
Untangling Irregular Actin Cytoskeleton Architectures in Tomograms of the Cell with Struwwel Tracer


Abstract
:1. Introduction
- Among the cryo-ET-related actin tracing methods, some are only applicable for the tracing of well-ordered filaments [4,6,7]. Among these, one noteworthy tool is our Spaghetti Tracer approach [7]. Spaghetti Tracer introduced a paradigm shift in the tracing of semi-regular actin filaments because it is a dynamic-programming-based method at the voxel level that does not require an expensive missing wedge correction, template convolution, or deconvolution. Therefore, it yields a substantial improvement in time efficiency over template convolution [8,10] or deconvolution methods [4], enabling fast and accurate tracing of such filament arrangements. (The accuracy of Spaghetti Tracer was validated in a rigorous statistical analysis, achieving F1-scores of 0.86–0.95 on phantom tomograms under experimental conditions.) The success of Spaghetti Tracer motivated us to extend its capabilities to randomly oriented actin filaments in the present work.
- There are very few earlier algorithms that are agnostic of the relative orientations and distances of the actin filaments, so that they can trace central lines of irregular filaments individually without leveraging the information of adjacent filaments or requiring a mean direction. Volume Tracer [8] utilized an expensive genetic-algorithm-based search employing a population of cylindrical templates (combined with a bi-directional tracing) to detect randomly oriented filaments in Dictyostelium discoideum filopodia. The genetic algorithm was implemented as part of our group’s free, open-source Situs and Sculptor packages, but it required extensive computational time on the order of several days when applied to a complete tomogram, without guaranteeing convergence (leading to false negatives when a user is impatient). Co-author Rigort also developed a similar template-matching method independently [10]. This approach was implemented in Amira, a commercial software requiring a paid license and limiting any algorithmic modifications by third parties or end users.
- Recently, a number of deep-learning-based approaches have been proposed for the segmentation of diverse biological assemblies, including actin [18,19]. For example, Chen et al. [18] presented a deep-learning-based segmentation approach for a voxel-level classification of shapes of interest in the tomogram and integrated the approach into the EMAN2 [20] package. However, these segmentation tools are generic in nature and are not specifically designed for filamentous shape structures. Besides, they require users to annotate training data and fine-tune the deep learning model, which could be a laborious process. These segmentation approaches only provide a voxel-level density segmentation, without any tracing of central lines. Recent studies that used these segmentation methods subsequently required separate approaches, such as the above template matching, for the additional tracing [11,12,13].
2. Results and Discussion
2.1. Statistical Evaluation of Tracing Accuracy in Simulated Tomograms
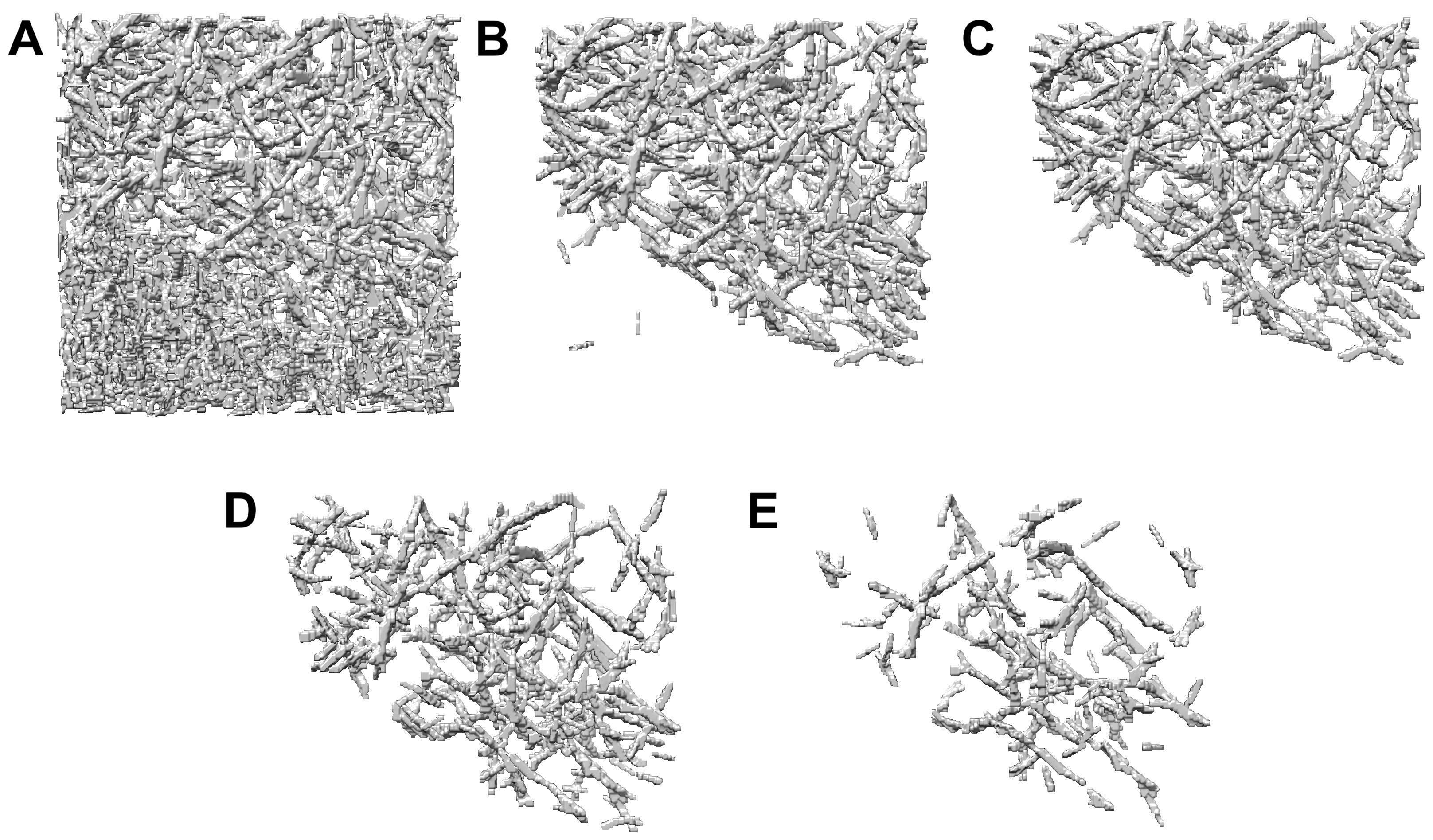
2.2. Measuring Filament Center Lines in a Previously Untraced Experimental Tomogram
2.3. Computation Time and Manual Intervention
2.4. Software Implementation and Dissemination
3. Materials and Methods
3.1. Simulated Phantom Tomograms
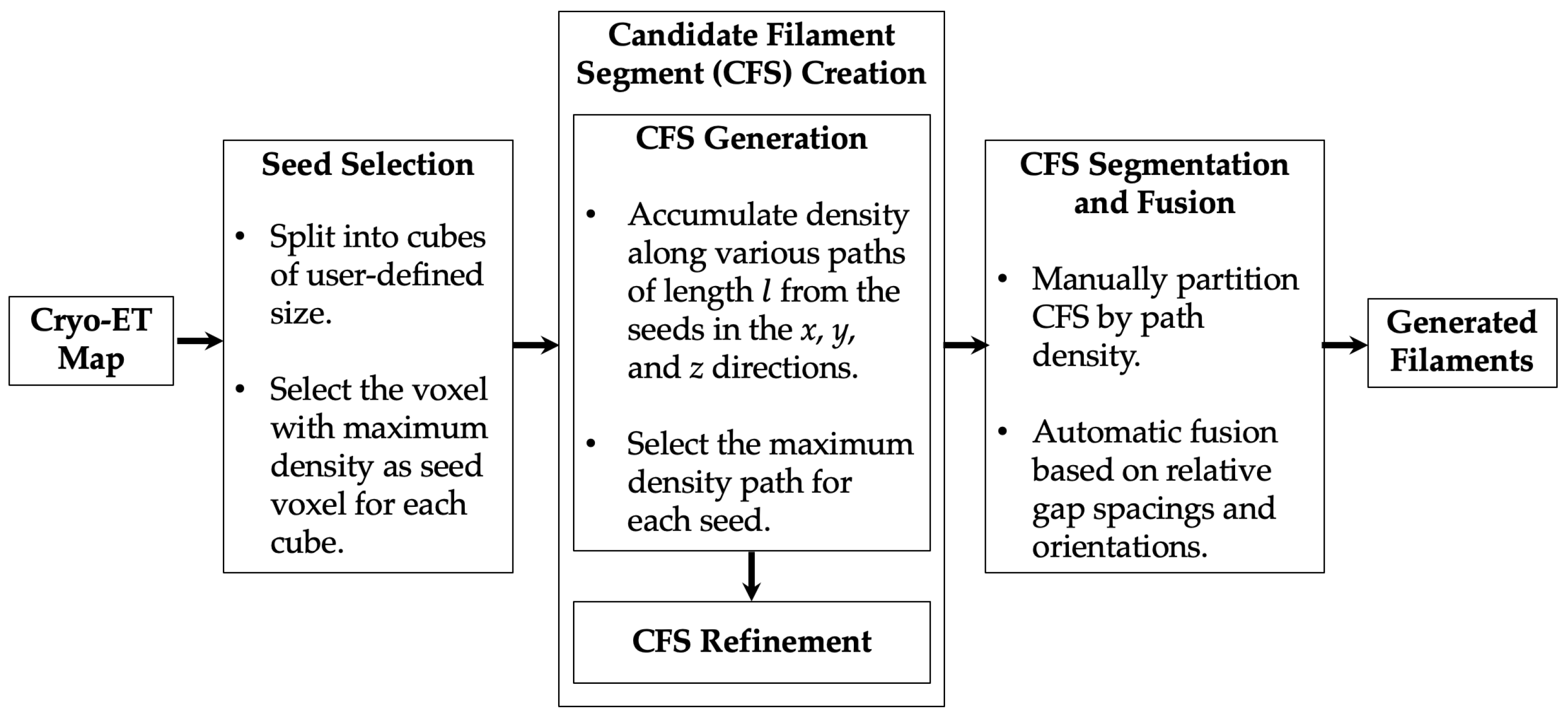
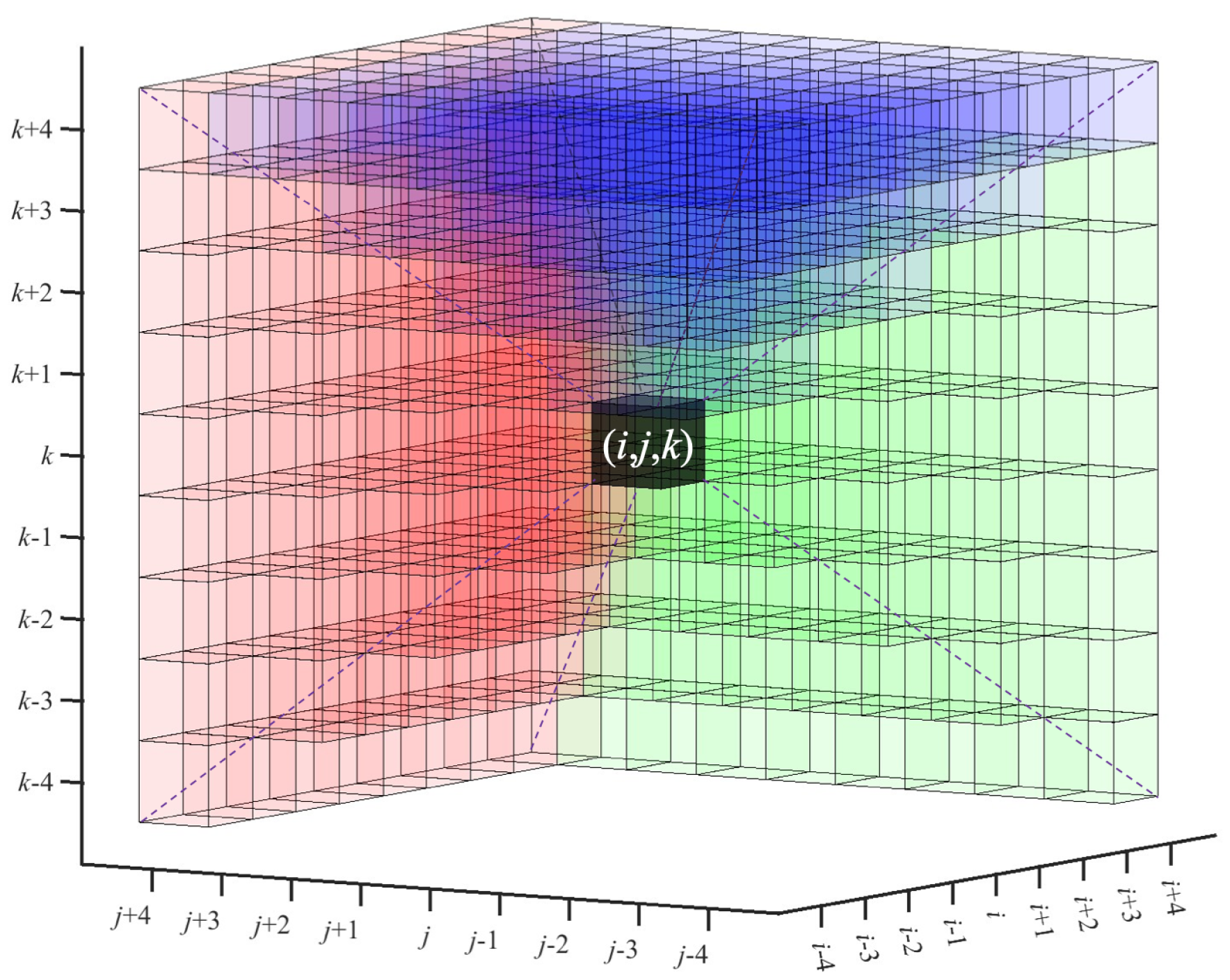
3.2. Automatic Seed Selection
3.3. CFS Creation
3.3.1. CFS Generation
3.3.2. CFS Refinement
3.4. CFS Segmentation
3.5. CFS Fusion
3.5.1. Fusion Based on Physical Proximity
3.5.2. Fusion by Extension
4. Conclusions
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Schoenenberger, C.A.; Mannherz, H.G.; Jockusch, B.M. Actin: From structural plasticity to functional diversity. Eur. J. Cell Biol. 2011, 90, 797–804. [Google Scholar] [CrossRef]
- Rodrigues-Oliveira, T.; Wollweber, F.; Ponce-Toledo, R.I.; Xu, J.; Rittmann, S.K.M.; Klingl, A.; Pilhofer, M.; Schleper, C. Actin cytoskeleton and complex cell architecture in an Asgard archaeon. Nature 2023, 613, 332–339. [Google Scholar] [CrossRef] [PubMed]
- Imachi, H.; Nobu, M.K.; Nakahara, N.; Morono, Y.; Ogawara, M.; Takaki, Y.; Takano, Y.; Uematsu, K.; Ikuta, T.; Ito, M.; et al. Isolation of an archaeon at the prokaryote–eukaryote interface. Nature 2020, 577, 519–525. [Google Scholar] [CrossRef] [PubMed]
- Kovacs, J.; Song, J.; Auer, M.; He, J.; Hunter, W.; Wriggers, W. Correction of missing-wedge artifacts in filamentous tomograms by template-based constrained deconvolution. J. Chem. Inf. Model. 2020, 60, 2626–2633. [Google Scholar] [CrossRef]
- Rogge, H.; Artelt, N.; Endlich, N.; Endlich, K. Automated segmentation and quantification of actin stress fibres undergoing experimentally induced changes. J. Microsc. 2017, 268, 129–140. [Google Scholar] [CrossRef]
- Sazzed, S.; Song, J.; Kovacs, J.A.; Wriggers, W.; Auer, M.; He, J. Tracing actin filament bundles in three-dimensional electron tomography density maps of hair cell stereocilia. Molecules 2018, 23, 882. [Google Scholar] [CrossRef] [PubMed]
- Sazzed, S.; Scheible, P.; He, J.; Wriggers, W. Spaghetti Tracer: A Framework for Tracing Semiregular Filamentous Densities in 3D Tomograms. Biomolecules 2022, 12, 1022. [Google Scholar] [CrossRef]
- Rusu, M.; Starosolski, Z.; Wahle, M.; Rigort, A.; Wriggers, W. Automated tracing of filaments in 3D electron tomography reconstructions using Sculptor and Situs. J. Struct. Biol. 2012, 178, 121–128. [Google Scholar] [CrossRef]
- Song, J.; Patterson, R.; Metlagel, Z.; Krey, J.F.; Hao, S.; Wang, L.; Ng, B.; Sazzed, S.; Kovacs, J.; Wriggers, W.; et al. A cryo-tomography-based volumetric model of the actin core of mouse vestibular hair cell stereocilia lacking plastin 1. J. Struct. Biol. 2020, 210, 107461. [Google Scholar] [CrossRef]
- Rigort, A.; Günther, D.; Hegerl, R.; Baum, D.; Weber, B.; Prohaska, S.; Medalia, O.; Baumeister, W.; Hege, H.C. Automated segmentation of electron tomograms for a quantitative description of actin filament networks. J. Struct. Biol. 2012, 177, 135–144. [Google Scholar] [CrossRef]
- Dimchev, G.; Amiri, B.; Fäßler, F.; Falcke, M.; Schur, F.K. Computational toolbox for ultrastructural quantitative analysis of filament networks in cryo-ET data. J. Struct. Biol. 2021, 213, 107808. [Google Scholar] [CrossRef] [PubMed]
- Martins, B.; Sorrentino, S.; Chung, W.L.; Tatli, M.; Medalia, O.; Eibauer, M. Unveiling the polarity of actin filaments by cryo-electron tomography. Structure 2021, 29, 488–498. [Google Scholar] [CrossRef] [PubMed]
- Schneider, J.; Jasnin, M. Capturing actin assemblies in cells using in situ cryo-electron tomography. Eur. J. Cell Biol. 2022, 101, 151224. [Google Scholar] [CrossRef] [PubMed]
- Redemann, S.; Baumgart, J.; Lindow, N.; Shelley, M.; Nazockdast, E.; Kratz, A.; Prohaska, S.; Brugués, J.; Fürthauer, S.; Müller-Reichert, T.C. elegans chromosomes connect to centrosomes by anchoring into the spindle network. Nat. Commun. 2017, 8, 1–13. [Google Scholar] [CrossRef]
- Loss, L.A.; Bebis, G.; Chang, H.; Auer, M.; Sarkar, P.; Parvin, B. Automatic segmentation and quantification of filamentous structures in electron tomography. In Proceedings of the ACM Conference on Bioinformatics, Computational Biology and Biomedicine, Orlando, FL, USA, 7–10 October 2012; pp. 170–177. [Google Scholar]
- Alioscha-Perez, M.; Benadiba, C.; Goossens, K.; Kasas, S.; Dietler, G.; Willaert, R.; Sahli, H. A robust actin filaments image analysis framework. PLoS Comput. Biol. 2016, 12, e1005063. [Google Scholar] [CrossRef]
- Smith, M.B.; Li, H.; Shen, T.; Huang, X.; Yusuf, E.; Vavylonis, D. Segmentation and tracking of cytoskeletal filaments using open active contours. Cytoskeleton 2010, 67, 693–705. [Google Scholar] [CrossRef]
- Chen, M.; Dai, W.; Sun, S.Y.; Jonasch, D.; He, C.Y.; Schmid, M.F.; Chiu, W.; Ludtke, S.J. Convolutional neural networks for automated annotation of cellular cryo-electron tomograms. Nat. Methods 2017, 14, 983–985. [Google Scholar] [CrossRef]
- Image and Data Analysis Facility, Core Reseach Facilities of DZNE. Yapic. 2022. Available online: https://yapic.github.io/yapic/ (accessed on 5 July 2023).
- Tang, G.; Peng, L.; Baldwin, P.R.; Mann, D.S.; Jiang, W.; Rees, I.; Ludtke, S.J. EMAN2: An extensible image processing suite for electron microscopy. J. Struct. Biol. 2007, 157, 38–46. [Google Scholar] [CrossRef]
- Mu, Y.; Sazzed, S.; Alshammari, M.; Sun, J.; He, J. A tool for segmentation of secondary structures in 3D cryo-EM density map components using deep convolutional neural networks. Front. Bioinform. 2021, 1, 51. [Google Scholar] [CrossRef]
- Pettersen, E.F.; Goddard, T.D.; Huang, C.C.; Couch, G.S.; Greenblatt, D.M.; Meng, E.C.; Ferrin, T.E. UCSF Chimera—A visualization system for exploratory research and analysis. J. Comput. Chem. 2004, 25, 1605–1612. [Google Scholar] [CrossRef]
- Fäßler, F.; Dimchev, G.; Hodirnau, V.V.; Wan, W.; Schur, F.K. Cryo-electron tomography structure of Arp2/3 complex in cells reveals new insights into the branch junction. Nat. Commun. 2020, 11, 6437. [Google Scholar] [CrossRef] [PubMed]
- Lawson, C.L.; Patwardhan, A.; Baker, M.L.; Hryc, C.; Garcia, E.S.; Hudson, B.P.; Lagerstedt, I.; Ludtke, S.J.; Pintilie, G.; Sala, R.; et al. EMDataBank unified data resource for 3DEM. Nucleic Acids Res. 2016, 44, D396–D403. [Google Scholar] [CrossRef] [PubMed]
- Scheible, P.; Sazzed, S.; He, J.; Wriggers, W. TomoSim: Simulation of filamentous cryo-electron tomograms. In Proceedings of the 2021 IEEE International Conference on Bioinformatics and Biomedicine (BIBM), Houston, TX, USA, 9–12 December 2021; pp. 2560–2565. [Google Scholar]
- Humphrey, W.; Dalke, A.; Schulten, K. VMD: Visual molecular dynamics. J. Mol. Graph. 1996, 14, 33–38. [Google Scholar] [CrossRef] [PubMed]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Noise Level | Precision | Recall | F1-Score |
|---|---|---|---|
| 0.35 | 0.97 | 0.85 | 0.90 |
| 0.50 | 0.97 | 0.81 | 0.88 |
| 0.65 | 0.96 | 0.84 | 0.89 |
| 0.80 | 0.95 | 0.79 | 0.87 |
| 0.95 | 0.95 | 0.78 | 0.85 |
| Parameter Name | strwtrc Argument | Description | Default Value | Program Stages |
|---|---|---|---|---|
| Required Parameter | ||||
| Threshold | -thr | Threshold for partitioning the CFS by the normalized path density. | N/A (user-defined based on the pruning map) | CFS segmentation |
| Optional Parameters | ||||
| Length | -len | Length (infinity norm) of the CFS in voxel units. Internally, this also defines the spacing of the cubic grid for placing seed points (half this value; see the text) and the extension length of the CFS (same value). | 10 | Automatic seed selection, CFS generation, and CFS fusion |
| Gap Spacing | -gap | Maximum gap spacing, in voxels, tolerated while fusing adjacent CFSs. | 10 | CFS fusion |
| Fusion Angle | -ang | Maximum angle, in degrees, tolerated while fusing adjacent CFSs. | 30 | CFS fusion |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Sazzed, S.; Scheible, P.; He, J.; Wriggers, W. Untangling Irregular Actin Cytoskeleton Architectures in Tomograms of the Cell with Struwwel Tracer. Int. J. Mol. Sci. 2023, 24, 17183. https://doi.org/10.3390/ijms242417183
Sazzed S, Scheible P, He J, Wriggers W. Untangling Irregular Actin Cytoskeleton Architectures in Tomograms of the Cell with Struwwel Tracer. International Journal of Molecular Sciences. 2023; 24(24):17183. https://doi.org/10.3390/ijms242417183
Chicago/Turabian StyleSazzed, Salim, Peter Scheible, Jing He, and Willy Wriggers. 2023. "Untangling Irregular Actin Cytoskeleton Architectures in Tomograms of the Cell with Struwwel Tracer" International Journal of Molecular Sciences 24, no. 24: 17183. https://doi.org/10.3390/ijms242417183
APA StyleSazzed, S., Scheible, P., He, J., & Wriggers, W. (2023). Untangling Irregular Actin Cytoskeleton Architectures in Tomograms of the Cell with Struwwel Tracer. International Journal of Molecular Sciences, 24(24), 17183. https://doi.org/10.3390/ijms242417183