Death Dis. 2018, 9, 763. [Google Scholar] [CrossRef] [PubMed]
- Ren, W.; Zhang, X.; Li, W.; Feng, Q.; Feng, H.; Tong, Y.; Rong, H.; Wang, W.; Zhang, D.; Zhang, Z.; et al. Circulating and tumor-infiltrating arginase 1-expressing cells in gastric adenocarcinoma patients were mainly immature and monocytic Myeloid-derived suppressor cells. Sci. Rep. 2020, 10, 8056. [Google Scholar] [CrossRef]
- Wang, H.; Franco, F.; Tsui, Y.C.; Xie, X.; Trefny, M.P.; Zappasodi, R.; Mohmood, S.R.; Fernandez-Garcia, J.; Tsai, C.H.; Schulze, I.; et al. CD36-mediated metabolic adaptation supports regulatory T cell survival and function in tumors. Nat. Immunol. 2020, 21, 298–308. [Google Scholar] [CrossRef]
- Wu, L.; Zhang, X.; Zheng, L.; Zhao, H.; Yan, G.; Zhang, Q.; Zhou, Y.; Lei, J.; Zhang, J.; Wang, J.; et al. RIPK3 Orchestrates Fatty Acid Metabolism in Tumor-Associated Macrophages and Hepatocarcinogenesis. Cancer Immunol. Res. 2020, 8, 710–721. [Google Scholar] [CrossRef]
- Veglia, F.; Tyurin, V.A.; Blasi, M.; De Leo, A.; Kossenkov, A.V.; Donthireddy, L.; To, T.K.J.; Schug, Z.; Basu, S.; Wang, F.; et al. Fatty acid transport protein 2 reprograms neutrophils in cancer. Nature 2019, 569, 73–78. [Google Scholar] [CrossRef] [PubMed]
- Nakamura, H.; Takada, K. Reactive oxygen species in cancer: Current findings and future directions. Cancer Sci. 2021, 112, 3945–3952. [Google Scholar] [CrossRef]
- Yu, X.; Lao, Y.; Teng, X.L.; Li, S.; Zhou, Y.; Wang, F.; Guo, X.; Deng, S.; Chang, Y.; Wu, X.; et al. SENP3 maintains the stability and function of regulatory T cells via BACH2 deSUMOylation. Nat. Commun. 2018, 9, 3157. [Google Scholar] [CrossRef]
- Corzo, C.A.; Cotter, M.J.; Cheng, P.; Cheng, F.; Kusmartsev, S.; Sotomayor, E.; Padhya, T.; McCaffrey, T.V.; McCaffrey, J.C.; Gabrilovich, D.I. Mechanism regulating reactive oxygen species in tumor-induced myeloid-derived suppressor cells. J. Immunol. 2009, 182, 5693–5701. [Google Scholar] [CrossRef]
- Singer, K.; Kastenberger, M.; Gottfried, E.; Hammerschmied, C.G.; Buttner, M.; Aigner, M.; Seliger, B.; Walter, B.; Schlosser, H.; Hartmann, A.; et al. Warburg phenotype in renal cell carcinoma: High expression of glucose-transporter 1 (GLUT-1) correlates with low CD8(+) T-cell infiltration in the tumor. Int. J. Cancer 2011, 128, 2085–2095. [Google Scholar] [CrossRef]
- Fu, Q.; Xu, L.; Wang, Y.; Jiang, Q.; Liu, Z.; Zhang, J.; Zhou, Q.; Zeng, H.; Tong, S.; Wang, T.; et al. Tumor-associated Macrophage-derived Interleukin-23 Interlinks Kidney Cancer Glutamine Addiction with Immune Evasion. Eur. Urol. 2019, 75, 752–763. [Google Scholar] [CrossRef]
- Duan, Q.; Li, H.; Gao, C.; Zhao, H.; Wu, S.; Wu, H.; Wang, C.; Shen, Q.; Yin, T. High glucose promotes pancreatic cancer cells to escape from immune surveillance via AMPK-Bmi1-GATA2-MICA/B pathway. J. Exp. Clin. Cancer Res. 2019, 38, 192. [Google Scholar] [CrossRef]
- Husain, Z.; Huang, Y.; Seth, P.; Sukhatme, V.P. Tumor-derived lactate modifies antitumor immune response: Effect on myeloid-derived suppressor cells and NK cells. J. Immunol. 2013, 191, 1486–1495. [Google Scholar] [CrossRef] [PubMed]
- Brand, A.; Singer, K.; Koehl, G.E.; Kolitzus, M.; Schoenhammer, G.; Thiel, A.; Matos, C.; Bruss, C.; Klobuch, S.; Peter, K.; et al. LDHA-Associated Lactic Acid Production Blunts Tumor Immunosurveillance by T and NK Cells. Cell Metab. 2016, 24, 657–671. [Google Scholar] [CrossRef] [PubMed]
- Xie, M.; Fu, X.G.; Jiang, K. Notch1/TAZ axis promotes aerobic glycolysis and immune escape in lung cancer. Cell Death Dis. 2021, 12, 832. [Google Scholar] [CrossRef] [PubMed]
- Park, A.; Yang, Y.; Lee, Y.; Kim, M.S.; Park, Y.J.; Jung, H.; Kim, T.D.; Lee, H.G.; Choi, I.; Yoon, S.R. Indoleamine-2,3-Dioxygenase in Thyroid Cancer Cells Suppresses Natural Killer Cell Function by Inhibiting NKG2D and NKp46 Expression via STAT Signaling Pathways. J. Clin. Med. 2019, 8, 842. [Google Scholar] [CrossRef]
- Takahashi, H.; Kawabata-Iwakawa, R.; Ida, S.; Mito, I.; Tada, H.; Chikamatsu, K. Upregulated glycolysis correlates with tumor progression and immune evasion in head and neck squamous cell carcinoma. Sci. Rep. 2021, 11, 17789. [Google Scholar] [CrossRef]
- Brown, T.P.; Bhattacharjee, P.; Ramachandran, S.; Sivaprakasam, S.; Ristic, B.; Sikder, M.O.F.; Ganapathy, V. The lactate receptor GPR81 promotes breast cancer growth via a paracrine mechanism involving antigen-presenting cells in the tumor microenvironment. Oncogene 2020, 39, 3292–3304. [Google Scholar] [CrossRef]
- Gottfried, E.; Kunz-Schughart, L.A.; Ebner, S.; Mueller-Klieser, W.; Hoves, S.; Andreesen, R.; Mackensen, A.; Kreutz, M. Tumor-derived lactic acid modulates dendritic cell activation and antigen expression. Blood 2006, 107, 2013–2021. [Google Scholar] [CrossRef] [PubMed]
- Herber, D.L.; Cao, W.; Nefedova, Y.; Novitskiy, S.V.; Nagaraj, S.; Tyurin, V.A.; Corzo, A.; Cho, H.I.; Celis, E.; Lennox, B.; et al. Lipid accumulation and dendritic cell dysfunction in cancer. Nat. Med. 2010, 16, 880–886. [Google Scholar] [CrossRef]
- Xu, F.; Wang, Z.; Zhang, H.; Chen, J.; Wang, X.; Cui, L.; Xie, C.; Li, M.; Wang, F.; Zhou, P.; et al. Mevalonate Blockade in Cancer Cells Triggers CLEC9A(+) Dendritic Cell-Mediated Antitumor Immunity. Cancer Res. 2021, 81, 4514–4528. [Google Scholar] [CrossRef]
- Jiang, L.; Fang, X.; Wang, H.; Li, D.; Wang, X. Ovarian Cancer-Intrinsic Fatty Acid Synthase Prevents Anti-tumor Immunity by Disrupting Tumor-Infiltrating Dendritic Cells. Front. Immunol. 2018, 9, 2927. [Google Scholar] [CrossRef] [PubMed]
- Vander Heiden, M.G.; Cantley, L.C.; Thompson, C.B. Understanding the Warburg effect: The metabolic requirements of cell proliferation. Science 2009, 324, 1029–1033. [Google Scholar] [CrossRef]
- Zhang, J.; Peng, Y.; He, Y.; Xiao, Y.; Wang, Q.; Zhao, Y.; Zhang, T.; Wu, C.; Xie, Y.; Zhou, J.; et al. GPX1-associated prognostic signature predicts poor survival in patients with acute myeloid leukemia and involves in immunosuppression. Biochim. Biophys. Acta Mol. Basis Dis. 2022, 1868, 166268. [Google Scholar] [CrossRef]
- Ahmed, K.M.; Veeramachaneni, R.; Deng, D.; Putluri, N.; Putluri, V.; Cardenas, M.F.; Wheeler, D.A.; Decker, W.K.; Frederick, A.I.; Kazi, S.; et al. Glutathione peroxidase 2 is a metabolic driver of the tumor immune microenvironment and immune checkpoint inhibitor response. J. Immunother. Cancer 2022, 10, e004752. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.; Li, Y.; Zou, J.; Lai, C.T.; Zeng, T.; Peng, J.; Zou, W.D.; Cao, B.; Liu, D.; Zhu, L.Y.; et al. Comprehensive analysis of the glutathione S-transferase Mu (GSTM) gene family in ovarian cancer identifies prognostic and expression significance. Front. Oncol. 2022, 12, 968547. [Google Scholar] [CrossRef] [PubMed]
- Townsend, D.M.; Tew, K.D. The role of glutathione-S-transferase in anti-cancer drug resistance. Oncogene 2003, 22, 7369–7375. [Google Scholar] [CrossRef] [PubMed]
- Ahmed, Y.B.; Al-Bzour, A.N.; Ababneh, O.E.; Abushukair, H.M.; Saeed, A. Genomic and Transcriptomic Predictors of Response to Immune Checkpoint Inhibitors in Melanoma Patients: A Machine Learning Approach. Cancers 2022, 14, 5605. [Google Scholar] [CrossRef] [PubMed]
- Ucche, S.; Yokoyama, S.; Mojic, M.; Oki, K.; Ohshima, C.; Tsuihiji, H.; Takasaki, I.; Tahara, H.; Hayakawa, Y. GSTA4 Governs Melanoma Immune Resistance and Metastasis. Mol. Cancer Res. 2023, 21, 76–85. [Google Scholar] [CrossRef] [PubMed]
- Qiao, T.; Xiong, Y.; Feng, Y.; Guo, W.; Zhou, Y.; Zhao, J.; Jiang, T.; Shi, C.; Han, Y. Inhibition of LDH-A by Oxamate Enhances the Efficacy of Anti-PD-1 Treatment in an NSCLC Humanized Mouse Model. Front. Oncol. 2021, 11, 632364. [Google Scholar] [CrossRef] [PubMed]
- Gross, M.; Chen, J.; Englert, J.; Janes, J.; Leone, R.; MacKinnon, A.; Parlati, F.; Rodriquez, M.; Shwonek, P.; Powell, J. Abstract 2329: Glutaminase inhibition with CB-839 enhances anti-tumor activity of PD-1 and PD-L1 antibodies by overcoming a metabolic checkpoint blocking T cell activation. Cancer Res. 2016, 76, 2329. [Google Scholar] [CrossRef]
- Varghese, S.; Pramanik, S.; Williams, L.J.; Hodges, H.R.; Hudgens, C.W.; Fischer, G.M.; Luo, C.K.; Knighton, B.; Tan, L.; Lorenzi, P.L.; et al. The Glutaminase Inhibitor CB-839 (Telaglenastat) Enhances the Antimelanoma Activity of T-Cell-Mediated Immunotherapies. Mol. Cancer Ther. 2021, 20, 500–511. [Google Scholar] [CrossRef] [PubMed]
- Spranger, S.; Koblish, H.K.; Horton, B.; Scherle, P.A.; Newton, R.; Gajewski, T.F. Mechanism of tumor rejection with doublets of CTLA-4, PD-1/PD-L1, or IDO blockade involves restored IL-2 production and proliferation of CD8(+) T cells directly within the tumor microenvironment. J. Immunother. Cancer 2014, 2, 3. [Google Scholar] [CrossRef]
- Liu, X.; Shin, N.; Koblish, H.K.; Yang, G.; Wang, Q.; Wang, K.; Leffet, L.; Hansbury, M.J.; Thomas, B.; Rupar, M.; et al. Selective inhibition of IDO1 effectively regulates mediators of antitumor immunity. Blood 2010, 115, 3520–3530. [Google Scholar] [CrossRef]
- Mitchell, T.C.; Hamid, O.; Smith, D.C.; Bauer, T.M.; Wasser, J.S.; Olszanski, A.J.; Luke, J.J.; Balmanoukian, A.S.; Schmidt, E.V.; Zhao, Y.; et al. Epacadostat Plus Pembrolizumab in Patients With Advanced Solid Tumors: Phase I Results From a Multicenter, Open-Label Phase I/II Trial (ECHO-202/KEYNOTE-037). J. Clin. Oncol. 2018, 36, 3223–3230. [Google Scholar] [CrossRef] [PubMed]
- Long, G.V.; Dummer, R.; Hamid, O.; Gajewski, T.F.; Caglevic, C.; Dalle, S.; Arance, A.; Carlino, M.S.; Grob, J.J.; Kim, T.M.; et al. Epacadostat plus pembrolizumab versus placebo plus pembrolizumab in patients with unresectable or metastatic melanoma (ECHO-301/KEYNOTE-252): A phase 3, randomised, double-blind study. Lancet Oncol. 2019, 20, 1083–1097. [Google Scholar] [CrossRef] [PubMed]
- Steggerda, S.M.; Bennett, M.K.; Chen, J.; Emberley, E.; Huang, T.; Janes, J.R.; Li, W.; MacKinnon, A.L.; Makkouk, A.; Marguier, G.; et al. Inhibition of arginase by CB-1158 blocks myeloid cell-mediated immune suppression in the tumor microenvironment. J. Immunother. Cancer 2017, 5, 101. [Google Scholar] [CrossRef] [PubMed]
- Al-Husein, B.A.; Dawah, B.; Bani-Hani, S.; Al Bashir, S.M.; Al-Sawalmeh, K.M.; Ayoub, N.M. Immunomodulatory effect of statins on Regulatory T Lymphocytes in human colorectal cancer is determined by the stage of disease. Oncotarget 2018, 9, 35752–35761. [Google Scholar] [CrossRef] [PubMed]
- Jiang, W.; Hu, J.W.; He, X.R.; Jin, W.L.; He, X.Y. Statins: A repurposed drug to fight cancer. J. Exp. Clin. Cancer Res. 2021, 40, 241. [Google Scholar] [CrossRef] [PubMed]
- Mao, W.; Cai, Y.; Chen, D.; Jiang, G.; Xu, Y.; Chen, R.; Wang, F.; Wang, X.; Zheng, M.; Zhao, X.; et al. Statin shapes inflamed tumor microenvironment and enhances immune checkpoint blockade in non-small cell lung cancer. JCI Insight 2022, 7, e161940. [Google Scholar] [CrossRef]
- Rossi, A.; Filetti, M.; Taurelli Salimbeni, B.; Piras, M.; Rizzo, F.; Giusti, R.; Marchetti, P. Statins and immunotherapy: Togetherness makes strength The potential effect of statins on immunotherapy for NSCLC. Cancer Rep. 2021, 4, e1368. [Google Scholar] [CrossRef]
- Xia, Y.; Xie, Y.; Yu, Z.; Xiao, H.; Jiang, G.; Zhou, X.; Yang, Y.; Li, X.; Zhao, M.; Li, L.; et al. The Mevalonate Pathway Is a Druggable Target for Vaccine Adjuvant Discovery. Cell 2018, 175, 1059–1073.E21. [Google Scholar] [CrossRef]
- Cantini, L.; Pecci, F.; Hurkmans, D.P.; Belderbos, R.A.; Lanese, A.; Copparoni, C.; Aerts, S.; Cornelissen, R.; Dumoulin, D.W.; Fiordoliva, I.; et al. High-intensity statins are associated with improved clinical activity of PD-1 inhibitors in malignant pleural mesothelioma and advanced non-small cell lung cancer patients. Eur. J. Cancer 2021, 144, 41–48. [Google Scholar] [CrossRef]
- Failing, J.J.; Finnes, H.D.; Kottschade, L.A.; Allred, J.B.; Markovic, S.N. Effects of commonly used chronic medications on the outcomes of ipilimumab therapy in patients with metastatic melanoma. Melanoma Res. 2016, 26, 609–615. [Google Scholar] [CrossRef]
- Santoni, M.; Massari, F.; Matrana, M.R.; Basso, U.; De Giorgi, U.; Aurilio, G.; Buti, S.; Incorvaia, L.; Rizzo, M.; Martignetti, A.; et al. Statin use improves the efficacy of nivolumab in patients with advanced renal cell carcinoma. Eur. J. Cancer 2022, 172, 191–198. [Google Scholar] [CrossRef] [PubMed]
- Danzi, F.; Pacchiana, R.; Mafficini, A.; Scupoli, M.T.; Scarpa, A.; Donadelli, M.; Fiore, A. To metabolomics and beyond: A technological portfolio to investigate cancer metabolism. Signal Transduct. Target. Ther. 2023, 8, 137. [Google Scholar] [CrossRef] [PubMed]
- Franciosa, G.; Kverneland, A.H.; Jensen, A.W.P.; Donia, M.; Olsen, J.V. Proteomics to study cancer immunity and improve treatment. Semin. Immunopathol. 2023, 45, 241–251. [Google Scholar] [CrossRef]
- Schwaiger-Haber, M.; Stancliffe, E.; Anbukumar, D.S.; Sells, B.; Yi, J.; Cho, K.; Adkins-Travis, K.; Chheda, M.G.; Shriver, L.P.; Patti, G.J. Using mass spectrometry imaging to map fluxes quantitatively in the tumor ecosystem. Nat. Commun. 2023, 14, 2876. [Google Scholar] [CrossRef] [PubMed]
- Ding, B.; Ye, Z.; Yin, H.; Hong, X.Y.; Feng, S.W.; Xu, J.Y.; Shen, Y. Comprehensive single-cell analysis reveals heterogeneity of fibroblast subpopulations in ovarian cancer tissue microenvironment. Heliyon 2024, 10, e27873. [Google Scholar] [CrossRef] [PubMed]
- Huang, Y.; Mohanty, V.; Dede, M.; Tsai, K.; Daher, M.; Li, L.; Rezvani, K.; Chen, K. Characterizing cancer metabolism from bulk and single-cell RNA-seq data using METAFlux. Nat. Commun. 2023, 14, 4883. [Google Scholar] [CrossRef]
- Wang, L.; Li, S.; Li, X.; Zhuo, G.; Zhang, Q.; Liu, G.; Pan, Y. Single cell analysis unveils the commonality and heterogeneity between nasopharyngeal and oropharyngeal carcinoma. Neoplasia 2024, 50, 100980. [Google Scholar] [CrossRef]


{kind=link}
{kind=link}
| Molecule | Function | Reference |
|---|---|---|
| GLUT | Increase tumor growth and invasion | [8] |
| HK2 | Tumor progression, immune cell infiltration | [10] |
| SLC1A5 | Tumor proliferation and survival | [12] |
| FASN | Tumor growth and proliferation | [17] |
| STAT3, c-Myc | Metabolic switch to glycolysis | [19] |
| Lactate | Activate ERK/STAT3 signaling, activating HIF-1α | [29,30] |
| IDO | Inhibit effector T cell function, increase Treg recruitment, activating MDSCs | [33,34] |
| ARG1 | Correlation with increase in MDSCs | [39] |
| ARG2 | Suppress CD4+ T cells, increase Treg recruitment | [37] |
| GPx1 | Elevation of MDSCs and TAM | [60] |
| GPx2 | Poor responsiveness to anti-CTLA-4 mAb | [61] |
| GSTM2, 3, 4 | Promoting immune escape | [62] |
| GSTA4 | Resistance to IFN-γ, increase metastatic ability | [65] |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ucche, S.; Hayakawa, Y. Immunological Aspects of Cancer Cell Metabolism. Int. J. Mol. Sci. 2024, 25, 5288. https://doi.org/10.3390/ijms25105288
Ucche S, Hayakawa Y. Immunological Aspects of Cancer Cell Metabolism. International Journal of Molecular Sciences. 2024; 25(10):5288. https://doi.org/10.3390/ijms25105288
Chicago/Turabian StyleUcche, Sisca, and Yoshihiro Hayakawa. 2024. "Immunological Aspects of Cancer Cell Metabolism" International Journal of Molecular Sciences 25, no. 10: 5288. https://doi.org/10.3390/ijms25105288
APA StyleUcche, S., & Hayakawa, Y. (2024). Immunological Aspects of Cancer Cell Metabolism. International Journal of Molecular Sciences, 25(10), 5288. https://doi.org/10.3390/ijms25105288