
The Mechanisms of Altered Blood–Brain Barrier Permeability in CD19 CAR T–Cell Recipients


Abstract
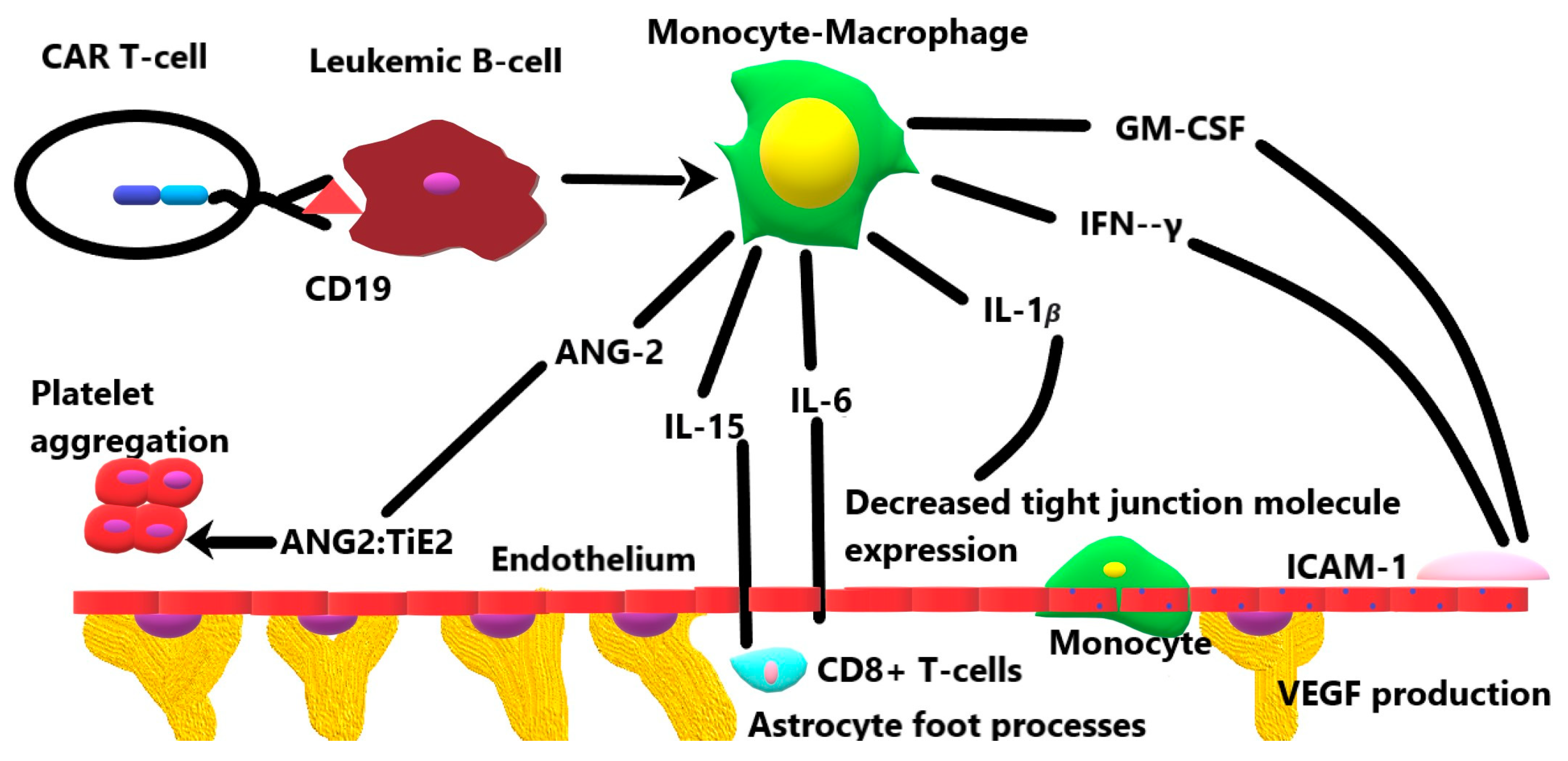
1. Introduction
2. The Blood–Brain Barrier
3. The Role of Cytokines in the Breakdown of the Blood–Brain Barrier
3.1. Granulocyte-Monocyte Colony-Stimulating Factor
3.2. Interferon-γ
3.3. Interleukin-1β
3.4. Interleukin-6
3.5. Interleukin-10
3.6. Interleukin-15
3.7. Angiopoeitin-2
4. Clinical and Imaging Features of ICANS
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Wang, K.; Wei, G.; Liu, D. CD19: A biomarker for B cell development, lymphoma diagnosis and therapy. Exp. Hematol. Oncol. 2012, 1, 36. [Google Scholar] [CrossRef] [PubMed]
- Larson, R.C.; Maus, M.V. Recent advances and discoveries in the mechanisms and functions of CAR T cells. Nat. Rev. Cancer 2021, 21, 145–161. [Google Scholar] [CrossRef] [PubMed]
- Doan, A.; Pulsipher, M.A. Hypogammaglobulinemia due to CAR T-cell therapy. Pediatr. Blood Cancer 2018, 65, 10. [Google Scholar] [CrossRef] [PubMed]
- Parker, K.R.; Migliorini, D.; Perkey, E.; Yost, K.E.; Bhaduri, A.; Bagga, P.; Haris, M.; Wilson, N.E.; Liu, F.; Gabunia, K.; et al. Single-Cell Analyses Identify Brain Mural Cells Expressing CD19 as Potential Off-Tumor Targets for CAR-T Immunotherapies. Cell 2020, 183, 126–142 e117. [Google Scholar] [CrossRef] [PubMed]
- Jess, J.; Yates, B.; Dulau-Florea, A.; Parker, K.; Inglefield, J.; Lichtenstein, D.; Schischlik, F.; Ongkeko, M.; Wang, Y.; Shahani, S.; et al. CD22 CAR T-cell associated hematologic toxicities, endothelial activation and relationship to neurotoxicity. J. Immunother. Cancer 2023, 11, e005898. [Google Scholar] [CrossRef] [PubMed]
- Gust, J.; Hay, K.A.; Hanafi, L.A.; Li, D.; Myerson, D.; Gonzalez-Cuyar, L.F.; Yeung, C.; Liles, W.C.; Wurfel, M.; Lopez, J.A.; et al. Endothelial Activation and Blood-Brain Barrier Disruption in Neurotoxicity after Adoptive Immunotherapy with CD19 CAR-T Cells. Cancer Discov. 2017, 7, 1404–1419. [Google Scholar] [CrossRef] [PubMed]
- Gust, J.; Taraseviciute, A.; Turtle, C.J. Neurotoxicity Associated with CD19-Targeted CAR-T Cell Therapies. CNS Drugs 2018, 32, 1091–1101. [Google Scholar] [CrossRef] [PubMed]
- Lee, D.W.; Santomasso, B.D.; Locke, F.L.; Ghobadi, A.; Turtle, C.J.; Brudno, J.N.; Maus, M.V.; Park, J.H.; Mead, E.; Pavletic, S.; et al. ASTCT Consensus Grading for Cytokine Release Syndrome and Neurologic Toxicity Associated with Immune Effector Cells. Biol. Blood Marrow Transplant. 2019, 25, 625–638. [Google Scholar] [CrossRef]
- Risau, W. Molecular biology of blood-brain barrier ontogenesis and function. Acta Neurochir. Suppl. 1994, 60, 109–112. [Google Scholar] [CrossRef]
- Daneman, R.; Zhou, L.; Kebede, A.A.; Barres, B.A. Pericytes are required for blood-brain barrier integrity during embryogenesis. Nature 2010, 468, 562–566. [Google Scholar] [CrossRef]
- Gerhardt, H.; Liebner, S.; Redies, C.; Wolburg, H. N-cadherin expression in endothelial cells during early angiogenesis in the eye and brain of the chicken: Relation to blood-retina and blood-brain barrier development. Eur. J. Neurosci. 1999, 11, 1191–1201. [Google Scholar] [CrossRef] [PubMed]
- Saija, A.; Princi, P.; Lanza, M.; Scalese, M.; Aramnejad, E.; De Sarro, A. Systemic cytokine administration can affect blood-brain barrier permeability in the rat. Life Sci. 1995, 56, 775–784. [Google Scholar] [CrossRef] [PubMed]
- Armulik, A.; Genove, G.; Mae, M.; Nisancioglu, M.H.; Wallgard, E.; Niaudet, C.; He, L.; Norlin, J.; Lindblom, P.; Strittmatter, K.; et al. Pericytes regulate the blood-brain barrier. Nature 2010, 468, 557–561. [Google Scholar] [CrossRef] [PubMed]
- Armulik, A.; Genove, G.; Betsholtz, C. Pericytes: Developmental, physiological, and pathological perspectives, problems, and promises. Dev. Cell 2011, 21, 193–215. [Google Scholar] [CrossRef] [PubMed]
- Paolinelli, R.; Corada, M.; Orsenigo, F.; Dejana, E. The molecular basis of the blood brain barrier differentiation and maintenance. Is it still a mystery? Pharmacol. Res. 2011, 63, 165–171. [Google Scholar] [CrossRef] [PubMed]
- Guerit, S.; Fidan, E.; Macas, J.; Czupalla, C.J.; Figueiredo, R.; Vijikumar, A.; Yalcin, B.H.; Thom, S.; Winter, P.; Gerhardt, H.; et al. Astrocyte-derived Wnt growth factors are required for endothelial blood-brain barrier maintenance. Prog. Neurobiol. 2021, 199, 101937. [Google Scholar] [CrossRef]
- Alvarez, J.I.; Dodelet-Devillers, A.; Kebir, H.; Ifergan, I.; Fabre, P.J.; Terouz, S.; Sabbagh, M.; Wosik, K.; Bourbonniere, L.; Bernard, M.; et al. The Hedgehog pathway promotes blood-brain barrier integrity and CNS immune quiescence. Science 2011, 334, 1727–1731. [Google Scholar] [CrossRef]
- Gauthier, J.; Turtle, C.J. Insights into cytokine release syndrome and neurotoxicity after CD19-specific CAR-T cell therapy. Curr. Res. Transl. Med. 2018, 66, 50–52. [Google Scholar] [CrossRef]
- Gust, J.; Ponce, R.; Liles, W.C.; Garden, G.A.; Turtle, C.J. Cytokines in CAR T Cell-Associated Neurotoxicity. Front. Immunol. 2020, 11, 577027. [Google Scholar] [CrossRef]
- Croxford, A.L.; Spath, S.; Becher, B. GM-CSF in Neuroinflammation: Licensing Myeloid Cells for Tissue Damage. Trends Immunol. 2015, 36, 651–662. [Google Scholar] [CrossRef]
- Vogel, D.Y.; Kooij, G.; Heijnen, P.D.; Breur, M.; Peferoen, L.A.; van der Valk, P.; de Vries, H.E.; Amor, S.; Dijkstra, C.D. GM-CSF promotes migration of human monocytes across the blood brain barrier. Eur. J. Immunol. 2015, 45, 1808–1819. [Google Scholar] [CrossRef] [PubMed]
- Koizumi, K.; Poulaki, V.; Doehmen, S.; Welsandt, G.; Radetzky, S.; Lappas, A.; Kociok, N.; Kirchhof, B.; Joussen, A.M. Contribution of TNF-alpha to leukocyte adhesion, vascular leakage, and apoptotic cell death in endotoxin-induced uveitis in vivo. Investig. Ophthalmol. Vis. Sci. 2003, 44, 2184–2191. [Google Scholar] [CrossRef] [PubMed]
- Matsuyama, S.; Yamamoto, R.; Murakami, K.; Takahashi, N.; Nishi, R.; Ishii, A.; Nio-Kobayashi, J.; Abe, N.; Tanaka, K.; Jiang, J.J.; et al. GM-CSF Promotes the Survival of Peripheral-Derived Myeloid Cells in the Central Nervous System for Pain-Induced Relapse of Neuroinflammation. J. Immunol. 2023, 211, 34–42. [Google Scholar] [CrossRef] [PubMed]
- Sterner, R.M.; Sakemura, R.; Cox, M.J.; Yang, N.; Khadka, R.H.; Forsman, C.L.; Hansen, M.J.; Jin, F.; Ayasoufi, K.; Hefazi, M.; et al. GM-CSF inhibition reduces cytokine release syndrome and neuroinflammation but enhances CAR-T cell function in xenografts. Blood 2019, 133, 697–709. [Google Scholar] [CrossRef] [PubMed]
- Cohen, A.D.; Garfall, A.L.; Stadtmauer, E.A.; Melenhorst, J.J.; Lacey, S.F.; Lancaster, E.; Vogl, D.T.; Weiss, B.M.; Dengel, K.; Nelson, A.; et al. B cell maturation antigen-specific CAR T cells are clinically active in multiple myeloma. J. Clin. Investig. 2019, 129, 2210–2221. [Google Scholar] [CrossRef] [PubMed]
- Santomasso, B.D.; Park, J.H.; Salloum, D.; Riviere, I.; Flynn, J.; Mead, E.; Halton, E.; Wang, X.; Senechal, B.; Purdon, T.; et al. Clinical and Biological Correlates of Neurotoxicity Associated with CAR T-cell Therapy in Patients with B-cell Acute Lymphoblastic Leukemia. Cancer Discov. 2018, 8, 958–971. [Google Scholar] [CrossRef]
- Shalabi, H.; Martin, S.; Yates, B.; Wolters, P.L.; Kaplan, C.; Smith, H.; Sesi, C.R.; Jess, J.; Toledo-Tamula, M.A.; Struemph, K.; et al. Neurotoxicity following CD19/CD28zeta CAR T-cells in children and young adults with B-cell malignancies. Neuro-Oncology 2022, 24, 1584–1597. [Google Scholar] [CrossRef]
- Gust, J.; Finney, O.C.; Li, D.; Brakke, H.M.; Hicks, R.M.; Futrell, R.B.; Gamble, D.N.; Rawlings-Rhea, S.D.; Khalatbari, H.K.; Ishak, G.E.; et al. Glial injury in neurotoxicity after pediatric CD19-directed chimeric antigen receptor T cell therapy. Ann. Neurol. 2019, 86, 42–54. [Google Scholar] [CrossRef]
- Melrose, J.; Tsurushita, N.; Liu, G.; Berg, E.L. IFN-gamma inhibits activation-induced expression of E- and P-selectin on endothelial cells. J. Immunol. 1998, 161, 2457–2464. [Google Scholar] [CrossRef]
- Li, M.; Cuff, C.F.; Pestka, J.J. T-2 toxin impairment of enteric reovirus clearance in the mouse associated with suppressed immunoglobulin and IFN-gamma responses. Toxicol. Appl. Pharmacol. 2006, 214, 318–325. [Google Scholar] [CrossRef]
- Mangalam, A.K.; Luo, N.; Luckey, D.; Papke, L.; Hubbard, A.; Wussow, A.; Smart, M.; Giri, S.; Rodriguez, M.; David, C. Absence of IFN-gamma increases brain pathology in experimental autoimmune encephalomyelitis-susceptible DRB1*0301.DQ8 HLA transgenic mice through secretion of proinflammatory cytokine IL-17 and induction of pathogenic monocytes/microglia into the central nervous system. J. Immunol. 2014, 193, 4859–4870. [Google Scholar] [CrossRef] [PubMed]
- Zhang, H.; Lv, X.; Kong, Q.; Tan, Y. IL-6/IFN-gamma double knockdown CAR-T cells reduce the release of multiple cytokines from PBMCs in vitro. Hum. Vaccin. Immunother. 2022, 18, 1–14. [Google Scholar] [CrossRef] [PubMed]
- Benallegue, N.; Kebir, H.; Kapoor, R.; Crockett, A.; Li, C.; Cheslow, L.; Abdel-Hakeem, M.S.; Gesualdi, J.; Miller, M.C.; Wherry, E.J.; et al. The hedgehog pathway suppresses neuropathogenesis in CD4 T cell-driven inflammation. Brain 2021, 144, 1670–1683. [Google Scholar] [CrossRef] [PubMed]
- Park, J.; Choi, H.M.; Yang, H.I.; Yoo, M.C.; Kim, K.S. Increased expression of IL-1 receptors in response to IL-1beta may produce more IL-6, IL-8, VEGF, and PGE(2) in senescent synovial cells induced in vitro than in presenescent cells. Rheumatol. Int. 2012, 32, 2005–2010. [Google Scholar] [CrossRef] [PubMed]
- Dong, H.J.; Shang, C.Z.; Peng, D.W.; Xu, J.; Xu, P.X.; Zhan, L.; Wang, P. Curcumin attenuates ischemia-like injury induced IL-1beta elevation in brain microvascular endothelial cells via inhibiting MAPK pathways and nuclear factor-kappaB activation. Neurol. Sci. 2014, 35, 1387–1392. [Google Scholar] [CrossRef] [PubMed]
- Norelli, M.; Camisa, B.; Barbiera, G.; Falcone, L.; Purevdorj, A.; Genua, M.; Sanvito, F.; Ponzoni, M.; Doglioni, C.; Cristofori, P.; et al. Monocyte-derived IL-1 and IL-6 are differentially required for cytokine-release syndrome and neurotoxicity due to CAR T cells. Nat. Med. 2018, 24, 739–748. [Google Scholar] [CrossRef] [PubMed]
- Kochenderfer, J.N.; Somerville, R.P.T.; Lu, T.; Shi, V.; Bot, A.; Rossi, J.; Xue, A.; Goff, S.L.; Yang, J.C.; Sherry, R.M.; et al. Lymphoma Remissions Caused by Anti-CD19 Chimeric Antigen Receptor T Cells Are Associated With High Serum Interleukin-15 Levels. J. Clin. Oncol. 2017, 35, 1803–1813. [Google Scholar] [CrossRef]
- Neelapu, S.S.; Locke, F.L.; Bartlett, N.L.; Lekakis, L.J.; Miklos, D.B.; Jacobson, C.A.; Braunschweig, I.; Oluwole, O.O.; Siddiqi, T.; Lin, Y.; et al. Axicabtagene Ciloleucel CAR T-Cell Therapy in Refractory Large B-Cell Lymphoma. N. Engl. J. Med. 2017, 377, 2531–2544. [Google Scholar] [CrossRef]
- Krizanac-Bengez, L.; Kapural, M.; Parkinson, F.; Cucullo, L.; Hossain, M.; Mayberg, M.R.; Janigro, D. Effects of transient loss of shear stress on blood-brain barrier endothelium: Role of nitric oxide and IL-6. Brain Res. 2003, 977, 239–246. [Google Scholar] [CrossRef]
- Takeshita, Y.; Fujikawa, S.; Serizawa, K.; Fujisawa, M.; Matsuo, K.; Nemoto, J.; Shimizu, F.; Sano, Y.; Tomizawa-Shinohara, H.; Miyake, S.; et al. New BBB Model Reveals That IL-6 Blockade Suppressed the BBB Disorder, Preventing Onset of NMOSD. Neurol.-Neuroimmunol. Neuroinflamm. 2021, 8. [Google Scholar] [CrossRef]
- Erta, M.; Giralt, M.; Jimenez, S.; Molinero, A.; Comes, G.; Hidalgo, J. Astrocytic IL-6 Influences the Clinical Symptoms of EAE in Mice. Brain Sci. 2016, 6, e1076. [Google Scholar] [CrossRef] [PubMed]
- Mumm, J.B.; Emmerich, J.; Zhang, X.; Chan, I.; Wu, L.; Mauze, S.; Blaisdell, S.; Basham, B.; Dai, J.; Grein, J.; et al. IL-10 elicits IFNgamma-dependent tumor immune surveillance. Cancer Cell 2011, 20, 781–796. [Google Scholar] [CrossRef] [PubMed]
- Tilg, H.; van Montfrans, C.; van den Ende, A.; Kaser, A.; van Deventer, S.J.; Schreiber, S.; Gregor, M.; Ludwiczek, O.; Rutgeerts, P.; Gasche, C.; et al. Treatment of Crohn’s disease with recombinant human interleukin 10 induces the proinflammatory cytokine interferon gamma. Gut 2002, 50, 191–195. [Google Scholar] [CrossRef]
- Cianciulli, A.; Dragone, T.; Calvello, R.; Porro, C.; Trotta, T.; Lofrumento, D.D.; Panaro, M.A. IL-10 plays a pivotal role in anti-inflammatory effects of resveratrol in activated microglia cells. Int. Immunopharmacol. 2015, 24, 369–376. [Google Scholar] [CrossRef] [PubMed]
- Pan, W.; Yu, C.; Hsuchou, H.; Khan, R.S.; Kastin, A.J. Cerebral microvascular IL15 is a novel mediator of TNF action. J. Neurochem. 2009, 111, 819–827. [Google Scholar] [CrossRef] [PubMed]
- Wu, X.; Pan, W.; Stone, K.P.; Zhang, Y.; Hsuchou, H.; Kastin, A.J. Expression and signaling of novel IL15Ralpha splicing variants in cerebral endothelial cells of the blood-brain barrier. J. Neurochem. 2010, 114, 122–129. [Google Scholar] [CrossRef]
- Saikali, P.; Antel, J.P.; Pittet, C.L.; Newcombe, J.; Arbour, N. Contribution of astrocyte-derived IL-15 to CD8 T cell effector functions in multiple sclerosis. J. Immunol. 2010, 185, 5693–5703. [Google Scholar] [CrossRef]
- Brickler, T.R.; Hazy, A.; Guilhaume Correa, F.; Dai, R.; Kowalski, E.J.A.; Dickerson, R.; Chen, J.; Wang, X.; Morton, P.D.; Whittington, A.; et al. Angiopoietin/Tie2 Axis Regulates the Age-at-Injury Cerebrovascular Response to Traumatic Brain Injury. J. Neurosci. 2018, 38, 9618–9634. [Google Scholar] [CrossRef]
- Higgins, S.J.; Purcell, L.A.; Silver, K.L.; Tran, V.; Crowley, V.; Hawkes, M.; Conroy, A.L.; Opoka, R.O.; Hay, J.G.; Quaggin, S.E.; et al. Dysregulation of angiopoietin-1 plays a mechanistic role in the pathogenesis of cerebral malaria. Sci. Transl. Med. 2016, 8, 358ra128. [Google Scholar] [CrossRef]
- Fu, C.C.; Wang, R.J.; Wu, D.P. Interpretation of ASTCT Consensus Responses by Chimeric Antigen Receptor T Cell Therapy CRS/ICANS—Review. Zhongguo Shi Yan Xue Ye Xue Za Zhi 2021, 29, 1982–1986. [Google Scholar]
- Saw, J.L.; Sidiqi, M.H.; Ruff, M.; Hocker, S.; Alkhateeb, H.; Ansell, S.M.; Bennani, N.N.; Dingli, D.; Hayman, S.R.; Johnston, P.B.; et al. Acute seizures and status epilepticus in immune effector cell associated neurotoxicity syndrome (ICANS). Blood Cancer J. 2022, 12, 62. [Google Scholar] [CrossRef] [PubMed]
- Schuster, S.J.; Svoboda, J.; Chong, E.A.; Nasta, S.D.; Mato, A.R.; Anak, O.; Brogdon, J.L.; Pruteanu-Malinici, I.; Bhoj, V.; Landsburg, D.; et al. Chimeric Antigen Receptor T Cells in Refractory B-Cell Lymphomas. N. Engl. J. Med. 2017, 377, 2545–2554. [Google Scholar] [CrossRef] [PubMed]
- Smith, D.A.; Kikano, E.; Tirumani, S.H.; de Lima, M.; Caimi, P.; Ramaiya, N.H. Imaging-based Toxicity and Response Pattern Assessment Following CAR T-Cell Therapy. Radiology 2022, 302, 438–445. [Google Scholar] [CrossRef] [PubMed]
- Lapidus, A.H.; Anderson, M.A.; Harrison, S.J.; Dickinson, M.; Kalincik, T.; Lasocki, A. Neuroimaging findings in immune effector cell associated neurotoxicity syndrome after chimeric antigen receptor T-cell therapy. Leuk. Lymphoma 2022, 63, 2364–2374. [Google Scholar] [CrossRef]
- Pinto, S.N.; Liu, C.J.; Nelson, M.D., Jr.; Bluml, S.; Livingston, D.; Tamrazi, B. Neuroimaging of complications arising after CD19 chimeric antigen receptor T-cell therapy: A review. J. Neuroimaging 2023, 33, 703–715. [Google Scholar] [CrossRef]

{kind=link}
| Neurotoxicity Domain | Immune Effector Cell–Associated Neurotoxicity Syndrome Grades | |||
|---|---|---|---|---|
| 1 | 2 | 3 | 4 | |
| ICE * score | 7–9 | 3–6 | 0–2 | 0 |
| CAPD † score | 1–8 | 1–8 | ≥9 | Unable to assess |
| Level of consciousness | Awakens spontaneously | Awakens to voice | Awakens to tactile stimulus | Stupor or coma |
| Motor weakness | None | None | None | Hemiparesis/paraparesis |
| Seizure | None | None | Any seizure that resolves rapidly or nonconvulsive seizure that resolves with intervention | Prolonged or life-threatening seizure lasting > 5 min, or repetitive seizures without return to baseline |
| Intracranial hypertension/cerebral edema | None | None | Focal/local edema on neuroimaging | Decerebrate/decorticate posturing, cranial nerve VI palsy, papilledema, Cushing’s triad, diffuse cerebral edema on imaging. |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Pinto, S.N.; Krenciute, G. The Mechanisms of Altered Blood–Brain Barrier Permeability in CD19 CAR T–Cell Recipients. Int. J. Mol. Sci. 2024, 25, 644. https://doi.org/10.3390/ijms25010644
Pinto SN, Krenciute G. The Mechanisms of Altered Blood–Brain Barrier Permeability in CD19 CAR T–Cell Recipients. International Journal of Molecular Sciences. 2024; 25(1):644. https://doi.org/10.3390/ijms25010644
Chicago/Turabian StylePinto, Soniya N., and Giedre Krenciute. 2024. "The Mechanisms of Altered Blood–Brain Barrier Permeability in CD19 CAR T–Cell Recipients" International Journal of Molecular Sciences 25, no. 1: 644. https://doi.org/10.3390/ijms25010644
APA StylePinto, S. N., & Krenciute, G. (2024). The Mechanisms of Altered Blood–Brain Barrier Permeability in CD19 CAR T–Cell Recipients. International Journal of Molecular Sciences, 25(1), 644. https://doi.org/10.3390/ijms25010644