

Oxidative Stress Linking Obesity and Cancer: Is Obesity a ‘Radical Trigger’ to Cancer?



{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
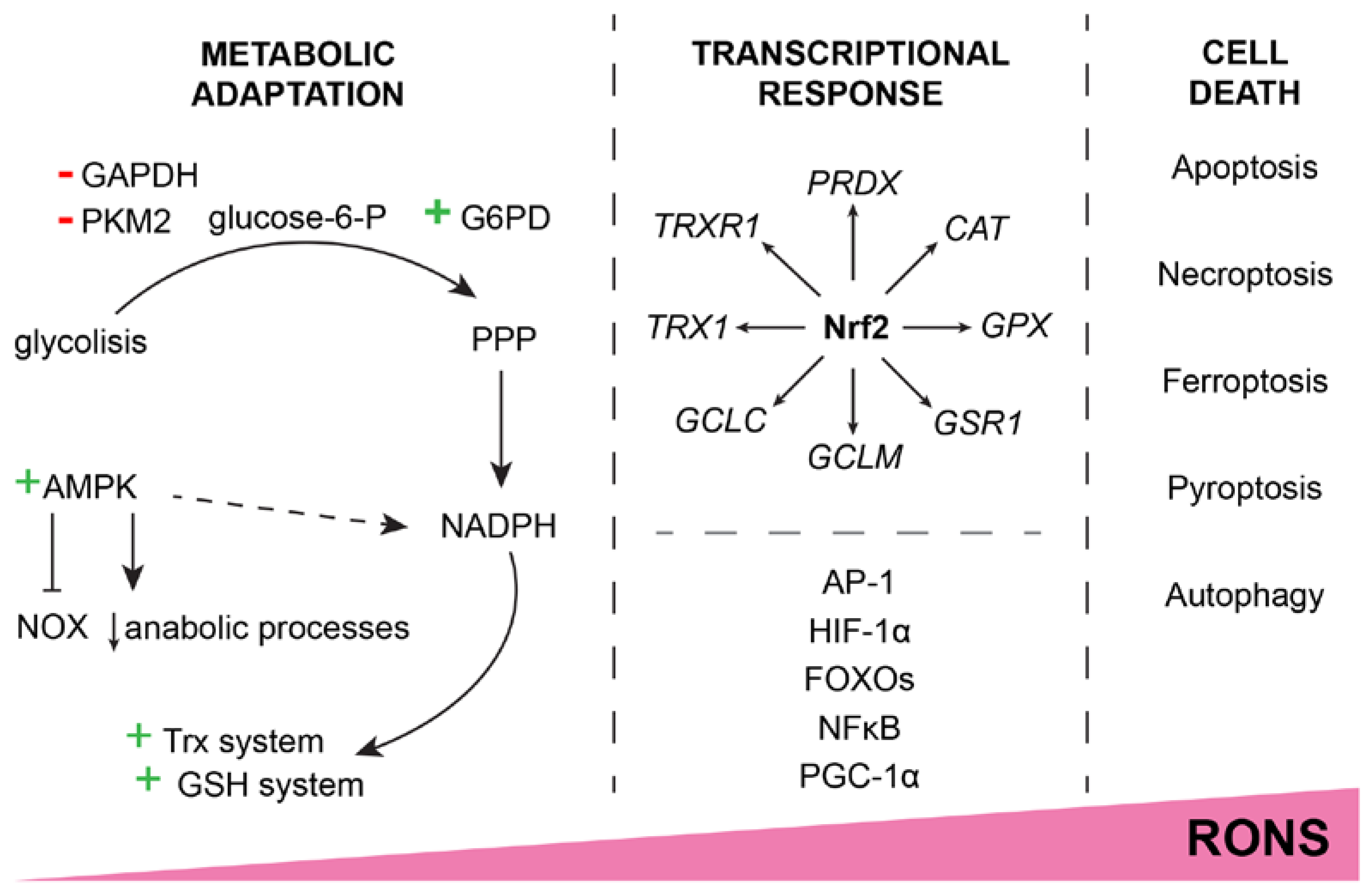
2. The “Goldilocks Paradox” of Reactive Species
3. Metabolic Pathology in Obesity Brings Systemic Havoc
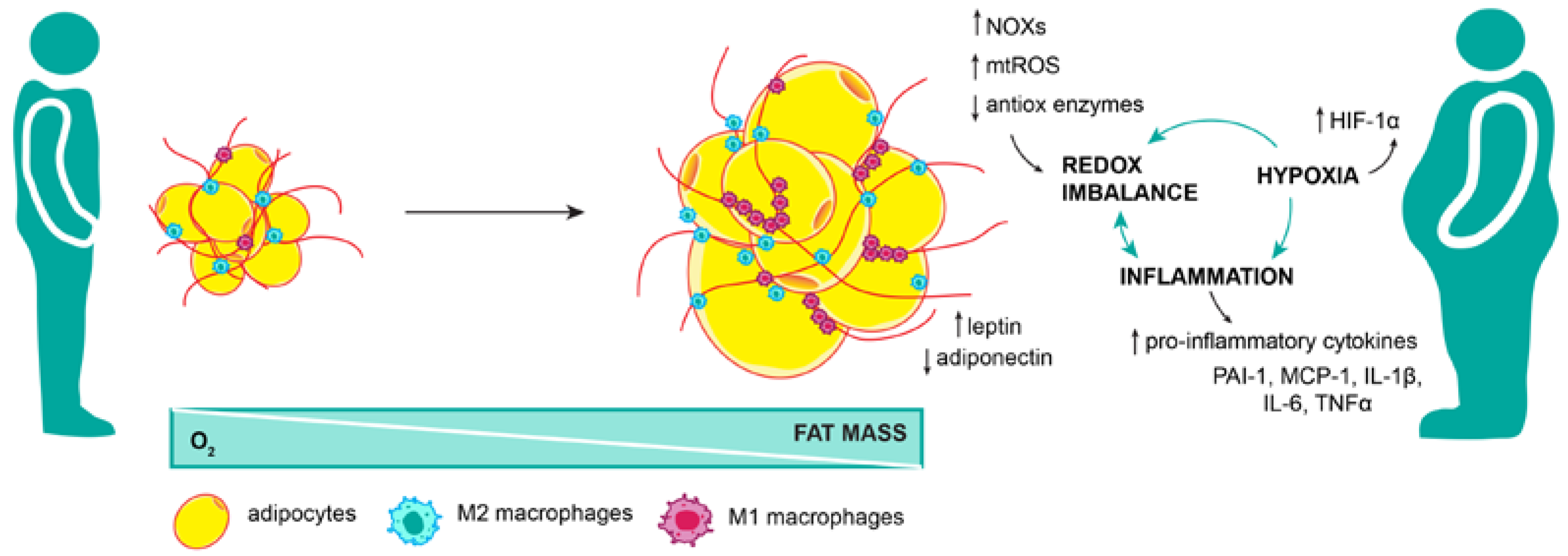
3.1. Metabolic Environment in Obesity
3.2. Obesity-Induced Oxidative Stress Causes DNA Instability
4. So It Begins: Obesity-Related Cell Transformation and Tumor Development
4.1. Examples of How Obesity-Driven Oxidative Stress Can Be a “Trump Card” to Cancer Development
4.2. The Clandestine Connection between Obesity and Cancer
5. Discussion
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Pigeyre, M.; Yazdi, F.T.; Kaur, Y.; Meyre, D. Recent progress in genetics, epigenetics and metagenomics unveils the pathophysiology of human obesity. Clin. Sci. 2016, 130, 943–986. [Google Scholar] [CrossRef] [PubMed]
- WHO. Obesity and Overweight. Available online: https://www.who.int/en/news-room/fact-sheets/detail/obesity-and-overweight (accessed on 30 March 2023).
- Pineda, E.; Sanchez-Romero, L.M.; Brown, M.; Jaccard, A.; Jewell, J.; Galea, G.; Webber, L.; Breda, J. Forecasting Future Trends in Obesity across Europe: The Value of Improving Surveillance. Obes. Facts 2018, 11, 360–371. [Google Scholar] [CrossRef] [PubMed]
- Ward, Z.J.; Bleich, S.N.; Cradock, A.L.; Barrett, J.L.; Giles, C.M.; Flax, C.; Long, M.W.; Gortmaker, S.L. Projected U.S. State-Level Prevalence of Adult Obesity and Severe Obesity. N. Engl. J. Med. 2019, 381, 2440–2450. [Google Scholar] [CrossRef]
- Kaggwa, M.M.; Favina, A.; Najjuka, S.M.; Zeba, Z.; Mamun, M.A.; Bongomin, F. Excessive eating and weight gain: A rare post-acute COVID-19 syndrome. Diabetes Metab. Syndr. 2021, 15, 102252. [Google Scholar] [CrossRef]
- Restrepo, B.J. Obesity Prevalence Among U.S. Adults during the COVID-19 Pandemic. Am. J. Prev. Med. 2022, 63, 102–106. [Google Scholar] [CrossRef] [PubMed]
- Nyberg, S.T.; Batty, G.D.; Pentti, J.; Virtanen, M.; Alfredsson, L.; Fransson, E.I.; Goldberg, M.; Heikkila, K.; Jokela, M.; Knutsson, A.; et al. Obesity and loss of disease-free years owing to major non-communicable diseases: A multicohort study. Lancet Public Health 2018, 3, e490–e497. [Google Scholar] [CrossRef]
- Zatonska, K.; Psikus, P.; Basiak-Rasala, A.; Stepnicka, Z.; Gawel-Dabrowska, D.; Wolyniec, M.; Gibka, J.; Szuba, A.; Poltyn-Zaradna, K. Obesity and Chosen Non-Communicable Diseases in PURE Poland Cohort Study. Int. J. Environ. Res. Public Health 2021, 18, 2701. [Google Scholar] [CrossRef]
- Tabassum, D.P.; Polyak, K. Tumorigenesis: It takes a village. Nat. Rev. Cancer 2015, 15, 473–483. [Google Scholar] [CrossRef]
- Cheung, E.C.; Vousden, K.H. The role of ROS in tumour development and progression. Nat. Rev. Cancer 2022, 22, 280–297. [Google Scholar] [CrossRef] [PubMed]
- Flavahan, W.A. Epigenetic plasticity, selection, and tumorigenesis. Biochem. Soc. Trans. 2020, 48, 1609–1621. [Google Scholar] [CrossRef] [PubMed]
- GBD 2019 Cancer Risk Factors Collaborators. The global burden of cancer attributable to risk factors, 2010–2019: A systematic analysis for the Global Burden of Disease Study 2019. Lancet 2022, 400, 563–591. [Google Scholar] [CrossRef]
- Lauby-Secretan, B.; Scoccianti, C.; Loomis, D.; Grosse, Y.; Bianchini, F.; Straif, K.; International Agency for Research on Cancer Handbook Working Group. Body Fatness and Cancer—Viewpoint of the IARC Working Group. N. Engl. J. Med. 2016, 375, 794–798. [Google Scholar] [CrossRef] [PubMed]
- Bray, F.; Ferlay, J.; Soerjomataram, I.; Siegel, R.L.; Torre, L.A.; Jemal, A. Global cancer statistics 2018: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA Cancer J. Clin. 2018, 68, 394–424. [Google Scholar] [CrossRef] [PubMed]
- Smittenaar, C.R.; Petersen, K.A.; Stewart, K.; Moitt, N. Cancer incidence and mortality projections in the UK until 2035. Br. J. Cancer 2016, 115, 1147–1155. [Google Scholar] [CrossRef] [PubMed]
- Harris, B.H.L.; Macaulay, V.M.; Harris, D.A.; Klenerman, P.; Karpe, F.; Lord, S.R.; Harris, A.L.; Buffa, F.M. Obesity: A perfect storm for carcinogenesis. Cancer Metastasis Rev. 2022, 41, 491–515. [Google Scholar] [CrossRef]
- Jovanovic, M.; Podolski-Renic, A.; Krasavin, M.; Pesic, M. The Role of the Thioredoxin Detoxification System in Cancer Progression and Resistance. Front. Mol. Biosci. 2022, 9, 883297. [Google Scholar] [CrossRef]
- Antunes, F.; Brito, P.M. Quantitative biology of hydrogen peroxide signaling. Redox Biol. 2017, 13, 1–7. [Google Scholar] [CrossRef]
- Murad, F. Nitric oxide signaling: Would you believe that a simple free radical could be a second messenger, autacoid, paracrine substance, neurotransmitter, and hormone? Recent Prog. Horm. Res. 1998, 53, 43–59; discussion 59–60. [Google Scholar]
- Holmstrom, K.M.; Finkel, T. Cellular mechanisms and physiological consequences of redox-dependent signalling. Nat. Rev. Mol. Cell Biol. 2014, 15, 411–421. [Google Scholar] [CrossRef]
- Fernando, V.; Zheng, X.; Walia, Y.; Sharma, V.; Letson, J.; Furuta, S. S-Nitrosylation: An Emerging Paradigm of Redox Signaling. Antioxidants 2019, 8, 404. [Google Scholar] [CrossRef]
- Kuehne, A.; Emmert, H.; Soehle, J.; Winnefeld, M.; Fischer, F.; Wenck, H.; Gallinat, S.; Terstegen, L.; Lucius, R.; Hildebrand, J.; et al. Acute Activation of Oxidative Pentose Phosphate Pathway as First-Line Response to Oxidative Stress in Human Skin Cells. Mol. Cell 2015, 59, 359–371. [Google Scholar] [CrossRef] [PubMed]
- Ge, T.; Yang, J.; Zhou, S.; Wang, Y.; Li, Y.; Tong, X. The Role of the Pentose Phosphate Pathway in Diabetes and Cancer. Front. Endocrinol. 2020, 11, 365. [Google Scholar] [CrossRef] [PubMed]
- Geldon, S.; Fernandez-Vizarra, E.; Tokatlidis, K. Redox-Mediated Regulation of Mitochondrial Biogenesis, Dynamics, and Respiratory Chain Assembly in Yeast and Human Cells. Front. Cell Dev. Biol. 2021, 9, 720656. [Google Scholar] [CrossRef]
- van der Reest, J.; Lilla, S.; Zheng, L.; Zanivan, S.; Gottlieb, E. Proteome-wide analysis of cysteine oxidation reveals metabolic sensitivity to redox stress. Nat. Commun. 2018, 9, 1581. [Google Scholar] [CrossRef]
- Jeon, S.M.; Chandel, N.S.; Hay, N. AMPK regulates NADPH homeostasis to promote tumour cell survival during energy stress. Nature 2012, 485, 661–665. [Google Scholar] [CrossRef]
- Pouremamali, F.; Pouremamali, A.; Dadashpour, M.; Soozangar, N.; Jeddi, F. An update of Nrf2 activators and inhibitors in cancer prevention/promotion. Cell Commun. Signal. 2022, 20, 100. [Google Scholar] [CrossRef] [PubMed]
- Rather, G.M.; Pramono, A.A.; Szekely, Z.; Bertino, J.R.; Tedeschi, P.M. In cancer, all roads lead to NADPH. Pharmacol. Ther. 2021, 226, 107864. [Google Scholar] [CrossRef]
- Ghosh, N.; Das, A.; Chaffee, S.; Roy, S.; Sen, C.K. Reactive Oxygen Species, Oxidative Damage and Cell Death. In Immunity and Inflammation in Health and Disease: Emerging Roles of Nutraceuticals and Functional Foods in Immune Support; Academic Press: Cambridge, MA, USA, 2018; pp. 45–55. [Google Scholar]
- Seaman, D.R. Weight gain as a consequence of living a modern lifestyle: A discussion of barriers to effective weight control and how to overcome them. J. Chiropr. Humanit. 2013, 20, 27–35. [Google Scholar] [CrossRef]
- Kompella, P.; Vasquez, K.M. Obesity and cancer: A mechanistic overview of metabolic changes in obesity that impact genetic instability. Mol. Carcinog. 2019, 58, 1531–1550. [Google Scholar] [CrossRef]
- Gray, S.L.; Vidal-Puig, A.J. Adipose tissue expandability in the maintenance of metabolic homeostasis. Nutr. Rev. 2007, 65, S7–S12. [Google Scholar] [CrossRef]
- Hauck, A.K.; Bernlohr, D.A. Oxidative stress and lipotoxicity. J. Lipid Res. 2016, 57, 1976–1986. [Google Scholar] [CrossRef]
- Nono Nankam, P.A.; Nguelefack, T.B.; Goedecke, J.H.; Bluher, M. Contribution of Adipose Tissue Oxidative Stress to Obesity-Associated Diabetes Risk and Ethnic Differences: Focus on Women of African Ancestry. Antioxidants 2021, 10, 622. [Google Scholar] [CrossRef] [PubMed]
- Choromanska, B.; Mysliwiec, P.; Luba, M.; Wojskowicz, P.; Mysliwiec, H.; Choromanska, K.; Dadan, J.; Zendzian-Piotrowska, M.; Zalewska, A.; Maciejczyk, M. Bariatric Surgery Normalizes Protein Glycoxidation and Nitrosative Stress in Morbidly Obese Patients. Antioxidants 2020, 9, 1087. [Google Scholar] [CrossRef]
- Krisanits, B.A.; Woods, P.; Nogueira, L.M.; Woolfork, D.D.; Lloyd, C.E.; Baldwin, A.; Frye, C.C.; Peterson, K.D.; Cosh, S.D.; Guo, Q.J.; et al. Non-enzymatic glycoxidation linked with nutrition enhances the tumorigenic capacity of prostate cancer epithelia through AGE mediated activation of RAGE in cancer associated fibroblasts. Transl. Oncol. 2022, 17, 101350. [Google Scholar] [CrossRef]
- Quoc Lam, B.; Shrivastava, S.K.; Shrivastava, A.; Shankar, S.; Srivastava, R.K. The Impact of obesity and diabetes mellitus on pancreatic cancer: Molecular mechanisms and clinical perspectives. J. Cell. Mol. Med. 2020, 24, 7706–7716. [Google Scholar] [CrossRef] [PubMed]
- Ahmad, S.; Khan, M.Y.; Rafi, Z.; Khan, H.; Siddiqui, Z.; Rehman, S.; Shahab, U.; Khan, M.S.; Saeed, M.; Alouffi, S.; et al. Oxidation, glycation and glycoxidation-The vicious cycle and lung cancer. Semin. Cancer Biol. 2018, 49, 29–36. [Google Scholar] [CrossRef]
- Arsova-Sarafinovska, Z.; Eken, A.; Matevska, N.; Erdem, O.; Sayal, A.; Savaser, A.; Banev, S.; Petrovski, D.; Dzikova, S.; Georgiev, V.; et al. Increased oxidative/nitrosative stress and decreased antioxidant enzyme activities in prostate cancer. Clin. Biochem. 2009, 42, 1228–1235. [Google Scholar] [CrossRef]
- Pietrocola, F.; Bravo-San Pedro, J.M. Targeting Autophagy to Counteract Obesity-Associated Oxidative Stress. Antioxidants 2021, 10, 102. [Google Scholar] [CrossRef] [PubMed]
- Usman, M.; Volpi, E.V. DNA damage in obesity: Initiator, promoter and predictor of cancer. Mutat. Res. Rev. Mutat. Res. 2018, 778, 23–37. [Google Scholar] [CrossRef]
- Furukawa, S.; Fujita, T.; Shimabukuro, M.; Iwaki, M.; Yamada, Y.; Nakajima, Y.; Nakayama, O.; Makishima, M.; Matsuda, M.; Shimomura, I. Increased oxidative stress in obesity and its impact on metabolic syndrome. J. Clin. Investig. 2004, 114, 1752–1761. [Google Scholar] [CrossRef] [PubMed]
- Kovacevic, S.; Brkljacic, J.; Vojnovic Milutinovic, D.; Gligorovska, L.; Bursac, B.; Elakovic, I.; Djordjevic, A. Fructose Induces Visceral Adipose Tissue Inflammation and Insulin Resistance Even Without Development of Obesity in Adult Female but Not in Male Rats. Front. Nutr. 2021, 8, 749328. [Google Scholar] [CrossRef]
- Bedard, K.; Krause, K.H. The NOX family of ROS-generating NADPH oxidases: Physiology and pathophysiology. Physiol. Rev. 2007, 87, 245–313. [Google Scholar] [CrossRef]
- Matsuzawa-Nagata, N.; Takamura, T.; Ando, H.; Nakamura, S.; Kurita, S.; Misu, H.; Ota, T.; Yokoyama, M.; Honda, M.; Miyamoto, K.; et al. Increased oxidative stress precedes the onset of high-fat diet-induced insulin resistance and obesity. Metab. Clin. Exp. 2008, 57, 1071–1077. [Google Scholar] [CrossRef] [PubMed]
- Yu, T.; Li, L.; Liu, W.; Ya, B.; Cheng, H.; Xin, Q. Silencing of NADPH Oxidase 4 Attenuates Hypoxia Resistance in Neuroblastoma Cells SH-SY5Y by Inhibiting PI3K/Akt-Dependent Glycolysis. Oncol. Res. 2019, 27, 525–532. [Google Scholar] [CrossRef] [PubMed]
- Gao, X.; Sun, J.; Huang, C.; Hu, X.; Jiang, N.; Lu, C. RNAi-mediated silencing of NOX4 inhibited the invasion of gastric cancer cells through JAK2/STAT3 signaling. Am. J. Transl. Res. 2017, 9, 4440–4449. [Google Scholar] [PubMed]
- Konate, M.M.; Antony, S.; Doroshow, J.H. Inhibiting the Activity of NADPH Oxidase in Cancer. Antioxid. Redox Signal. 2020, 33, 435–454. [Google Scholar] [CrossRef]
- Calle, E.E.; Kaaks, R. Overweight, obesity and cancer: Epidemiological evidence and proposed mechanisms. Nat. Rev. Cancer 2004, 4, 579–591. [Google Scholar] [CrossRef]
- Ogunwobi, O.; Mutungi, G.; Beales, I.L. Leptin stimulates proliferation and inhibits apoptosis in Barrett’s esophageal adenocarcinoma cells by cyclooxygenase-2-dependent, prostaglandin-E2-mediated transactivation of the epidermal growth factor receptor and c-Jun NH2-terminal kinase activation. Endocrinology 2006, 147, 4505–4516. [Google Scholar] [CrossRef] [PubMed]
- Renehan, A.G.; Frystyk, J.; Flyvbjerg, A. Obesity and cancer risk: The role of the insulin-IGF axis. Trends Endocrinol. Metab. 2006, 17, 328–336. [Google Scholar] [CrossRef]
- Barb, D.; Williams, C.J.; Neuwirth, A.K.; Mantzoros, C.S. Adiponectin in relation to malignancies: A review of existing basic research and clinical evidence. Am. J. Clin. Nutr. 2007, 86, s858–s866. [Google Scholar] [CrossRef]
- Ohshima, H.; Tatemichi, M.; Sawa, T. Chemical basis of inflammation-induced carcinogenesis. Arch. Biochem. Biophys. 2003, 417, 3–11. [Google Scholar] [CrossRef] [PubMed]
- Morris, R.M.; Mortimer, T.O.; O’Neill, K.L. Cytokines: Can Cancer Get the Message? Cancers 2022, 14, 2178. [Google Scholar] [CrossRef]
- Bernstein, C.N.; Blanchard, J.F.; Kliewer, E.; Wajda, A. Cancer risk in patients with inflammatory bowel disease: A population-based study. Cancer 2001, 91, 854–862. [Google Scholar] [CrossRef] [PubMed]
- Shawki, S.M.; Meshaal, S.S.; El Dash, A.S.; Zayed, N.A.; Hanna, M.O. Increased DNA damage in hepatitis C virus-related hepatocellular carcinoma. DNA Cell Biol. 2014, 33, 884–890. [Google Scholar] [CrossRef]
- Tezal, M.; Grossi, S.G.; Genco, R.J. Is periodontitis associated with oral neoplasms? J. Periodontol. 2005, 76, 406–410. [Google Scholar] [CrossRef]
- Bloch-Damti, A.; Bashan, N. Proposed mechanisms for the induction of insulin resistance by oxidative stress. Antioxid. Redox Signal. 2005, 7, 1553–1567. [Google Scholar] [CrossRef] [PubMed]
- Kovacevic, S.; Nestorov, J.; Matic, G.; Elakovic, I. Fructose-enriched diet induces inflammation and reduces antioxidative defense in visceral adipose tissue of young female rats. Eur. J. Nutr. 2017, 56, 151–160. [Google Scholar] [CrossRef] [PubMed]
- Kojta, I.; Chacinska, M.; Blachnio-Zabielska, A. Obesity, Bioactive Lipids, and Adipose Tissue Inflammation in Insulin Resistance. Nutrients 2020, 12, 1305. [Google Scholar] [CrossRef]
- Hirosumi, J.; Tuncman, G.; Chang, L.; Gorgun, C.Z.; Uysal, K.T.; Maeda, K.; Karin, M.; Hotamisligil, G.S. A central role for JNK in obesity and insulin resistance. Nature 2002, 420, 333–336. [Google Scholar] [CrossRef] [PubMed]
- Li, B.D.; Khosravi, M.J.; Berkel, H.J.; Diamandi, A.; Dayton, M.A.; Smith, M.; Yu, H. Free insulin-like growth factor-I and breast cancer risk. Int. J. Cancer 2001, 91, 736–739. [Google Scholar]
- Fonteneau, G.; Redding, A.; Hoag-Lee, H.; Sim, E.S.; Heinrich, S.; Gaida, M.M.; Grabocka, E. Stress Granules Determine the Development of Obesity-Associated Pancreatic Cancer. Cancer Discov. 2022, 12, 1984–2005. [Google Scholar] [CrossRef] [PubMed]
- Renehan, A.G.; O’Connell, J.; O’Halloran, D.; Shanahan, F.; Potten, C.S.; O’Dwyer, S.T.; Shalet, S.M. Acromegaly and colorectal cancer: A comprehensive review of epidemiology, biological mechanisms, and clinical implications. Horm. Metab. Res. 2003, 35, 712–725. [Google Scholar] [CrossRef]
- Caldon, C.E.; Sutherland, R.L.; Musgrove, E. Cell cycle proteins in epithelial cell differentiation: Implications for breast cancer. Cell Cycle 2010, 9, 1918–1928. [Google Scholar] [CrossRef]
- Bernstein, L. The risk of breast, endometrial and ovarian cancer in users of hormonal preparations. Basic Clin. Pharmacol. Toxicol. 2006, 98, 288–296. [Google Scholar] [CrossRef]
- Choromanska, B.; Mysliwiec, P.; Luba, M.; Wojskowicz, P.; Mysliwiec, H.; Choromanska, K.; Dadan, J.; Zalewska, A.; Maciejczyk, M. The Impact of Hypertension and Metabolic Syndrome on Nitrosative Stress and Glutathione Metabolism in Patients with Morbid Obesity. Oxidative Med. Cell. Longev. 2020, 2020, 1057570. [Google Scholar] [CrossRef] [PubMed]
- Jakubiak, G.K.; Osadnik, K.; Lejawa, M.; Osadnik, T.; Golawski, M.; Lewandowski, P.; Pawlas, N. “Obesity and Insulin Resistance” Is the Component of the Metabolic Syndrome Most Strongly Associated with Oxidative Stress. Antioxidants 2021, 11, 79. [Google Scholar] [CrossRef]
- Jang, Y.; Kim, O.Y.; Ryu, H.J.; Kim, J.Y.; Song, S.H.; Ordovas, J.M.; Lee, J.H. Visceral fat accumulation determines postprandial lipemic response, lipid peroxidation, DNA damage, and endothelial dysfunction in nonobese Korean men. J. Lipid Res. 2003, 44, 2356–2364. [Google Scholar] [CrossRef] [PubMed]
- Ali, I.; Hogberg, J.; Hsieh, J.H.; Auerbach, S.; Korhonen, A.; Stenius, U.; Silins, I. Gender differences in cancer susceptibility: Role of oxidative stress. Carcinogenesis 2016, 37, 985–992. [Google Scholar] [CrossRef]
- Kim, S.Y. Oxidative stress and gender disparity in cancer. Free Radic. Res. 2022, 56, 90–105. [Google Scholar] [CrossRef]
- Tiberi, J.; Cesarini, V.; Stefanelli, R.; Canterini, S.; Fiorenza, M.T.; La Rosa, P. Sex differences in antioxidant defence and the regulation of redox homeostasis in physiology and pathology. Mech. Ageing Dev. 2023, 211, 111802. [Google Scholar] [CrossRef]
- Maga, G.; Crespan, E.; Wimmer, U.; van Loon, B.; Amoroso, A.; Mondello, C.; Belgiovine, C.; Ferrari, E.; Locatelli, G.; Villani, G.; et al. Replication protein A and proliferating cell nuclear antigen coordinate DNA polymerase selection in 8-oxo-guanine repair. Proc. Natl. Acad. Sci. USA 2008, 105, 20689–20694. [Google Scholar] [CrossRef]
- McMurray, F.; Patten, D.A.; Harper, M.E. Reactive Oxygen Species and Oxidative Stress in Obesity-Recent Findings and Empirical Approaches. Obesity 2016, 24, 2301–2310. [Google Scholar] [CrossRef]
- Avkin, S.; Livneh, Z. Efficiency, specificity and DNA polymerase-dependence of translesion replication across the oxidative DNA lesion 8-oxoguanine in human cells. Mutat. Res. 2002, 510, 81–90. [Google Scholar] [CrossRef]
- Lea, I.A.; Jackson, M.A.; Li, X.; Bailey, S.; Peddada, S.D.; Dunnick, J.K. Genetic pathways and mutation profiles of human cancers: Site- and exposure-specific patterns. Carcinogenesis 2007, 28, 1851–1858. [Google Scholar] [CrossRef] [PubMed]
- Nishigori, C.; Hattori, Y.; Toyokuni, S. Role of reactive oxygen species in skin carcinogenesis. Antioxid. Redox Signal. 2004, 6, 561–570. [Google Scholar] [CrossRef]
- Brancato, B.; Munnia, A.; Cellai, F.; Ceni, E.; Mello, T.; Bianchi, S.; Catarzi, S.; Risso, G.G.; Galli, A.; Peluso, M.E. 8-Oxo-7,8-dihydro-2′-deoxyguanosine and other lesions along the coding strand of the exon 5 of the tumour suppressor gene P53 in a breast cancer case-control study. DNA Res. Int. J. Rapid Publ. Rep. Genes Genomes 2016, 23, 395–402. [Google Scholar] [CrossRef] [PubMed]
- Valinluck, V.; Tsai, H.H.; Rogstad, D.K.; Burdzy, A.; Bird, A.; Sowers, L.C. Oxidative damage to methyl-CpG sequences inhibits the binding of the methyl-CpG binding domain (MBD) of methyl-CpG binding protein 2 (MeCP2). Nucleic Acids Res. 2004, 32, 4100–4108. [Google Scholar] [CrossRef]
- Sebekova, K.; Somoza, V.; Jarcuskova, M.; Heidland, A.; Podracka, L. Plasma advanced glycation end products are decreased in obese children compared with lean controls. Int. J. Pediatr. Obes. 2009, 4, 112–118. [Google Scholar] [CrossRef]
- Karbownik-Lewinska, M.; Szosland, J.; Kokoszko-Bilska, A.; Stepniak, J.; Zasada, K.; Gesing, A.; Lewinski, A. Direct contribution of obesity to oxidative damage to macromolecules. Neuroendocrinol. Lett. 2012, 33, 453–461. [Google Scholar]
- Jors, A.; Lund, M.A.V.; Jespersen, T.; Hansen, T.; Poulsen, H.E.; Holm, J.C. Urinary markers of nucleic acid oxidation increase with age, obesity and insulin resistance in Danish children and adolescents. Free Radic. Biol. Med. 2020, 155, 81–86. [Google Scholar] [CrossRef] [PubMed]
- Valko, M.; Rhodes, C.J.; Moncol, J.; Izakovic, M.; Mazur, M. Free radicals, metals and antioxidants in oxidative stress-induced cancer. Chem. Biol. Interact. 2006, 160, 1–40. [Google Scholar] [CrossRef]
- Burcham, P.C. Genotoxic lipid peroxidation products: Their DNA damaging properties and role in formation of endogenous DNA adducts. Mutagenesis 1998, 13, 287–305. [Google Scholar] [CrossRef]
- Kalezic, A.; Udicki, M.; Srdic Galic, B.; Aleksic, M.; Korac, A.; Jankovic, A.; Korac, B. Redox profile of breast tumor and associated adipose tissue in premenopausal women—Interplay between obesity and malignancy. Redox Biol. 2021, 41, 101939. [Google Scholar] [CrossRef]
- VanderVeen, L.A.; Hashim, M.F.; Shyr, Y.; Marnett, L.J. Induction of frameshift and base pair substitution mutations by the major DNA adduct of the endogenous carcinogen malondialdehyde. Proc. Natl. Acad. Sci. USA 2003, 100, 14247–14252. [Google Scholar] [CrossRef]
- Niedernhofer, L.J.; Daniels, J.S.; Rouzer, C.A.; Greene, R.E.; Marnett, L.J. Malondialdehyde, a product of lipid peroxidation, is mutagenic in human cells. J. Biol. Chem. 2003, 278, 31426–31433. [Google Scholar] [CrossRef]
- Hu, W.; Feng, Z.; Eveleigh, J.; Iyer, G.; Pan, J.; Amin, S.; Chung, F.L.; Tang, M.S. The major lipid peroxidation product, trans-4-hydroxy-2-nonenal, preferentially forms DNA adducts at codon 249 of human p53 gene, a unique mutational hotspot in hepatocellular carcinoma. Carcinogenesis 2002, 23, 1781–1789. [Google Scholar] [CrossRef]
- Stone, M.P.; Cho, Y.J.; Huang, H.; Kim, H.Y.; Kozekov, I.D.; Kozekova, A.; Wang, H.; Minko, I.G.; Lloyd, R.S.; Harris, T.M.; et al. Interstrand DNA cross-links induced by α,β-unsaturated aldehydes derived from lipid peroxidation and environmental sources. Acc. Chem. Res. 2008, 41, 793–804. [Google Scholar] [CrossRef]
- Lee, H.W.; Wang, H.T.; Weng, M.W.; Hu, Y.; Chen, W.S.; Chou, D.; Liu, Y.; Donin, N.; Huang, W.C.; Lepor, H.; et al. Acrolein- and 4-Aminobiphenyl-DNA adducts in human bladder mucosa and tumor tissue and their mutagenicity in human urothelial cells. Oncotarget 2014, 5, 3526–3540. [Google Scholar] [CrossRef]
- Fang, L.; Chen, D.; Yu, C.; Li, H.; Brocato, J.; Huang, L.; Jin, C. Mechanisms Underlying Acrolein-Mediated Inhibition of Chromatin Assembly. Mol. Cell. Biol. 2016, 36, 2995–3008. [Google Scholar] [CrossRef]
- Nakamura, H.; Takada, K. Reactive oxygen species in cancer: Current findings and future directions. Cancer Sci. 2021, 112, 3945–3952. [Google Scholar] [CrossRef]
- Tennant, D.A.; Duran, R.V.; Boulahbel, H.; Gottlieb, E. Metabolic transformation in cancer. Carcinogenesis 2009, 30, 1269–1280. [Google Scholar] [CrossRef]
- Szatrowski, T.P.; Nathan, C.F. Production of large amounts of hydrogen peroxide by human tumor cells. Cancer Res. 1991, 51, 794–798. [Google Scholar]
- Tomlinson, I.P.; Novelli, M.R.; Bodmer, W.F. The mutation rate and cancer. Proc. Natl. Acad. Sci. USA 1996, 93, 14800–14803. [Google Scholar] [CrossRef] [PubMed]
- Sporn, M.B.; Liby, K.T. NRF2 and cancer: The good, the bad and the importance of context. Nat. Rev. Cancer 2012, 12, 564–571. [Google Scholar] [CrossRef]
- DeNicola, G.M.; Karreth, F.A.; Humpton, T.J.; Gopinathan, A.; Wei, C.; Frese, K.; Mangal, D.; Yu, K.H.; Yeo, C.J.; Calhoun, E.S.; et al. Oncogene-induced Nrf2 transcription promotes ROS detoxification and tumorigenesis. Nature 2011, 475, 106–109. [Google Scholar] [CrossRef] [PubMed]
- Corcoran, A.; Cotter, T.G. Redox regulation of protein kinases. FEBS J. 2013, 280, 1944–1965. [Google Scholar] [CrossRef] [PubMed]
- Hayes, J.D.; Dinkova-Kostova, A.T.; Tew, K.D. Oxidative Stress in Cancer. Cancer Cell 2020, 38, 167–197. [Google Scholar] [CrossRef]
- Fox, D.B.; Garcia, N.M.G.; McKinney, B.J.; Lupo, R.; Noteware, L.C.; Newcomb, R.; Liu, J.; Locasale, J.W.; Hirschey, M.D.; Alvarez, J.V. NRF2 activation promotes the recurrence of dormant tumour cells through regulation of redox and nucleotide metabolism. Nat. Metab. 2020, 2, 318–334. [Google Scholar] [CrossRef]
- Gopalakrishna, R.; Jaken, S. Protein kinase C signaling and oxidative stress. Free Radic. Biol. Med. 2000, 28, 1349–1361. [Google Scholar] [CrossRef]
- Johnson, D.E.; O’Keefe, R.A.; Grandis, J.R. Targeting the IL-6/JAK/STAT3 signalling axis in cancer. Nat. Rev. Clin. Oncol. 2018, 15, 234–248. [Google Scholar] [CrossRef]
- Kobayashi, E.H.; Suzuki, T.; Funayama, R.; Nagashima, T.; Hayashi, M.; Sekine, H.; Tanaka, N.; Moriguchi, T.; Motohashi, H.; Nakayama, K.; et al. Nrf2 suppresses macrophage inflammatory response by blocking proinflammatory cytokine transcription. Nat. Commun. 2016, 7, 11624. [Google Scholar] [CrossRef]
- Smith, U. Abdominal obesity: A marker of ectopic fat accumulation. J. Clin. Investig. 2015, 125, 1790–1792. [Google Scholar] [CrossRef]
- Kozawa, J.; Shimomura, I. Ectopic Fat Accumulation in Pancreas and Heart. J. Clin. Med. 2021, 10, 1326. [Google Scholar] [CrossRef]
- Neeland, I.J.; Ross, R.; Despres, J.P.; Matsuzawa, Y.; Yamashita, S.; Shai, I.; Seidell, J.; Magni, P.; Santos, R.D.; Arsenault, B.; et al. Visceral and ectopic fat, atherosclerosis, and cardiometabolic disease: A position statement. Lancet Diabetes Endocrinol. 2019, 7, 715–725. [Google Scholar] [CrossRef]
- Snel, M.; Jonker, J.T.; Schoones, J.; Lamb, H.; de Roos, A.; Pijl, H.; Smit, J.W.; Meinders, A.E.; Jazet, I.M. Ectopic fat and insulin resistance: Pathophysiology and effect of diet and lifestyle interventions. Int. J. Endocrinol. 2012, 2012, 983814. [Google Scholar] [CrossRef]
- Britton, K.A.; Massaro, J.M.; Murabito, J.M.; Kreger, B.E.; Hoffmann, U.; Fox, C.S. Body fat distribution, incident cardiovascular disease, cancer, and all-cause mortality. J. Am. Coll. Cardiol. 2013, 62, 921–925. [Google Scholar] [CrossRef]
- Bhardwaj, P.; Au, C.C.; Benito-Martin, A.; Ladumor, H.; Oshchepkova, S.; Moges, R.; Brown, K.A. Estrogens and breast cancer: Mechanisms involved in obesity-related development, growth and progression. J. Steroid Biochem. Mol. Biol. 2019, 189, 161–170. [Google Scholar] [CrossRef]
- Kolb, R.; Zhang, W. Obesity and Breast Cancer: A Case of Inflamed Adipose Tissue. Cancers 2020, 12, 1686. [Google Scholar] [CrossRef]
- Engin, A.B.; Engin, A.; Gonul, I.I. The effect of adipocyte-macrophage crosstalk in obesity-related breast cancer. J. Mol. Endocrinol. 2019, 62, R201–R222. [Google Scholar] [CrossRef]
- Futreal, P.A.; Liu, Q.; Shattuck-Eidens, D.; Cochran, C.; Harshman, K.; Tavtigian, S.; Bennett, L.M.; Haugen-Strano, A.; Swensen, J.; Miki, Y.; et al. BRCA1 mutations in primary breast and ovarian carcinomas. Science 1994, 266, 120–122. [Google Scholar] [CrossRef]
- Yi, Y.W.; Kang, H.J.; Bae, I. BRCA1 and Oxidative Stress. Cancers 2014, 6, 771–795. [Google Scholar] [CrossRef] [PubMed]
- Gorrini, C.; Baniasadi, P.S.; Harris, I.S.; Silvester, J.; Inoue, S.; Snow, B.; Joshi, P.A.; Wakeham, A.; Molyneux, S.D.; Martin, B.; et al. BRCA1 interacts with Nrf2 to regulate antioxidant signaling and cell survival. J. Exp. Med. 2013, 210, 1529–1544. [Google Scholar] [CrossRef] [PubMed]
- Marengo, A.; Rosso, C.; Bugianesi, E. Liver Cancer: Connections with Obesity, Fatty Liver, and Cirrhosis. Annu. Rev. Med. 2016, 67, 103–117. [Google Scholar] [CrossRef] [PubMed]
- Kucukoglu, O.; Sowa, J.P.; Mazzolini, G.D.; Syn, W.K.; Canbay, A. Hepatokines and adipokines in NASH-related hepatocellular carcinoma. J. Hepatol. 2021, 74, 442–457. [Google Scholar] [CrossRef]
- Anstee, Q.M.; Reeves, H.L.; Kotsiliti, E.; Govaere, O.; Heikenwalder, M. From NASH to HCC: Current concepts and future challenges. Nat. Rev. Gastroenterol. Hepatol. 2019, 16, 411–428. [Google Scholar] [CrossRef]
- Gupta, A.; Das, A.; Majumder, K.; Arora, N.; Mayo, H.G.; Singh, P.P.; Beg, M.S.; Singh, S. Obesity is Independently Associated With Increased Risk of Hepatocellular Cancer-related Mortality: A Systematic Review and Meta-Analysis. Am. J. Clin. Oncol. 2018, 41, 874–881. [Google Scholar] [CrossRef]
- Frederiks, W.M.; Vizan, P.; Bosch, K.S.; Vreeling-Sindelarova, H.; Boren, J.; Cascante, M. Elevated activity of the oxidative and non-oxidative pentose phosphate pathway in (pre)neoplastic lesions in rat liver. Int. J. Exp. Pathol. 2008, 89, 232–240. [Google Scholar] [CrossRef]
- Broadfield, L.A.; Duarte, J.A.G.; Schmieder, R.; Broekaert, D.; Veys, K.; Planque, M.; Vriens, K.; Karasawa, Y.; Napolitano, F.; Fujita, S.; et al. Fat Induces Glucose Metabolism in Nontransformed Liver Cells and Promotes Liver Tumorigenesis. Cancer Res. 2021, 81, 1988–2001. [Google Scholar] [CrossRef]
- Brahma, M.K.; Gilglioni, E.H.; Zhou, L.; Trepo, E.; Chen, P.; Gurzov, E.N. Oxidative stress in obesity-associated hepatocellular carcinoma: Sources, signaling and therapeutic challenges. Oncogene 2021, 40, 5155–5167. [Google Scholar] [CrossRef]
- Wang, Z.; Li, Z.; Ye, Y.; Xie, L.; Li, W. Oxidative Stress and Liver Cancer: Etiology and Therapeutic Targets. Oxidative Med. Cell. Longev. 2016, 2016, 7891574. [Google Scholar] [CrossRef]
- Xu, M.; Jung, X.; Hines, O.J.; Eibl, G.; Chen, Y. Obesity and Pancreatic Cancer: Overview of Epidemiology and Potential Prevention by Weight Loss. Pancreas 2018, 47, 158–162. [Google Scholar] [CrossRef] [PubMed]
- Johnston, P.C.; Thompson, J.; McKee, A.; Hamill, C.; Wallace, I. Diabetes and Chronic Pancreatitis: Considerations in the Holistic Management of an Often Neglected Disease. J. Diabetes Res. 2019, 2019, 2487804. [Google Scholar] [CrossRef] [PubMed]
- Protter, D.S.W.; Parker, R. Principles and Properties of Stress Granules. Trends Cell Biol. 2016, 26, 668–679. [Google Scholar] [CrossRef]
- Biswas, S.K. Does the Interdependence between Oxidative Stress and Inflammation Explain the Antioxidant Paradox? Oxidative Med. Cell. Longev. 2016, 2016, 5698931. [Google Scholar] [CrossRef]
- Levin, B. Inflammatory bowel disease and colon cancer. Cancer 1992, 70, 1313–1316. [Google Scholar] [CrossRef]
- Terzic, J.; Grivennikov, S.; Karin, E.; Karin, M. Inflammation and colon cancer. Gastroenterology 2010, 138, 2101–2114.e5. [Google Scholar] [CrossRef] [PubMed]
- Li, X.; Wei, X.; Sun, Y.; Du, J.; Li, X.; Xun, Z.; Li, Y.C. High-fat diet promotes experimental colitis by inducing oxidative stress in the colon. Am. J. Physiol. Gastrointest. Liver Physiol. 2019, 317, G453–G462. [Google Scholar] [CrossRef]
- Agus, A.; Denizot, J.; Thevenot, J.; Martinez-Medina, M.; Massier, S.; Sauvanet, P.; Bernalier-Donadille, A.; Denis, S.; Hofman, P.; Bonnet, R.; et al. Western diet induces a shift in microbiota composition enhancing susceptibility to Adherent-Invasive E. coli infection and intestinal inflammation. Sci. Rep. 2016, 6, 19032. [Google Scholar] [CrossRef] [PubMed]
- Christ, A.; Lauterbach, M.; Latz, E. Western Diet and the Immune System: An Inflammatory Connection. Immunity 2019, 51, 794–811. [Google Scholar] [CrossRef]
- Okayasu, I.; Hatakeyama, S.; Yamada, M.; Ohkusa, T.; Inagaki, Y.; Nakaya, R. A novel method in the induction of reliable experimental acute and chronic ulcerative colitis in mice. Gastroenterology 1990, 98, 694–702. [Google Scholar] [CrossRef]
- Tanaka, T.; Kohno, H.; Suzuki, R.; Yamada, Y.; Sugie, S.; Mori, H. A novel inflammation-related mouse colon carcinogenesis model induced by azoxymethane and dextran sodium sulfate. Cancer Sci. 2003, 94, 965–973. [Google Scholar] [CrossRef]
- De Robertis, M.; Massi, E.; Poeta, M.L.; Carotti, S.; Morini, S.; Cecchetelli, L.; Signori, E.; Fazio, V.M. The AOM/DSS murine model for the study of colon carcinogenesis: From pathways to diagnosis and therapy studies. J. Carcinog. 2011, 10, 9. [Google Scholar] [CrossRef]
- Snider, A.J.; Bialkowska, A.B.; Ghaleb, A.M.; Yang, V.W.; Obeid, L.M.; Hannun, Y.A. Murine Model for Colitis-Associated Cancer of the Colon. Methods Mol. Biol. 2016, 1438, 245–254. [Google Scholar] [CrossRef] [PubMed]
- Malesza, I.J.; Malesza, M.; Walkowiak, J.; Mussin, N.; Walkowiak, D.; Aringazina, R.; Bartkowiak-Wieczorek, J.; Madry, E. High-Fat, Western-Style Diet, Systemic Inflammation, and Gut Microbiota: A Narrative Review. Cells 2021, 10, 3164. [Google Scholar] [CrossRef] [PubMed]
- Arthur, J.C.; Perez-Chanona, E.; Muhlbauer, M.; Tomkovich, S.; Uronis, J.M.; Fan, T.J.; Campbell, B.J.; Abujamel, T.; Dogan, B.; Rogers, A.B.; et al. Intestinal inflammation targets cancer-inducing activity of the microbiota. Science 2012, 338, 120–123. [Google Scholar] [CrossRef] [PubMed]
- Tilg, H.; Adolph, T.E.; Gerner, R.R.; Moschen, A.R. The Intestinal Microbiota in Colorectal Cancer. Cancer Cell 2018, 33, 954–964. [Google Scholar] [CrossRef]
- Cheng, Y.; Ling, Z.; Li, L. The Intestinal Microbiota and Colorectal Cancer. Front. Immunol. 2020, 11, 615056. [Google Scholar] [CrossRef]
- Yu, J.; Feng, Q.; Wong, S.H.; Zhang, D.; Liang, Q.Y.; Qin, Y.; Tang, L.; Zhao, H.; Stenvang, J.; Li, Y.; et al. Metagenomic analysis of faecal microbiome as a tool towards targeted non-invasive biomarkers for colorectal cancer. Gut 2017, 66, 70–78. [Google Scholar] [CrossRef]
- Kostic, A.D.; Gevers, D.; Pedamallu, C.S.; Michaud, M.; Duke, F.; Earl, A.M.; Ojesina, A.I.; Jung, J.; Bass, A.J.; Tabernero, J.; et al. Genomic analysis identifies association of Fusobacterium with colorectal carcinoma. Genome Res. 2012, 22, 292–298. [Google Scholar] [CrossRef]
- Weihrauch-Bluher, S.; Schwarz, P.; Klusmann, J.H. Childhood obesity: Increased risk for cardiometabolic disease and cancer in adulthood. Metab. Clin. Exp. 2019, 92, 147–152. [Google Scholar] [CrossRef]
- O’Rourke, K. Overweight children have an increased risk of cancer in adulthood. Cancer 2022, 128, 3143–3144. [Google Scholar] [CrossRef] [PubMed]
- Sachdeva, P.; Ghosh, S.; Ghosh, S.; Han, S.; Banerjee, J.; Bhaskar, R.; Sinha, J.K. Childhood Obesity: A Potential Key Factor in the Development of Glioblastoma Multiforme. Life 2022, 12, 1673. [Google Scholar] [CrossRef]
- Garcia-Estevez, L.; Cortes, J.; Perez, S.; Calvo, I.; Gallegos, I.; Moreno-Bueno, G. Obesity and Breast Cancer: A Paradoxical and Controversial Relationship Influenced by Menopausal Status. Front. Oncol. 2021, 11, 705911. [Google Scholar] [CrossRef]
- Stupp, R.; Mason, W.P.; van den Bent, M.J.; Weller, M.; Fisher, B.; Taphoorn, M.J.; Belanger, K.; Brandes, A.A.; Marosi, C.; Bogdahn, U.; et al. Radiotherapy plus concomitant and adjuvant temozolomide for glioblastoma. N. Engl. J. Med. 2005, 352, 987–996. [Google Scholar] [CrossRef]
- Poon, M.T.C.; Sudlow, C.L.M.; Figueroa, J.D.; Brennan, P.M. Longer-term (>/= 2 years) survival in patients with glioblastoma in population-based studies pre- and post-2005: A systematic review and meta-analysis. Sci. Rep. 2020, 10, 11622. [Google Scholar] [CrossRef] [PubMed]
- Fisher, J.P.; Adamson, D.C. Current FDA-Approved Therapies for High-Grade Malignant Gliomas. Biomedicines 2021, 9, 324. [Google Scholar] [CrossRef]
- Howell, A.E.; Robinson, J.W.; Wootton, R.E.; McAleenan, A.; Tsavachidis, S.; Ostrom, Q.T.; Bondy, M.; Armstrong, G.; Relton, C.; Haycock, P.; et al. Testing for causality between systematically identified risk factors and glioma: A Mendelian randomization study. BMC Cancer 2020, 20, 508. [Google Scholar] [CrossRef]
- Kovacevic, S.; Nestorov, J.; Matic, G.; Elakovic, I. Chronic Stress Combined with a Fructose Diet Reduces Hypothalamic Insulin Signaling and Antioxidative Defense in Female Rats. Neuroendocrinology 2019, 108, 278–290. [Google Scholar] [CrossRef]
- Mullins, C.A.; Gannaban, R.B.; Khan, M.S.; Shah, H.; Siddik, M.A.B.; Hegde, V.K.; Reddy, P.H.; Shin, A.C. Neural Underpinnings of Obesity: The Role of Oxidative Stress and Inflammation in the Brain. Antioxidants 2020, 9, 1018. [Google Scholar] [CrossRef] [PubMed]
- Helseth, A.; Tretli, S. Pre-morbid height and weight as risk factors for development of central nervous system neoplasms. Neuroepidemiology 1989, 8, 277–282. [Google Scholar] [CrossRef]
- Jones, L.W.; Ali-Osman, F.; Lipp, E.; Marcello, J.E.; McCarthy, B.; McCoy, L.; Rice, T.; Wrensch, M.; Il’yasova, D. Association between body mass index and mortality in patients with glioblastoma mutliforme. Cancer Causes Control 2010, 21, 2195–2201. [Google Scholar] [CrossRef] [PubMed]
- Moore, S.C.; Rajaraman, P.; Dubrow, R.; Darefsky, A.S.; Koebnick, C.; Hollenbeck, A.; Schatzkin, A.; Leitzmann, M.F. Height, body mass index, and physical activity in relation to glioma risk. Cancer Res. 2009, 69, 8349–8355. [Google Scholar] [CrossRef]
- Little, R.B.; Madden, M.H.; Thompson, R.C.; Olson, J.J.; Larocca, R.V.; Pan, E.; Browning, J.E.; Egan, K.M.; Nabors, L.B. Anthropometric factors in relation to risk of glioma. Cancer Causes Control 2013, 24, 1025–1031. [Google Scholar] [CrossRef]
- Kemerdere, R.; Kacira, T.; Hanimoglu, H.; Kucur, M.; Tanriverdi, T.; Canbaz, B. Tissue and plasma thioredoxin reductase expressions in patients with glioblastoma multiforme. J. Neurol. Surgery. Part A Cent. Eur. Neurosurg. 2013, 74, 234–238. [Google Scholar] [CrossRef]
- Erdi, F.; Kaya, B.; Esen, H.; Karatas, Y.; Findik, S.; Keskin, F.; Feyzioglu, B.; Kalkan, E. New Clues in the Malignant Progression of Glioblastoma: Can the Thioredoxin System Play a Role? Turk. Neurosurg. 2018, 28, 7–12. [Google Scholar] [CrossRef]
- Yao, A.; Storr, S.J.; Al-Hadyan, K.; Rahman, R.; Smith, S.; Grundy, R.; Paine, S.; Martin, S.G. Thioredoxin System Protein Expression Is Associated with Poor Clinical Outcome in Adult and Paediatric Gliomas and Medulloblastomas. Mol. Neurobiol. 2020, 57, 2889–2901. [Google Scholar] [CrossRef] [PubMed]
- Jovanovic, M.; Zhukovsky, D.; Podolski-Renic, A.; Zalubovskis, R.; Dar’in, D.; Sharoyko, V.; Tennikova, T.; Pesic, M.; Krasavin, M. Further exploration of DVD-445 as a lead thioredoxin reductase (TrxR) inhibitor for cancer therapy: Optimization of potency and evaluation of anticancer potential. Eur. J. Med. Chem. 2020, 191, 112119. [Google Scholar] [CrossRef]
- Jovanovic, M.; Zhukovsky, D.; Podolski-Renic, A.; Domraceva, I.; Zalubovskis, R.; Sencanski, M.; Glisic, S.; Sharoyko, V.; Tennikova, T.; Dar’in, D.; et al. Novel electrophilic amides amenable by the Ugi reaction perturb thioredoxin system via thioredoxin reductase 1 (TrxR1) inhibition: Identification of DVD-445 as a new lead compound for anticancer therapy. Eur. J. Med. Chem. 2019, 181, 111580. [Google Scholar] [CrossRef]
- Zhong, Q.; Lin, R.; Nong, Q. Adiposity and Serum Selenium in U.S. Adults. Nutrients 2018, 10, 727. [Google Scholar] [CrossRef]
- Alasfar, F.; Ben-Nakhi, M.; Khoursheed, M.; Kehinde, E.O.; Alsaleh, M. Selenium is significantly depleted among morbidly obese female patients seeking bariatric surgery. Obes. Surg. 2011, 21, 1710–1713. [Google Scholar] [CrossRef]
- Ortega, R.M.; Rodriguez-Rodriguez, E.; Aparicio, A.; Jimenez-Ortega, A.I.; Palmeros, C.; Perea, J.M.; Navia, B.; Lopez-Sobaler, A.M. Young children with excess of weight show an impaired selenium status. Int. J. Vitam. Nutr. Res. 2012, 82, 121–129. [Google Scholar] [CrossRef] [PubMed]
- Rayman, M.P. The importance of selenium to human health. Lancet 2000, 356, 233–241. [Google Scholar] [CrossRef] [PubMed]
- Tinkov, A.A.; Ajsuvakova, O.P.; Filippini, T.; Zhou, J.C.; Lei, X.G.; Gatiatulina, E.R.; Michalke, B.; Skalnaya, M.G.; Vinceti, M.; Aschner, M.; et al. Selenium and Selenoproteins in Adipose Tissue Physiology and Obesity. Biomolecules 2020, 10, 658. [Google Scholar] [CrossRef]
- Krohn, K.; Maier, J.; Paschke, R. Mechanisms of disease: Hydrogen peroxide, DNA damage and mutagenesis in the development of thyroid tumors. Nat. Clin. Pract. Endocrinol. Metab. 2007, 3, 713–720. [Google Scholar] [CrossRef] [PubMed]
- Xing, M. Oxidative stress: A new risk factor for thyroid cancer. Endocr. Relat. Cancer 2012, 19, C7–C11. [Google Scholar] [CrossRef] [PubMed]
- Ameziane El Hassani, R.; Buffet, C.; Leboulleux, S.; Dupuy, C. Oxidative stress in thyroid carcinomas: Biological and clinical significance. Endocr.-Relat. Cancer 2019, 26, R131–R143. [Google Scholar] [CrossRef]
- Dogan, R.; Dogan, E.E.; Guler, E.M.; Senturk, E.; Yenigun, A.; Celik, I.; Aksoy, F.; Ozturan, O. Oxidative stress values of tumor core, edge, and healthy thyroid tissue in thyroid masses. Eur. Arch. Oto-Rhino-Laryngol. 2021, 278, 2953–2960. [Google Scholar] [CrossRef]
- Metere, A.; Frezzotti, F.; Graves, C.E.; Vergine, M.; De Luca, A.; Pietraforte, D.; Giacomelli, L. A possible role for selenoprotein glutathione peroxidase (GPx1) and thioredoxin reductases (TrxR1) in thyroid cancer: Our experience in thyroid surgery. Cancer Cell Int. 2018, 18, 7. [Google Scholar] [CrossRef]
- Cammarota, F.; Fiscardi, F.; Esposito, T.; de Vita, G.; Salvatore, M.; Laukkanen, M.O. Clinical relevance of thyroid cell models in redox research. Cancer Cell Int. 2015, 15, 113. [Google Scholar] [CrossRef] [PubMed]
- de Oliveira Maia, M.; Batista, B.A.M.; Sousa, M.P.; de Souza, L.M.; Maia, C.S.C. Selenium and thyroid cancer: A systematic review. Nutr. Cancer 2020, 72, 1255–1263. [Google Scholar] [CrossRef] [PubMed]
- Oliveros, E.; Somers, V.K.; Sochor, O.; Goel, K.; Lopez-Jimenez, F. The concept of normal weight obesity. Prog. Cardiovasc. Dis. 2014, 56, 426–433. [Google Scholar] [CrossRef]
- Ruderman, N.; Chisholm, D.; Pi-Sunyer, X.; Schneider, S. The metabolically obese, normal-weight individual revisited. Diabetes 1998, 47, 699–713. [Google Scholar] [CrossRef] [PubMed]
- Lyman, G.H.; Sparreboom, A. Chemotherapy dosing in overweight and obese patients with cancer. Nat. Rev. Clin. Oncol. 2013, 10, 451–459. [Google Scholar] [CrossRef]
- Bhardwaj, N.J.; Chae, K.; Sheng, J.Y.; Yeh, H.C. Clinical interventions to break the obesity and cancer link: A narrative review. Cancer Metastasis Rev. 2022, 41, 719–735. [Google Scholar] [CrossRef] [PubMed]
- O’Connell, F.; O’Sullivan, J. Help or hindrance: The obesity paradox in cancer treatment response. Cancer Lett. 2021, 522, 269–280. [Google Scholar] [CrossRef]
- Sheng, X.; Parmentier, J.H.; Tucci, J.; Pei, H.; Cortez-Toledo, O.; Dieli-Conwright, C.M.; Oberley, M.J.; Neely, M.; Orgel, E.; Louie, S.G.; et al. Adipocytes Sequester and Metabolize the Chemotherapeutic Daunorubicin. Mol. Cancer Res. 2017, 15, 1704–1713. [Google Scholar] [CrossRef]
- Coelho, P.; Silva, L.; Faria, I.; Vieria, M.; Monteiro, A.; Pinto, G.; Prudencio, C.; Fernandes, R.; Soares, R. Adipocyte Secretome Increases Radioresistance of Malignant Melanocytes by Improving Cell Survival and Decreasing Oxidative Status. Radiat. Res. 2017, 187, 581–588. [Google Scholar] [CrossRef]
- Okunogbe, A.; Nugent, R.; Spencer, G.; Powis, J.; Ralston, J.; Wilding, J. Economic impacts of overweight and obesity: Current and future estimates for 161 countries. BMJ Glob. Health 2022, 7, e009773. [Google Scholar] [CrossRef]
- Luo, J.; Hendryx, M.; Manson, J.E.; Figueiredo, J.C.; LeBlanc, E.S.; Barrington, W.; Rohan, T.E.; Howard, B.V.; Reding, K.; Ho, G.Y.; et al. Intentional Weight Loss and Obesity-Related Cancer Risk. JNCI Cancer Spectr. 2019, 3, pkz054. [Google Scholar] [CrossRef]
- Chlebowski, R.T.; Luo, J.; Anderson, G.L.; Barrington, W.; Reding, K.; Simon, M.S.; Manson, J.E.; Rohan, T.E.; Wactawski-Wende, J.; Lane, D.; et al. Weight loss and breast cancer incidence in postmenopausal women. Cancer 2019, 125, 205–212. [Google Scholar] [CrossRef]
- Moy, F.M.; Greenwood, D.C.; Cade, J.E. Associations of clothing size, adiposity and weight change with risk of postmenopausal breast cancer in the UK Women’s Cohort Study (UKWCS). BMJ Open 2018, 8, e022599. [Google Scholar] [CrossRef] [PubMed]
- Kanikowska, D.; Kanikowska, A.; Swora-Cwynar, E.; Grzymislawski, M.; Sato, M.; Breborowicz, A.; Witowski, J.; Korybalska, K. Moderate Caloric Restriction Partially Improved Oxidative Stress Markers in Obese Humans. Antioxidants 2021, 10, 1018. [Google Scholar] [CrossRef] [PubMed]
- Schwingshackl, L.; Hoffmann, G. Does a Mediterranean-Type Diet Reduce Cancer Risk? Curr. Nutr. Rep. 2016, 5, 9–17. [Google Scholar] [CrossRef]
- El Assar, M.; Alvarez-Bustos, A.; Sosa, P.; Angulo, J.; Rodriguez-Manas, L. Effect of Physical Activity/Exercise on Oxidative Stress and Inflammation in Muscle and Vascular Aging. Int. J. Mol. Sci. 2022, 23, 8713. [Google Scholar] [CrossRef]
- Thirupathi, A.; Pinho, R.A.; Ugbolue, U.C.; He, Y.; Meng, Y.; Gu, Y. Effect of Running Exercise on Oxidative Stress Biomarkers: A Systematic Review. Front. Physiol. 2020, 11, 610112. [Google Scholar] [CrossRef]
- Powers, S.K.; Deminice, R.; Ozdemir, M.; Yoshihara, T.; Bomkamp, M.P.; Hyatt, H. Exercise-induced oxidative stress: Friend or foe? J. Sport Health Sci. 2020, 9, 415–425. [Google Scholar] [CrossRef]
- Wiggins, T.; Antonowicz, S.S.; Markar, S.R. Cancer Risk Following Bariatric Surgery-Systematic Review and Meta-analysis of National Population-Based Cohort Studies. Obes. Surg. 2019, 29, 1031–1039. [Google Scholar] [CrossRef]
- Chakhtoura, M.; Haber, R.; Ghezzawi, M.; Rhayem, C.; Tcheroyan, R.; Mantzoros, C.S. Pharmacotherapy of obesity: An update on the available medications and drugs under investigation. EClinicalMedicine 2023, 58, 101882. [Google Scholar] [CrossRef] [PubMed]
- Anderson, A.S.; Renehan, A.G.; Saxton, J.M.; Bell, J.; Cade, J.; Cross, A.J.; King, A.; Riboli, E.; Sniehotta, F.; Treweek, S.; et al. Cancer prevention through weight control-where are we in 2020? Br. J. Cancer 2021, 124, 1049–1056. [Google Scholar] [CrossRef]
- Redeker, C.; Wardle, J.; Wilder, D.; Hiom, S.; Miles, A. The launch of Cancer Research UK’s ‘Reduce the Risk’campaign: Baseline measurements of public awareness of cancer risk factors in 2004. Eur. J. Cancer 2009, 45, 827–836. [Google Scholar] [CrossRef]
- Consedine, N.S.; Magai, C.; Conway, F.; Neugut, A.I. Obesity and awareness of obesity as risk factors for breast cancer in six ethnic groups. Obes. Res. 2004, 12, 1680–1689. [Google Scholar] [CrossRef] [PubMed]
- American Institute for Cancer Research. The AICR 2013 Cancer Risk Awareness Survey Report; American Institute for Cancer Research: Washington, DC, USA, 2013. [Google Scholar]
- Silverman, K.R.; Ohman-Strickland, P.A.; Christian, A.H. Perceptions of cancer risk: Differences by weight status. J. Cancer Educ. 2017, 32, 357–363. [Google Scholar] [CrossRef] [PubMed]
- Burkbauer, L.; Goldbach, M.; Huang, C.; Lewandowski, J.; Krouse, R.; Allison, K.; Tchou, J. Awareness of link between obesity and breast cancer risk is associated with willingness to participate in weight loss intervention. Breast Cancer Res. Treat. 2022, 194, 541–550. [Google Scholar] [CrossRef] [PubMed]




Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Jovanović, M.; Kovačević, S.; Brkljačić, J.; Djordjevic, A. Oxidative Stress Linking Obesity and Cancer: Is Obesity a ‘Radical Trigger’ to Cancer? Int. J. Mol. Sci. 2023, 24, 8452. https://doi.org/10.3390/ijms24098452
Jovanović M, Kovačević S, Brkljačić J, Djordjevic A. Oxidative Stress Linking Obesity and Cancer: Is Obesity a ‘Radical Trigger’ to Cancer? International Journal of Molecular Sciences. 2023; 24(9):8452. https://doi.org/10.3390/ijms24098452
Chicago/Turabian StyleJovanović, Mirna, Sanja Kovačević, Jelena Brkljačić, and Ana Djordjevic. 2023. "Oxidative Stress Linking Obesity and Cancer: Is Obesity a ‘Radical Trigger’ to Cancer?" International Journal of Molecular Sciences 24, no. 9: 8452. https://doi.org/10.3390/ijms24098452
APA StyleJovanović, M., Kovačević, S., Brkljačić, J., & Djordjevic, A. (2023). Oxidative Stress Linking Obesity and Cancer: Is Obesity a ‘Radical Trigger’ to Cancer? International Journal of Molecular Sciences, 24(9), 8452. https://doi.org/10.3390/ijms24098452