
Regulators of G-Protein Signaling (RGS) in Sporadic and Colitis-Associated Colorectal Cancer



Abstract
1. Introduction
2. Genetic Mutation Patterns in Sporadic CRC and CAC
3. Role of Inflammation and Immune System in CRC Oncogenesis
3.1. Inflammation Preceding Carcinogenesis
3.2. Tumor-Associated Inflammation
3.3. Inflammation Induced by CRC Treatment
4. G-Protein Coupled Receptors
5. Selected GPCR in IBD and Their Connections to Carcinogenesis
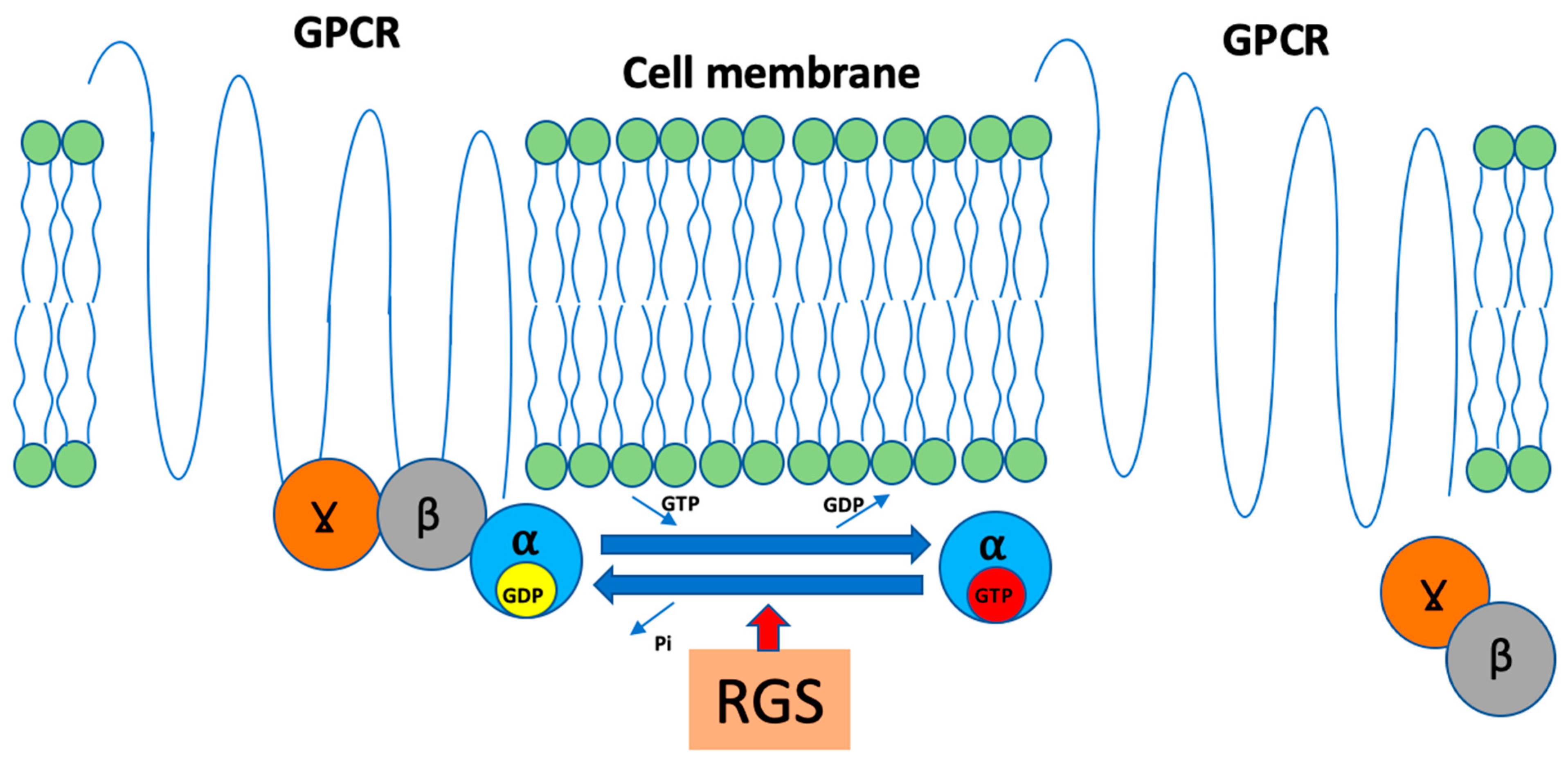
6. Regulators of G-Protein Signaling and AXIN
7. RGS in IBD, CAC and Sporadic CRC Carcinogenesis
7.1. R4 Family: RGS1, RGS2, RGS4, RGS13 and RGS16
7.2. R7 Family: RGS6, RGS7, RGS9-2 and RGS11
7.3. R12 Family—RGS10
7.4. RZ Family: RGS17, RGS19, RGS20
7.5. Atypical RGS—AXIN
8. Future Perspectives
9. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- American Cancer Society Key Statistics for Colorectal Cancer. Available online: https://www.cancer.org/cancer/colon-rectal-cancer/about/key-statistics.html (accessed on 31 October 2023).
- International Agency for Research on Cancer; World Health Organization. CANCER TODAY. Data Visualization Tools for Exploring the Global Cancer Burden in 2020. Available online: https://gco.iarc.fr/today/home (accessed on 31 October 2023).
- Dekker, E.; Tanis, P.J.; Vleugels, J.L.A.; Kasi, P.M.; Wallace, M.B. Colorectal Cancer. Lancet 2019, 394, 1467–1480. [Google Scholar] [CrossRef] [PubMed]
- Zielińska, M.; Szymaszkiewicz, A.; Jacenik, D.; Schodel, L.; Sałaga, M.; Zatorski, H.; Kordek, R.; Becker, C.; Krajewska, W.M.; Fichna, J. Cyclic Derivative of Morphiceptin Dmt-Cyclo-(D-Lys-Phe-D-Pro-Asp)-NH2(P-317), a Mixed Agonist of MOP and KOP Opioid Receptors, Exerts Anti-Inflammatory and Anti-Tumor Activity in Colitis and Colitis-Associated Colorectal Cancer in Mice. Eur. J. Pharmacol. 2020, 885, 173463. [Google Scholar] [CrossRef] [PubMed]
- Andersen, N.N.; Jess, T. Has the Risk of Colorectal Cancer in Inflammatory Bowel Disease Decreased? World J. Gastroenterol. 2013, 19, 7561–7568. [Google Scholar] [CrossRef] [PubMed]
- Li, J.; Ma, X.; Chakravarti, D.; Shalapour, S.; Depinho, R.A. Genetic and Biological Hallmarks of Colorectal Cancer. Genes Dev. 2021, 35, 787–820. [Google Scholar] [CrossRef] [PubMed]
- Schatoff, E.M.; Leach, B.I.; Dow, L.E. WNT Signaling and Colorectal Cancer. Curr. Color. Cancer Rep. 2017, 13, 101–110. [Google Scholar] [CrossRef] [PubMed]
- Muzny, D.M.; Bainbridge, M.N.; Chang, K.; Dinh, H.H.; Drummond, J.A.; Fowler, G.; Kovar, C.L.; Lewis, L.R.; Morgan, M.B.; Newsham, I.F.; et al. Comprehensive Molecular Characterization of Human Colon and Rectal Cancer. Nature 2012, 487, 330–337. [Google Scholar] [CrossRef]
- Świerczyński, M.; Szymaszkiewicz, A.; Fichna, J.; Zielińska, M. New Insights into Molecular Pathways in Colorectal Cancer: Adiponectin, Interleukin-6 and Opioid Signaling. Biochim. Et Biophys. Acta (BBA)—Rev. Cancer 2021, 1875, 188460. [Google Scholar] [CrossRef] [PubMed]
- Dulai, P.S.; Sandborn, W.J.; Gupta, S. Colorectal Cancer and Dysplasia in Inflammatory Bowel Disease: A Review of Disease Epidemiology, Pathophysiology, and Management. Cancer Prev. Res. 2016, 9, 887–894. [Google Scholar] [CrossRef]
- Schmitt, M.; Greten, F.R. The Inflammatory Pathogenesis of Colorectal Cancer. Nat. Rev. Immunol. 2021, 21, 653–667. [Google Scholar] [CrossRef]
- Singh, N.; Baby, D.; Rajguru, J.; Patil, P.; Thakkannavar, S.; Pujari, V. Inflammation and Cancer. Ann. Afr. Med. 2019, 18, 121. [Google Scholar] [CrossRef]
- Świerczyński, M.; Fichna, J. Inflamasom Jako Czynnik Sprawczy i Ochronny w Patogenezie Nieswoistych Chorób Zapalnych Jelit. Postep. Biochem. 2021, 67, 44–53. [Google Scholar] [CrossRef] [PubMed]
- Schmitt, H.; Neurath, M.F.; Atreya, R. Role of the IL23/IL17 Pathway in Crohn’s Disease. Front. Immunol. 2021, 12, 622934. [Google Scholar] [CrossRef] [PubMed]
- Hanauer, S.B. Inflammatory Bowel Disease: Epidemiology, Pathogenesis, and Therapeutic Opportunities. Inflamm. Bowel Dis. 2006, 12, S3–S9. [Google Scholar] [CrossRef] [PubMed]
- Ananthakrishnan, A.N. Epidemiology and Risk Factors for IBD. Nat. Rev. Gastroenterol. Hepatol. 2015, 12, 205–217. [Google Scholar] [CrossRef] [PubMed]
- Zeng, J.; Tang, Z.-H.; Liu, S.; Guo, S.-S. Clinicopathological Significance of Overexpression of Interleukin-6 in Colorectal Cancer. World J. Gastroenterol. 2017, 23, 1780–1786. [Google Scholar] [CrossRef] [PubMed]
- Grivennikov, S.I.; Wang, K.; Mucida, D.; Stewart, C.A.; Schnabl, B.; Jauch, D.; Taniguchi, K.; Yu, G.-Y.; Osterreicher, C.H.; Hung, K.E.; et al. Adenoma-Linked Barrier Defects and Microbial Products Drive IL-23/IL-17-Mediated Tumour Growth. Nature 2012, 491, 254–258. [Google Scholar] [CrossRef] [PubMed]
- Long, A.G.; Lundsmith, E.T.; Hamilton, K.E. Inflammation and Colorectal Cancer. Curr. Color. Cancer Rep. 2017, 13, 341–351. [Google Scholar] [CrossRef]
- Dong, S.; Liang, S.; Cheng, Z.; Zhang, X.; Luo, L.; Li, L.; Zhang, W.; Li, S.; Xu, Q.; Zhong, M.; et al. ROS/PI3K/Akt and Wnt/β-Catenin Signalings Activate HIF-1α-Induced Metabolic Reprogramming to Impart 5-Fluorouracil Resistance in Colorectal Cancer. J. Exp. Clin. Cancer Res. 2022, 41, 15. [Google Scholar] [CrossRef]
- Roth, M.T.; Das, S. Pembrolizumab in Unresectable or Metastatic MSI-High Colorectal Cancer: Safety and Efficacy. Expert Rev. Anticancer Ther. 2021, 21, 229–238. [Google Scholar] [CrossRef]
- Hill, M.; Segovia, M.; Russo, S.; Girotti, M.R.; Rabinovich, G.A. The Paradoxical Roles of Inflammation during PD-1 Blockade in Cancer. Trends Immunol. 2020, 41, 982–993. [Google Scholar] [CrossRef]
- Lee, L.; Gupta, M.; Sahasranaman, S. Immune Checkpoint Inhibitors: An Introduction to the next-Generation Cancer Immunotherapy. J. Clin. Pharmacol. 2016, 56, 157–169. [Google Scholar] [CrossRef] [PubMed]
- Chaudhary, P.K.; Kim, S. An Insight into GPCR and G-Proteins as Cancer Drivers. Cells 2021, 10, 3288. [Google Scholar] [CrossRef] [PubMed]
- Lundstrom, K. An Overview on GPCRs and Drug Discovery: Structure-Based Drug Design and Structural Biology on GPCRs. In G Protein-Coupled Receptors in Drug Discovery; Leifert, W.R., Ed.; Humana Press: Totowa, NJ, USA, 2009; pp. 51–66. ISBN 978-1-60327-317-6. [Google Scholar]
- Kobilka, B.K. G Protein Coupled Receptor Structure and Activation. Biochim. Biophys. Acta 2007, 1768, 794–807. [Google Scholar] [CrossRef] [PubMed]
- Sobczak, M.; Sałaga, M.; Storr, M.A.; Fichna, J. Physiology, Signaling, and Pharmacology of Opioid Receptors and Their Ligands in the Gastrointestinal Tract: Current Concepts and Future Perspectives. J. Gastroenterol. 2014, 49, 24–45. [Google Scholar] [CrossRef] [PubMed]
- Roy, S.; Balasubramanian, S.; Sumandeep, S.; Charboneau, R.; Wang, J.; Melnyk, D.; Beilman, G.J.; Vatassery, R.; Barke, R.A. Morphine Directs T Cells toward T(H2) Differentiation. Surgery 2001, 130, 304–309. [Google Scholar] [CrossRef] [PubMed]
- Plein, L.M.; Rittner, H.L. Opioids and the Immune System—Friend or Foe. Br. J. Pharmacol. 2018, 175, 2717–2725. [Google Scholar] [CrossRef]
- Ninković, J.; Roy, S. Role of the Mu-Opioid Receptor in Opioid Modulation of Immune Function. Amino Acids 2013, 45, 9–24. [Google Scholar] [CrossRef]
- Pol, O.; Palacio, J.R.; Puig, M.M. The Expression of Delta- and Kappa-Opioid Receptor Is Enhanced during Intestinal Inflammation in Mice. J. Pharmacol. Exp. Ther. 2003, 306, 455–462. [Google Scholar] [CrossRef]
- Salaga, M.; Storr, M.; Martemyanov, K.A.; Fichna, J. RGS Proteins as Targets in the Treatment of Intestinal Inflammation and Visceral Pain: New Insights and Future Perspectives. BioEssays 2016, 38, 344–354. [Google Scholar] [CrossRef]
- Sacerdote, P. Effects of in Vitro and in Vivo Opioids on the Production of IL-12 and IL-10 by Murine Macrophages. Ann. N. Y. Acad. Sci. 2003, 992, 129–140. [Google Scholar] [CrossRef]
- Walker, J.S. Anti-Inflammatory Effects of Opioids. Adv. Exp. Med. Biol. 2003, 521, 148–160. [Google Scholar] [PubMed]
- Keränen, U.; Kiviluoto, T.; Järvinen, H.; Bäck, N.; Kivilaakso, E.; Soinila, S. Changes in Substance P-Immunoreactive Innervation of Human Colon Associated with Ulcerative Colitis. Dig. Dis. Sci. 1995, 40, 2250–2258. [Google Scholar] [CrossRef] [PubMed]
- Patel, M.; Valaiyaduppu Subas, S.; Ghani, M.R.; Busa, V.; Dardeir, A.; Marudhai, S.; Cancarevic, I. Role of Substance P in the Pathophysiology of Inflammatory Bowel Disease and Its Correlation With the Degree of Inflammation. Cureus 2020, 12, e11027. [Google Scholar] [CrossRef] [PubMed]
- Beck, T.C.; Hapstack, M.A.; Beck, K.R.; Dix, T.A. Therapeutic Potential of Kappa Opioid Agonists. Pharmaceuticals 2019, 12, 95. [Google Scholar] [CrossRef] [PubMed]
- Nagata, K.; Nagase, H.; Okuzumi, A.; Nishiyama, C. Delta Opioid Receptor Agonists Ameliorate Colonic Inflammation by Modulating Immune Responses. Front. Immunol. 2021, 12, 730706. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Rajput, A.; Jin, N.; Wang, J. Mechanisms of Immunosuppression in Colorectal Cancer. Cancers 2020, 12, 3850. [Google Scholar] [CrossRef] [PubMed]
- Wu, Q.; Chen, X.; Wang, J.; Sun, P.; Weng, M.; Chen, W.; Sun, Z.; Zhu, M.; Miao, C. Nalmefene Attenuates Malignant Potential in Colorectal Cancer Cell via Inhibition of Opioid Receptor. Acta Biochim. Biophys. Sin. 2018, 50, 156–163. [Google Scholar] [CrossRef]
- Howlett, A.C. The Cannabinoid Receptors. Prostaglandins Other Lipid Mediat. 2002, 68–69, 619–631. [Google Scholar] [CrossRef]
- Singh, U.P.; Singh, N.P.; Singh, B.; Price, R.L.; Nagarkatti, M.; Nagarkatti, P.S. Cannabinoid Receptor-2 (CB2) Agonist Ameliorates Colitis in IL-10(-/-) Mice by Attenuating the Activation of T Cells and Promoting Their Apoptosis. Toxicol. Appl. Pharmacol. 2012, 258, 256–267. [Google Scholar] [CrossRef]
- Massa, F.; Marsicano, G.; Hermann, H.; Cannich, A.; Monory, K.; Cravatt, B.F.; Ferri, G.-L.; Sibaev, A.; Storr, M.; Lutz, B. The Endogenous Cannabinoid System Protects against Colonic Inflammation. J. Clin. Investig. 2004, 113, 1202–1209. [Google Scholar] [CrossRef]
- D’Argenio, G.; Valenti, M.; Scaglione, G.; Cosenza, V.; Sorrentini, I.; Di Marzo, V. Up-Regulation of Anandamide Levels as an Endogenous Mechanism and a Pharmacological Strategy to Limit Colon Inflammation. FASEB J. 2006, 20, 568–570. [Google Scholar] [CrossRef] [PubMed]
- Sałaga, M.; Mokrowiecka, A.; Zakrzewski, P.K.; Cygankiewicz, A.; Leishman, E.; Sobczak, M.; Zatorski, H.; Małecka-Panas, E.; Kordek, R.; Storr, M.; et al. Experimental Colitis in Mice Is Attenuated by Changes in the Levels of Endocannabinoid Metabolites Induced by Selective Inhibition of Fatty Acid Amide Hydrolase (FAAH). J. Crohns Colitis 2014, 8, 998–1009. [Google Scholar] [CrossRef] [PubMed]
- Malfitano, A.M.; Ciaglia, E.; Gangemi, G.; Gazzerro, P.; Laezza, C.; Bifulco, M. Update on the Endocannabinoid System as an Anticancer Target. Expert Opin. Ther. Targets 2011, 15, 297–308. [Google Scholar] [CrossRef] [PubMed]
- Wang, D.; Wang, H.; Ning, W.; Backlund, M.G.; Dey, S.K.; DuBois, R.N. Loss of Cannabinoid Receptor 1 Accelerates Intestinal Tumor Growth. Cancer Res. 2008, 68, 6468–6476. [Google Scholar] [CrossRef] [PubMed]
- Izzo, A.A.; Aviello, G.; Petrosino, S.; Orlando, P.; Marsicano, G.; Lutz, B.; Borrelli, F.; Capasso, R.; Nigam, S.; Capasso, F.; et al. Increased Endocannabinoid Levels Reduce the Development of Precancerous Lesions in the Mouse Colon. J. Mol. Med. 2008, 86, 89–98. [Google Scholar] [CrossRef] [PubMed]
- Nomura, D.K.; Long, J.Z.; Niessen, S.; Hoover, H.S.; Ng, S.-W.; Cravatt, B.F. Monoacylglycerol Lipase Regulates a Fatty Acid Network That Promotes Cancer Pathogenesis. Cell 2010, 140, 49–61. [Google Scholar] [CrossRef] [PubMed]
- Pagano, C.; Navarra, G.; Coppola, L.; Bifulco, M.; Laezza, C. Molecular Mechanism of Cannabinoids in Cancer Progression. Int. J. Mol. Sci. 2021, 22, 3680. [Google Scholar] [CrossRef]
- Roberto, D.; Klotz, L.H.; Venkateswaran, V. Cannabinoid WIN 55,212-2 Induces Cell Cycle Arrest and Apoptosis, and Inhibits Proliferation, Migration, Invasion, and Tumor Growth in Prostate Cancer in a Cannabinoid-Receptor 2 Dependent Manner. Prostate 2019, 79, 151–159. [Google Scholar] [CrossRef]
- McCorvy, J.D.; Roth, B.L. Structure and Function of Serotonin G Protein-Coupled Receptors. Pharmacol. Ther. 2015, 150, 129–142. [Google Scholar] [CrossRef]
- Salaga, M.; Binienda, A.; Piscitelli, F.; Mokrowiecka, A.; Cygankiewicz, A.I.; Verde, R.; Malecka-Panas, E.; Kordek, R.; Krajewska, W.M.; Di Marzo, V.; et al. Systemic Administration of Serotonin Exacerbates Abdominal Pain and Colitis via Interaction with the Endocannabinoid System. Biochem. Pharmacol. 2019, 161, 37–51. [Google Scholar] [CrossRef]
- Zhou, Y.; Zhi, F. Lower Level of Bacteroides in the Gut Microbiota Is Associated with Inflammatory Bowel Disease: A Meta-Analysis. Biomed Res. Int. 2016, 5828959. [Google Scholar] [CrossRef] [PubMed]
- Margolis, K.G.; Stevanovic, K.; Li, Z.; Yang, Q.M.; Oravecz, T.; Zambrowicz, B.; Jhaver, K.G.; Diacou, A.; Gershon, M.D. Pharmacological Reduction of Mucosal but Not Neuronal Serotonin Opposes Inflammation in Mouse Intestine. Gut 2014, 63, 928–937. [Google Scholar] [CrossRef] [PubMed]
- Kwon, Y.H.; Wang, H.; Denou, E.; Ghia, J.-E.; Rossi, L.; Fontes, M.E.; Bernier, S.P.; Shajib, M.S.; Banskota, S.; Collins, S.M.; et al. Modulation of Gut Microbiota Composition by Serotonin Signaling Influences Intestinal Immune Response and Susceptibility to Colitis. Cell. Mol. Gastroenterol. Hepatol. 2019, 7, 709–728. [Google Scholar] [CrossRef] [PubMed]
- Kidd, M.; Gustafsson, B.I.; Drozdov, I.; Modlin, I.M. IL1beta- and LPS-Induced Serotonin Secretion Is Increased in EC Cells Derived from Crohn’s Disease. Neurogastroenterol. Motil. 2009, 21, 439–450. [Google Scholar] [CrossRef] [PubMed]
- Manzella, C.R.; Jayawardena, D.; Pagani, W.; Li, Y.; Alrefai, W.A.; Bauer, J.; Jung, B.; Weber, C.R.; Gill, R.K. Serum Serotonin Differentiates Between Disease Activity States in Crohn’s Patients. Inflamm. Bowel Dis. 2020, 26, 1607–1618. [Google Scholar] [CrossRef] [PubMed]
- Ala, M. Tryptophan Metabolites Modulate Inflammatory Bowel Disease and Colorectal Cancer by Affecting Immune System. Int. Rev. Immunol. 2022, 41, 326–345. [Google Scholar] [CrossRef] [PubMed]
- Zhu, P.; Lu, T.; Chen, Z.; Liu, B.; Fan, D.; Li, C.; Wu, J.; He, L.; Zhu, X.; Du, Y.; et al. 5-Hydroxytryptamine Produced by Enteric Serotonergic Neurons Initiates Colorectal Cancer Stem Cell Self-Renewal and Tumorigenesis. Neuron 2022, 110, 2268–2282.e4. [Google Scholar] [CrossRef]
- Ahlers-Dannen, K.E.; Alqinyah, M.; Bodle, C.; Bou Dagher, J.; Chakravarti, B.; Choudhuri, S.P.; Druey, K.M.; Fisher, R.A.; Gerber, K.J.; Hepler, J.R.; et al. Regulators of G Protein Signaling (RGS) Proteins in GtoPdb v.2021.2. IUPHAR/BPS Guide Pharmacol. CITE 2021, 2020. [Google Scholar] [CrossRef]
- Verstockt, B.; Verstockt, S.; Veny, M.; Dehairs, J.; Arnauts, K.; Van Assche, G.; De Hertogh, G.; Vermeire, S.; Salas, A.; Ferrante, M. Expression Levels of 4 Genes in Colon Tissue Might Be Used to Predict Which Patients Will Enter Endoscopic Remission After Vedolizumab Therapy for Inflammatory Bowel Diseases. Clin. Gastroenterol. Hepatol. 2020, 18, 1142–1151.e10. [Google Scholar] [CrossRef]
- Zhu, F.; Qin, Y.; Wang, Y.; Zhang, F.; Xu, Z.; Dai, F.; Chu, W.; Wang, Y.; Zhou, G. Critical Roles of RGS16 in the Mucosal Inflammation of Ulcerative Colitis. Eur. J. Gastroenterol. Hepatol. 2022, 34, 993–999. [Google Scholar] [CrossRef]
- Alam, N.A.; Gorman, P.; Jaeger, E.E.M.; Kelsell, D.; Leigh, I.M.; Ratnavel, R.; Murdoch, M.E.; Houlston, R.S.; Aaltonen, L.A.; Roylance, R.R.; et al. Germline Deletions of EXO1 Do Not Cause Colorectal Tumors and Lesions Which Are Null for EXO1 Do Not Have Microsatellite Instability. Cancer Genet. Cytogenet. 2003, 147, 121–127. [Google Scholar] [CrossRef] [PubMed]
- Houser, M.C.; Caudle, W.M.; Chang, J.; Kannarkat, G.T.; Yang, Y.; Kelly, S.D.; Oliver, D.; Joers, V.; Shannon, K.M.; Keshavarzian, A.; et al. Experimental Colitis Promotes Sustained, Sex-Dependent, T-Cell-Associated Neuroinflammation and Parkinsonian Neuropathology. Acta Neuropathol. Commun. 2021, 9, 139. [Google Scholar] [CrossRef] [PubMed]
- Suzuki, K.; Kakuta, Y.; Naito, T.; Takagawa, T.; Hanai, H.; Araki, H.; Sasaki, Y.; Sakuraba, H.; Sasaki, M.; Hisamatsu, T.; et al. Genetic Background of Mesalamine-Induced Fever and Diarrhea in Japanese Patients with Inflammatory Bowel Disease. Inflamm. Bowel Dis. 2022, 28, 21–31. [Google Scholar] [CrossRef] [PubMed]
- Dhukhwa, A.; Al Aameri, R.F.H.; Sheth, S.; Mukherjea, D.; Rybak, L.; Ramkumar, V. Regulator of G Protein Signaling 17 Represents a Novel Target for Treating Cisplatin Induced Hearing Loss. Sci. Rep. 2021, 11, 8116. [Google Scholar] [CrossRef] [PubMed]
- Vallée, A.; Lecarpentier, Y. Crosstalk Between Peroxisome Proliferator-Activated Receptor Gamma and the Canonical WNT/β-Catenin Pathway in Chronic Inflammation and Oxidative Stress During Carcinogenesis. Front. Immunol. 2018, 9, 745. [Google Scholar] [CrossRef] [PubMed]
- Behrens, J.; Jerchow, B.-A.; Würtele, M.; Grimm, J.; Asbrand, C.; Wirtz, R.; Kühl, M.; Wedlich, D.; Birchmeier, W. Functional Interaction of an Axin Homolog, Conductin, with β-Catenin, APC, and GSK3β. Science 1998, 280, 596–599. [Google Scholar] [CrossRef] [PubMed]
- Xie, Z.; Li, Z.; Guo, L.; Ye, C.; Li, J.; Yu, X.; Yang, H.; Wang, Y.; Chen, C.; Zhang, D.; et al. Regulator of G Protein Signaling Proteins Differentially Modulate Signaling of Mu and Delta Opioid Receptors. Eur. J. Pharmacol. 2007, 565, 45–53. [Google Scholar] [CrossRef]
- Kim, Y.; Ghil, S. Regulators of G-Protein Signaling, RGS2 and RGS4, Inhibit Protease-Activated Receptor 4-Mediated Signaling by Forming a Complex with the Receptor and Gα in Live Cells. Cell Commun. Signal. 2020, 18, 86. [Google Scholar] [CrossRef]
- Hu, W.; Li, F.; Mahavadi, S.; Murthy, K.S. Upregulation of RGS4 Expression by IL-1beta in Colonic Smooth Muscle Is Enhanced by ERK1/2 and P38 MAPK and Inhibited by the PI3K/Akt/GSK3beta Pathway. Am. J. Physiol. Cell Physiol. 2009, 296, C1310–C1320. [Google Scholar] [CrossRef][Green Version]
- Georgoussi, Z.; Leontiadis, L.; Mazarakou, G.; Merkouris, M.; Hyde, K.; Hamm, H. Selective Interactions between G Protein Subunits and RGS4 with the C-Terminal Domains of the μ- and δ-Opioid Receptors Regulate Opioid Receptor Signaling. Cell Signal. 2006, 18, 771–782. [Google Scholar] [CrossRef]
- Yoon, S.Y.; Woo, J.; Park, J.O.; Choi, E.J.; Shin, H.S.; Roh, D.H.; Kim, K.S. Intrathecal RGS4 Inhibitor, CCG50014, Reduces Nociceptive Responses and Enhances Opioid-Mediated Analgesic Effects in the Mouse Formalin Test. Anesth. Analg. 2015, 120, 671–677. [Google Scholar] [CrossRef]
- Sutor, S.; Heilmann, J.; Seifert, R. Impact of Fusion to Gα(I2) and Co-Expression with RGS Proteins on Pharmacological Properties of Human Cannabinoid Receptors CB1R and CB2R. J. Pharm. Pharmacol. 2011, 63, 1043–1055. [Google Scholar] [CrossRef] [PubMed]
- Hu, W.; Li, F.; Mahavadi, S.; Murthy, K.S. Interleukin-1β up-Regulates RGS4 through the Canonical IKK2/IκBα/NF-ΚB Pathway in Rabbit Colonic Smooth Muscle. Biochem. J. 2008, 412, 34–43. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Zhang, H.; Jiang, P.; Zhang, C.; Lee, S.; Wang, W.; Zou, H. PAR4 Overexpression Promotes Colorectal Cancer Cell Proliferation and Migration. Oncol. Lett. 2018, 16, 5745–5752. [Google Scholar] [CrossRef] [PubMed]
- Jiang, Z.; Wang, Z.; Xu, Y.; Wang, B.; Huang, W.; Cai, S. Analysis of RGS2 Expression and Prognostic Significance in Stage II and III Colorectal Cancer. Biosci. Rep. 2010, 30, 383–390. [Google Scholar] [CrossRef] [PubMed]
- Weisshaar, N.; Wu, J.; Ming, Y.; Madi, A.; Hotz-Wagenblatt, A.; Ma, S.; Mieg, A.; Hering, M.; Zettl, F.; Mohr, K.; et al. Rgs16 Promotes Antitumor CD8+ T Cell Exhaustion. Sci. Immunol. 2022, 7, eabh1873. [Google Scholar] [CrossRef]
- Sade-Feldman, M.; Yizhak, K.; Bjorgaard, S.L.; Ray, J.P.; de Boer, C.G.; Jenkins, R.W.; Lieb, D.J.; Chen, J.H.; Frederick, D.T.; Barzily-Rokni, M.; et al. Defining T Cell States Associated with Response to Checkpoint Immunotherapy in Melanoma. Cell 2018, 175, 998–1013.e20. [Google Scholar] [CrossRef]
- Miyoshi, N.; Ishii, H.; Sekimoto, M.; Doki, Y.; Mori, M. RGS16 Is a Marker for Prognosis in Colorectal Cancer. Ann. Surg. Oncol. 2009, 16, 3507–3514. [Google Scholar] [CrossRef]
- Hwang, I.-Y.; Hwang, K.-S.; Park, C.; Harrison, K.A.; Kehrl, J.H. Rgs13 Constrains Early B Cell Responses and Limits Germinal Center Sizes. PLoS ONE 2013, 8, e60139. [Google Scholar] [CrossRef]
- Luo, Y.; Qin, S.L.; Yu, M.H.; Mu, Y.F.; Wang, Z.S.; Zhong, M. Prognostic Value of Regulator of G-Protein Signaling 6 in Colorectal Cancer. Biomed. Pharmacother. 2015, 76, 147–152. [Google Scholar] [CrossRef]
- Huang, J.; Stewart, A.; Maity, B.; Hagen, J.; Fagan, R.L.; Yang, J.; Quelle, D.E.; Brenner, C.; Fisher, R.A. RGS6 Suppresses Ras-Induced Cellular Transformation by Facilitating Tip60-Mediated Dnmt1 Degradation and Promoting Apoptosis. Oncogene 2014, 33, 3604–3611. [Google Scholar] [CrossRef] [PubMed]
- Saeed, O.; Lopez-Beltran, A.; Fisher, K.W.; Scarpelli, M.; Montironi, R.; Cimadamore, A.; Massari, F.; Santoni, M.; Cheng, L. RAS Genes in Colorectal Carcinoma: Pathogenesis, Testing Guidelines and Treatment Implications. J. Clin. Pathol. 2019, 72, 135–139. [Google Scholar] [CrossRef] [PubMed]
- Martinez-Cardús, A.; Martinez-Balibrea, E.; Bandrés, E.; Malumbres, R.; Ginés, A.; Manzano, J.L.; Taron, M.; Garcia-Foncillas, J.; Abad, A. Pharmacogenomic Approach for the Identification of Novel Determinants of Acquired Resistance to Oxaliplatin in Colorectal Cancer. Mol. Cancer Ther. 2009, 8, 194–202. [Google Scholar] [CrossRef] [PubMed]
- Chang, Y.-F.; Huang, Y.-Q.; Wu, K.-M.; Jou, A.F.-J.; Shih, N.-Y.; Ho, J.-A.A. Diagnosing the RGS11 Lung Cancer Biomarker: The Integration of Competitive Immunoassay and Isothermal Nucleic Acid Exponential Amplification Reaction. Anal. Chem. 2019, 91, 3327–3335. [Google Scholar] [CrossRef]
- Ghavami, A.; Hunt, R.A.; Olsen, M.A.; Zhang, J.; Smith, D.L.; Kalgaonkar, S.; Rahman, Z.; Young, K.H. Differential Effects of Regulator of G Protein Signaling (RGS) Proteins on Serotonin 5-HT1A, 5-HT2A, and Dopamine D2 Receptor-Mediated Signaling and Adenylyl Cyclase Activity. Cell Signal. 2004, 16, 711–721. [Google Scholar] [CrossRef]
- Caldiran, F.Y.; Cacan, E. RGS10 Suppression by DNA Methylation Is Associated with Low Survival Rates in Colorectal Carcinoma. Pathol. Res. Pract. 2022, 236, 154007. [Google Scholar] [CrossRef]
- Rodríguez-Muñoz, M.; de la Torre-Madrid, E.; Sánchez-Blázquez, P.; Wang, J.B.; Garzón, J. NMDAR-NNOS Generated Zinc Recruits PKCgamma to the HINT1-RGS17 Complex Bound to the C Terminus of Mu-Opioid Receptors. Cell Signal. 2008, 20, 1855–1864. [Google Scholar] [CrossRef]
- Wang, Y.; Tong, Y.; Tso, P.H.; Wong, Y.H. Regulator of G Protein Signaling 19 Suppresses Ras-Induced Neoplastic Transformation and Tumorigenesis. Cancer Lett. 2013, 339, 33–41. [Google Scholar] [CrossRef]
- Katoh, M. Functional Proteomics, Human Genetics and Cancer Biology of GIPC Family Members. Exp. Mol. Med. 2013, 45, e26. [Google Scholar] [CrossRef]
- Gao, H.; Ma, L.; Zou, Q.; Hu, B.; Cai, K.; Sun, Y.; Lu, L.; Ren, D. Unraveling Dynamic Interactions between Tumor-Associated Macrophages and Consensus Molecular Subtypes in Colorectal Cancer: An Integrative Analysis of Single-Cell and Bulk RNA Transcriptome. Heliyon 2023, 9, e19224. [Google Scholar] [CrossRef]
- Yang, L.; Lee, M.M.K.; Leung, M.M.H.; Wong, Y.H. Regulator of G Protein Signaling 20 Enhances Cancer Cell Aggregation, Migration, Invasion and Adhesion. Cell Signal. 2016, 28, 1663–1672. [Google Scholar] [CrossRef] [PubMed]
- Wang, S.L.; Dong, X.W.; Zhao, F.; Li, C.X. MiR-203 Inhibits Cell Proliferation, Invasion, and Migration of Ovarian Cancer through Regulating RGS17. J. Biol. Regul. Homeost. Agents 2021, 35, 1109–1115. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Li, L.; Lin, J.; Hu, X.; Li, B.; Xue, A.; Shen, Y.; Jiang, J.; Zhang, M.; Xie, J.; et al. Deregulation of RGS17 Expression Promotes Breast Cancer Progression. J. Cancer 2015, 6, 767–775. [Google Scholar] [CrossRef] [PubMed]
- Su, W.-Z.; Ren, L.-F. MiRNA-199 Inhibits Malignant Progression of Lung Cancer through Mediating RGS17. Eur. Rev. Med. Pharmacol. Sci. 2019, 23, 3390–3400. [Google Scholar] [CrossRef] [PubMed]
- Zhang, L.-S.; Ma, H.-G.; Sun, F.-H.; Zhao, W.-C.; Li, G. MiR-203 Inhibits the Malignant Behavior of Prostate Cancer Cells by Targeting RGS17. Eur. Rev. Med. Pharmacol. Sci. 2019, 23, 5667–5674. [Google Scholar] [CrossRef] [PubMed]
- Yu, Q.; Zhang, N.; Jiang, Y.; Huang, Y.; Lian, Y.-Y.; Liu, T.; Li, N.; Guan, G. RGS17 Inhibits Tumorigenesis and Improves 5-Fluorouracil Sensitivity in Nasopharyngeal Carcinoma. Onco Targets Ther. 2018, 11, 7591–7600. [Google Scholar] [CrossRef] [PubMed]
- Mazzoni, S.M.; Fearon, E.R. AXIN1 and AXIN2 Variants in Gastrointestinal Cancers. Cancer Lett. 2014, 355, 1–8. [Google Scholar] [CrossRef]
- Cha, P.H.; Cho, Y.H.; Lee, S.K.; Lee, J.; Jeong, W.J.; Moon, B.S.; Yun, J.H.; Yang, J.S.; Choi, S.; Yoon, J.; et al. Small-Molecule Binding of the Axin RGS Domain Promotes β-Catenin and Ras Degradation. Nat. Chem. Biol. 2016, 12, 593–600. [Google Scholar] [CrossRef]
- Li, W.; Zhang, N.; Jin, C.; Long, M.D.; Rajabi, H.; Yasumizu, Y.; Fushimi, A.; Yamashita, N.; Hagiwara, M.; Zheng, R.; et al. MUC1-C Drives Stemness in Progression of Colitis to Colorectal Cancer. JCI Insight 2020, 5, e137112. [Google Scholar] [CrossRef]
- Kufe, D.W. MUC1-C in Chronic Inflammation and Carcinogenesis; Emergence as a Target for Cancer Treatment. Carcinogenesis 2020, 41, 1173–1183. [Google Scholar] [CrossRef]
- Yan, K.S.; Chia, L.A.; Li, X.; Ootani, A.; Su, J.; Lee, J.Y.; Su, N.; Luo, Y.; Heilshorn, S.C.; Amieva, M.R.; et al. The Intestinal Stem Cell Markers Bmi1 and Lgr5 Identify Two Functionally Distinct Populations. Proc. Natl. Acad. Sci. USA 2012, 109, 466–471. [Google Scholar] [CrossRef] [PubMed]
- Barker, N.; Ridgway, R.A.; van Es, J.H.; van de Wetering, M.; Begthel, H.; van den Born, M.; Danenberg, E.; Clarke, A.R.; Sansom, O.J.; Clevers, H. Crypt Stem Cells as the Cells-of-Origin of Intestinal Cancer. Nature 2009, 457, 608–611. [Google Scholar] [CrossRef] [PubMed]
- De Sousa e Melo, F.; Kurtova, A.V.; Harnoss, J.M.; Kljavin, N.; Hoeck, J.D.; Hung, J.; Anderson, J.E.; Storm, E.E.; Modrusan, Z.; Koeppen, H.; et al. A Distinct Role for Lgr5+ Stem Cells in Primary and Metastatic Colon Cancer. Nature 2017, 543, 676–680. [Google Scholar] [CrossRef] [PubMed]
- Cacan, E. Epigenetic Regulation of RGS2 (Regulator of G-Protein Signaling 2) in Chemoresistant Ovarian Cancer Cells. J. Chemother. 2017, 29, 173–178. [Google Scholar] [CrossRef]
- Tso, P.H.; Yung, L.Y.; Wang, Y.; Wong, Y.H. RGS19 Stimulates Cell Proliferation by Deregulating Cell Cycle Control and Enhancing Akt Signaling. Cancer Lett. 2011, 309, 199–208. [Google Scholar] [CrossRef]
- Steffan, J.J.; Dykes, S.S.; Coleman, D.T.; Adams, L.K.; Rogers, D.; Carroll, J.L.; Williams, B.J.; Cardelli, J.A. Supporting a Role for the GTPase Rab7 in Prostate Cancer Progression. PLoS ONE 2014, 9, e87882. [Google Scholar] [CrossRef]



{kind=link}
{kind=link}
| Feature | UC | CD | Reference |
|---|---|---|---|
| Anatomical location | Large intestine, from left-sided colitis to pancolitis, with the possibility of backwash ileitis | Any part of the GI tract may be involved with ‘skip areas’ between inflammatory sites | [14,15,16] |
| Depth | Limited to mucosa and submucosa | The whole width of the GI tract wall can be affected by fistulae formation | |
| Immune cells phenotype and key cytokines | Th2 IL-4, IL-5, IL-10, IL-13 | Th1/Th17 IL-2, TNF, IFN-γ, IL-17, IL-23 | |
| CAC relative risk ratio | RR = 2.75 (95% CI: 1.91–3.97) Well-studied, generally considered higher than in CD | RR = 2.5 (95% CI: 1.3–4.7) Less studied, generally considered lower than in UC | [5] |
| RGS Family | Representatives | Connection with IBD and IBD-Related GPCR | References |
|---|---|---|---|
| R4 | RGS: 1, 2, 3, 4, 5, 8, 13, 16, 18, 21 | OR: RGS1, RGS2, RGS4 CB: RGS4 5-HTR: RGS4 Other links with IBD: RGS13, RGS16 | [32,62,63] [32,64] [32,65] |
| R7 | RGS: 6, 7, 9–2, 11 | OR: RGS6, RGS7, RGS11 CB: RGS7 5-HTR: RGS6 Other links with CRC: RGS7 | |
| R12 | RGS: 10, 12, 14 | OR: RGS10 5-HTR: RGS10, RGS12 Other links with IBD: RGS10 | |
| RZ | RGS: 17, 19, 20 | OR: RGS17, RGS19 RGS20 CB: RGS17 Other links with IBD: RGS17 | [32,66,67] |
| Atypical RGS | AXIN1, AXIN2 | Prevents pro-inflammatory effects of Wnt/β-catenin pathway | [68] |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Swierczynski, M.; Kasprzak, Z.; Makaro, A.; Salaga, M. Regulators of G-Protein Signaling (RGS) in Sporadic and Colitis-Associated Colorectal Cancer. Int. J. Mol. Sci. 2024, 25, 577. https://doi.org/10.3390/ijms25010577
Swierczynski M, Kasprzak Z, Makaro A, Salaga M. Regulators of G-Protein Signaling (RGS) in Sporadic and Colitis-Associated Colorectal Cancer. International Journal of Molecular Sciences. 2024; 25(1):577. https://doi.org/10.3390/ijms25010577
Chicago/Turabian StyleSwierczynski, Mikolaj, Zuzanna Kasprzak, Adam Makaro, and Maciej Salaga. 2024. "Regulators of G-Protein Signaling (RGS) in Sporadic and Colitis-Associated Colorectal Cancer" International Journal of Molecular Sciences 25, no. 1: 577. https://doi.org/10.3390/ijms25010577
APA StyleSwierczynski, M., Kasprzak, Z., Makaro, A., & Salaga, M. (2024). Regulators of G-Protein Signaling (RGS) in Sporadic and Colitis-Associated Colorectal Cancer. International Journal of Molecular Sciences, 25(1), 577. https://doi.org/10.3390/ijms25010577