
Back-Up Base Excision DNA Repair in Human Cells Deficient in the Major AP Endonuclease, APE1

 , and
, and
Abstract
1. Introduction
2. Results
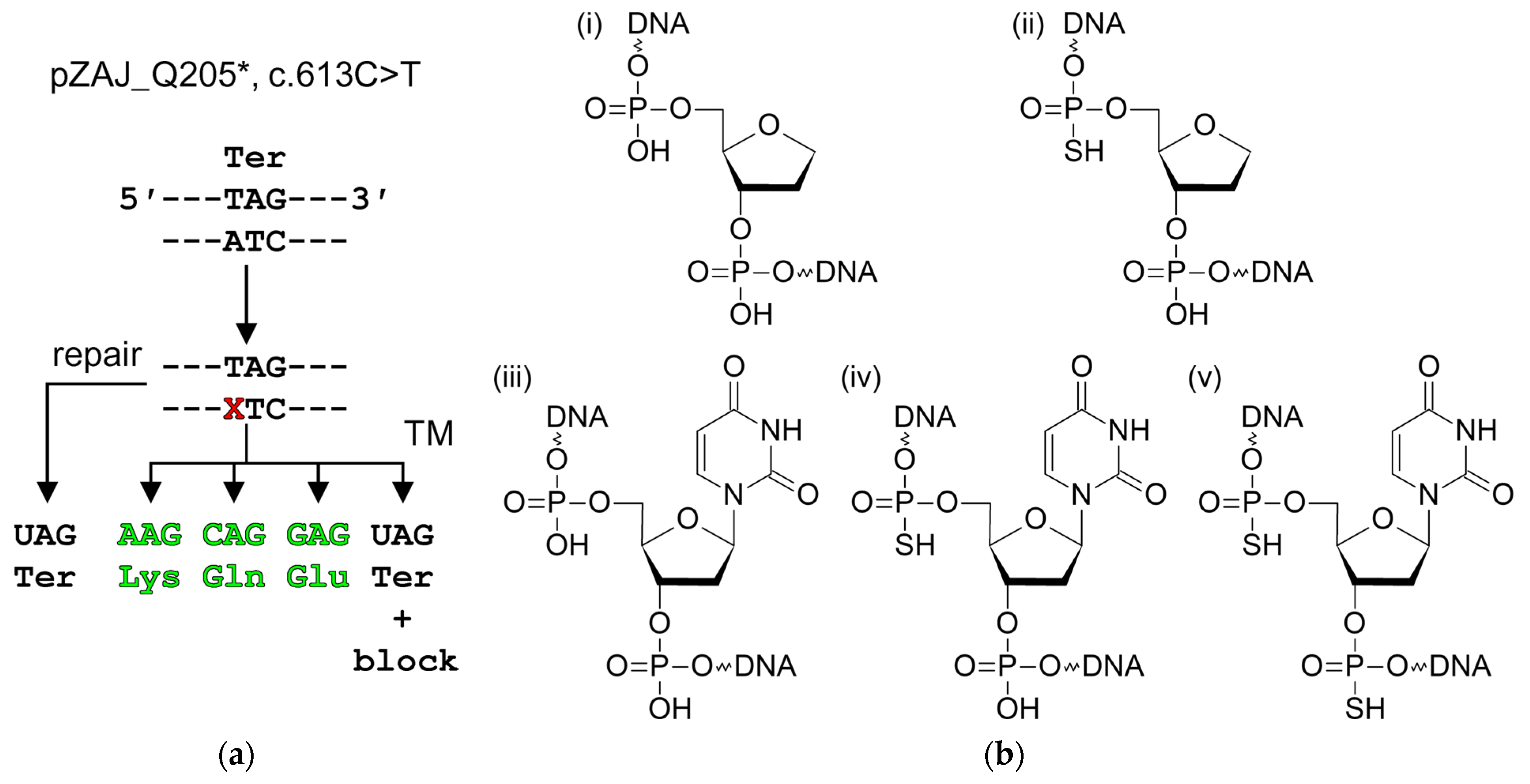
2.1. Plasmid Reporter Systems to Study Base Excision Repair in Cellulo
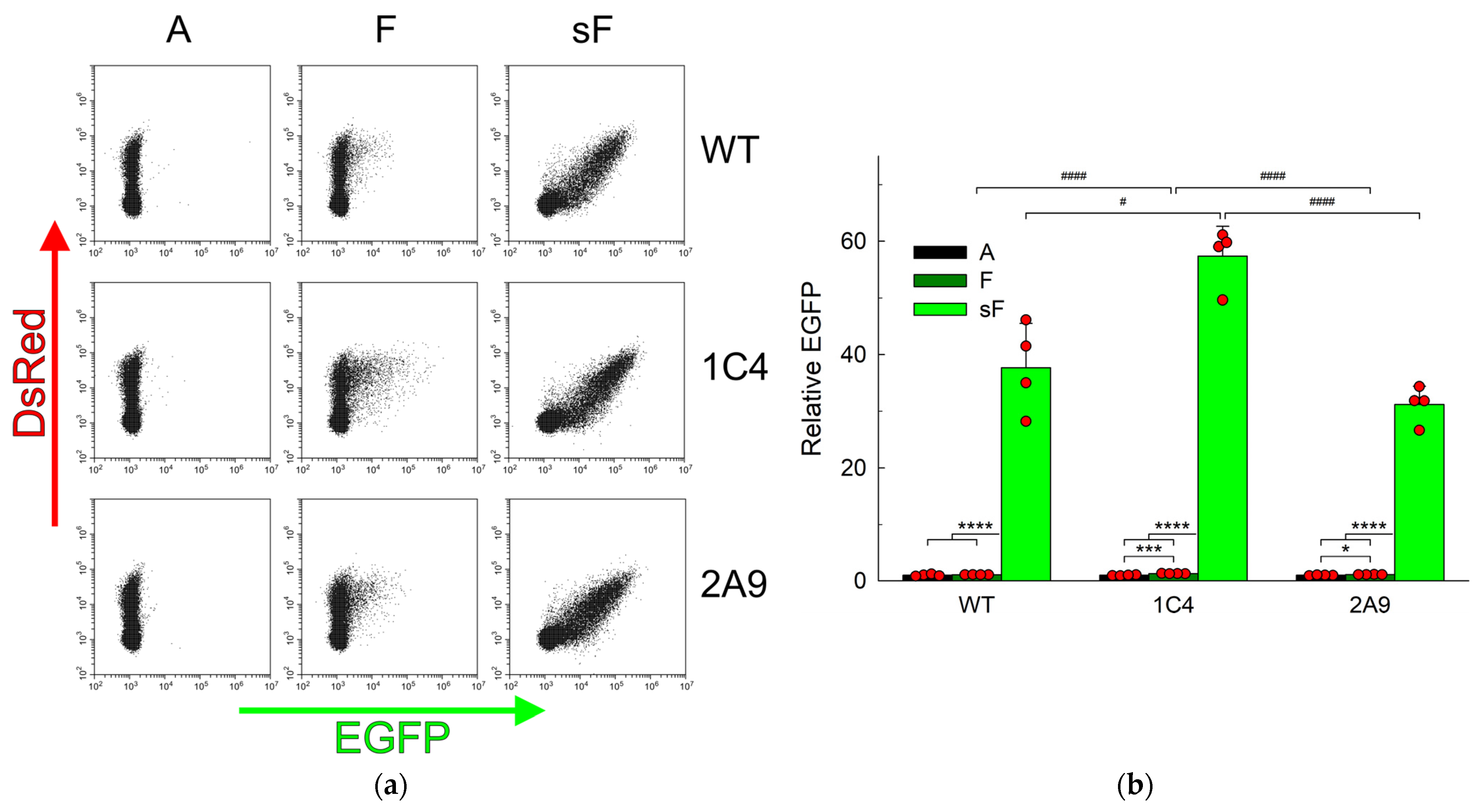
2.2. AP Sites Are Repaired in APEX1 Knockout Cells
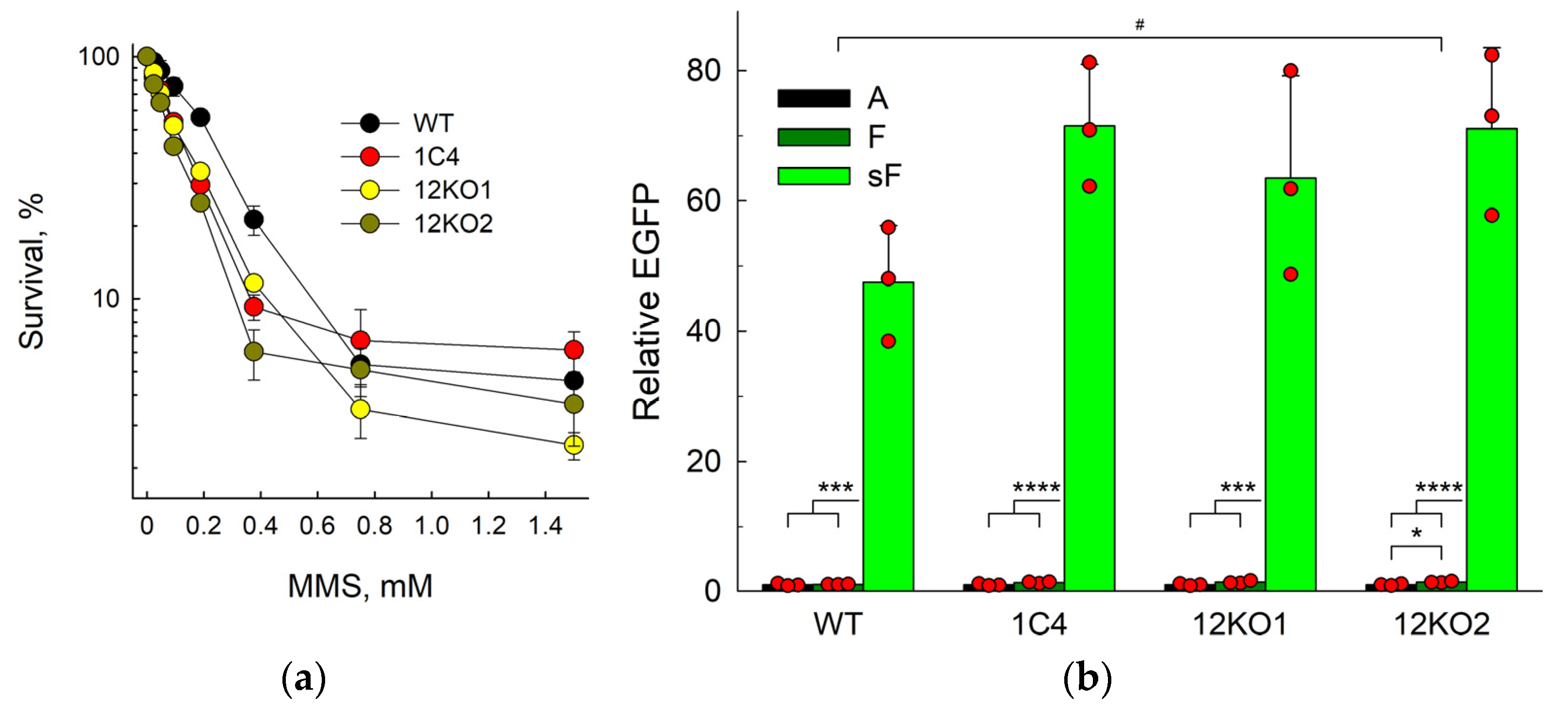
2.3. The Backup Repair in APEX1 Knockout Cells Is Independent of APE2
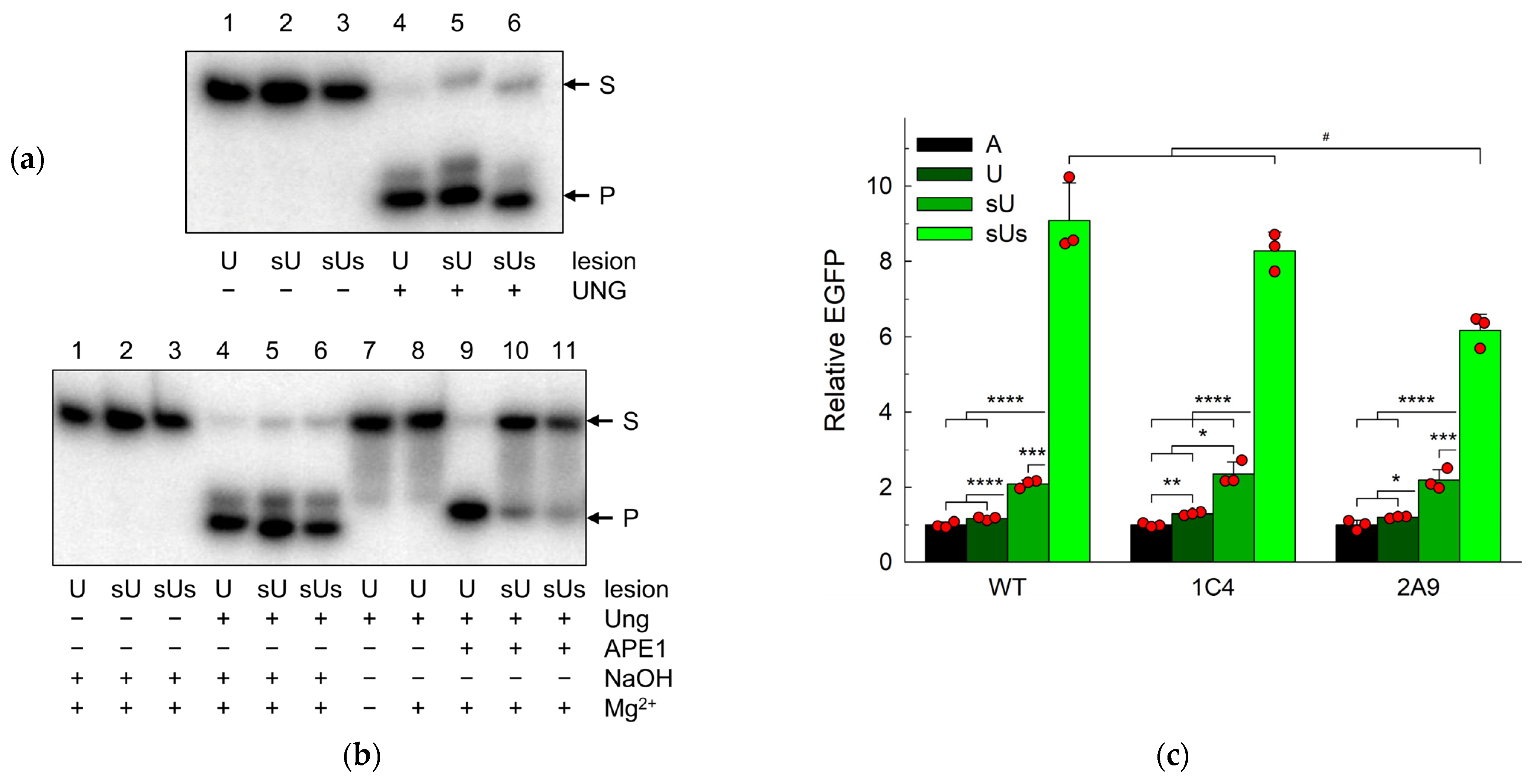
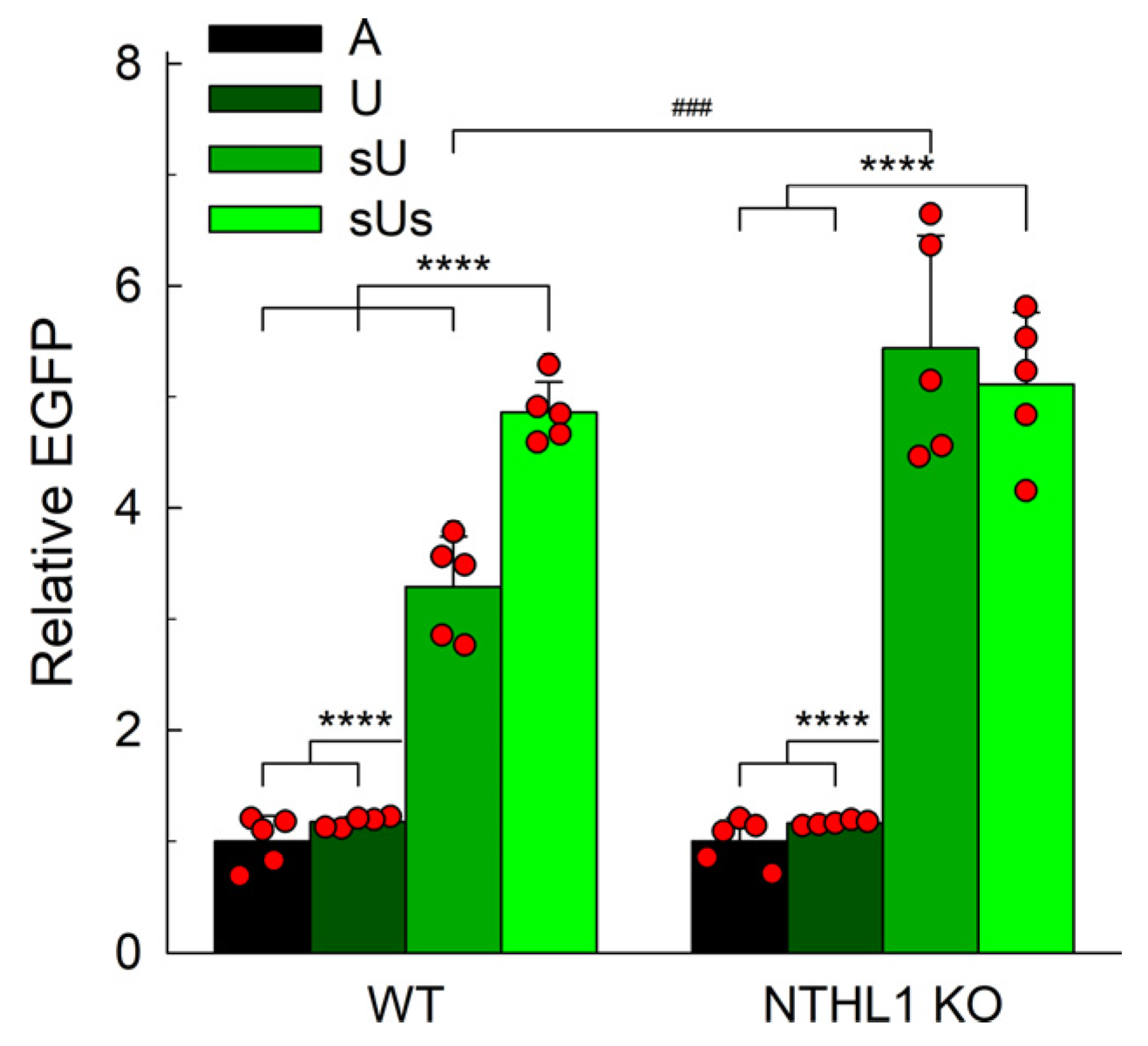
2.4. Uracil in DNA Is Repaired in APEX1 Knockout Cells
2.5. NTHL1 Contributes to the Backup Uracil Repair
3. Discussion
4. Materials and Methods
4.1. Enzymes, Oligonucleotides, Plasmids, and Cells
4.2. Enzyme Assays
4.3. Generation of APEX1KO APEX2KO Cells
4.4. Damaged Plasmids
4.5. Transcriptional Mutagenesis Experiments
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Lindahl, T.; Nyberg, B. Rate of depurination of native deoxyribonucleic acid. Biochemistry 1972, 11, 3610–3618. [Google Scholar] [CrossRef] [PubMed]
- Atamna, H.; Cheung, I.; Ames, B.N. A method for detecting abasic sites in living cells: Age-dependent changes in base excision repair. Proc. Natl. Acad. Sci. USA 2000, 97, 686–691. [Google Scholar] [CrossRef] [PubMed]
- Boiteux, S.; Guillet, M. Abasic sites in DNA: Repair and biological consequences in Saccharomyces cerevisiae. DNA Repair 2004, 3, 1–12. [Google Scholar] [CrossRef] [PubMed]
- Thompson, P.S.; Cortez, D. New insights into abasic site repair and tolerance. DNA Repair 2020, 90, 102866. [Google Scholar] [CrossRef]
- Zharkov, D.O. Base excision DNA repair. Cell. Mol. Life Sci. 2008, 65, 1544–1565. [Google Scholar] [CrossRef]
- Beard, W.A.; Horton, J.K.; Prasad, R.; Wilson, S.H. Eukaryotic base excision repair: New approaches shine light on mechanism. Annu. Rev. Biochem. 2019, 88, 137–162. [Google Scholar] [CrossRef]
- Wozniak, K.J.; Simmons, L.A. Bacterial DNA excision repair pathways. Nat. Rev. Microbiol. 2022, 20, 465–477. [Google Scholar] [CrossRef]
- Fortini, P.; Dogliotti, E. Base damage and single-strand break repair: Mechanisms and functional significance of short- and long-patch repair subpathways. DNA Repair 2007, 6, 398–409. [Google Scholar] [CrossRef]
- Tell, G.; Fantini, D.; Quadrifoglio, F. Understanding different functions of mammalian AP endonuclease (APE1) as a promising tool for cancer treatment. Cell. Mol. Life Sci. 2010, 67, 3589–3608. [Google Scholar] [CrossRef]
- Whitaker, A.M.; Freudenthal, B.D. APE1: A skilled nucleic acid surgeon. DNA Repair 2018, 71, 93–100. [Google Scholar] [CrossRef]
- Xanthoudakis, S.; Miao, G.; Wang, F.; Pan, Y.-C.E.; Curran, T. Redox activation of Fos–Jun DNA binding activity is mediated by a DNA repair enzyme. EMBO J. 1992, 11, 3323–3335. [Google Scholar] [CrossRef] [PubMed]
- Kuninger, D.T.; Izumi, T.; Papaconstantinou, J.; Mitra, S. Human AP-endonuclease 1 and hnRNP-L interact with a nCaRE-like repressor element in the AP-endonuclease 1 promoter. Nucleic Acids Res. 2002, 30, 823–829. [Google Scholar] [CrossRef] [PubMed]
- Bhakat, K.K.; Izumi, T.; Yang, S.-H.; Hazra, T.K.; Mitra, S. Role of acetylated human AP-endonuclease (APE1/Ref-1) in regulation of the parathyroid hormone gene. EMBO J. 2003, 22, 6299–6309. [Google Scholar] [CrossRef] [PubMed]
- Hanson, S.; Kim, E.; Deppert, W. Redox factor 1 (Ref-1) enhances specific DNA binding of p53 by promoting p53 tetramerization. Oncogene 2005, 24, 1641–1647. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Seemann, S.; Hainaut, P. Roles of thioredoxin reductase 1 and APE/Ref-1 in the control of basal p53 stability and activity. Oncogene 2005, 24, 3853–3863. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Sengupta, S.; Mantha, A.K.; Mitra, S.; Bhakat, K.K. Human AP endonuclease (APE1/Ref-1) and its acetylation regulate YB-1-p300 recruitment and RNA polymerase II loading in the drug-induced activation of multidrug resistance gene MDR1. Oncogene 2011, 30, 482–493. [Google Scholar] [CrossRef] [PubMed]
- Hajkova, P.; Jeffries, S.J.; Lee, C.; Miller, N.; Jackson, S.P.; Surani, M.A. Genome-wide reprogramming in the mouse germ line entails the base excision repair pathway. Science 2010, 329, 78–82. [Google Scholar] [CrossRef]
- Weber, A.R.; Krawczyk, C.; Robertson, A.B.; Kuśnierczyk, A.; Vågbø, C.B.; Schuermann, D.; Klungland, A.; Schär, P. Biochemical reconstitution of TET1–TDG–BER-dependent active DNA demethylation reveals a highly coordinated mechanism. Nat. Commun. 2016, 7, 10806. [Google Scholar] [CrossRef]
- Yamamori, T.; DeRicco, J.; Naqvi, A.; Hoffman, T.A.; Mattagajasingh, I.; Kasuno, K.; Jung, S.-B.; Kim, C.-S.; Irani, K. SIRT1 deacetylates APE1 and regulates cellular base excision repair. Nucleic Acids Res. 2010, 38, 832–845. [Google Scholar] [CrossRef]
- Hwang, B.-J.; Jin, J.; Gao, Y.; Shi, G.; Madabushi, A.; Yan, A.; Guan, X.; Zalzman, M.; Nakajima, S.; Lan, L.; et al. SIRT6 protein deacetylase interacts with MYH DNA glycosylase, APE1 endonuclease, and Rad9–Rad1–Hus1 checkpoint clamp. BMC Mol. Biol. 2015, 16, 12. [Google Scholar] [CrossRef]
- Madlener, S.; Ströbel, T.; Vose, S.; Saydam, O.; Price, B.D.; Demple, B.; Saydam, N. Essential role for mammalian apurinic/apyrimidinic (AP) endonuclease Ape1/Ref-1 in telomere maintenance. Proc. Natl. Acad. Sci. USA 2013, 110, 17844–17849. [Google Scholar] [CrossRef] [PubMed]
- Li, M.; Yang, X.; Lu, X.; Dai, N.; Zhang, S.; Cheng, Y.; Zhang, L.; Yang, Y.; Liu, Y.; Yang, Z.; et al. APE1 deficiency promotes cellular senescence and premature aging features. Nucleic Acids Res. 2018, 46, 5664–5677. [Google Scholar] [CrossRef] [PubMed]
- Vascotto, C.; Fantini, D.; Romanello, M.; Cesaratto, L.; Deganuto, M.; Leonardi, A.; Radicella, J.P.; Kelley, M.R.; D’Ambrosio, C.; Scaloni, A.; et al. APE1/Ref-1 interacts with NPM1 within nucleoli and plays a role in the rRNA quality control process. Mol. Cell. Biol. 2009, 29, 1834–1854. [Google Scholar] [CrossRef] [PubMed]
- Malfatti, M.C.; Balachander, S.; Antoniali, G.; Koh, K.D.; Saint-Pierre, C.; Gasparutto, D.; Chon, H.; Crouch, R.J.; Storici, F.; Tell, G. Abasic and oxidized ribonucleotides embedded in DNA are processed by human APE1 and not by RNase H2. Nucleic Acids Res. 2017, 45, 11193–11212. [Google Scholar] [CrossRef] [PubMed]
- Barnes, T.; Kim, W.-C.; Mantha, A.K.; Kim, S.-E.; Izumi, T.; Mitra, S.; Lee, C.H. Identification of Apurinic/apyrimidinic endonuclease 1 (APE1) as the endoribonuclease that cleaves c-myc mRNA. Nucleic Acids Res. 2009, 37, 3946–3958. [Google Scholar] [CrossRef] [PubMed]
- Antoniali, G.; Serra, F.; Lirussi, L.; Tanaka, M.; D’Ambrosio, C.; Zhang, S.; Radovic, S.; Dalla, E.; Ciani, Y.; Scaloni, A.; et al. Mammalian APE1 controls miRNA processing and its interactome is linked to cancer RNA metabolism. Nat. Commun. 2017, 8, 797. [Google Scholar] [CrossRef]
- Fan, Z.; Beresford, P.J.; Zhang, D.; Xu, Z.; Novina, C.D.; Yoshida, A.; Pommier, Y.; Lieberman, J. Cleaving the oxidative repair protein Ape1 enhances cell death mediated by granzyme A. Nat. Immunol. 2003, 4, 145–153. [Google Scholar] [CrossRef]
- Xanthoudakis, S.; Smeyne, R.J.; Wallace, J.D.; Curran, T. The redox/DNA repair protein, Ref-1, is essential for early embryonic development in mice. Proc. Natl. Acad. Sci. USA 1996, 93, 8919–8923. [Google Scholar] [CrossRef]
- Ludwig, D.L.; MacInnes, M.A.; Takiguchi, Y.; Purtymun, P.E.; Henrie, M.; Flannery, M.; Meneses, J.; Pedersen, R.A.; Chen, D.J. A murine AP-endonuclease gene-targeted deficiency with post-implantation embryonic progression and ionizing radiation sensitivity. Mutat. Res. 1998, 409, 17–29. [Google Scholar] [CrossRef]
- Meira, L.B.; Devaraj, S.; Kisby, G.E.; Burns, D.K.; Daniel, R.L.; Hammer, R.E.; Grundy, S.; Jialal, I.; Friedberg, E.C. Heterozygosity for the mouse Apex gene results in phenotypes associated with oxidative stress. Cancer Res. 2001, 61, 5552–5557. [Google Scholar]
- Wang, Y.; Shupenko, C.C.; Melo, L.F.; Strauss, P.R. DNA repair protein involved in heart and blood development. Mol. Cell. Biol. 2006, 26, 9083–9093. [Google Scholar] [CrossRef] [PubMed]
- Han, D.; Schomacher, L.; Schüle, K.M.; Mallick, M.; Musheev, M.U.; Karaulanov, E.; Krebs, L.; von Seggern, A.; Niehrs, C. NEIL1 and NEIL2 DNA glycosylases protect neural crest development against mitochondrial oxidative stress. eLife 2019, 8, e49044. [Google Scholar] [CrossRef] [PubMed]
- Stetler, R.A.; Gao, Y.; Leak, R.K.; Weng, Z.; Shi, Y.; Zhang, L.; Pu, H.; Zhang, F.; Hu, X.; Hassan, S.; et al. APE1/Ref-1 facilitates recovery of gray and white matter and neurological function after mild stroke injury. Proc. Natl. Acad. Sci. USA 2016, 113, E3558–E3567. [Google Scholar] [CrossRef] [PubMed]
- Dumitrache, L.C.; Shimada, M.; Downing, S.M.; Kwak, Y.D.; Li, Y.; Illuzzi, J.L.; Russell, H.R.; Wilson, D.M., III; McKinnon, P.J. Apurinic endonuclease-1 preserves neural genome integrity to maintain homeostasis and thermoregulation and prevent brain tumors. Proc. Natl. Acad. Sci. USA 2018, 115, E12285–E12294. [Google Scholar] [CrossRef] [PubMed]
- Masani, S.; Han, L.; Yu, K. Apurinic/apyrimidinic endonuclease 1 is the essential nuclease during immunoglobulin class switch recombination. Mol. Cell. Biol. 2013, 33, 1468–1473. [Google Scholar] [CrossRef] [PubMed]
- Chen, T.; Liu, C.; Lu, H.; Yin, M.; Shao, C.; Hu, X.; Wu, J.; Wang, Y. The expression of APE1 in triple-negative breast cancer and its effect on drug sensitivity of olaparib. Tumor Biol. 2017, 39, 1010428317713390. [Google Scholar] [CrossRef] [PubMed]
- Izumi, T.; Brown, D.B.; Naidu, C.V.; Bhakat, K.K.; MacInnes, M.A.; Saito, H.; Chen, D.J.; Mitra, S. Two essential but distinct functions of the mammalian abasic endonuclease. Proc. Natl. Acad. Sci. USA 2005, 102, 5739–5743. [Google Scholar] [CrossRef] [PubMed]
- Malfatti, M.C.; Gerratana, L.; Dalla, E.; Isola, M.; Damante, G.; Di Loreto, C.; Puglisi, F.; Tell, G. APE1 and NPM1 protect cancer cells from platinum compounds cytotoxicity and their expression pattern has a prognostic value in TNBC. J. Exp. Clin. Cancer Res. 2019, 38, 309. [Google Scholar] [CrossRef]
- Kim, D.V.; Kulishova, L.M.; Torgasheva, N.A.; Melentyev, V.S.; Dianov, G.L.; Medvedev, S.P.; Zakian, S.M.; Zharkov, D.O. Mild phenotype of knockouts of the major apurinic/apyrimidinic endonuclease APEX1 in a non-cancer human cell line. PLoS ONE 2021, 16, e0257473. [Google Scholar] [CrossRef]
- Stivers, J.T.; Jiang, Y.L. A mechanistic perspective on the chemistry of DNA repair glycosylases. Chem. Rev. 2003, 103, 2729–2760. [Google Scholar] [CrossRef]
- Dizdaroglu, M.; Coskun, E.; Jaruga, P. Repair of oxidatively induced DNA damage by DNA glycosylases: Mechanisms of action, substrate specificities and excision kinetics. Mutat. Res. 2017, 771, 99–127. [Google Scholar] [CrossRef] [PubMed]
- Pommier, Y.; Huang, S.-y.N.; Gao, R.; Das, B.B.; Murai, J.; Marchand, C. Tyrosyl-DNA-phosphodiesterases (TDP1 and TDP2). DNA Repair 2014, 19, 114–129. [Google Scholar] [CrossRef] [PubMed]
- Burkovics, P.; Szukacsov, V.; Unk, I.; Haracska, L. Human Ape2 protein has a 3′–5′ exonuclease activity that acts preferentially on mismatched base pairs. Nucleic Acids Res. 2006, 34, 2508–2515. [Google Scholar] [CrossRef] [PubMed]
- Lebedeva, N.A.; Rechkunova, N.I.; El-Khamisy, S.F.; Lavrik, O.I. Tyrosyl-DNA phosphodiesterase 1 initiates repair of apurinic/apyrimidinic site. Biochimie 2012, 94, 1749–1753. [Google Scholar] [CrossRef] [PubMed]
- Wiederhold, L.; Leppard, J.B.; Kedar, P.; Karimi-Busheri, F.; Rasouli-Nia, A.; Weinfeld, M.; Tomkinson, A.E.; Izumi, T.; Prasad, R.; Wilson, S.H.; et al. AP endonuclease-independent DNA base excision repair in human cells. Mol. Cell 2004, 15, 209–220. [Google Scholar] [CrossRef] [PubMed]
- Kanno, S.-I.; Kuzuoka, H.; Sasao, S.; Hong, Z.; Lan, L.; Nakajima, S.; Yasui, A. A novel human AP endonuclease with conserved zinc-finger-like motifs involved in DNA strand break responses. EMBO J. 2007, 26, 2094–2103. [Google Scholar] [CrossRef] [PubMed]
- Dorival, J.; Eichman, B.F. Human and bacterial TatD enzymes exhibit apurinic/apyrimidinic (AP) endonuclease activity. Nucleic Acids Res. 2023, 51, 2838–2849. [Google Scholar] [CrossRef]
- Huang, J.-C.; Hsu, D.S.; Kazantsev, A.; Sancar, A. Substrate spectrum of human excinuclease: Repair of abasic sites, methylated bases, mismatches, and bulky adducts. Proc. Natl. Acad. Sci. USA 1994, 91, 12213–12217. [Google Scholar] [CrossRef]
- Torres-Ramos, C.A.; Johnson, R.E.; Prakash, L.; Prakash, S. Evidence for the involvement of nucleotide excision repair in the removal of abasic sites in yeast. Mol. Cell. Biol. 2000, 20, 3522–3528. [Google Scholar] [CrossRef]
- Kim, N.; Jinks-Robertson, S. Abasic sites in the transcribed strand of yeast DNA are removed by transcription-coupled nucleotide excision repair. Mol. Cell. Biol. 2010, 30, 3206–3215. [Google Scholar] [CrossRef]
- Kitsera, N.; Rodriguez-Alvarez, M.; Emmert, S.; Carell, T.; Khobta, A. Nucleotide excision repair of abasic DNA lesions. Nucleic Acids Res. 2019, 47, 8537–8547. [Google Scholar] [CrossRef]
- Wittschieben, B.Ø.; Iwai, S.; Wood, R.D. DDB1-DDB2 (xeroderma pigmentosum group E) protein complex recognizes a cyclobutane pyrimidine dimer, mismatches, apurinic/apyrimidinic sites, and compound lesions in DNA. J. Biol. Chem. 2005, 280, 39982–39989. [Google Scholar] [CrossRef]
- Scrima, A.; Koníčková, R.; Czyzewski, B.K.; Kawasaki, Y.; Jeffrey, P.D.; Groisman, R.; Nakatani, Y.; Iwai, S.; Pavletich, N.P.; Thomä, N.H. Structural basis of UV DNA-damage recognition by the DDB1-DDB2 complex. Cell 2008, 135, 1213–1223. [Google Scholar] [CrossRef]
- Jang, S.; Kumar, N.; Beckwitt, E.C.; Kong, M.; Fouquerel, E.; Rapić-Otrin, V.; Prasad, R.; Watkins, S.C.; Khuu, C.; Majumdar, C.; et al. Damage sensor role of UV-DDB during base excision repair. Nat. Struct. Mol. Biol. 2019, 26, 695–703. [Google Scholar] [CrossRef]
- Klein, H.L.; Bačinskaja, G.; Che, J.; Cheblal, A.; Elango, R.; Epshtein, A.; Fitzgerald, D.M.; Gómez-González, B.; Khan, S.R.; Kumar, S.; et al. Guidelines for DNA recombination and repair studies: Cellular assays of DNA repair pathways. Microb. Cell 2019, 6, 1–64. [Google Scholar] [CrossRef]
- Owiti, N.A.; Nagel, Z.D.; Engelward, B.P. Fluorescence sheds light on DNA damage, DNA repair, and mutations. Trends Cancer 2021, 7, 240–248. [Google Scholar] [CrossRef]
- Lühnsdorf, B.; Kitsera, N.; Warken, D.; Lingg, T.; Epe, B.; Khobta, A. Generation of reporter plasmids containing defined base modifications in the DNA strand of choice. Anal. Biochem. 2012, 425, 47–53. [Google Scholar] [CrossRef]
- Raetz, A.G.; Xie, Y.; Kundu, S.; Brinkmeyer, M.K.; Chang, C.; David, S.S. Cancer-associated variants and a common polymorphism of MUTYH exhibit reduced repair of oxidative DNA damage using a GFP-based assay in mammalian cells. Carcinogenesis 2012, 33, 2301–2309. [Google Scholar] [CrossRef]
- Chaim, I.A.; Gardner, A.; Wu, J.; Iyama, T.; Wilson, D.M., III; Samson, L.D. A novel role for transcription-coupled nucleotide excision repair for the in vivo repair of 3,N4-ethenocytosine. Nucleic Acids Res. 2017, 45, 3242–3252. [Google Scholar] [CrossRef][Green Version]
- Chaim, I.A.; Nagel, Z.D.; Jordan, J.J.; Mazzucato, P.; Ngo, L.P.; Samson, L.D. In vivo measurements of interindividual differences in DNA glycosylases and APE1 activities. Proc. Natl. Acad. Sci. USA 2017, 114, E10379–E10388. [Google Scholar] [CrossRef]
- Rodriguez-Alvarez, M.; Kim, D.; Khobta, A. EGFP reporters for direct and sensitive detection of mutagenic bypass of DNA lesions. Biomolecules 2020, 10, 902. [Google Scholar] [CrossRef]
- Wilson, D.M., III; Takeshita, M.; Grollman, A.P.; Demple, B. Incision activity of human apurinic endonuclease (Ape) at abasic site analogs in DNA. J. Biol. Chem. 1995, 270, 16002–16007. [Google Scholar] [CrossRef]
- Mundle, S.T.; Delaney, J.C.; Essigmann, J.M.; Strauss, P.R. Enzymatic mechanism of human apurinic/apyrimidinic endonuclease against a THF AP site model substrate. Biochemistry 2009, 48, 19–26. [Google Scholar] [CrossRef]
- Iwai, S.; Maeda, M.; Shirai, M.; Shimada, Y.; Osafune, T.; Murata, T.; Ohtsuka, E. Reaction mechanism of T4 endonuclease V determined by analysis using modified oligonucleotide duplexes. Biochemistry 1995, 34, 4601–4609. [Google Scholar] [CrossRef]
- Allgayer, J.; Kitsera, N.; Bartelt, S.; Epe, B.; Khobta, A. Widespread transcriptional gene inactivation initiated by a repair intermediate of 8-oxoguanine. Nucleic Acids Res. 2016, 44, 7267–7280. [Google Scholar] [CrossRef]
- Li, H.; Endutkin, A.V.; Bergonzo, C.; Fu, L.; Grollman, A.P.; Zharkov, D.O.; Simmerling, C. DNA deformation-coupled recognition of 8-oxoguanine: Conformational kinetic gating in human DNA glycosylase. J. Am. Chem. Soc. 2017, 139, 2682–2692. [Google Scholar] [CrossRef]
- Demple, B.; Sung, J.-S. Molecular and biological roles of Ape1 protein in mammalian base excision repair. DNA Repair 2005, 4, 1442–1449. [Google Scholar] [CrossRef]
- McNeill, D.R.; Whitaker, A.M.; Stark, W.J.; Illuzzi, J.L.; McKinnon, P.J.; Freudenthal, B.D.; Wilson, D.M., III. Functions of the major abasic endonuclease (APE1) in cell viability and genotoxin resistance. Mutagenesis 2020, 35, 27–38. [Google Scholar] [CrossRef]
- Wang, W.; Walmacq, C.; Chong, J.; Kashlev, M.; Wang, D. Structural basis of transcriptional stalling and bypass of abasic DNA lesion by RNA polymerase II. Proc. Natl. Acad. Sci. USA 2018, 115, E2538–E2545. [Google Scholar] [CrossRef]
- Mengwasser, K.E.; Adeyemi, R.O.; Leng, Y.; Choi, M.Y.; Clairmont, C.; D’Andrea, A.D.; Elledge, S.J. Genetic screens reveal FEN1 and APEX2 as BRCA2 synthetic lethal targets. Mol. Cell 2019, 73, 885–899. [Google Scholar] [CrossRef]
- Álvarez-Quilón, A.; Wojtaszek, J.L.; Mathieu, M.-C.; Patel, T.; Appel, C.D.; Hustedt, N.; Rossi, S.E.; Wallace, B.D.; Setiaputra, D.; Adam, S.; et al. Endogenous DNA 3′ blocks are vulnerabilities for BRCA1 and BRCA2 deficiency and are reversed by the APE2 nuclease. Mol. Cell 2020, 78, 1152–1165. [Google Scholar] [CrossRef]
- Visnes, T.; Doseth, B.; Pettersen, H.S.; Hagen, L.; Sousa, M.M.L.; Akbari, M.; Otterlei, M.; Kavli, B.; Slupphaug, G.; Krokan, H.E. Uracil in DNA and its processing by different DNA glycosylases. Philos. Trans. R. Soc. B Biol. Sci. 2009, 364, 563–568. [Google Scholar] [CrossRef]
- Kubota, Y.; Nash, R.A.; Klungland, A.; Schär, P.; Barnes, D.E.; Lindahl, T. Reconstitution of DNA base excision-repair with purified human proteins: Interaction between DNA polymerase β and the XRCC1 protein. EMBO J. 1996, 15, 6662–6670. [Google Scholar] [CrossRef]
- Nicholl, I.D.; Nealon, K.; Kenny, M.K. Reconstitution of human base excision repair with purified proteins. Biochemistry 1997, 36, 7557–7566. [Google Scholar] [CrossRef]
- Akbari, M.; Otterlei, M.; Peña-Diaz, J.; Aas, P.A.; Kavli, B.; Liabakk, N.B.; Hagen, L.; Imai, K.; Durandy, A.; Slupphaug, G.; et al. Repair of U/G and U/A in DNA by UNG2-associated repair complexes takes place predominantly by short-patch repair both in proliferating and growth-arrested cells. Nucleic Acids Res. 2004, 32, 5486–5498. [Google Scholar] [CrossRef]
- Zharkov, D.O.; Rosenquist, T.A.; Gerchman, S.E.; Grollman, A.P. Substrate specificity and reaction mechanism of murine 8-oxoguanine-DNA glycosylase. J. Biol. Chem. 2000, 275, 28607–28617. [Google Scholar] [CrossRef]
- Hill, J.W.; Hazra, T.K.; Izumi, T.; Mitra, S. Stimulation of human 8-oxoguanine-DNA glycosylase by AP-endonuclease: Potential coordination of the initial steps in base excision repair. Nucleic Acids Res. 2001, 29, 430–438. [Google Scholar] [CrossRef]
- Saitoh, T.; Shinmura, K.; Yamaguchi, S.; Tani, M.; Seki, S.; Murakami, H.; Nojima, Y.; Yokota, J. Enhancement of OGG1 protein AP lyase activity by increase of APEX protein. Mutat. Res. 2001, 486, 31–40. [Google Scholar] [CrossRef]
- Vidal, A.E.; Hickson, I.D.; Boiteux, S.; Radicella, J.P. Mechanism of stimulation of the DNA glycosylase activity of hOGG1 by the major human AP endonuclease: Bypass of the AP lyase activity step. Nucleic Acids Res. 2001, 29, 1285–1292. [Google Scholar] [CrossRef]
- Das, A.; Wiederhold, L.; Leppard, J.B.; Kedar, P.; Prasad, R.; Wang, H.; Boldogh, I.; Karimi-Busheri, F.; Weinfeld, M.; Tomkinson, A.E.; et al. NEIL2-initiated, APE-independent repair of oxidized bases in DNA: Evidence for a repair complex in human cells. DNA Repair 2006, 5, 1439–1448. [Google Scholar] [CrossRef]
- Zharkov, D.O.; Shoham, G.; Grollman, A.P. Structural characterization of the Fpg family of DNA glycosylases. DNA Repair 2003, 2, 839–862. [Google Scholar] [CrossRef]
- Kuznetsov, N.A.; Zharkov, D.O.; Koval, V.V.; Buckle, M.; Fedorova, O.S. Reversible chemical step and rate-limiting enzyme regeneration in the reaction catalyzed by formamidopyrimidine-DNA-glycosylase. Biochemistry 2009, 48, 11335–11343. [Google Scholar] [CrossRef]
- Aspinwall, R.; Rothwell, D.G.; Roldan-Arjona, T.; Anselmino, C.; Ward, C.J.; Cheadle, J.P.; Sampson, J.R.; Lindahl, T.; Harris, P.C.; Hickson, I.D. Cloning and characterization of a functional human homolog of Escherichia coli endonuclease III. Proc. Natl. Acad. Sci. USA 1997, 94, 109–114. [Google Scholar] [CrossRef]
- Ikeda, S.; Biswas, T.; Roy, R.; Izumi, T.; Boldogh, I.; Kurosky, A.; Sarker, A.H.; Seki, S.; Mitra, S. Purification and characterization of human NTH1, a homolog of Escherichia coli endonuclease III: Direct identification of Lys-212 as the active nucleophilic residue. J. Biol. Chem. 1998, 273, 21585–21593. [Google Scholar] [CrossRef]
- Sarmini, L.; Meabed, M.; Emmanouil, E.; Atsaves, G.; Robeska, E.; Karwowski, B.T.; Campalans, A.; Gimisis, T.; Khobta, A. Requirement of transcription-coupled nucleotide excision repair for the removal of a specific type of oxidatively induced DNA damage. Nucleic Acids Res. 2023, 51, 4982–4994. [Google Scholar] [CrossRef]
- Khobta, A.; Lingg, T.; Schulz, I.; Warken, D.; Kitsera, N.; Epe, B. Mouse CSB protein is important for gene expression in the presence of a single-strand break in the non-transcribed DNA strand. DNA Repair 2010, 9, 985–993. [Google Scholar] [CrossRef]
- Kitsera, N.; Stathis, D.; Lühnsdorf, B.; Müller, H.; Carell, T.; Epe, B.; Khobta, A. 8-Oxo-7,8-dihydroguanine in DNA does not constitute a barrier to transcription, but is converted into transcription-blocking damage by OGG1. Nucleic Acids Res. 2011, 39, 5926–5934. [Google Scholar] [CrossRef]
- Allgayer, J.; Kitsera, N.; von der Lippen, C.; Epe, B.; Khobta, A. Modulation of base excision repair of 8-oxoguanine by the nucleotide sequence. Nucleic Acids Res. 2013, 41, 8559–8571. [Google Scholar] [CrossRef]
- Lühnsdorf, B.; Epe, B.; Khobta, A. Excision of uracil from transcribed DNA negatively affects gene expression. J. Biol. Chem. 2014, 289, 22008–22018. [Google Scholar] [CrossRef]
- Kitsera, N.; Allgayer, J.; Parsa, E.; Geier, N.; Rossa, M.; Carell, T.; Khobta, A. Functional impacts of 5-hydroxymethylcytosine, 5-formylcytosine, and 5-carboxycytosine at a single hemi-modified CpG dinucleotide in a gene promoter. Nucleic Acids Res. 2017, 45, 11033–11042. [Google Scholar] [CrossRef]
- Eckstein, F. Phosphorothioates, essential components of therapeutic oligonucleotides. Nucleic Acid Ther. 2014, 24, 374–387. [Google Scholar] [CrossRef]
- Matsumoto, Y.; Kim, K.; Bogenhagen, D.F. Proliferating cell nuclear antigen-dependent abasic site repair in Xenopus laevis oocytes: An alternative pathway of base excision DNA repair. Mol. Cell. Biol. 1994, 14, 6187–6197. [Google Scholar]
- Matsumoto, Y.; Kim, K.; Hurwitz, J.; Gary, R.; Levin, D.S.; Tomkinson, A.E.; Park, M.S. Reconstitution of proliferating cell nuclear antigen-dependent repair of apurinic/apyrimidinic sites with purified human proteins. J. Biol. Chem. 1999, 274, 33703–33708. [Google Scholar] [CrossRef]
- Woodrick, J.; Gupta, S.; Camacho, S.; Parvathaneni, S.; Choudhury, S.; Cheema, A.; Bai, Y.; Khatkar, P.; Erkizan, H.V.; Sami, F.; et al. A new sub-pathway of long-patch base excision repair involving 5′ gap formation. EMBO J. 2017, 36, 1605–1622. [Google Scholar] [CrossRef]
- Schomacher, L.; Han, D.; Musheev, M.U.; Arab, K.; Kienhöfer, S.; von Seggern, A.; Niehrs, C. Neil DNA glycosylases promote substrate turnover by Tdg during DNA demethylation. Nat. Struct. Mol. Biol. 2016, 23, 116–124. [Google Scholar] [CrossRef]
- Rahimoff, R.; Kosmatchev, O.; Kirchner, A.; Pfaffeneder, T.; Spada, F.; Brantl, V.; Müller, M.; Carell, T. 5-Formyl- and 5-carboxydeoxycytidines do not cause accumulation of harmful repair intermediates in stem cells. J. Am. Chem. Soc. 2017, 139, 10359–10364. [Google Scholar] [CrossRef]
- Barbado, C.; Córdoba-Cañero, D.; Ariza, R.R.; Roldán-Arjona, T. Nonenzymatic release of N7-methylguanine channels repair of abasic sites into an AP endonuclease-independent pathway in Arabidopsis. Proc. Natl. Acad. Sci. USA 2018, 115, E916–E924. [Google Scholar] [CrossRef]
- Osman, F.; Bjørås, M.; Alseth, I.; Morland, I.; McCready, S.; Seeberg, E.; Tsaneva, I. A new Schizosaccharomyces pombe base excision repair mutant, nth1, reveals overlapping pathways for repair of DNA base damage. Mol. Microbiol. 2003, 48, 465–480. [Google Scholar] [CrossRef]
- Alseth, I.; Korvald, H.; Osman, F.; Seeberg, E.; Bjørås, M. A general role of the DNA glycosylase Nth1 in the abasic sites cleavage step of base excision repair in Schizosaccharomyces pombe. Nucleic Acids Res. 2004, 32, 5119–5125. [Google Scholar] [CrossRef]
- Sugimoto, T.; Igawa, E.; Tanihigashi, H.; Matsubara, M.; Ide, H.; Ikeda, S. Roles of base excision repair enzymes Nth1p and Apn2p from Schizosaccharomyces pombe in processing alkylation and oxidative DNA damage. DNA Repair 2005, 4, 1270–1280. [Google Scholar] [CrossRef]
- Nilsen, L.; Forstrøm, R.J.; Bjørås, M.; Alseth, I. AP endonuclease independent repair of abasic sites in Schizosaccharomyces pombe. Nucleic Acids Res. 2012, 40, 2000–2009. [Google Scholar] [CrossRef]
- Grin, I.R.; Mechetin, G.V.; Kasymov, R.D.; Diatlova, E.A.; Yudkina, A.V.; Shchelkunov, S.N.; Gileva, I.P.; Denisova, A.A.; Stepanov, G.A.; Chilov, G.G.; et al. A new class of uracil–DNA glycosylase inhibitors active against human and vaccinia virus enzyme. Molecules 2021, 26, 6668. [Google Scholar] [CrossRef]
- Ran, F.A.; Hsu, P.D.; Wright, J.; Agarwala, V.; Scott, D.A.; Zhang, F. Genome engineering using the CRISPR-Cas9 system. Nat. Protoc. 2013, 8, 2281–2308. [Google Scholar] [CrossRef]
- Brinkman, E.K.; Chen, T.; Amendola, M.; van Steensel, B. Easy quantitative assessment of genome editing by sequence trace decomposition. Nucleic Acids Res. 2014, 42, e168. [Google Scholar] [CrossRef]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Oligo ID | Sequence (5′→3′) | Modification |
|---|---|---|
| Construction of lesion-containing plasmids | ||
| ts.613_A | TCAGGGCGGACTAGGTGC | |
| ts.613_F | TCAGGGCGGACTXGGTGC | X = F |
| ts.613_sF | TCAGGGCGGACTXGGTGC | X = sF |
| ts.613_U | TCAGGGCGGACTXGGTGC | X = U |
| ts.613_sU | TCAGGGCGGACTXGGTGC | X = sU |
| ts.613_sUs | TCAGGGCGGACTXGGTGC | X = sUs |
| Q205_compl | GCACCTAGTCCGCCCTGA | |
| sgRNA cloning for APEX2 knockout | ||
| APEX2_top | CACCGATTCGGAGACCCCTGCAAG | |
| APEX2_bot | AAACCTTGCAGGGGTCTCCGAATC | |
| TA-cloning and TIDE for APEX2 knockout | ||
| APEX2_fwd1 | AGGAAGCAGTTCGCTCGC | |
| APEX2_rev1 | CTGAGGGGAGATAAGAGGGTGAA | |
| APEX2_fwd2 | CTTTGCTTCCTTCAGCGTCC | |
| APEX2_rev2 | TTCGGGGGTTTGACTTGG | |
| Real-time RT-PCR for APEX2 mRNA | ||
| APEX2_rt_fwd | CTGGAACATCAATGGGATTCGG | |
| APEX2_rt_rev | CCAGCTCGTCCAAAATGCG | |
| GAPDH_rt_fwd | ACATCGCTCAGACACCAT | |
| GAPDH_rt_rev | TGTAGTTGAGGTCAATGAAGG | |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kim, D.V.; Diatlova, E.A.; Zharkov, T.D.; Melentyev, V.S.; Yudkina, A.V.; Endutkin, A.V.; Zharkov, D.O. Back-Up Base Excision DNA Repair in Human Cells Deficient in the Major AP Endonuclease, APE1. Int. J. Mol. Sci. 2024, 25, 64. https://doi.org/10.3390/ijms25010064
Kim DV, Diatlova EA, Zharkov TD, Melentyev VS, Yudkina AV, Endutkin AV, Zharkov DO. Back-Up Base Excision DNA Repair in Human Cells Deficient in the Major AP Endonuclease, APE1. International Journal of Molecular Sciences. 2024; 25(1):64. https://doi.org/10.3390/ijms25010064
Chicago/Turabian StyleKim, Daria V., Evgeniia A. Diatlova, Timofey D. Zharkov, Vasily S. Melentyev, Anna V. Yudkina, Anton V. Endutkin, and Dmitry O. Zharkov. 2024. "Back-Up Base Excision DNA Repair in Human Cells Deficient in the Major AP Endonuclease, APE1" International Journal of Molecular Sciences 25, no. 1: 64. https://doi.org/10.3390/ijms25010064
APA StyleKim, D. V., Diatlova, E. A., Zharkov, T. D., Melentyev, V. S., Yudkina, A. V., Endutkin, A. V., & Zharkov, D. O. (2024). Back-Up Base Excision DNA Repair in Human Cells Deficient in the Major AP Endonuclease, APE1. International Journal of Molecular Sciences, 25(1), 64. https://doi.org/10.3390/ijms25010064