
iPSC-Derived Endothelial Cells Reveal LDLR Dysfunction and Dysregulated Gene Expression Profiles in Familial Hypercholesterolemia

 , , , , ,
, , , , ,  ,
, 
Abstract
1. Introduction
2. Results
2.1. Generation and Characterization of Endothelial Derivatives from iPSCs
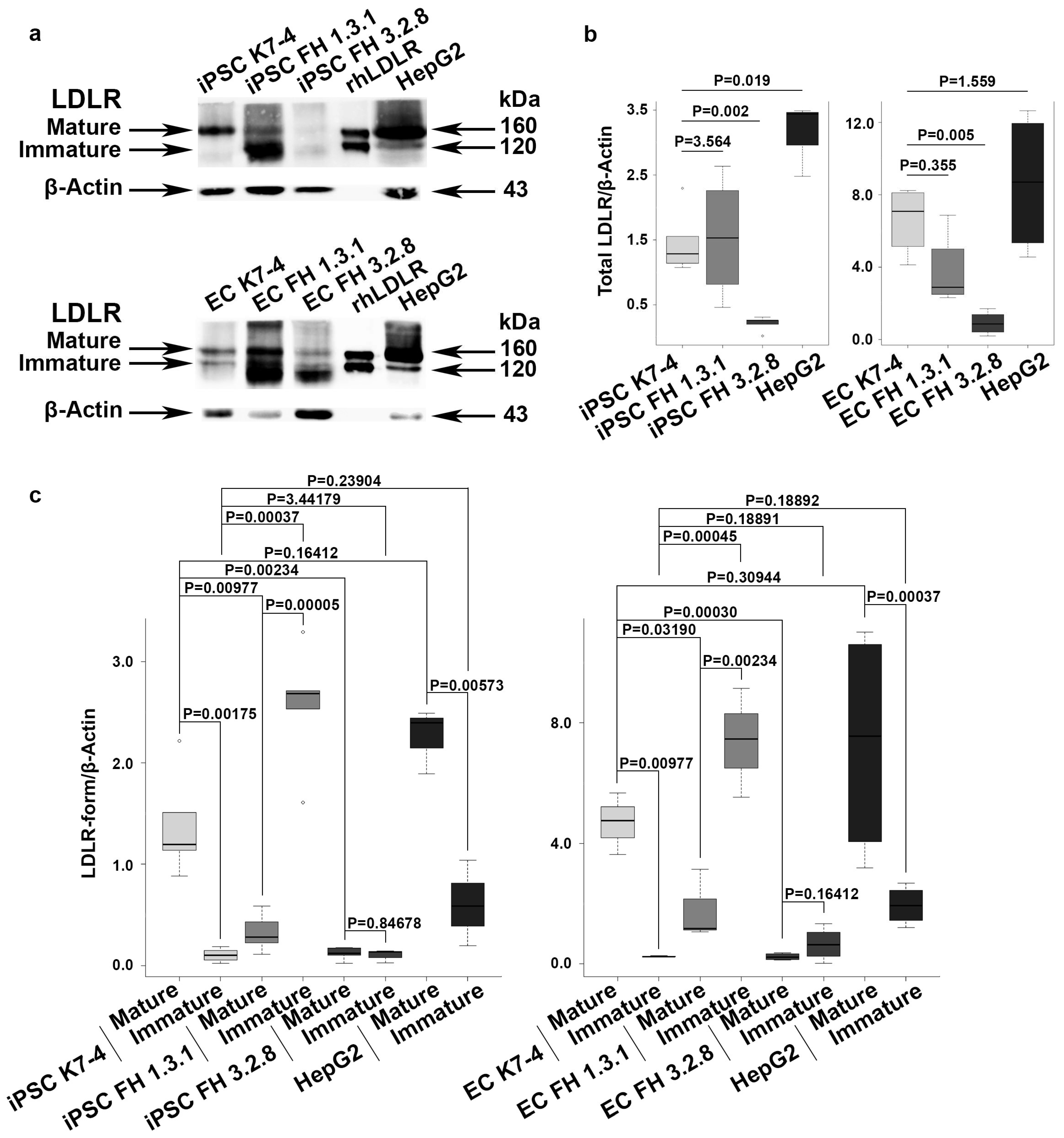
2.2. iPSCs and iPSC-Derived Endothelial Cells (iPSC-ECs) from Patients with FH Have Reduced Levels of Mature LDLR
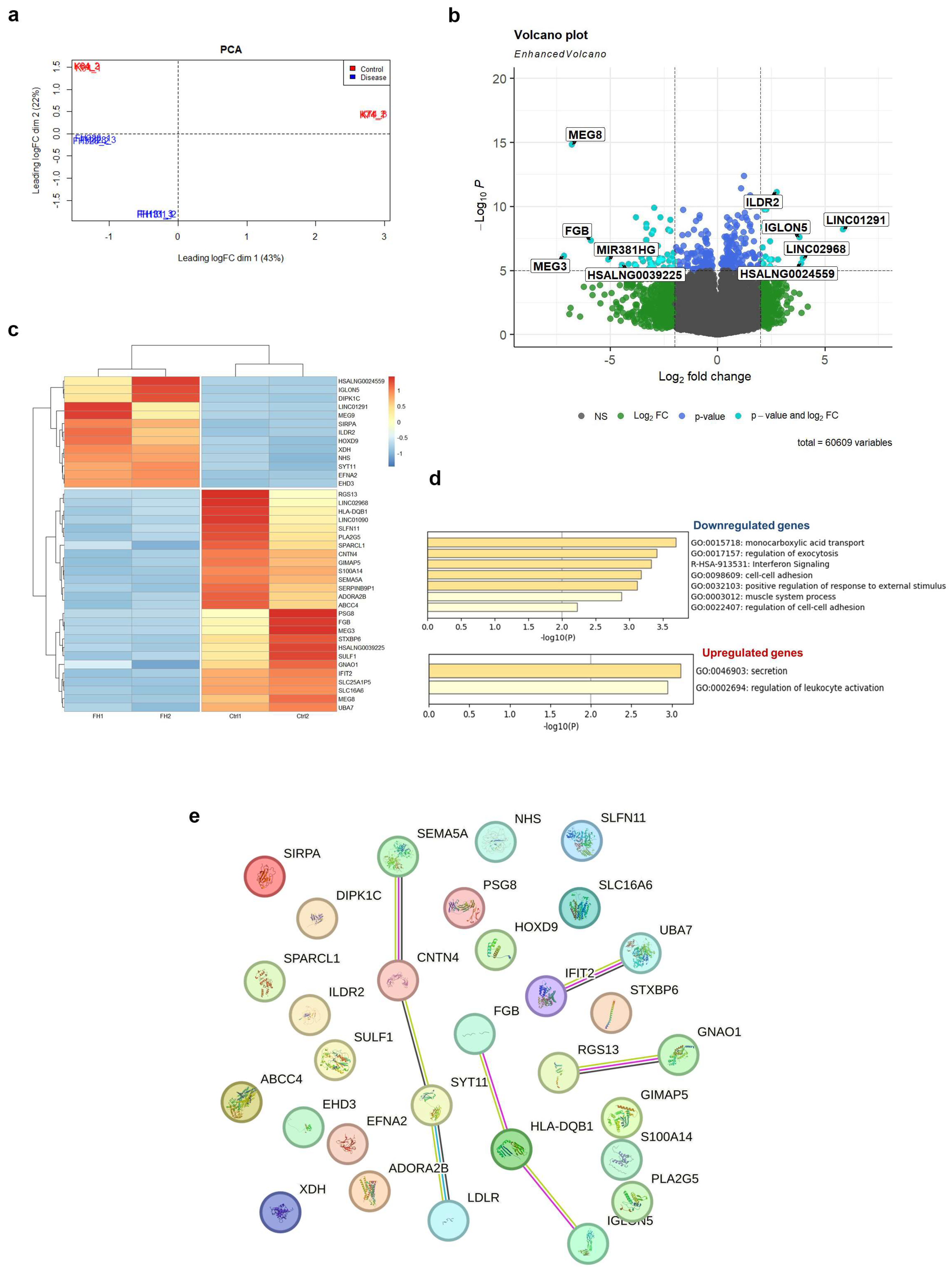
2.3. Transcriptome Profiling Reveals Dysregulation of Several Signaling Pathways in LDLR Mutant iPSC-ECs
3. Discussion
4. Materials and Methods
4.1. Ethical Statements
4.2. iPSC Lines and Their Cultivation
4.3. Directed Differentiation of iPSCs into Endothelial Derivatives
4.4. Flow Cytometry
4.5. Immunostaining
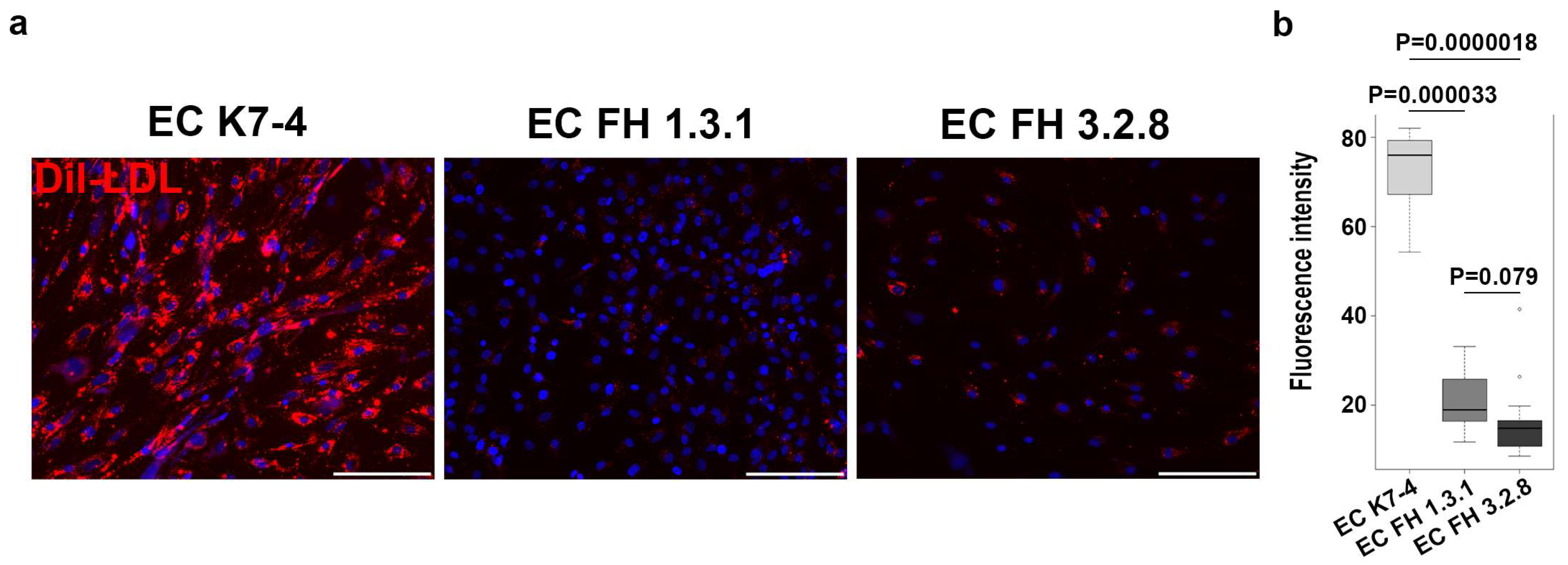
4.6. Functional Evaluation of Endothelial Cells
4.7. Immunoblotting
4.8. RNA-seq
4.9. Statistics
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Goldstein, J.L.; Brown, M.S. The LDL Receptor. Arterioscler. Thromb. Vasc. Biol. 2009, 29, 431–438. [Google Scholar] [CrossRef] [PubMed]
- Alves, A.C.; Azevedo, S.; Benito-Vicente, A.; Graça, R.; Galicia-Garcia, U.; Barros, P.; Jordan, P.; Martin, C.; Bourbon, M. LDLR Variants Functional Characterization: Contribution to Variant Classification. Atherosclerosis 2021, 329, 14–21. [Google Scholar] [CrossRef] [PubMed]
- Benito-Vicente, A.; Uribe, K.B.; Jebari, S.; Galicia-Garcia, U.; Ostolaza, H.; Martin, C. Familial Hypercholesterolemia: The Most Frequent Cholesterol Metabolism Disorder Caused Disease. Int. J. Mol. Sci. 2018, 19, 3426. [Google Scholar] [CrossRef] [PubMed]
- Beheshti, S.O.; Madsen, C.M.; Varbo, A.; Nordestgaard, B.G. Worldwide Prevalence of Familial Hypercholesterolemia: Meta-Analyses of 11 Million Subjects. J. Am. Coll. Cardiol. 2020, 75, 2553–2566. [Google Scholar] [CrossRef] [PubMed]
- Kastelein, J.J.P.; Reeskamp, L.F.; Hovingh, G.K. Familial Hypercholesterolemia: The Most Common Monogenic Disorder in Humans. J. Am. Coll. Cardiol. 2020, 75, 2567–2569. [Google Scholar] [CrossRef]
- GWAS Catalog. Available online: https://www.ebi.ac.uk/gwas/ (accessed on 1 November 2023).
- Kobiyama, K.; Ley, K. Atherosclerosis a Chronic Inflammatory Disease with an Autoimmune Component. Circ. Res. 2018, 123, 1118–1120. [Google Scholar] [CrossRef] [PubMed]
- Botts, S.R.; Fish, J.E.; Howe, K.L. Dysfunctional Vascular Endothelium as a Driver of Atherosclerosis: Emerging Insights into Pathogenesis and Treatment. Front. Pharmacol. 2021, 12, 787541. [Google Scholar] [CrossRef]
- Gimbrone, M.A.; García-Cardeña, G. Endothelial Cell Dysfunction and the Pathobiology of Atherosclerosis. Circ. Res. 2016, 118, 620–636. [Google Scholar] [CrossRef]
- Hu, P.; Dharmayat, K.I.; Stevens, C.A.T.; Sharabiani, M.T.A.; Jones, R.S.; Watts, G.F.; Genest, J.; Ray, K.K.; Vallejo-Vaz, A.J. Prevalence of Familial Hypercholesterolemia among the General Population and Patients with Atherosclerotic Cardiovascular Disease: A Systematic Review and Meta-Analysis. Circulation 2020, 141, 1742–1759. [Google Scholar] [CrossRef]
- Watts, G.F.; Gidding, S.S.; Hegele, R.A.; Raal, F.J.; Sturm, A.C.; Jones, L.K.; Sarkies, M.N.; Al-Rasadi, K.; Blom, D.J.; Daccord, M.; et al. International Atherosclerosis Society Guidance for Implementing Best Practice in the Care of Familial Hypercholesterolaemia. Nat. Rev. Cardiol. 2023, 20, 845–869. [Google Scholar] [CrossRef]
- Ganjali, S.; Keshavarz, R.; Hosseini, S.; Mansouri, A.; Mannarino, M.R.; Pirro, M.; Jamialahmadi, T.; Sahebkar, A. Evaluation of Oxidative Stress Status in Familial Hypercholesterolemia. J. Clin. Med. 2021, 10, 5867. [Google Scholar] [CrossRef] [PubMed]
- Mussbacher, M.; Schossleitner, K.; Kral-Pointner, J.B.; Salzmann, M.; Schrammel, A.; Schmid, J.A. More than Just a Monolayer: The Multifaceted Role of Endothelial Cells in the Pathophysiology of Atherosclerosis. Curr. Atheroscler. Rep. 2022, 24, 483–492. [Google Scholar] [CrossRef] [PubMed]
- Nielsen, M.H.; Irvine, H.; Vedel, S.; Raungaard, B.; Beck-Nielsen, H.; Handberg, A. Elevated Atherosclerosis-Related Gene Expression, Monocyte Activation and Microparticle-Release Are Related to Increased Lipoprotein-Associated Oxidative Stress in Familial Hypercholesterolemia. PLoS ONE 2015, 10, e0121516. [Google Scholar] [CrossRef]
- Prajapati, R.; Agrawal, V. Familial Hypercholesterolemia Supravalvular Aortic Stenosis and Extensive Atherosclerosis. Indian Heart J. 2018, 70, 575–577. [Google Scholar] [CrossRef] [PubMed]
- Ten Kate, G.J.R.; Bos, S.; Dedic, A.; Neefjes, L.A.; Kurata, A.; Langendonk, J.G.; Liem, A.; Moelker, A.; Krestin, G.P.; De Feyter, P.J.; et al. Increased Aortic Valve Calcification in Familial Hypercholesterolemia Prevalence, Extent, and Associated Risk Factors. J. Am. Coll. Cardiol. 2015, 66, 2687–2695. [Google Scholar] [CrossRef] [PubMed]
- Chowdhury, U.K.; Chauhan, A.; Hasija, S.; Jena, J.K.; Sankhyan, L.K.; Phulware, R. Aortic Root Enlargement and Aortic Valve Replacement for Calcified Supravalvular and Valvular Aortic Stenosis in Homozygous Familial Hypercholesterolemia: A Case Report. World J. Pediatr. Congenit. Heart Surg. 2020, 11, NP221–NP225. [Google Scholar] [CrossRef] [PubMed]
- Pérez De Isla, L.; Watts, G.F.; Alonso, R.; Díaz-Díaz, J.L.; Muñiz-Grijalvo, O.; Zambón, D.; Fuentes, F.; De Andrés, R.; Padró, T.; López-Miranda, J.; et al. Lipoprotein(a), LDL-Cholesterol, and Hypertension: Predictors of the Need for Aortic Valve Replacement in Familial Hypercholesterolaemia. Eur. Heart J. 2021, 42, 2201–2211. [Google Scholar] [CrossRef]
- Medvedev, S.P.; Shevchenko, A.I.; Zakian, S.M. Induced Pluripotent Stem Cells: Problems and Advantages When Applying Them in Regenerative Medicine. Acta Naturae 2010, 2, 18–28. [Google Scholar] [CrossRef]
- Kennedy, C.C.; Brown, E.E.; Abutaleb, N.O.; Truskey, G.A. Development and Application of Endothelial Cells Derived From Pluripotent Stem Cells in Microphysiological Systems Models. Front. Cardiovasc. Med. 2021, 8, 625016. [Google Scholar] [CrossRef]
- Lin, Y.; Gil, C.H.; Yoder, M.C. Differentiation, Evaluation, and Application of Human Induced Pluripotent Stem Cell? Derived Endothelial Cells. Arterioscler. Thromb. Vasc. Biol. 2017, 37, 2014–2025. [Google Scholar] [CrossRef]
- Ong, S.B.; Lee, W.H.; Shao, N.Y.; Ismail, N.I.; Katwadi, K.; Lim, M.M.; Kwek, X.Y.; Michel, N.A.; Li, J.; Newson, J.; et al. Calpain Inhibition Restores Autophagy and Prevents Mitochondrial Fragmentation in a Human IPSC Model of Diabetic Endotheliopathy. Stem Cell Rep. 2019, 12, 597–610. [Google Scholar] [CrossRef] [PubMed]
- Atchison, L.; Abutaleb, N.O.; Snyder-Mounts, E.; Gete, Y.; Ladha, A.; Ribar, T.; Cao, K.; Truskey, G.A. IPSC-Derived Endothelial Cells Affect Vascular Function in a Tissue-Engineered Blood Vessel Model of Hutchinson-Gilford Progeria Syndrome. Stem Cell Rep. 2020, 14, 325–337. [Google Scholar] [CrossRef] [PubMed]
- Kelleher, J.; Dickinson, A.; Cain, S.; Hu, Y.; Bates, N.; Harvey, A.; Ren, J.; Zhang, W.; Moreton, F.C.; Muir, K.W.; et al. Patient-Specific IPSC Model of a Genetic Vascular Dementia Syndrome Reveals Failure of Mural Cells to Stabilize Capillary Structures. Stem Cell Rep. 2019, 13, 817–831. [Google Scholar] [CrossRef] [PubMed]
- Jiang, B.; Wang, X.; Rivera-Bolanos, N.; Ameer, G.A. Generation of Autologous Vascular Endothelial Cells for Patients with Peripheral Artery Disease. J. Cardiovasc. Transl. Res. 2023. ahead of print. [Google Scholar] [CrossRef]
- Caron, J.; Pène, V.; Tolosa, L.; Villaret, M.; Luce, E.; Fourrier, A.; Heslan, J.M.; Saheb, S.; Bruckert, E.; Gómez-Lechón, M.J.; et al. Low-Density Lipoprotein Receptor-Deficient Hepatocytes Differentiated from Induced Pluripotent Stem Cells Allow Familial Hypercholesterolemia Modeling, CRISPR/Cas-Mediated Genetic Correction, and Productive Hepatitis C Virus Infection. Stem Cell Res. Ther. 2019, 10, 221. [Google Scholar] [CrossRef]
- Omer, L.; Hudson, E.A.; Zheng, S.; Hoying, J.B.; Shan, Y.; Boyd, N.L. CRISPR Correction of a Homozygous Low-Density Lipoprotein Receptor Mutation in Familial Hypercholesterolemia Induced Pluripotent Stem Cells. Hepatol. Commun. 2017, 1, 886–898. [Google Scholar] [CrossRef] [PubMed]
- Omer, L.; Hindi, L.; Militello, G.; Stivers, K.B.; Tien, K.C.; Boyd, N.L. Familial Hypercholesterolemia Class II Low-Density Lipoprotein Receptor Response to Statin Treatment. DMM Dis. Model. Mech. 2020, 13, dmm042911. [Google Scholar] [CrossRef]
- Okada, H.; Nakanishi, C.; Yoshida, S.; Shimojima, M.; Yokawa, J.; Mori, M.; Tada, H.; Yoshimuta, T.; Hayashi, K.; Yamano, T.; et al. Function and Immunogenicity of Gene-Corrected IPSC-Derived Hepatocyte-Like Cells in Restoring Low Density Lipoprotein Uptake in Homozygous Familial Hypercholesterolemia. Sci. Rep. 2019, 9, 4695. [Google Scholar] [CrossRef]
- Ramakrishnan, V.M.; Yang, J.Y.; Tien, K.T.; McKinley, T.R.; Bocard, B.R.; Maijub, J.G.; Burchell, P.O.; Williams, S.K.; Morris, M.E.; Hoying, J.B.; et al. Restoration of Physiologically Responsive Low-Density Lipoprotein Receptor-Mediated Endocytosis in Genetically Deficient Induced Pluripotent Stem Cells. Sci. Rep. 2015, 5, 13231. [Google Scholar] [CrossRef]
- De Lorenzo, A.; Moreira, A.S.B.; Muccillo, F.B.; Assad, M.; Tibirica, E.V. Microvascular Function and Endothelial Progenitor Cells in Patients with Severe Hypercholesterolemia and the Familial Hypercholesterolemia Phenotype. Cardiology 2017, 137, 231–236. [Google Scholar] [CrossRef]
- Pajkowski, M.; Dudziak, M.; Chlebus, K.; Hellmann, M. Assessment of Microvascular Function and Pharmacological Regulation in Genetically Confirmed Familial Hypercholesterolemia. Microvasc. Res. 2021, 138, 104216. [Google Scholar] [CrossRef] [PubMed]
- Vuorio, A.; Kovanen, P.T.; Raal, F.J. Coronary Microcirculatory Dysfunction in Hypercholesterolemic Patients with COVID-19: Potential Benefit from Cholesterol-Lowering Treatment. Ann. Med. 2023, 55, 2199218. [Google Scholar] [CrossRef] [PubMed]
- Catar, R.; Chen, L.; Zhao, H.; Wu, D.; Kamhieh-Milz, J.; Lücht, C.; Zickler, D.; Krug, A.W.; Ziegler, C.G.; Morawietz, H.; et al. Native and Oxidized Low-Density Lipoproteins Increase the Expression of the LDL Receptor and the LOX-1 Receptor, Respectively, in Arterial Endothelial Cells. Cells 2022, 11, 204. [Google Scholar] [CrossRef] [PubMed]
- Luchetti, F.; Crinelli, R.; Nasoni, M.G.; Benedetti, S.; Palma, F.; Fraternale, A.; Iuliano, L. LDL Receptors, Caveolae and Cholesterol in Endothelial Dysfunction: OxLDLs Accomplices or Victims? Br. J. Pharmacol. 2021, 178, 3104–3114. [Google Scholar] [CrossRef] [PubMed]
- Zhang, X.; Sessa, W.C.; Fernández-Hernando, C. Endothelial Transcytosis of Lipoproteins in Atherosclerosis. Front. Cardiovasc. Med. 2018, 5, 130. [Google Scholar] [CrossRef] [PubMed]
- Dehouck, B.; Fenart, L.; Dehouck, M.P.; Pierce, A.; Torpier, G.; Cecchelli, R. A New Function for the LDL Receptor: Transcytosis of LDL across the Blood-Brain Barrier. J. Cell Biol. 1997, 138, 877–889. [Google Scholar] [CrossRef]
- Zakharova, I.S.; Shevchenko, A.I.; Tmoyan, N.A.; Elisaphenko, E.A.; Kalinin, A.P.; Sleptcov, A.A.; Nazarenko, M.S.; Ezhov, M.V.; Kukharchuk, V.V.; Parfyonova, Y.V.; et al. Induced Pluripotent Stem Cell Line ICGi037-A, Obtained by Reprogramming Peripheral Blood Mononuclear Cells from a Patient with Familial Hypercholesterolemia Due to Heterozygous p.Trp443Arg Mutations in LDLR. Stem Cell Res. 2022, 60, 102703. [Google Scholar] [CrossRef]
- Zakharova, I.S.; Shevchenko, A.I.; Tmoyan, N.A.; Elisaphenko, E.A.; Zubkova, E.S.; Sleptcov, A.A.; Nazarenko, M.S.; Ezhov, M.V.; Kukharchuk, V.V.; Parfyonova, Y.V.; et al. Induced Pluripotent Stem Cell Line ICGi036-A Generated by Reprogramming Peripheral Blood Mononuclear Cells from a Patient with Familial Hypercholesterolemia Caused Due to Compound Heterozygous p.Ser177Leu/p.Cys352Arg Mutations in LDLR. Stem Cell Res. 2022, 59, 102653. [Google Scholar] [CrossRef]
- Nazarenko, M.S.; Sleptcov, A.A.; Zarubin, A.A.; Salakhov, R.R.; Shevchenko, A.I.; Tmoyan, N.A.; Elisaphenko, E.A.; Zubkova, E.S.; Zheltysheva, N.V.; Ezhov, M.V.; et al. Calling and Phasing of Single-Nucleotide and Structural Variants of the LDLR Gene Using Oxford Nanopore MinION. Int. J. Mol. Sci. 2023, 24, 4471. [Google Scholar] [CrossRef]
- Aden, D.P.; Fogel, A.; Plotkin, S.; Damjanov, I.; Knowles, B.B. Controlled Synthesis of HBsAg in a Differentiated Human Liver Carcinoma-Derived Cell Line. Nature 1979, 282, 615–616. [Google Scholar] [CrossRef]
- Oommen, D.; Kizhakkedath, P.; Jawabri, A.A.; Varghese, D.S.; Ali, B.R. Proteostasis Regulation in the Endoplasmic Reticulum: An Emerging Theme in the Molecular Pathology and Therapeutic Management of Familial Hypercholesterolemia. Front. Genet. 2020, 11, 570355. [Google Scholar] [CrossRef] [PubMed]
- Thormaehlen, A.S.; Schuberth, C.; Won, H.H.; Blattmann, P.; Joggerst-Thomalla, B.; Theiss, S.; Asselta, R.; Duga, S.; Merlini, P.A.; Ardissino, D.; et al. Systematic Cell-Based Phenotyping of Missense Alleles Empowers Rare Variant Association Studies: A Case for LDLR and Myocardial Infarction. PLoS Genet. 2015, 11, e1004855. [Google Scholar] [CrossRef]
- Galicia-Garcia, U.; Benito-Vicente, A.; Uribe, K.B.; Jebari, S.; Larrea-Sebal, A.; Alonso-Estrada, R.; Aguilo-Arce, J.; Ostolaza, H.; Palacios, L.; Martin, C. Mutation Type Classification and Pathogenicity Assignment of Sixteen Missense Variants Located in the EGF-Precursor Homology Domain of the LDLR. Sci. Rep. 2020, 10, 1727. [Google Scholar] [CrossRef] [PubMed]
- Semenova, A.E.; Sergienko, I.V.; García-Giustiniani, D.; Monserrat, L.; Popova, A.B.; Nozadze, D.N.; Ezhov, M.V. Verification of Underlying Genetic Cause in a Cohort of Russian Patients with Familial Hypercholesterolemia Using Targeted next Generation Sequencing. J. Cardiovasc. Dev. Dis. 2020, 7, 16. [Google Scholar] [CrossRef] [PubMed]
- Meshkov, A.; Ershova, A.; Kiseleva, A.; Zotova, E.; Sotnikova, E.; Petukhova, A.; Zharikova, A.; Malyshev, P.; Rozhkova, T.; Blokhina, A.; et al. The Ldlr, Apob, and Pcsk9 Variants of Index Patients with Familial Hypercholesterolemia in Russia. Genes 2021, 12, 66. [Google Scholar] [CrossRef] [PubMed]
- Lagace, T.A. PCSK9 and LDLR Degradation: Regulatory Mechanisms in Circulation and in Cells. Curr. Opin. Lipidol. 2014, 25, 387–393. [Google Scholar] [CrossRef] [PubMed]
- Xia, X.D.; Peng, Z.S.; Gu, H.M.; Wang, M.; Wang, G.Q.; Zhang, D.W. Regulation of PCSK9 Expression and Function: Mechanisms and Therapeutic Implications. Front. Cardiovasc. Med. 2021, 8, 764038. [Google Scholar] [CrossRef] [PubMed]
- Dahan, P.; Lu, V.; Nguyen, R.M.T.; Kennedy, S.A.L.; Teitell, M.A. Metabolism in Pluripotency: Both Driver and Passenger? J. Biol. Chem. 2019, 294, 5420–5429. [Google Scholar] [CrossRef]
- González-Becerra, K.; Ramos-Lopez, O.; Barrón-Cabrera, E.; Riezu-Boj, J.I.; Milagro, F.I.; Martínez-López, E.; Martínez, J.A. Fatty Acids, Epigenetic Mechanisms and Chronic Diseases: A Systematic Review. Lipids Health Dis. 2019, 18, 178. [Google Scholar] [CrossRef]
- Trefely, S.; Lovell, C.D.; Snyder, N.W.; Wellen, K.E. Compartmentalised Acyl-CoA Metabolism and Roles in Chromatin Regulation. Mol. Metab. 2020, 38, 100941. [Google Scholar] [CrossRef]
- Konishi, Y.; Shimizu, M. Transepithelial Transport of Ferulic Acid by Monocarboxylic Acid Transporter in Caco-2 Cell Monolayers. Biosci. Biotechnol. Biochem. 2003, 67, 856–862. [Google Scholar] [CrossRef] [PubMed]
- Chrzanowska-Wodnicka, M. Rap1 in Endothelial Biology. Curr. Opin. Hematol. 2017, 24, 248–255. [Google Scholar] [CrossRef] [PubMed]
- Remans, P.H.J.; Gringhuis, S.I.; van Laar, J.M.; Sanders, M.E.; Papendrecht-van der Voort, E.A.M.; Zwartkruis, F.J.T.; Levarht, E.W.N.; Rosas, M.; Coffer, P.J.; Breedveld, F.C.; et al. Rap1 Signaling Is Required for Suppression of Ras-Generated Reactive Oxygen Species and Protection Against Oxidative Stress in T Lymphocytes. J. Immunol. 2004, 173, 920–931. [Google Scholar] [CrossRef] [PubMed]
- Chung, H.Y.; Baek, B.S.; Song, S.H.; Kim, M.S.; Huh, J.I.; Shim, K.H.; Kim, K.W.; Lee, K.H. Xanthine Dehydrogenase/Xanthine Oxldase and Oxidative Stress. J. Am. Aging Assoc. 1997, 20, 127–140. [Google Scholar] [CrossRef]
- Singh, B.; Kosuru, R.; Lakshmikanthan, S.; Sorci-Thomas, M.G.; Zhang, D.X.; Sparapani, R.; Vasquez-Vivar, J.; Chrzanowska, M. Endothelial Rap1 (Ras-Association Proximate 1) Restricts Inflammatory Signaling to Protect from the Progression of Atherosclerosis. Arterioscler. Thromb. Vasc. Biol. 2021, 41, 638–650. [Google Scholar] [CrossRef]
- Van Gils, J.M.; Ramkhelawon, B.; Fernandes, L.; Stewart, M.C.; Guo, L.; Seibert, T.; Menezes, G.B.; Cara, D.C.; Chow, C.; Kinane, T.B.; et al. Endothelial Expression of Guidance Cues in Vessel Wall Homeostasis Dysregulation under Proatherosclerotic Conditions. Arterioscler. Thromb. Vasc. Biol. 2013, 33, 911–919. [Google Scholar] [CrossRef] [PubMed]
- Shimojo, M.; Madara, J.; Pankow, S.; Liu, X.; Yates, J.; Südhof, T.C.; Maximov, A. Synaptotagmin-11 Mediates a Vesicle Trafficking Pathway That Is Essential for Development and Synaptic Plasticity. Genes Dev. 2019, 33, 365–376. [Google Scholar] [CrossRef]
- Oguro-Ando, A.; Bamford, R.A.; Sital, W.; Sprengers, J.J.; Zuko, A.; Matser, J.M.; Oppelaar, H.; Sarabdjitsingh, A.; Joëls, M.; Burbach, J.P.H.; et al. Cntn4, a Risk Gene for Neuropsychiatric Disorders, Modulates Hippocampal Synaptic Plasticity and Behavior. Transl. Psychiatry 2021, 11, 106. [Google Scholar] [CrossRef]
- Du, C.; Wang, Y.; Zhang, F.; Yan, S.; Guan, Y.; Gong, X.; Zhang, T.; Cui, X.; Wang, X.; Zhang, C.X. Synaptotagmin-11 Inhibits Cytokine Secretion and Phagocytosis in Microglia. Glia 2017, 65, 1656–1667. [Google Scholar] [CrossRef]
- Wang, K.; Huang, X.-T.; Miao, Y.-P.; Bai, X.-L.; Jin, F. MiR-148a-3p Attenuates Apoptosis and Inflammation by Targeting CNTN4 in Atherosclerosis. Ann. Transl. Med. 2022, 10, 1201. [Google Scholar] [CrossRef]
- Jeon, B.-N.; Kim, S.; Kim, Y.; Yu, H.; Kim, H.; Ha, Y.; Kim, Y.Y.; Park, C.; Kim, G.; Cha, M.; et al. CNTN4/APP Axis of Cancer Cells and T-Cells. PREPRINT (Version 1). Res. Sq. 2023. preprint. [Google Scholar] [CrossRef]
- Huang, Q.; Gan, Y.; Yu, Z.; Wu, H.; Zhong, Z. Endothelial to Mesenchymal Transition: An Insight in Atherosclerosis. Front. Cardiovasc. Med. 2021, 8, 734550. [Google Scholar] [CrossRef] [PubMed]
- Widlansky, M.E.; Gokce, N.; Keaney, J.F.; Vita, J.A. The Clinical Implications of Endothelial Dysfunction. J. Am. Coll. Cardiol. 2003, 42, 1149–1160. [Google Scholar] [CrossRef] [PubMed]
- Tabas, I.; García-Cardeña, G.; Owens, G.K. Recent Insights into the Cellular Biology of Atherosclerosis. J. Cell Biol. 2015, 209, 13–22. [Google Scholar] [CrossRef] [PubMed]
- Mahdi, A.; Wodaje, T.; Kövamees, O.; Tengbom, J.; Zhao, A.; Jiao, T.; Henricsson, M.; Yang, J.; Zhou, Z.; Nieminen, A.I.; et al. The Red Blood Cell as a Mediator of Endothelial Dysfunction in Patients with Familial Hypercholesterolemia and Dyslipidemia. J. Intern. Med. 2023, 293, 228–245. [Google Scholar] [CrossRef] [PubMed]
- Daiber, A.; Steven, S.; Weber, A.; Shuvaev, V.V.; Muzykantov, V.R.; Laher, I.; Li, H.; Lamas, S.; Münzel, T. Targeting Vascular (Endothelial) Dysfunction. Br. J. Pharmacol. 2017, 174, 1591–1619. [Google Scholar] [CrossRef] [PubMed]
- Little, P.J.; Askew, C.D.; Xu, S.; Kamato, D. Endothelial Dysfunction and Cardiovascular Disease: History and Analysis of the Clinical Utility of the Relationship. Biomedicines 2021, 9, 699. [Google Scholar] [CrossRef] [PubMed]
- Vlahos, A.P.; Naka, K.K.; Bechlioulis, A.; Theoharis, P.; Vakalis, K.; Moutzouri, E.; Miltiadous, G.; Michalis, L.K.; Siamopoulou-Mavridou, A.; Elisaf, M.; et al. Endothelial Dysfunction, but Not Structural Atherosclerosis, Is Evident Early in Children with Heterozygous Familial Hypercholesterolemia. Pediatr. Cardiol. 2014, 35, 63–70. [Google Scholar] [CrossRef]
- Malakhova, A.A.; Grigor’eva, E.V.; Pavlova, S.V.; Malankhanova, T.B.; Valetdinova, K.R.; Vyatkin, Y.V.; Khabarova, E.A.; Rzaev, J.A.; Zakian, S.M.; Medvedev, S.P. Generation of Induced Pluripotent Stem Cell Lines ICGi021-A and ICGi022-A from Peripheral Blood Mononuclear Cells of Two Healthy Individuals from Siberian Population. Stem Cell Res. 2020, 48, 101952. [Google Scholar] [CrossRef]
- Gu, M. Efficient Differentiation of Human Pluripotent Stem Cells to Endothelial Cells. Curr. Protoc. Hum. Genet. 2018, 98, e64. [Google Scholar] [CrossRef]
- Shevchenko, A.I.; Arssan, A.M.; Zakian, S.M.; Zakharova, I.S. Chemokine CCL2 Activates Hypoxia Response Factors Regulating Pluripotency and Directed Endothelial Differentiation of Human Pluripotent Stem Cells. Russ. J. Dev. Biol. 2023, 54, 134–146. [Google Scholar] [CrossRef]
- Zakharova, I.S.; Zhiven’, M.K.; Saaya, S.B.; Shevchenko, A.I.; Smirnova, A.M.; Strunov, A.; Karpenko, A.A.; Pokushalov, E.A.; Ivanova, L.N.; Makarevich, P.I.; et al. Endothelial and Smooth Muscle Cells Derived from Human Cardiac Explants Demonstrate Angiogenic Potential and Suitable for Design of Cell-Containing Vascular Grafts. J. Transl. Med. 2017, 15, 54. [Google Scholar] [CrossRef] [PubMed]
- Zakharova, I.; Saaya, S.; Shevchenko, A.; Stupnikova, A.; Zhiven’, M.; Laktionov, P.; Stepanova, A.; Romashchenko, A.; Yanshole, L.; Chernonosov, A.; et al. Mitomycin-Treated Endothelial and Smooth Muscle Cells Suitable for Safe Tissue Engineering Approaches. Front. Bioeng. Biotechnol. 2022, 10, 772981. [Google Scholar] [CrossRef] [PubMed]
- Vaskova, E.A.; Medvedev, S.P.; Sorokina, A.E.; Nemudryy, A.A.; Elisaphenko, E.A.; Zakharova, I.S.; Shevchenko, A.I.; Kizilova, E.A.; Zhelezova, A.I.; Evshin, I.S.; et al. Transcriptome Characteristics and X-Chromosome Inactivation Status in Cultured Rat Pluripotent Stem Cells. Stem Cells Dev. 2015, 24, 2912–2924. [Google Scholar] [CrossRef] [PubMed]
- Zudaire, E.; Gambardella, L.; Kurcz, C.; Vermeren, S. A Computational Tool for Quantitative Analysis of Vascular Networks. PLoS ONE 2011, 6, e27385. [Google Scholar] [CrossRef] [PubMed]
- Ewels, P.; Magnusson, M.; Lundin, S.; Käller, M. MultiQC: Summarize Analysis Results for Multiple Tools and Samples in a Single Report. Bioinformatics 2016, 32, 3047–3048. [Google Scholar] [CrossRef]
- Chen, Y.; Lun, A.T.L.; Smyth, G.K. From Reads to Genes to Pathways: Differential Expression Analysis of RNA-Seq Experiments Using Rsubread and the EdgeR Quasi-Likelihood Pipeline. F1000Research 2016, 5, 1438. [Google Scholar] [CrossRef] [PubMed]
- Zhou, Y.; Zhou, B.; Pache, L.; Chang, M.; Khodabakhshi, A.H.; Tanaseichuk, O.; Benner, C.; Chanda, S.K. Metascape Provides a Biologist-Oriented Resource for the Analysis of Systems-Level Datasets. Nat. Commun. 2019, 10, 1523. [Google Scholar] [CrossRef] [PubMed]




{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Name | Supplier | Catalogue Number | RRID | Isotype | Dilution |
|---|---|---|---|---|---|
| FACS | |||||
| CD144 + PerCP-Cy5.5 | BD | 561566 | AB_10715835 | mouse IgG1 | 1/20 |
| mouse IgG1 + PerCP-Cy5.5 | BD | 552834 | AB_394484 | mouse IgG1 | 1/50 |
| Immunofluorescence | |||||
| CD 31 | Cell Marque | 131M-96 | AB_1516765 | mouse IgG1 | 1/50 |
| Von Willebrand factor | Dako | A0082 | AB_2315602 | rabbit IgG | 1/200 |
| mouse IgG1 + Alexa 568 | Thermo Fisher Scientific | A21124 | AB_2535766 | goat IgG | 1/400 |
| rabbit IgG + Alexa 488 | Thermo Fisher Scientific | A110088 | AB_143165 | goat IgG | 1/400 |
| goat IgG + Alexa 488 | Thermo Fisher Scientific | A11055 | AB_2534102 | donkey IgG | 1/400 |
| Immunoblotting | |||||
| LDLR | R&D System | AF2148 | AB_2135126 | goat IgG | 1/1000 |
| ACTB | Abcam | ab8227 | AB_2305186 | rabbit IgG | 1/5000 |
| goat IgG + peroxidase | Jackson ImmunoResearch | 705-035-003 | AB_2340390 | donkey IgG | 1/5000 |
| rabbit IgG + peroxidase | Jackson ImmunoResearch | 711-035-152 | AB_10015282 | donkey IgG | 1/5000 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zakharova, I.S.; Shevchenko, A.I.; Arssan, M.A.; Sleptcov, A.A.; Nazarenko, M.S.; Zarubin, A.A.; Zheltysheva, N.V.; Shevchenko, V.A.; Tmoyan, N.A.; Saaya, S.B.; et al. iPSC-Derived Endothelial Cells Reveal LDLR Dysfunction and Dysregulated Gene Expression Profiles in Familial Hypercholesterolemia. Int. J. Mol. Sci. 2024, 25, 689. https://doi.org/10.3390/ijms25020689
Zakharova IS, Shevchenko AI, Arssan MA, Sleptcov AA, Nazarenko MS, Zarubin AA, Zheltysheva NV, Shevchenko VA, Tmoyan NA, Saaya SB, et al. iPSC-Derived Endothelial Cells Reveal LDLR Dysfunction and Dysregulated Gene Expression Profiles in Familial Hypercholesterolemia. International Journal of Molecular Sciences. 2024; 25(2):689. https://doi.org/10.3390/ijms25020689
Chicago/Turabian StyleZakharova, Irina S., Alexander I. Shevchenko, Mhd Amin Arssan, Aleksei A. Sleptcov, Maria S. Nazarenko, Aleksei A. Zarubin, Nina V. Zheltysheva, Vlada A. Shevchenko, Narek A. Tmoyan, Shoraan B. Saaya, and et al. 2024. "iPSC-Derived Endothelial Cells Reveal LDLR Dysfunction and Dysregulated Gene Expression Profiles in Familial Hypercholesterolemia" International Journal of Molecular Sciences 25, no. 2: 689. https://doi.org/10.3390/ijms25020689
APA StyleZakharova, I. S., Shevchenko, A. I., Arssan, M. A., Sleptcov, A. A., Nazarenko, M. S., Zarubin, A. A., Zheltysheva, N. V., Shevchenko, V. A., Tmoyan, N. A., Saaya, S. B., Ezhov, M. V., Kukharchuk, V. V., Parfyonova, Y. V., & Zakian, S. M. (2024). iPSC-Derived Endothelial Cells Reveal LDLR Dysfunction and Dysregulated Gene Expression Profiles in Familial Hypercholesterolemia. International Journal of Molecular Sciences, 25(2), 689. https://doi.org/10.3390/ijms25020689