
MDSPACE and MDTOMO Software for Extracting Continuous Conformational Landscapes from Datasets of Single Particle Images and Subtomograms Based on Molecular Dynamics Simulations: Latest Developments in ContinuousFlex Software Package



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Results
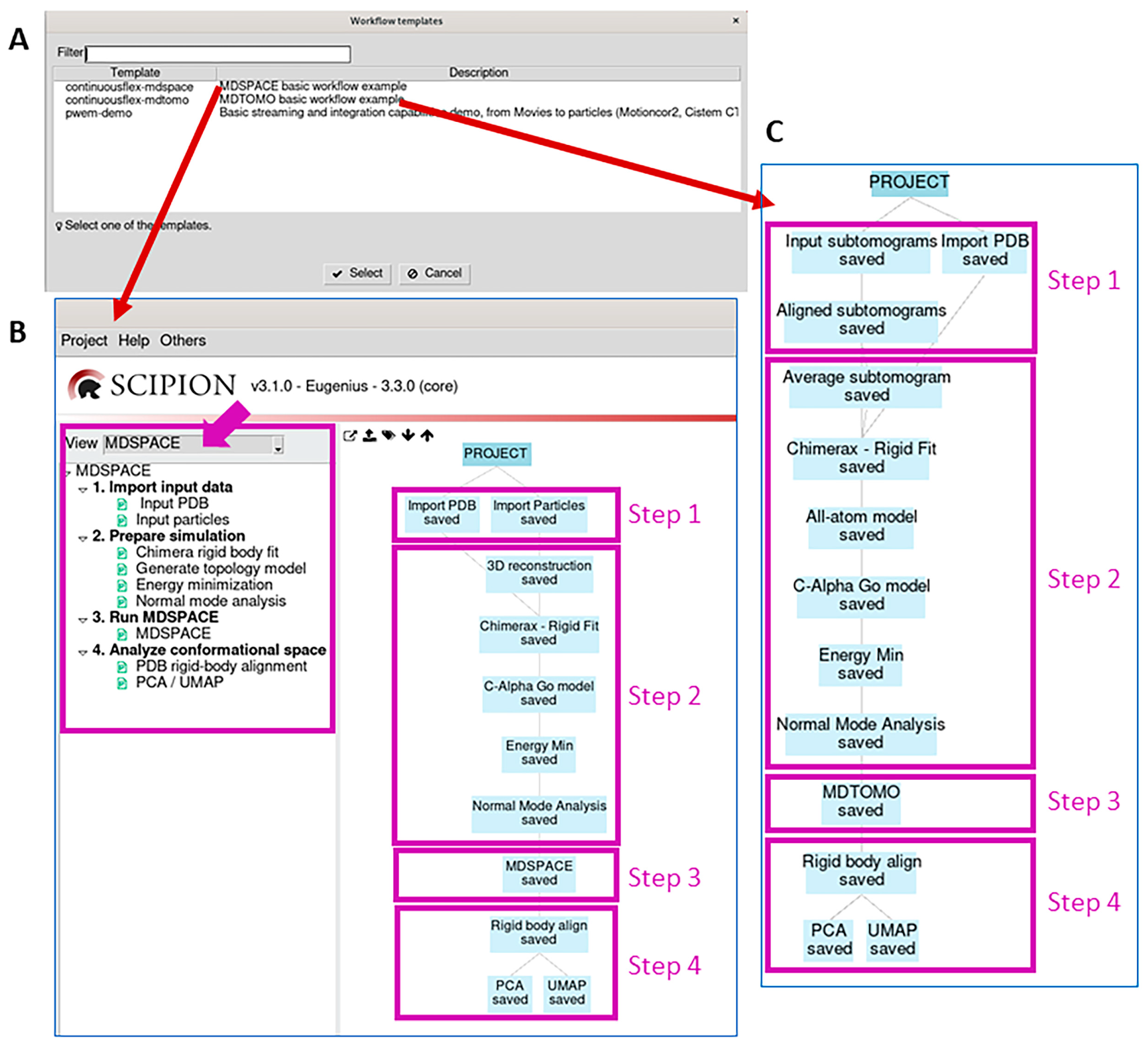
2.1. Import Input Data
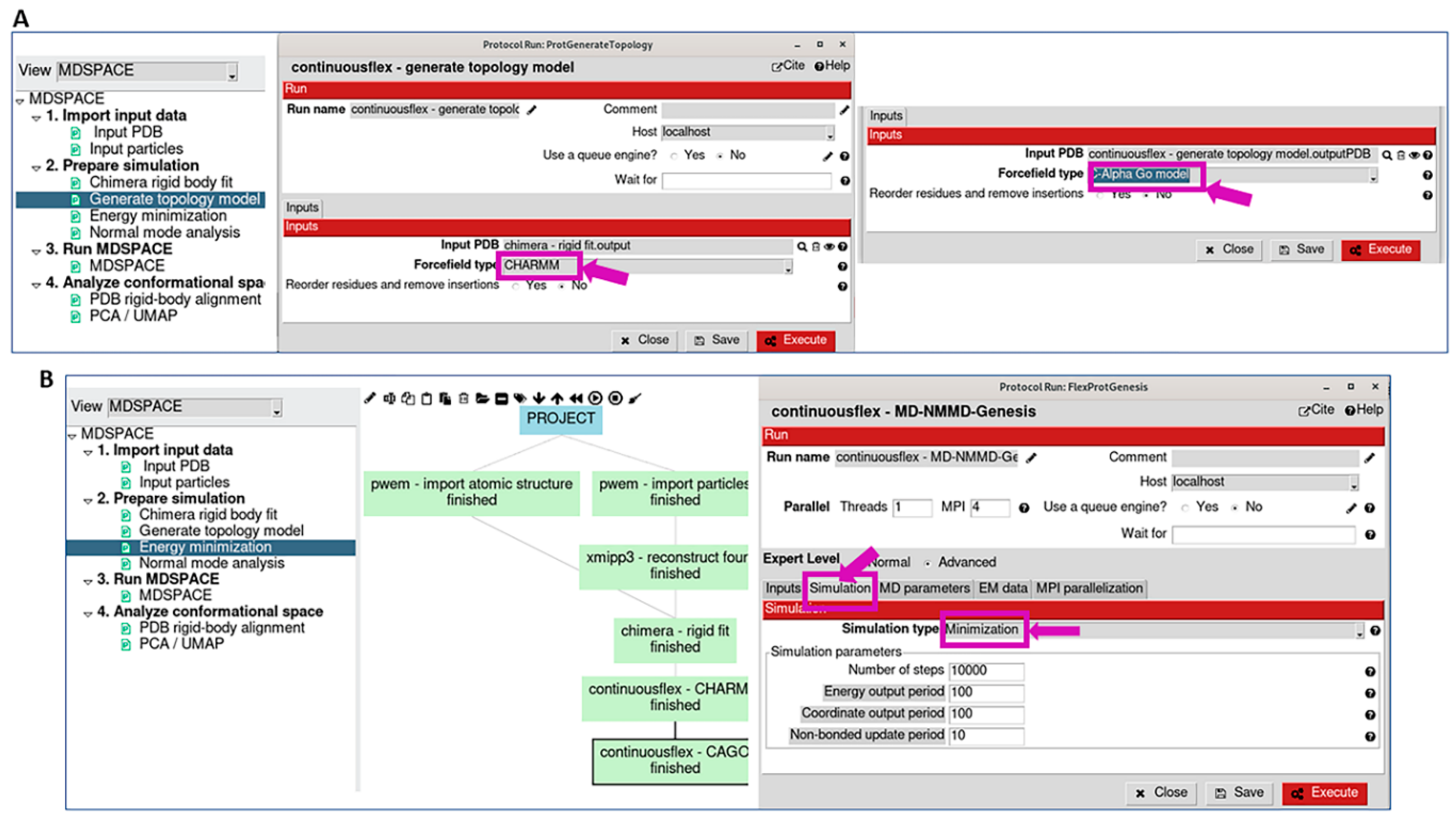
2.2. Prepare Simulation
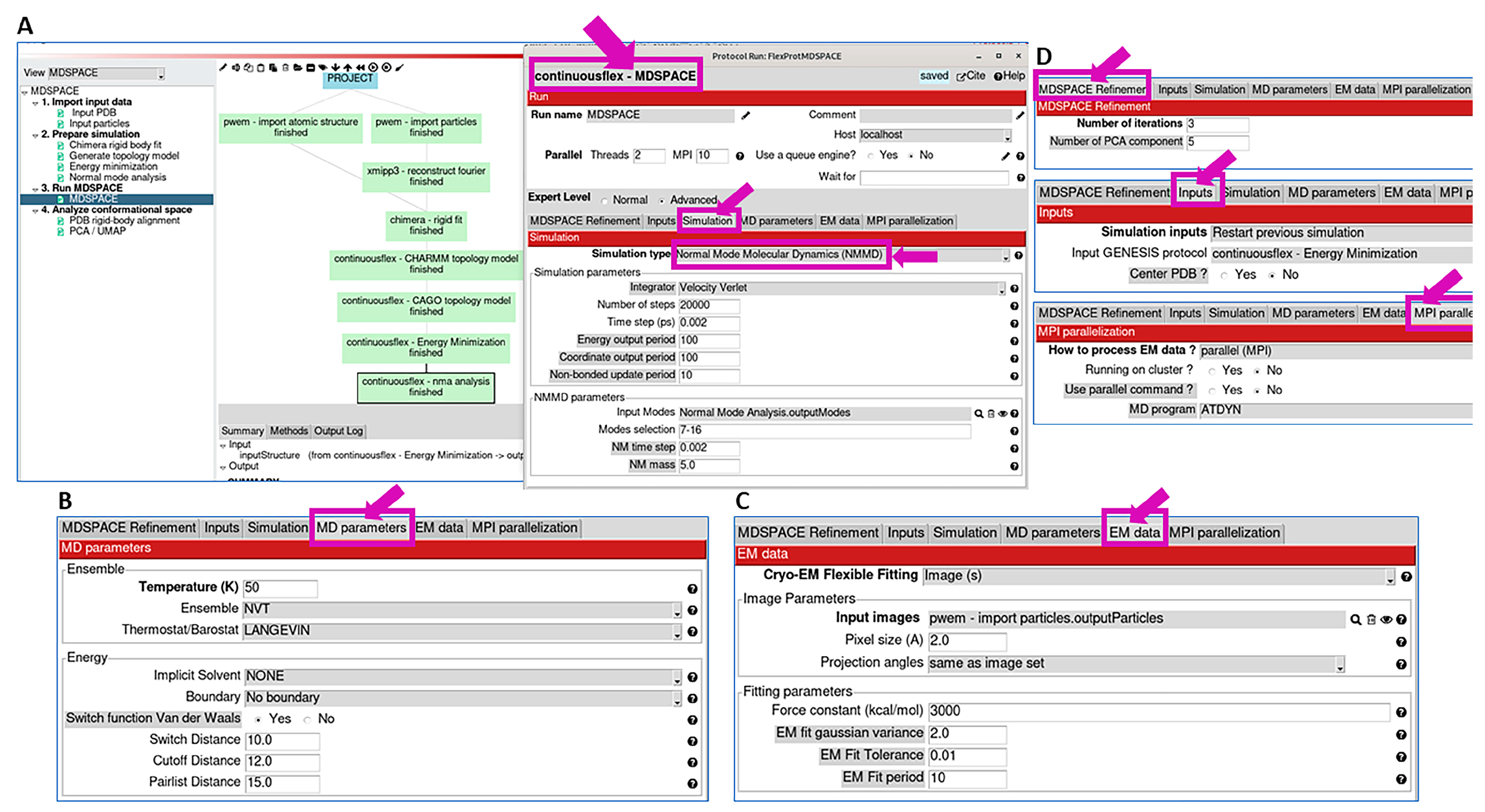
2.3. Run MDSPACE/MDTOMO
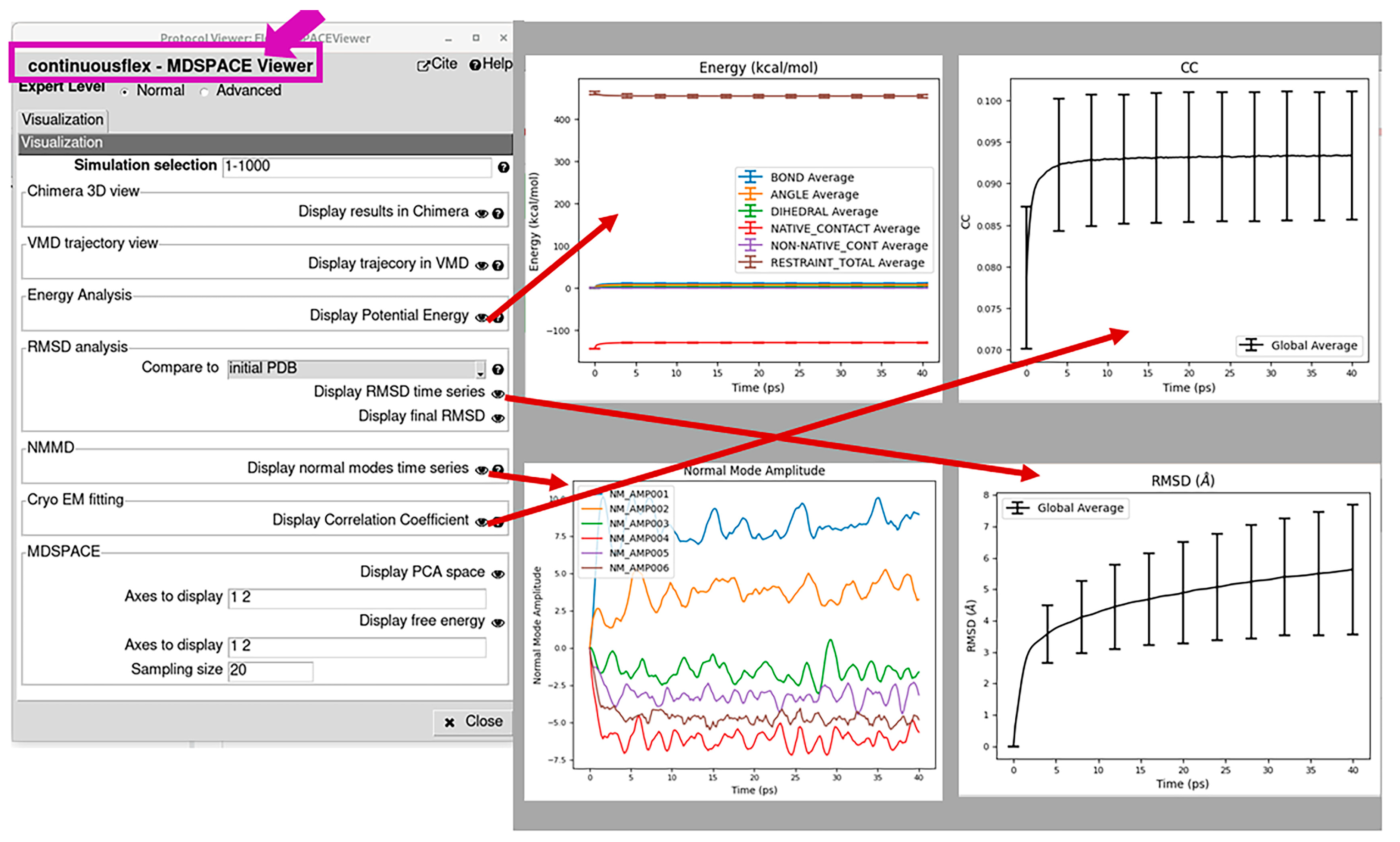
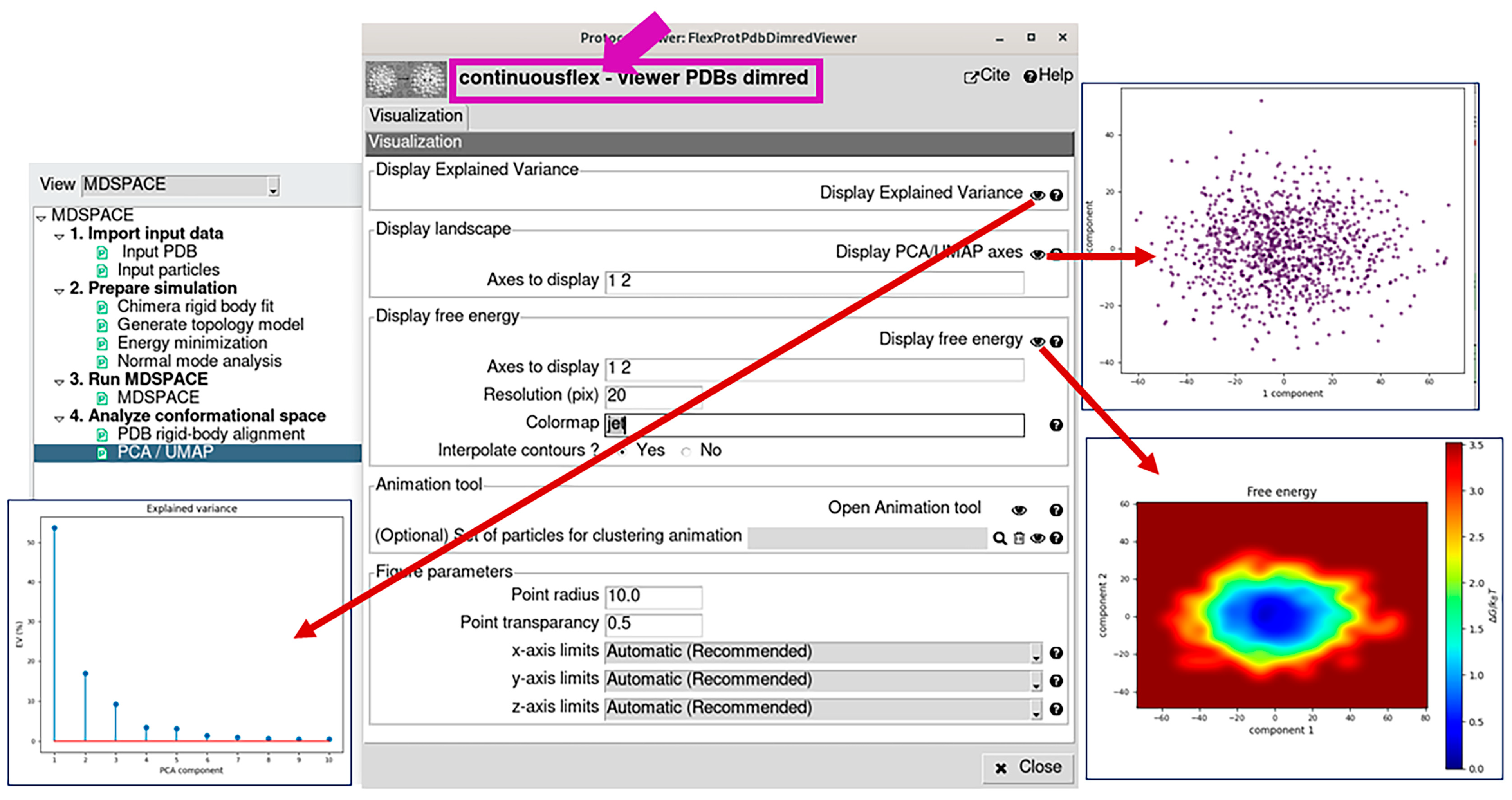
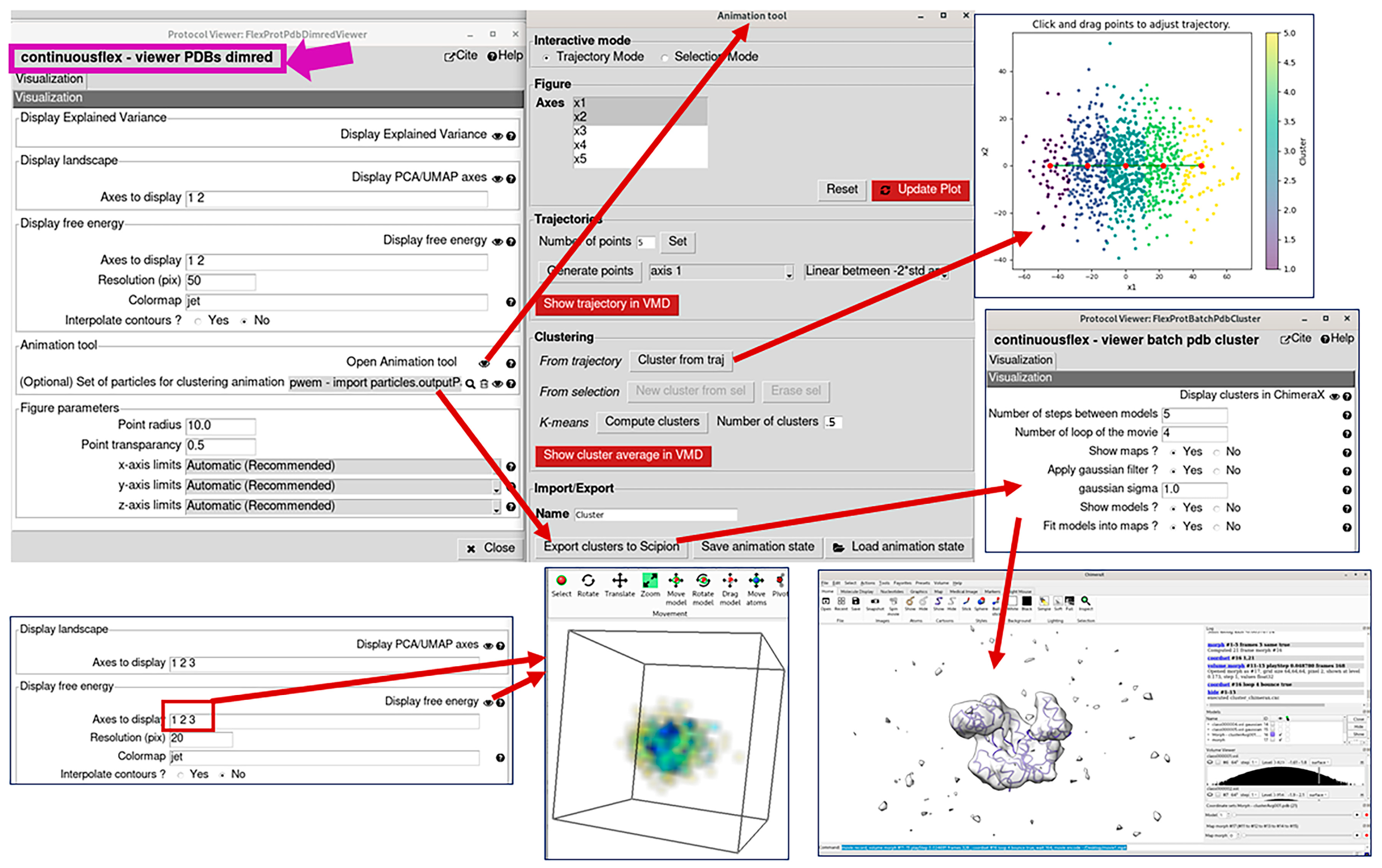
2.4. Analyze Conformational Space
3. Discussion
4. Materials and Methods
Author Contributions
Funding
Data Availability Statement
Conflicts of Interest
References
- Svidritskiy, E.; Brilot, A.F.; Koh, C.S.; Grigorieff, N.; Korostelev, A.A. Structures of yeast 80S ribosome-tRNA complexes in the rotated and nonrotated conformations. Structure 2014, 22, 1210–1218. [Google Scholar] [CrossRef] [PubMed]
- Zhou, A.; Rohou, A.; Schep, D.G.; Bason, J.V.; Montgomery, M.G.; Walker, J.E.; Grigorieff, N.; Rubinstein, J.L. Structure conformational states of the bovine mitochondrial ATP synthase by cryo-EM. eLife 2015, 4, e10180. [Google Scholar] [CrossRef] [PubMed]
- Bai, X.C.; Rajendra, E.; Yang, G.; Shi, Y.; Scheres, S.H. Sampling the conformational space of the catalytic subunit of human gamma-secretase. eLife 2015, 4, e11182. [Google Scholar] [CrossRef] [PubMed]
- Abeyrathne, P.D.; Koh, C.S.; Grant, T.; Grigorieff, N.; Korostelev, A.A. Ensemble cryo-EM uncovers inchworm-like translocation of a viral IRES through the ribosome. eLife 2016, 5, e14874. [Google Scholar] [CrossRef] [PubMed]
- Banerjee, S.; Bartesaghi, A.; Merk, A.; Rao, P.; Bulfer, S.L.; Yan, Y.; Green, N.; Mroczkowski, B.; Neitz, R.J.; Wipf, P.; et al. 2.3 A resolution cryo-EM structure of human p97 and mechanism of allosteric inhibition. Science 2016, 351, 871–875. [Google Scholar] [CrossRef]
- Hofmann, S.; Januliene, D.; Mehdipour, A.R.; Thomas, C.; Stefan, E.; Brüchert, S.; Kuhn, B.T.; Geertsma, E.R.; Hummer, G.; Tampé, R.; et al. Conformation space of a heterodimeric ABC exporter under turnover conditions. Nature 2019, 571, 580–583. [Google Scholar] [CrossRef]
- Nakane, T.; Kotecha, A.; Sente, A.; McMullan, G.; Masiulis, S.; Brown, P.M.G.E.; Grigoras, I.T.; Malinauskaite, L.; Malinauskas, T.; Miehling, J.; et al. Single-particle cryo-EM at atomic resolution. Nature 2020, 587, 152–156. [Google Scholar] [CrossRef]
- Kato, K.; Miyazaki, N.; Hamaguchi, T.; Nakajima, Y.; Akita, F.; Yonekura, K.; Shen, J.-R. High-resolution cryo-EM structure of photosystem II reveals damage from high-dose electron beams. Commun. Biol. 2021, 4, 382. [Google Scholar] [CrossRef]
- Schur, F.K.; Obr, M.; Hagen, W.J.; Wan, W.; Jakobi, A.J.; Kirkpatrick, J.M.; Sachse, C.; Kräusslich, H.-G.; Briggs, J.A.G. An atomic model of HIV-1 capsid-SP1 reveals structures regulating assembly and maturation. Science 2016, 353, 506–508. [Google Scholar] [CrossRef]
- Wan, W.; Kolesnikova, L.; Clarke, M.; Koehler, A.; Noda, T.; Becker, S.; Briggs, J.A.G. Structure and assembly of the Ebola virus nucleocapsid. Nature 2017, 551, 394–397. [Google Scholar] [CrossRef]
- Himes, B.A.; Zhang, P. emClarity: Software for high-resolution cryo-electron tomography and subtomogram averaging. Nat. Methods 2018, 15, 955–961. [Google Scholar] [CrossRef] [PubMed]
- von Kügelgen, A.; Tang, H.; Hardy, G.G.; Kureisaite-Ciziene, D.; Brun, Y.V.; Stansfeld, P.J.; Robinson, C.V.; Bharat, T.A. In Situ Structure of an Intact Lipopolysaccharide-Bound Bacterial Surface Layer. Cell 2020, 180, 348–358.e15. [Google Scholar] [CrossRef] [PubMed]
- Scheres, S.H.; Gao, H.; Valle, M.; Herman, G.T.; Eggermont, P.P.; Frank, J.; Carazo, J.-M. Disentangling conformational states of macromolecules in 3D-EM through likelihood optimization. Nat. Methods 2007, 4, 27–29. [Google Scholar] [CrossRef] [PubMed]
- Scheres, S.H. RELION: Implementation of a Bayesian approach to cryo-EM structure determination. J. Struct. Biol. 2012, 180, 519–530. [Google Scholar] [CrossRef]
- Lyumkis, D.; Brilot, A.F.; Theobald, D.L.; Grigorieff, N. Likelihood-based classification of cryo-EM images using FREALIGN. J. Struct. Biol. 2013, 183, 377–388. [Google Scholar] [CrossRef]
- Punjani, A.; Rubinstein, J.L.; Fleet, D.J.; Brubaker, M.A. cryoSPARC: Algorithms for rapid unsupervised cryo-EM structure determination. Nat. Methods 2017, 14, 290–296. [Google Scholar] [CrossRef]
- Kimanius, D.; Dong, L.; Sharov, G.; Nakane, T.; Scheres, S.H.W. New tools for automated cryo-EM single-particle analysis in RELION-4.0. Biochem. J. 2021, 478, 4169–4185. [Google Scholar] [CrossRef]
- Scheres, S.H.W.; Melero, R.; Valle, M.; Carazo, J.-M. Averaging of Electron Subtomograms and Random Conical Tilt Reconstructions through Likelihood Optimization. Structure 2009, 17, 1563–1572. [Google Scholar] [CrossRef]
- Stölken, M.; Beck, F.; Haller, T.; Hegerl, R.; Gutsche, I.; Carazo, J.-M.; Baumeister, W.; Scheres, S.H.; Nickell, S. Maximum likelihood based classification of electron tomographic data. J. Struct. Biol. 2011, 173, 77–85. [Google Scholar] [CrossRef]
- Bharat, T.A.M.; Scheres, S.H.W. Resolving macromolecular structures from electron cryo-tomography data using subtomogram averaging in RELION. Nat. Protoc. 2016, 11, 2054–2065. [Google Scholar] [CrossRef]
- Jin, Q.; Sorzano, C.O.S.; De La Rosa-Trevín, J.M.; Bilbao-Castro, J.R.; Núñez-Ramírez, R.; Llorca, O.; Tama, F.; Jonić, S. Iterative elastic 3D-to-2D alignment method using normal modes for studying structural dynamics of large macromolecular complexes. Structure 2014, 22, 496–506. [Google Scholar] [CrossRef] [PubMed]
- Dashti, A.; Schwander, P.; Langlois, R.; Fung, R.; Li, W.; Hosseinizadeh, A.; Liao, H.Y.; Pallesen, J.; Sharma, G.; Stupina, V.A.; et al. Trajectories of the ribosome as a Brownian nanomachine. Proc. Natl. Acad. Sci. USA 2014, 111, 17492–17497. [Google Scholar] [CrossRef] [PubMed]
- Moscovich, A.; Halevi, A.; Andén, J.; Singer, A. Cryo-EM reconstruction of continuous heterogeneity by Laplacian spectral volumes. Inverse Probl. 2020, 36, 024003. [Google Scholar] [CrossRef]
- Lederman, R.R.; Andén, J.; Singer, A. Hyper-molecules: On the representation and recovery of dynamical structures for applications in flexible macro-molecules in cryo-EM. Inverse Probl. 2020, 36, 044005. [Google Scholar] [CrossRef]
- Chen, M.; Ludtke, S.J. Deep learning-based mixed-dimensional Gaussian mixture model for characterizing variability in cryo-EM. Nat. Methods 2021, 18, 930–936. [Google Scholar] [CrossRef]
- Punjani, A. Fleet DJ 3D variability analysis: Resolving continuous flexibility discrete heterogeneity from single particle, cryo-EM. J. Struct. Biol. 2021, 213, 107702. [Google Scholar] [CrossRef]
- Zhong, E.D.; Bepler, T.; Berger, B.; Davis, J.H. CryoDRGN: Reconstruction of heterogeneous cryo-EM structures using neural networks. Nat. Methods 2021, 18, 176–185. [Google Scholar] [CrossRef]
- Zhong, E.D.; Lerer, A.; Davis, J.H.; Berger, B. CryoDRGN2, Ab initio neural reconstruction of 3D protein structures from real cryo-EM images. In Proceedings of the 2021 IEEE/CVF International Conference on Computer Vision (ICCV), Montreal, QC, Canada, 10–17 October 2021; pp. 4046–4055. [Google Scholar]
- Levy, A.; Wetzstein, G.; Martel, J.; Poitevin, F.; Zhong, E.D. Amortized Inference for Heterogeneous Reconstruction in Cryo-EM. Adv. Neural. Inf. Process. Syst. 2022, 35, 13038–13049. [Google Scholar]
- Hamitouche, I.; Jonic, S. DeepHEMNMA: ResNet-based hybrid analysis of continuous conformational heterogeneity in cryo-EM single particle images. Front. Mol. Biosci. 2022, 9, 965645. [Google Scholar] [CrossRef]
- Punjani, A. Fleet DJ 3DFlex: Determining structure motion of flexible proteins from, cryo-EM. Nat. Methods 2023, 20, 860–870. [Google Scholar] [CrossRef]
- Herreros, D.; Lederman, R.R.; Krieger, J.M.; Jiménez-Moreno, A.; Martínez, M.; Myška, D.; Strelak, D.; Filipovic, J.; Sorzano, C.O.; Carazo, J.M. Estimating conformational landscapes from Cryo-EM particles by 3D Zernike polynomials. Nat. Commun. 2023, 14, 154. [Google Scholar] [CrossRef] [PubMed]
- Vuillemot, R.; Mirzaei, A.; Harastani, M.; Hamitouche, I.; Fréchin, L.; Klaholz, B.P.; Miyashita, O.; Tama, F.; Rouiller, I.; Jonic, S. MDSPACE: Extracting Continuous Conformational Landscapes from Cryo-EM Single Particle Datasets Using 3D-to-2D Flexible Fitting based on Molecular Dynamics Simulation. J. Mol. Biol. 2023, 435, 167951. [Google Scholar] [CrossRef] [PubMed]
- Harastani, M.; Eltsov, M.; Leforestier, A.; Jonic, S. HEMNMA-3D: Cryo Electron Tomography Method Based on Normal Mode Analysis to Study Continuous Conformational Variability of Macromolecular Complexes. Front. Mol. Biosci. 2021, 8, 663121. [Google Scholar] [CrossRef] [PubMed]
- Vuillemot, R.; Rouiller, I.; Jonić, S. MDTOMO method for continuous conformational variability analysis in cryo electron subtomograms based on molecular dynamics simulations. Sci. Rep. 2023, 13, 10596. [Google Scholar] [CrossRef] [PubMed]
- Harastani, M.; Eltsov, M.; Leforestier, A.; Jonic, S. TomoFlow: Analysis of continuous conformational variability of macromolecules in cryogenic subtomograms based on 3D dense optical flow. J. Mol. Biol. 2022, 434, 167381. [Google Scholar] [CrossRef] [PubMed]
- Powell, B.M.; Davis, J.H. Learning structural heterogeneity from cryo-electron sub-tomograms with tomoDRGN. bioRxiv 2023. [Google Scholar] [CrossRef]
- Tagare, H.D.; Kucukelbir, A.; Sigworth, F.J.; Wang, H.; Rao, M. Directly reconstructing principal components of heterogeneous particles from cryo-EM images. J. Struct. Biol. 2015, 191, 245–262. [Google Scholar] [CrossRef]
- Katsevich, E.; Katsevich, A.; Singer, A. Covariance Matrix Estimation for the Cryo-EM Heterogeneity Problem. SIAM J. Imaging Sci. 2015, 8, 126–185. [Google Scholar] [CrossRef]
- Liao, H.Y.; Hashem, Y.; Frank, J. Efficient Estimation of Three-Dimensional Covariance and its Application in the Analysis of Heterogeneous Samples in Cryo-Electron Microscopy. Structure 2015, 23, 1129–1137. [Google Scholar] [CrossRef][Green Version]
- Marshall, N.F.; Mickelin, O.; Shi, Y.; Singer, A. Fast principal component analysis for cryo-electron microscopy images. Biol. Imaging 2023, 3, e2. [Google Scholar] [CrossRef]
- Tama, F.; Miyashita, O.; Brooks, C.L. Flexible Multi-scale Fitting of Atomic Structures into Low-resolution Electron Density Maps with Elastic Network Normal Mode Analysis. J. Mol. Biol. 2004, 337, 985–999. [Google Scholar] [CrossRef] [PubMed]
- Orzechowski, M.; Tama, F. Flexible fitting of high-resolution x-ray structures into cryoelectron microscopy maps using biased molecular dynamics simulations. Biophys. J. 2008, 95, 5692–5705. [Google Scholar] [CrossRef] [PubMed]
- Miyashita, O.; Kobayashi, C.; Mori, T.; Sugita, Y.; Tama, F. Flexible fitting to cryo-EM density map using ensemble molecular dynamics simulations. J. Comput. Chem. 2017, 38, 1447–1461. [Google Scholar] [CrossRef] [PubMed]
- Miyashita, O.; Tama, F. Hybrid Methods for Macromolecular Modeling by Molecular Mechanics Simulations with Experimental Data. Adv. Exp. Med. Biol. 2018, 1105, 199–217. [Google Scholar] [PubMed]
- Vuillemot, R.; Miyashita, O.; Tama, F.; Rouiller, I.; Jonic, S. NMMD: Efficient cryo-EM flexible fitting based on simultaneous Normal Mode and Molecular Dynamics atomic displacements. J. Mol. Biol. 2022, 434, 167483. [Google Scholar] [CrossRef] [PubMed]
- Harastani, M.; Vuillemot, R.; Hamitouche, I.; Barati Moghadam, N.; Jonic, S. ContinuousFlex: Software package for analyzing continuous conformational variability of macromolecules in cryo electron microscopy and tomography data. J. Struct. Biol. 2022, 214, 107906. [Google Scholar] [CrossRef] [PubMed]
- Conesa, P.; Fonseca, Y.C.; Jiménez de la Morena, J.; Sharov, G.; de la Rosa-Trevín, J.M.; Cuervo, A.; Mena, A.G.; de Francisco, B.R.; del Hoyo, D.; Herreros, D.; et al. Scipion3, A workflow engine for cryo-electron microscopy image processing and structural biology. Biol. Imaging 2023, 3, e13. [Google Scholar] [CrossRef]
- De la Rosa-Trevín, J.; Quintana, A.; Del Cano, L.; Zaldívar, A.; Foche, I.; Gutiérrez, J.; Gómez-Blanco, J.; Burguet-Castell, J.; Cuenca-Alba, J.; Abrishami, V.; et al. Scipion: A software framework toward integration, reproducibility and validation in 3D electron microscopy. J. Struct. Biol. 2016, 195, 93–99. [Google Scholar] [CrossRef]
- Jiménez de la Morena, J.; Conesa, P.; Fonseca, Y.C.; de Isidro-Gómez, F.P.; Herreros, D.; Fernández-Giménez, E.; Strelak, D.; Moebel, E.; Buchholz, T.; Jug, F.; et al. ScipionTomo: Towards cryo-electron tomography software integration, reproducibility, and validation. J. Struct. Biol. 2022, 214, 107872. [Google Scholar] [CrossRef]
- Herreros, D.; Krieger, J.M.; Fonseca, Y.; Conesa, P.; Harastani, M.; Vuillemot, R.; Hamitouche, I.; Gutiérrez, R.S.; Gragera, M.; Melero, R.; et al. Scipion Flexibility Hub: An integrative framework for advanced analysis of conformational heterogeneity in, cryoEM. Acta Crystallogr. Sect. D 2023, 79, 569–584. [Google Scholar] [CrossRef]
- Harastani, M.; Sorzano, C.O.S.; Jonić, S. Hybrid Electron Microscopy Normal Mode Analysis with Scipion. Protein Sci. 2020, 29, 223–236. [Google Scholar] [CrossRef] [PubMed]
- Pearson, K. LIII. On lines and planes of closest fit to systems of points in space. Lond. Edinb. Dublin Philos. Mag. J. Sci. 1901, 2, 559–572. [Google Scholar] [CrossRef]
- McInnes, L.; Healy, J.; Melville, J. UMAP: Uniform Manifold Approximation and Projection for Dimension Reduction. arXiv 2018, arXiv:1802.03426. [Google Scholar]
- Kobayashi, C.; Jung, J.; Matsunaga, Y.; Mori, T.; Ando, T.; Tamura, K.; Kamiya, M.; Sugita, Y. GENESIS 1.1, A hybrid-parallel molecular dynamics simulator with enhanced sampling algorithms on multiple computational platforms. J. Comput. Chem. 2017, 38, 2193–2206. [Google Scholar] [CrossRef] [PubMed]
- Huang, J.; MacKerell, A.D., Jr. CHARMM36 all-atom additive protein force field: Validation based on comparison to NMR data. J. Comput. Chem. 2013, 34, 2135–2145. [Google Scholar] [CrossRef]
- Karanicolas, J.; Brooks, C.L. Improved Gō-like Models Demonstrate the Robustness of Protein Folding Mechanisms Towards Non-native Interactions. J. Mol. Biol. 2003, 334, 309–325. [Google Scholar] [CrossRef]
- Noel, J.K.; Levi, M.; Raghunathan, M.; Lammert, H.; Hayes, R.L.; Onuchic, J.N.; Whitford, P.C. SMOG 2, A Versatile Software Package for Generating Structure-Based Models. PLoS Comput. Biol. 2016, 12, e1004794. [Google Scholar] [CrossRef]
- Suhre, K.; Sanejouand, Y.H. ElNemo: A normal mode web server for protein movement analysis and the generation of templates for molecular replacement. Nucleic Acids Res. 2004, 32, W610–W614. [Google Scholar] [CrossRef]
- Goddard, T.D.; Huang, C.C.; Meng, E.C.; Pettersen, E.F.; Couch, G.S.; Morris, J.H.; Ferrin, T.E. UCSF ChimeraX: Meeting modern challenges in visualization and analysis. Protein Sci. 2018, 27, 14–25. [Google Scholar] [CrossRef]
- Humphrey, W.; Dalke, A.; Schulten, K. VMD: Visual molecular dynamics. J. Mol. Graph. 1996, 14, 27–28, 33–38. [Google Scholar] [CrossRef]






Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Vuillemot, R.; Harastani, M.; Hamitouche, I.; Jonic, S. MDSPACE and MDTOMO Software for Extracting Continuous Conformational Landscapes from Datasets of Single Particle Images and Subtomograms Based on Molecular Dynamics Simulations: Latest Developments in ContinuousFlex Software Package. Int. J. Mol. Sci. 2024, 25, 20. https://doi.org/10.3390/ijms25010020
Vuillemot R, Harastani M, Hamitouche I, Jonic S. MDSPACE and MDTOMO Software for Extracting Continuous Conformational Landscapes from Datasets of Single Particle Images and Subtomograms Based on Molecular Dynamics Simulations: Latest Developments in ContinuousFlex Software Package. International Journal of Molecular Sciences. 2024; 25(1):20. https://doi.org/10.3390/ijms25010020
Chicago/Turabian StyleVuillemot, Rémi, Mohamad Harastani, Ilyes Hamitouche, and Slavica Jonic. 2024. "MDSPACE and MDTOMO Software for Extracting Continuous Conformational Landscapes from Datasets of Single Particle Images and Subtomograms Based on Molecular Dynamics Simulations: Latest Developments in ContinuousFlex Software Package" International Journal of Molecular Sciences 25, no. 1: 20. https://doi.org/10.3390/ijms25010020
APA StyleVuillemot, R., Harastani, M., Hamitouche, I., & Jonic, S. (2024). MDSPACE and MDTOMO Software for Extracting Continuous Conformational Landscapes from Datasets of Single Particle Images and Subtomograms Based on Molecular Dynamics Simulations: Latest Developments in ContinuousFlex Software Package. International Journal of Molecular Sciences, 25(1), 20. https://doi.org/10.3390/ijms25010020