

Transcriptome, Ectopic Expression and Genetic Population Analysis Identify Candidate Genes for Fiber Quality Improvement in Cotton
 , , ,
, , ,
Abstract
1. Introduction
2. Results
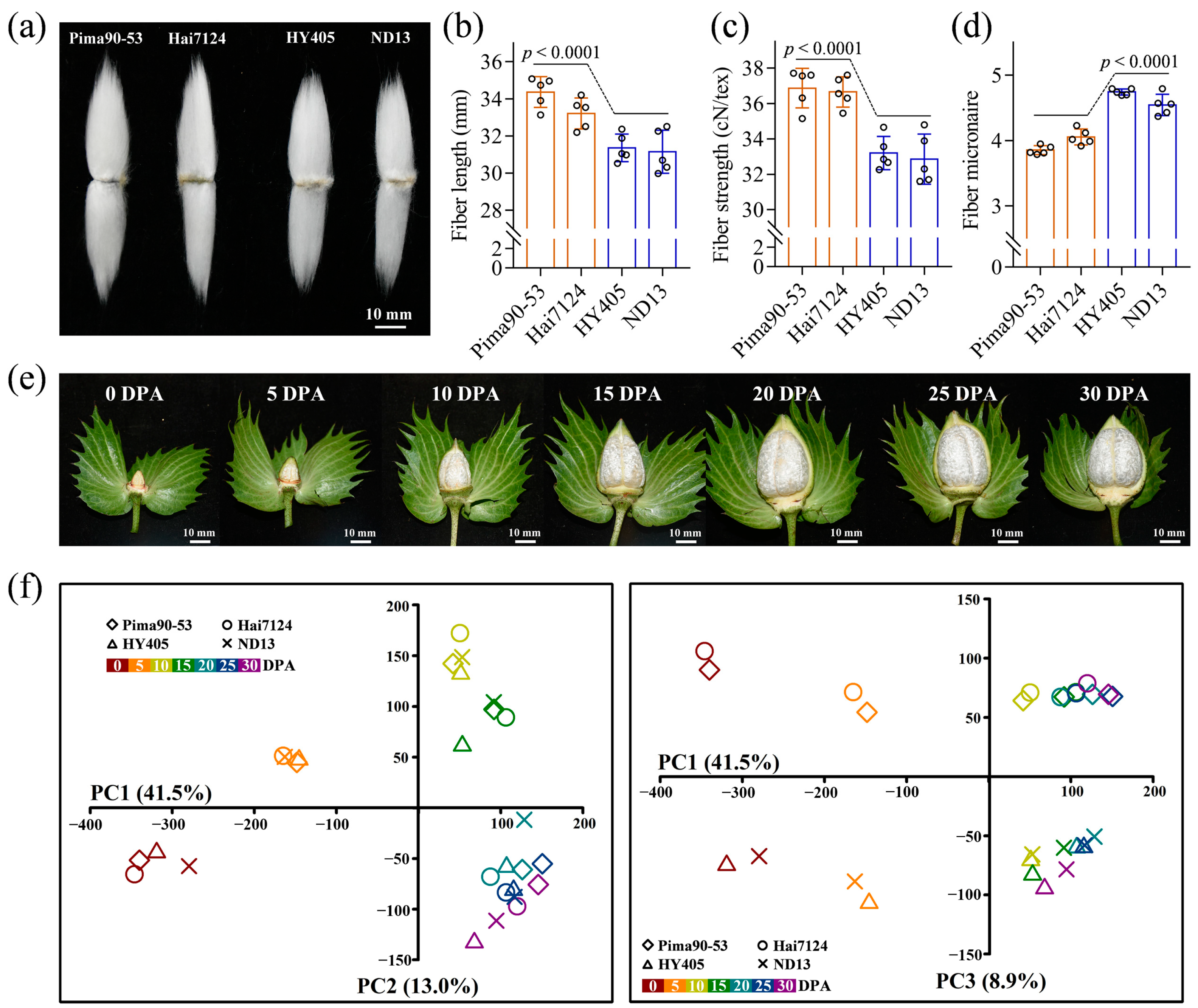
2.1. Overview of Fiber Transcriptomes
2.2. Preferentially Expressed Genes during Fiber Elongation in G. barbadense
2.3. Identification and Functional Analysis of Fiber Elongation-Related Genes
2.4. Preferentially Expressed Genes during SCW Biosynthesis in G. barbadense
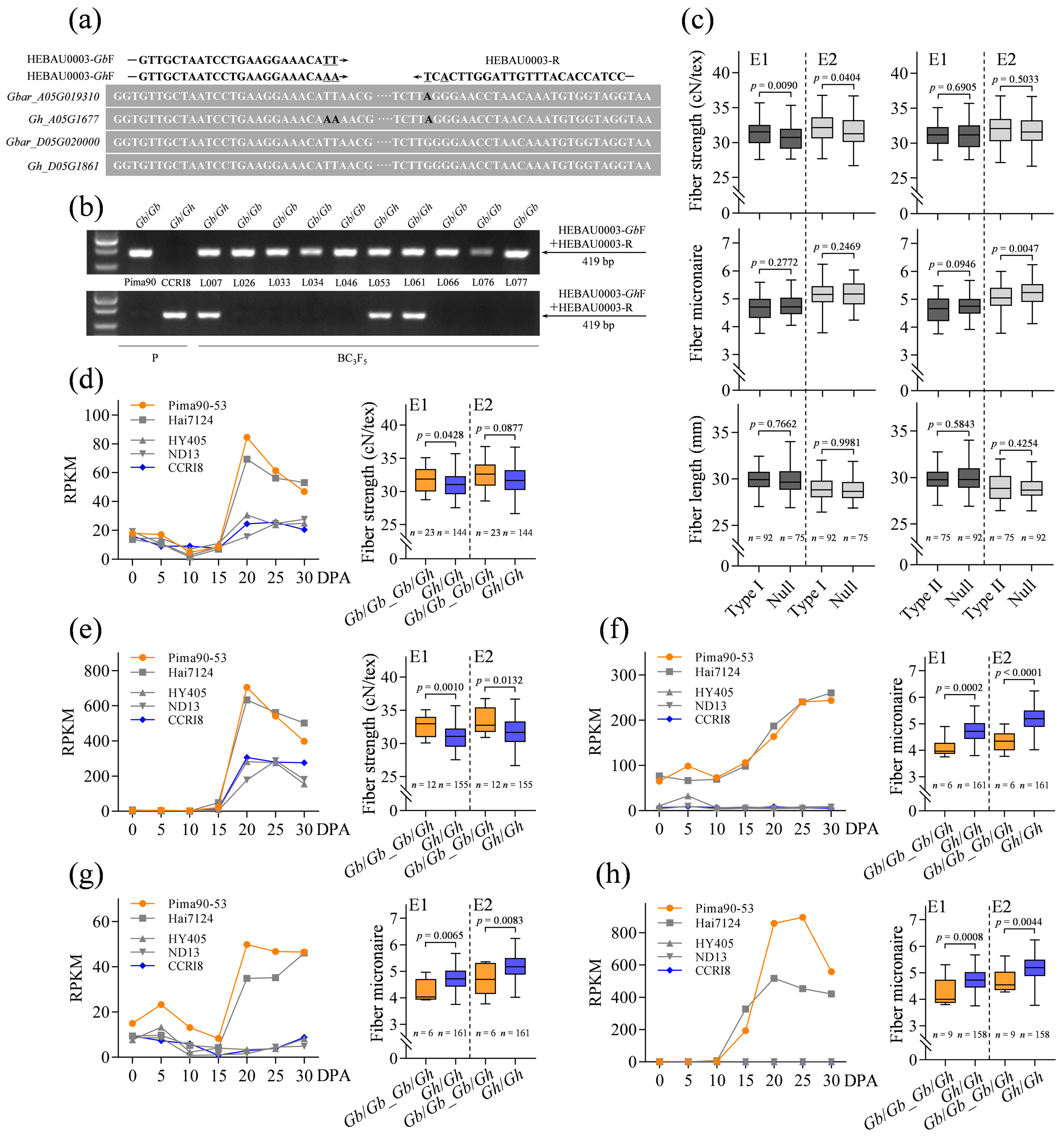
2.5. Identification and Functional Analysis of the SCW Biosynthesis-Related Genes
3. Discussion
4. Materials and Methods
4.1. Plant Materials and Growth Conditions
4.2. Transcriptome Sequencing
4.3. Functional Analysis of the Elongation-Related Candidate Genes
4.4. Functional Identification of the SCW Biosynthesis-Related Candidate Genes
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Chen, Z.J.; Scheffler, B.E.; Dennis, E.; Triplett, B.A.; Zhang, T.; Guo, W.; Chen, X.; Stelly, D.M.; Rabinowicz, P.D.; Town, C.D.; et al. Toward sequencing cotton (Gossypium) genomes. Plant Physiol. 2007, 145, 1303–1310. [Google Scholar] [CrossRef] [PubMed]
- Wang, M.; Tu, L.; Yuan, D.; Zhu, D.; Shen, C.; Li, J.; Liu, F.; Pei, L.; Wang, P.; Zhao, G.; et al. Reference genome sequences of two cultivated allotetraploid cottons Gossypium hirsutum and Gossypium barbadense. Nat. Genet. 2019, 51, 224–229. [Google Scholar] [CrossRef] [PubMed]
- Hu, Y.; Chen, J.; Fang, L.; Zhang, Z.; Ma, W.; Niu, Y.; Ju, L.; Deng, J.; Zhao, T.; Lian, J.; et al. Gossypium barbadense and Gossypium hirsutum genomes provide insights into the origin and evolution of allotetraploid cotton. Nat. Genet. 2019, 51, 739–748. [Google Scholar] [CrossRef] [PubMed]
- Malone, J.H.; Oliver, B. Microarrays, deep sequencing and the true measure of the transcriptome. BMC Biol. 2011, 9, 34. [Google Scholar] [CrossRef]
- Vega-Sánchez, M.E.; Gowda, M.; Wang, G.L. Tag-based approaches for deep transcriptome analysis in plants. Plant Sci. 2007, 173, 371–380. [Google Scholar] [CrossRef]
- Wang, Z.; Gerstein, M.; Snyder, M. RNA-Seq: A revolutionary tool for transcriptomics. Nat. Rev. Genet. 2009, 10, 57–63. [Google Scholar] [CrossRef]
- Nigam, D.; Kavita, P.; Tripathi, R.K.; Ranjan, A.; Goel, R.; Asif, M.; Shukla, A.; Singh, G.; Rana, D.; Sawant, S.V. Transcriptome dynamics during fibre development in contrasting genotypes of Gossypium hirsutum L. Plant Biotechnol. J. 2014, 12, 204–218. [Google Scholar] [CrossRef]
- Ma, J.; Jiang, Y.; Pei, W.; Wu, M.; Ma, Q.; Liu, J.; Song, J.; Jia, B.; Liu, S.; Wu, J.; et al. Expressed genes and their new alleles identification during fibre elongation reveal the genetic factors underlying improvements of fibre length in cotton. Plant Biotechnol. J. 2022, 20, 1940–1955. [Google Scholar] [CrossRef]
- Islam, M.S.; Fang, D.D.; Thyssen, G.N.; Delhom, C.D.; Liu, Y.; Kim, H.J. Comparative fiber property and transcriptome analyses reveal key genes potentially related to high fiber strength in cotton (Gossypium hirsutum L.) line MD52ne. BMC Plant Biol. 2016, 16, 36. [Google Scholar] [CrossRef]
- Zou, X.; Liu, A.; Zhang, Z.; Ge, Q.; Fan, S.; Gong, W.; Li, J.; Gong, J.; Shi, Y.; Tian, B.; et al. Co-expression network analysis and hub gene selection for high-quality fiber in upland cotton (Gossypium hirsutum) using RNA sequencing analysis. Genes 2019, 10, 119. [Google Scholar] [CrossRef]
- Qin, Y.; Sun, H.; Hao, P.; Wang, H.; Wang, C.; Ma, L.; Wei, H.; Yu, S. Transcriptome analysis reveals differences in the mechanisms of fiber initiation and elongation between long- and short-fiber cotton (Gossypium hirsutum L.) lines. BMC Genom. 2019, 20, 633. [Google Scholar] [CrossRef]
- Naoumkina, M.; Thyssen, G.N.; Fang, D.D.; Hinchliffe, D.J.; Florane, C.; Yeater, K.M.; Page, J.T.; Udall, J.A. The Li2 mutation results in reduced subgenome expression bias in elongating fibers of allotetraploid cotton (Gossypium hirsutum L.). PLoS ONE 2014, 9, e90830. [Google Scholar] [CrossRef]
- Naoumkina, M.; Thyssen, G.N.; Fang, D.D. RNA-seq analysis of short fiber mutants Ligon-lintless-1 (Li1) and -2 (Li2) revealed important role of aquaporins in cotton (Gossypium hirsutum L.) fiber elongation. BMC Plant Biol. 2015, 15, 65. [Google Scholar] [CrossRef]
- Liang, W.; Fang, L.; Xiang, D.; Hu, Y.; Feng, H.; Chang, L.; Zhang, T. Transcriptome analysis of short fiber mutant Ligon lintless-1 (Li1) reveals critical genes and key pathways in cotton fiber elongation and leaf development. PLoS ONE 2015, 10, e0143503. [Google Scholar] [CrossRef]
- Ma, Q.F.; Wu, C.H.; Wu, M.; Pei, W.F.; Li, X.L.; Wang, W.K.; Zhang, J.; Yu, J.W.; Yu, S.X. Integrative transcriptome, proteome, phosphoproteome and genetic mapping reveals new aspects in a fiberless mutant of cotton. Sci. Rep. 2016, 6, 24485. [Google Scholar] [CrossRef]
- Naoumkina, M.; Bechere, E.; Fang, D.D.; Thyssen, G.N.; Florane, C.B. Genome-wide analysis of gene expression of EMS-induced short fiber mutant Ligon lintless-y (liy) in cotton (Gossypium hirsutum L.). Genomics 2017, 109, 320–329. [Google Scholar] [CrossRef]
- Hu, H.; Wang, M.; Ding, Y.; Zhu, S.; Zhao, G.; Tu, L.; Zhang, X. Transcriptomic repertoires depict the initiation of lint and fuzz fibres in cotton (Gossypium hirsutum L.). Plant Biotechnol. J. 2018, 16, 1002–1012. [Google Scholar] [CrossRef]
- Salih, H.; Gong, W.; He, S.; Xia, W.; Odongo, M.R.; Du, X. Long non-coding RNAs and their potential functions in Ligon-lintless-1 mutant cotton during fiber development. BMC Genom. 2019, 20, 661. [Google Scholar] [CrossRef]
- Liu, X.; Moncuquet, P.; Zhu, Q.H.; Stiller, W.; Zhang, Z.; Wilson, I. Genetic identification and transcriptome analysis of lintless and fuzzless traits in Gossypium arboreum L. Int. J. Mol. Sci. 2020, 21, 1675. [Google Scholar] [CrossRef]
- Li, P.T.; Wang, M.; Lu, Q.W.; Ge, Q.; Rashid, M.H.O.; Liu, A.Y.; Gong, J.W.; Shang, H.H.; Gong, W.K.; Li, J.W.; et al. Comparative transcriptome analysis of cotton fiber development of Upland cotton (Gossypium hirsutum) and Chromosome Segment Substitution Lines from G. hirsutum × G. barbadense. BMC Genom. 2017, 18, 705. [Google Scholar] [CrossRef]
- Lu, Q.; Shi, Y.; Xiao, X.; Li, P.; Gong, J.; Gong, W.; Liu, A.; Shang, H.; Li, J.; Ge, Q.; et al. Transcriptome analysis suggests that chromosome introgression fragments from Sea Island cotton (Gossypium barbadense) increase fiber strength in Upland cotton (Gossypium hirsutum). G3-Genes Genom. Genet. 2017, 7, 3469–3479. [Google Scholar] [CrossRef] [PubMed]
- Song, Z.; Chen, Y.; Zhang, C.; Zhang, J.; Huo, X.; Gao, Y.; Pan, A.; Du, Z.; Zhou, J.; Zhao, Y.; et al. RNA-seq reveals hormone-regulated synthesis of non-cellulose polysaccharides associated with fiber strength in a single-chromosomal-fragment-substituted upland cotton line. Crop J. 2020, 8, 273–286. [Google Scholar] [CrossRef]
- Lacape, J.M.; Claverie, M.; Vidal, R.O.; Carazzolle, M.F.; Guimaraes Pereira, G.A.; Ruiz, M.; Pre, M.; Llewellyn, D.; Al-Ghazi, Y.; Jacobs, J.; et al. Deep sequencing reveals differences in the transcriptional landscapes of fibers from two cultivated species of cotton. PLoS ONE 2012, 7, e48855. [Google Scholar] [CrossRef] [PubMed]
- Tuttle, J.R.; Nah, G.; Duke, M.V.; Alexander, D.C.; Guan, X.; Song, Q.; Chen, Z.J.; Scheffler, B.E.; Haigler, C.H. Metabolomic and transcriptomic insights into how cotton fiber transitions to secondary wall synthesis, represses lignification and prolongs elongation. BMC Genom. 2015, 16, 477. [Google Scholar] [CrossRef]
- Zhang, D.; Zhang, T.; Sang, Z.; Guo, W. Comparative development of lint and fuzz using different cotton fiber-specific developmental mutants in Gossypium hirsutum. J. Integr. Plant Biol. 2007, 49, 1038–1046. [Google Scholar] [CrossRef]
- Haigler, C.H.; Betancur, L.; Stiff, M.R.; Tuttle, J.R. Cotton fiber, a powerful single-cell model for cell wall and cellulose research. Front. Plant Sci. 2012, 3, 104. [Google Scholar] [CrossRef]
- Jareczek, J.J.; Grover, C.E.; Wendel, J.F. Cotton fiber as a model for understanding shifts in cell development under domestication. Front. Plant Sci. 2023, 14, 1146802. [Google Scholar] [CrossRef]
- Schubert, A.M.; Benedict, C.R.; Gates, C.E.; Kohel, R.J. Growth and development of the lint fibers of Pima S-4 cotton. Crop Sci. 1976, 16, 539–543. [Google Scholar] [CrossRef]
- Zhang, T.; Hu, Y.; Jiang, W.; Fang, L.; Guan, X.; Chen, J.; Zhang, J.; Saski, C.A.; Scheffler, B.E.; Stelly, D.M.; et al. Sequencing of allotetraploid cotton (Gossypium hirsutum L. acc. TM-1) provides a resource for fiber improvement. Nat. Biotechnol. 2015, 33, 531–537. [Google Scholar] [CrossRef]
- Wang, X.C.; Li, Q.; Jin, X.; Xiao, G.H.; Liu, G.J.; Liu, N.J.; Qin, Y.M. Quantitative proteomics and transcriptomics reveal key metabolic processes associated with cotton fiber initiation. J. Proteom. 2015, 114, 16–27. [Google Scholar] [CrossRef]
- Townsend, B.J.; Poole, A.; Blake, C.J.; Llewellyn, D.J. Antisense suppression of a (+)-delta-cadinene synthase gene in cotton prevents the induction of this defense response gene during bacterial blight infection but not its constitutive expression. Plant Physiol. 2005, 138, 516–528. [Google Scholar] [CrossRef]
- Liu, Q.; Talbot, M.; Llewellyn, D.J. Pectin methylesterase and pectin remodelling differ in the fibre walls of two Gossypium species with very different fibre properties. PLoS ONE 2013, 8, e65131. [Google Scholar] [CrossRef]
- Meng, C.; Yan, Y.; Liu, Z.; Chen, L.; Zhang, Y.; Li, X.; Wu, L.; Zhang, G.; Wang, X.; Ma, Z. Systematic analysis of cotton non-specific lipid transfer protein family revealed a special group that is involved in fiber elongation. Front. Plant Sci. 2018, 9, 1285. [Google Scholar] [CrossRef]
- Lv, L.M.; Zuo, D.Y.; Wang, X.F.; Cheng, H.L.; Zhang, Y.P.; Wang, Q.L.; Song, G.L.; Ma, Z.Y. Genome-wide identification of the expansin gene family reveals that expansin genes are involved in fibre cell growth in cotton. BMC Plant Biol. 2020, 20, 223. [Google Scholar] [CrossRef]
- Liu, Z.; Wang, X.; Sun, Z.; Zhang, Y.; Meng, C.; Chen, B.; Wang, G.; Ke, H.; Wu, J.; Yan, Y.; et al. Evolution, expression and functional analysis of cultivated allotetraploid cotton DIR genes. BMC Plant Biol. 2021, 21, 89. [Google Scholar] [CrossRef]
- Biswas, K.K.; Ooura, C.; Higuchi, K.; Miyazaki, Y.; Van Nguyen, V.; Rahman, A.; Uchimiya, H.; Kiyosue, T.; Koshiba, T.; Tanaka, A.; et al. Genetic characterization of mutants resistant to the antiauxin p-chlorophenoxyisobutyric acid reveals that AAR3, a gene encoding a DCN1-like protein, regulates responses to the synthetic auxin 2,4-dichlorophenoxyacetic acid in Arabidopsis roots. Plant Physiol. 2007, 145, 773–785. [Google Scholar] [CrossRef]
- Kurz, T.; Ozlu, N.; Rudolf, F.; O’Rourke, S.M.; Luke, B.; Hofmann, K.; Hyman, A.A.; Bowerman, B.; Peter, M. The conserved protein DCN-1/Dcn1p is required for cullin neddylation in C. elegans and S. cerevisiae. Nature 2005, 435, 1257–1261. [Google Scholar] [CrossRef]
- Hayashi, K. The interaction and integration of auxin signaling components. Plant Cell Physiol. 2012, 53, 965–975. [Google Scholar] [CrossRef]
- Guan, X.; Yu, N.; Shangguan, X.; Wang, S.; Lu, S.; Wang, L.; Chen, X. Arabidopsis trichome research sheds light on cotton fiber development mechanisms. Chin. Sci. Bull. 2007, 52, 1734–1741. [Google Scholar] [CrossRef]
- Guan, X.; Pang, M.; Nah, G.; Shi, X.; Ye, W.; Stelly, D.M.; Chen, Z.J. miR828 and miR858 regulate homoeologous MYB2 gene functions in Arabidopsis trichome and cotton fibre development. Nat. Commun. 2014, 5, 3050. [Google Scholar] [CrossRef] [PubMed]
- Ma, Z.; He, S.; Wang, X.; Sun, J.; Zhang, Y.; Zhang, G.; Wu, L.; Li, Z.; Liu, Z.; Sun, G.; et al. Resequencing a core collection of upland cotton identifies genomic variation and loci influencing fiber quality and yield. Nat. Genet. 2018, 50, 803–813. [Google Scholar] [CrossRef] [PubMed]
- Yu, Y.; Wu, S.; Nowak, J.; Wang, G.; Han, L.; Feng, Z.; Mendrinna, A.; Ma, Y.; Wang, H.; Zhang, X.; et al. Live-cell imaging of the cytoskeleton in elongating cotton fibres. Nat. Plants 2019, 5, 498–504. [Google Scholar] [CrossRef] [PubMed]
- Gendreau, E.; Traas, J.; Desnos, T.; Grandjean, O.; Caboche, M.; Hofte, H. Cellular basis of hypocotyl growth in Arabidopsis thaliana. Plant Physiol. 1997, 114, 295–305. [Google Scholar] [CrossRef] [PubMed]
- Boron, A.K.; Vissenberg, K. The Arabidopsis thaliana hypocotyl, a model to identify and study control mechanisms of cellular expansion. Plant Cell Rep. 2014, 33, 697–706. [Google Scholar] [CrossRef] [PubMed]
- Fiume, E.; Guyon, V.; Remoue, C.; Magnani, E.; Miquel, M.; Grain, D.; Lepiniec, L. TWS1, a novel small protein, regulates various aspects of seed and plant development. Plant Physiol. 2016, 172, 1732–1745. [Google Scholar] [CrossRef]
- Kuang, B.; Zhao, X.; Zhou, C.; Zeng, W.; Ren, J.; Ebert, B.; Beahan, C.T.; Deng, X.; Zeng, Q.; Zhou, G.; et al. Role of UDP-glucuronic acid decarboxylase in xylan biosynthesis in Arabidopsis. Mol. Plant 2016, 9, 1119–1131. [Google Scholar] [CrossRef]
- Hoffmann, N.; Benske, A.; Betz, H.; Schuetz, M.; Samuels, A.L. Laccases and peroxidases co-localize in lignified secondary cell walls throughout stem development. Plant Physiol. 2020, 184, 806–822. [Google Scholar] [CrossRef]
- MacMillan, C.P.; Mansfield, S.D.; Stachurski, Z.H.; Evans, R.; Southerton, S.G. Fasciclin-like arabinogalactan proteins: Specialization for stem biomechanics and cell wall architecture in Arabidopsis and Eucalyptus. Plant J. 2010, 62, 689–703. [Google Scholar] [CrossRef]
- Grantham, N.J.; Wurman-Rodrich, J.; Terrett, O.M.; Lyczakowski, J.J.; Stott, K.; Iuga, D.; Simmons, T.J.; Durand-Tardif, M.; Brown, S.P.; Dupree, R.; et al. An even pattern of xylan substitution is critical for interaction with cellulose in plant cell walls. Nat. Plants 2017, 3, 859–865. [Google Scholar] [CrossRef]
- Arsovski, A.A.; Popma, T.M.; Haughn, G.W.; Carpita, N.C.; McCann, M.C.; Western, T.L. AtBXL1 encodes a bifunctional beta-D-xylosidase/alpha-L-arabinofuranosidase required for pectic arabinan modification in Arabidopsis mucilage secretory cells. Plant Physiol. 2009, 150, 1219–1234. [Google Scholar] [CrossRef]
- Glass, M.; Barkwill, S.; Unda, F.; Mansfield, S.D. Endo-beta-1,4-glucanases impact plant cell wall development by influencing cellulose crystallization. J. Integr. Plant Biol. 2015, 57, 396–410. [Google Scholar] [CrossRef]
- McFarlane, H.E.; Doring, A.; Persson, S. The cell biology of cellulose synthesis. Annu. Rev. Plant Biol. 2014, 65, 69–94. [Google Scholar] [CrossRef]
- Bai, W.Q.; Xiao, Y.H.; Zhao, J.; Song, S.Q.; Hu, L.; Zeng, J.Y.; Li, X.B.; Hou, L.; Luo, M.; Li, D.M.; et al. Gibberellin overproduction promotes sucrose synthase expression and secondary cell wall deposition in cotton fibers. PLoS ONE 2014, 9, e96537. [Google Scholar] [CrossRef]
- Singh, S.; Brocker, C.; Koppaka, V.; Chen, Y.; Jackson, B.C.; Matsumoto, A.; Thompson, D.C.; Vasiliou, V. Aldehyde dehydrogenases in cellular responses to oxidative/electrophilic stress. Free Radical Biol. Med. 2013, 56, 89–101. [Google Scholar] [CrossRef]
- Newman, J.W.; Morisseau, C.; Hammock, B.D. Epoxide hydrolases, their roles and interactions with lipid metabolism. Prog. Lipid Res. 2005, 44, 1–51. [Google Scholar] [CrossRef]
- Reyt, G.; Chao, Z.; Flis, P.; Salas-Gonzalez, I.; Castrillo, G.; Chao, D.Y.; Salt, D.E. Uclacyanin proteins are required for lignified nanodomain formation within casparian strips. Curr. Biol. 2020, 30, 4103–4111. [Google Scholar] [CrossRef]
- Zhu, W.; Gao, E.; Shaban, M.; Wang, Y.; Wang, H.; Nie, X.; Zhu, L. GhUMC1, a blue copper-binding protein, regulates lignin synthesis and cotton immune response. Biochem. Biophys. Res. Commun. 2018, 504, 75–81. [Google Scholar] [CrossRef]
- Waszczak, C.; Carmody, M.; Kangasjarvi, J. Reactive oxygen species in plant signaling. Annu. Rev. Plant Biol. 2018, 69, 209–236. [Google Scholar] [CrossRef]
- Cui, H.; Kong, D.; Wei, P.; Hao, Y.; Torii, K.U.; Lee, J.S.; Li, J. SPINDLY, ERECTA and its ligand STOMAGEN have a role in redox-mediated cortex proliferation in the Arabidopsis root. Mol. Plant 2014, 7, 1727–1739. [Google Scholar] [CrossRef]
- Mnich, E.; Bjarnholt, N.; Eudes, A.; Harholt, J.; Holland, C.; Jorgensen, B.; Larsen, F.H.; Liu, M.; Manat, R.; Meyer, A.S.; et al. Phenolic cross-links, building and de-constructing the plant cell wall. Nat. Prod. Rep. 2020, 37, 919–961. [Google Scholar]
- Gill, S.S.; Tuteja, N. Reactive oxygen species and antioxidant machinery in abiotic stress tolerance in crop plants. Plant Physiol. Biochem. 2010, 48, 909–930. [Google Scholar] [CrossRef]
- ul Haq, S.; Khan, A.; Ali, M.; Khattak, A.M.; Gai, W.X.; Zhang, H.X.; Wei, A.M.; Gong, Z.H. Heat shock proteins: Dynamic biomolecules to counter plant biotic and abiotic stresses. Int. J. Mol. Sci. 2019, 20, 5321. [Google Scholar] [CrossRef] [PubMed]
- Alekseeva, A.A.; Savin, S.S.; Tishkov, V.I. NAD+-dependent formate dehydrogenase from plants. Acta Nat. 2011, 3, 38–54. [Google Scholar] [CrossRef]
- Sauter, M.; Moffatt, B.; Saechao, M.C.; Hell, R.; Wirtz, M. Methionine salvage and S-adenosylmethionine, essential links between sulfur, ethylene and polyamine biosynthesis. Biochem. J. 2013, 451, 145–154. [Google Scholar] [CrossRef] [PubMed]
- Han, L.B.; Li, Y.B.; Wang, H.Y.; Wu, X.M.; Li, C.L.; Luo, M.; Wu, S.J.; Kong, Z.S.; Pei, Y.; Jiao, G.L.; et al. The dual functions of WLIM1a in cell elongation and secondary wall formation in developing cotton fibers. Plant Cell 2013, 25, 4421–4438. [Google Scholar] [CrossRef]
- Gao, Z.; Sun, W.; Wang, J.; Zhao, C.; Zuo, K. GhbHLH18 negatively regulates fiber strength and length by enhancing lignin biosynthesis in cotton fibers. Plant Sci. 2019, 286, 7–16. [Google Scholar] [CrossRef]
- Feurtado, J.A.; Huang, D.; Wicki-Stordeur, L.; Hemstock, L.E.; Potentier, M.S.; Tsang, E.W.; Cutler, A.J. The Arabidopsis C2H2 zinc finger INDETERMINATE DOMAIN1/ENHYDROUS promotes the transition to germination by regulating light and hormonal signaling during seed maturation. Plant Cell 2011, 23, 1772–1794. [Google Scholar] [CrossRef]
- Fukazawa, J.; Teramura, H.; Murakoshi, S.; Nasuno, K.; Nishida, N.; Ito, T.; Yoshida, M.; Kamiya, Y.; Yamaguchi, S.; Takahashi, Y. DELLAs function as coactivators of GAI-ASSOCIATED FACTOR1 in regulation of gibberellin homeostasis and signaling in Arabidopsis. Plant Cell 2014, 26, 2920–2938. [Google Scholar] [CrossRef]
- Mussig, C.; Lisso, J.; Coll-Garcia, D.; Altmann, T. Molecular analysis of brassinosteroid action. Plant Biol. 2006, 8, 291–296. [Google Scholar] [CrossRef]
- Sun, Y.; Veerabomma, S.; Fokar, M.; Abidi, N.; Hequet, E.; Payton, P.; Allen, R.D. Brassinosteroid signaling affects secondary cell wall deposition in cotton fibers. Ind. Crop Prod. 2015, 65, 334–342. [Google Scholar] [CrossRef]
- Borghi, L.; Kang, J.; Ko, D.; Lee, Y.; Martinoia, E. The role of ABCG-type ABC transporters in phytohormone transport. Biochem. Soc. Trans. 2015, 43, 924–930. [Google Scholar] [CrossRef]
- Zhong, R.; Cui, D.; Ye, Z.H. Secondary cell wall biosynthesis. New Phytol. 2019, 221, 1703–1723. [Google Scholar] [CrossRef]
- Xiao, G.; Zhao, P.; Zhang, Y. A pivotal role of hormones in regulating cotton fiber development. Front. Plant Sci. 2019, 10, 87. [Google Scholar] [CrossRef]
- Yu, J.Z.; Ulloa, M.; Hoffman, S.M.; Kohel, R.J.; Pepper, A.E.; Fang, D.D.; Percy, R.G.; Burke, J.J. Mapping genomic loci for cotton plant architecture, yield components and fiber properties in an interspecific (Gossypium hirsutum L. × G. barbadense L.) RIL population. Mol. Genet. Genom. 2014, 289, 1347–1367. [Google Scholar] [CrossRef]
- Lacape, J.M.; Llewellyn, D.; Jacobs, J.; Arioli, T.; Becker, D.; Calhoun, S.; Al-Ghazi, Y.; Liu, S.; Palai, O.; Georges, S.; et al. Meta-analysis of cotton fiber quality QTLs across diverse environments in a Gossypium hirsutum × G. barbadense RIL population. BMC Plant Biol. 2010, 10, 132. [Google Scholar] [CrossRef]
- Guo, X.; Runavot, J.L.; Bourot, S.; Meulewaeter, F.; Hernandez-Gomez, M.; Holland, C.; Harholt, J.; Willats, W.G.T.; Mravec, J.; Knox, P.; et al. Metabolism of polysaccharides in dynamic middle lamellae during cotton fibre development. Planta 2019, 249, 1565–1581. [Google Scholar] [CrossRef]
- Wang, X.F.; Ma, J.; Wang, W.S.; Zheng, Y.M.; Zhang, G.Y.; Liu, C.J.; Ma, Z.Y. Construction and characterization of the first bacterial artificial chromosome library for the cotton species Gossypium barbadense L. Genome 2006, 49, 1393–1398. [Google Scholar] [CrossRef]
- Kim, D.; Pertea, G.; Trapnell, C.; Pimentel, H.; Kelley, R.; Salzberg, S.L. TopHat2, accurate alignment of transcriptomes in the presence of insertions, deletions and gene fusions. Genome Biol. 2013, 14, R36. [Google Scholar] [CrossRef]
- Trapnell, C.; Williams, B.A.; Pertea, G.; Mortazavi, A.; Kwan, G.; van Baren, M.J.; Salzberg, S.L.; Wold, B.J.; Pachter, L. Transcript assembly and quantification by RNA-Seq reveals unannotated transcripts and isoform switching during cell differentiation. Nat. Biotechnol. 2010, 28, 511–515. [Google Scholar] [CrossRef]
- Anders, S.; Pyl, P.T.; Huber, W. HTSeq—A Python framework to work with high-throughput sequencing data. Bioinformatics 2015, 31, 166–169. [Google Scholar] [CrossRef]
- Robinson, M.D.; McCarthy, D.J.; Smyth, G.K. edgeR, a Bioconductor package for differential expression analysis of digital gene expression data. Bioinformatics 2010, 26, 139–140. [Google Scholar] [CrossRef] [PubMed]
- Wang, L.; Feng, Z.; Wang, X.; Wang, X.; Zhang, X. DEGseq, an R package for identifying differentially expressed genes from RNA-seq data. Bioinformatics 2010, 26, 136–138. [Google Scholar] [CrossRef] [PubMed]
- Xie, C.; Mao, X.; Huang, J.; Ding, Y.; Wu, J.; Dong, S.; Kong, L.; Gao, G.; Li, C.Y.; Wei, L. KOBAS 2.0, a web server for annotation and identification of enriched pathways and diseases. Nucleic Acids Res. 2011, 39, W316–W322. [Google Scholar] [CrossRef] [PubMed]
- Collins, T.J. ImageJ for microscopy. Biotechniques 2007, 43, 25–30. [Google Scholar] [CrossRef] [PubMed]
- Paterson, A.H.; Brubaker, C.L.; Wendel, J.F. A rapid method for extraction of cotton (Gossypium spp.) genomic DNA suitable for RFLP or PCR analysis. Plant Mol. Biol. Rep. 1993, 11, 122–127. [Google Scholar] [CrossRef]
- Ugozzoli, L.; Wallace, R.B. Allele-specific polymerase chain reaction. Methods 1991, 2, 42–48. [Google Scholar] [CrossRef]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Type | Reference a | Orthologous Genes b | Functional Annotation |
|---|---|---|---|
| IV-I | Gh_A03G0826 | Gbar_A03G010660 | GPI-anchored adhesin-like protein |
| Gh_A07G1193 | Gbar_A07G013810 | S-adenosylmethionine synthase | |
| Gh_A11G1313 | Gbar_A11G014750 | Stomagen | |
| Gh_A11G3137 | Gbar_A11G021290 | 3-hydroxyacyl-CoA dehydratase | |
| Gh_A13G0932 | Gbar_A13G012640 | SMALLER WITH VARIABLE BRANCHES | |
| Gh_D03G0078 | Gbar_D03G000870 | α-mannosidase | |
| Gh_D05G3507 | Gbar_D05G037750 | (+)-δ-cadinene synthase | |
| Gh_D08G1067 | Gbar_D08G011240 | BAHD acyltransferase | |
| Gh_D09G0438 | Gbar_D09G004860 | Subtilisin-like protease | |
| Gh_D11G1462 | Gbar_D11G015570 | Stomagen | |
| IV-II | Gh_A03G2157 | Gbar_A03G019080 | Nonspecific lipid-transfer protein |
| Gh_A05G1646 | Gbar_A05G018970 | β-mannanase | |
| Gh_A05G1812 | Gbar_A05G020690 | TWISTED SEED | |
| Gh_A07G0152 | Gbar_A07G001880 | Pectinesterase/pectinesterase inhibitor | |
| Gh_A07G0664 | Gbar_A07G007890 | α-expansin | |
| Gh_A08G0668 | Gbar_A08G007900 | BTB/POZ domain-containing protein | |
| Gh_A09G1368 | Gbar_A09G015680 | S-adenosylmethionine synthase | |
| Gh_A11G0547 | Gbar_A11G006130 | Serine protease inhibitor | |
| Gh_A12G1104 | Gbar_A12G013240 | Ribonuclease H-like protein | |
| Gh_D04G0006 | Gbar_D04G000050 | Pectinesterase/pectinesterase inhibitor | |
| Gh_D05G2968 | Gbar_D05G031740 | Importin-β | |
| Gh_D07G1581 | Gbar_D07G017340 | Acidic leucine-rich nuclear phosphoprotein | |
| Gh_D08G0151 | Gbar_D08G001620 | Dirigent protein | |
| Gh_D09G1136 | Gbar_D09G012750 | Alpha carbonic anhydrase | |
| Gh_D10G1363 | Gbar_D10G014320 | Methylenetetrahydrofolate reductase | |
| Gh_D11G0625 | Gbar_D11G006420 | Fatty acid amide hydrolase | |
| Gh_D11G3426 | Gbar_D11G024050 | Glutathione S-transferase | |
| Gh_D12G2436 | Gbar_D12G026630 | Heat shock protein | |
| IV-III | Gh_A02G1644 | Gbar_A02G018320 | α-mannosidase |
| Gh_A05G0923 | Gbar_A05G010310 | Heavy metal transport | |
| Gh_A05G3172 | Gbar_A05G037170 | ANTIAUXIN-RESISTANT | |
| Gh_A06G1116 | Gbar_A06G013440 | Cystathionine β-synthase | |
| Gh_A07G2014 | Gbar_A07G023310 | Formate dehydrogenase | |
| Gh_A10G0126 | Gbar_A10G001450 | Actin cytoskeleton-regulatory complex | |
| Gh_D04G0153 | Gbar_D04G001950 | Cytochrome P450 | |
| Gh_D04G1621 | Gbar_D04G017890 | Cytosolic cyclophilin | |
| Gh_D05G2178 | Gbar_D05G023090 | Sugar transport protein | |
| Gh_D08G2441 | Gbar_D08G025770 | Leucine-rich repeat (LRR) protein | |
| Gh_D09G1484 | Gbar_D09G016550 | Short-chain dehydrogenase | |
| Gh_D11G1941 | Gbar_D11G020770 | PLC-like phosphodiesterase | |
| Gh_D12G0289 | Gbar_D12G003220 | SAUR-like auxin-responsive protein | |
| Gh_D12G0652 | Gbar_D12G007360 | Membrane-associated kinase regulator |
| Reference a | Orthologous Genes b | Functional Annotation | Genetic Marker |
|---|---|---|---|
| Gh_A04G0686 | Gbar_A04G008430 | Soluble inorganic pyrophosphatase | HEBAU0001 |
| Gh_A05G0370 | Gbar_A05G004360 | F-box/LRR-repeat protein | HEBAU0002 |
| Gh_A05G1677 | Gbar_A05G019310 | C2H2-like zinc finger protein | HEBAU0003 |
| Gh_A05G1930 | Gbar_A05G022250 | EXORDIUM protein | NA |
| Gh_A05G2177 | Gbar_A05G025000 | Transmembrane protein | HEBAU0004 |
| Gh_A06G1590 | Gbar_A06G019480 | Leucine-rich repeat protein kinase | HEBAU0005 |
| Gh_A10G1937 | Gbar_A10G023160 | UDP-Xyl synthase | HEBAU0006 |
| Gh_A11G2936 | Gbar_A11G034900 | Laccase | HEBAU0007 |
| Gh_A12G0165 | Gbar_A12G001790 | Nucleobase–ascorbate transporter | HEBAU0008 |
| Gh_A13G0090 | GB_A13G0103 | Cysteine/histidine-rich DC1 domain | HEBAU0009 |
| Gh_A13G1919 | Gbar_A13G023580 | Fasciclin-like arabinogalactan protein | HEBAU0010 |
| Gh_D01G1187 | Gbar_D01G013340 | Protein of unknown function | HEBAU0011 |
| Gh_D02G0286 | GB_D02G0284 | Cytochrome P450 | HEBAU0012 |
| Gh_D02G1423 | Gbar_D02G015210 | Plasmodesmata-located protein | NA |
| Gh_D03G0537 | Gbar_D03G005540 | Cytochrome P450 | HEBAU0013 |
| Gh_D03G1074 | Gbar_D03G011820 | Feruloyl-CoA transferase | HEBAU0014 |
| Gh_D04G1519 | Gbar_D04G016960 | 2-nitropropane dioxygenase | HEBAU0015 |
| Gh_D05G1062 | Gbar_D05G011330 | LIGHT SENSITIVE HYPOCOTYLS | HEBAU0016 |
| Gh_D05G2936 | Gbar_D05G031310 | Phosphate transporter | HEBAU0017 |
| Gh_D06G2276 | Gbar_D06G000230 | β-tubulin | HEBAU0018 |
| Gh_D07G0285 | Gbar_D07G003310 | Clathrin assembly protein | NA |
| Gh_D09G0011 | Gbar_D09G000120 | Protein of unknown function | HEBAU0019 |
| Gh_D10G1036 | Gbar_D10G011020 | Xylan glucuronosyltransferase | HEBAU0020 |
| Gh_D10G1767 | Gbar_D10G018450 | β-xylosidase | HEBAU0021 |
| Gh_D12G2377 | Gbar_D12G025300 | Protein of unknown function | NA |
| Reference a | Orthologous Genes b | Functional Annotation | Genetic Marker |
|---|---|---|---|
| Gh_A03G1620 | Gbar_A03G020290 | Progesterone 5β-reductase | HEBAU0022 |
| Gh_A05G0912 | Gbar_A05G010210 | BEL1-like homeodomain protein | HEBAU0023 |
| Gh_A05G1599 | Gbar_A05G018470 | Endo-β-1, 4-glucanase | HEBAU0024 |
| Gh_A06G0882 | Gbar_A06G010210 | Transmembrane protein | HEBAU0025 |
| Gh_A06G1256 | Gbar_A06G015550 | Aldehyde dehydrogenase | HEBAU0026 |
| Gh_A07G0322 | Gbar_A07G004070 | Cellulose synthase | NA |
| Gh_A09G0389 | NA | Protein of unknown function | NA |
| Gh_A10G0145 | Gbar_A10G001660 | NTF2-like protein | NA |
| Gh_A10G0696 | Gbar_A10G008340 | ABC transporter | HEBAU0027 |
| Gh_A12G0108 | Gbar_A12G001190 | Protein of unknown function | HEBAU0028 |
| Gh_A12G1564 | Gbar_A12G018330 | SLH domain protein | HEBAU0029 |
| Gh_A12G2552 | Gbar_A12G010170 | Hydroxyproline-rich glycoprotein | HEBAU0030 |
| Gh_A13G0149 | Gbar_A13G001740 | Auxin influx transporter | HEBAU0031 |
| Gh_A13G0378 | Gbar_A13G004310 | Leucine-rich repeat (LRR) protein | HEBAU0032 |
| Gh_A13G1904 | Gbar_A13G023450 | Epoxide hydrolase | HEBAU0033 |
| Gh_A13G2039 | Gbar_A13G024860 | Networked (NET) family protein | HEBAU0034 |
| Gh_A13G2237 | NA | Protein of unknown function | NA |
| Gh_D03G0611 | Gbar_D03G007510 | Cellulose synthase | HEBAU0035 |
| Gh_D05G2536 | Gbar_D05G026930 | Blue copper protein | HEBAU0036 |
| Gh_D06G1911 | Gbar_D06G019950 | RING-type E3 ubiquitin ligase | HEBAU0037 |
| Gh_D08G1431 | Gbar_D08G015060 | Subtilisin-like protease | HEBAU0038 |
| Gh_D10G1810 | Gbar_D10G018890 | UDP-glycosyltransferase | NA |
| Gh_D12G0575 | Gbar_D12G006270 | Kinesin-like protein | HEBAU0039 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Liu, Z.; Sun, Z.; Ke, H.; Chen, B.; Gu, Q.; Zhang, M.; Wu, N.; Chen, L.; Li, Y.; Meng, C.; et al. Transcriptome, Ectopic Expression and Genetic Population Analysis Identify Candidate Genes for Fiber Quality Improvement in Cotton. Int. J. Mol. Sci. 2023, 24, 8293. https://doi.org/10.3390/ijms24098293
Liu Z, Sun Z, Ke H, Chen B, Gu Q, Zhang M, Wu N, Chen L, Li Y, Meng C, et al. Transcriptome, Ectopic Expression and Genetic Population Analysis Identify Candidate Genes for Fiber Quality Improvement in Cotton. International Journal of Molecular Sciences. 2023; 24(9):8293. https://doi.org/10.3390/ijms24098293
Chicago/Turabian StyleLiu, Zhengwen, Zhengwen Sun, Huifeng Ke, Bin Chen, Qishen Gu, Man Zhang, Nan Wu, Liting Chen, Yanbin Li, Chengsheng Meng, and et al. 2023. "Transcriptome, Ectopic Expression and Genetic Population Analysis Identify Candidate Genes for Fiber Quality Improvement in Cotton" International Journal of Molecular Sciences 24, no. 9: 8293. https://doi.org/10.3390/ijms24098293
APA StyleLiu, Z., Sun, Z., Ke, H., Chen, B., Gu, Q., Zhang, M., Wu, N., Chen, L., Li, Y., Meng, C., Wang, G., Wu, L., Zhang, G., Ma, Z., Zhang, Y., & Wang, X. (2023). Transcriptome, Ectopic Expression and Genetic Population Analysis Identify Candidate Genes for Fiber Quality Improvement in Cotton. International Journal of Molecular Sciences, 24(9), 8293. https://doi.org/10.3390/ijms24098293