

Vitamin D Modulates the Response of Patient-Derived Metastatic Melanoma Cells to Anticancer Drugs

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Results
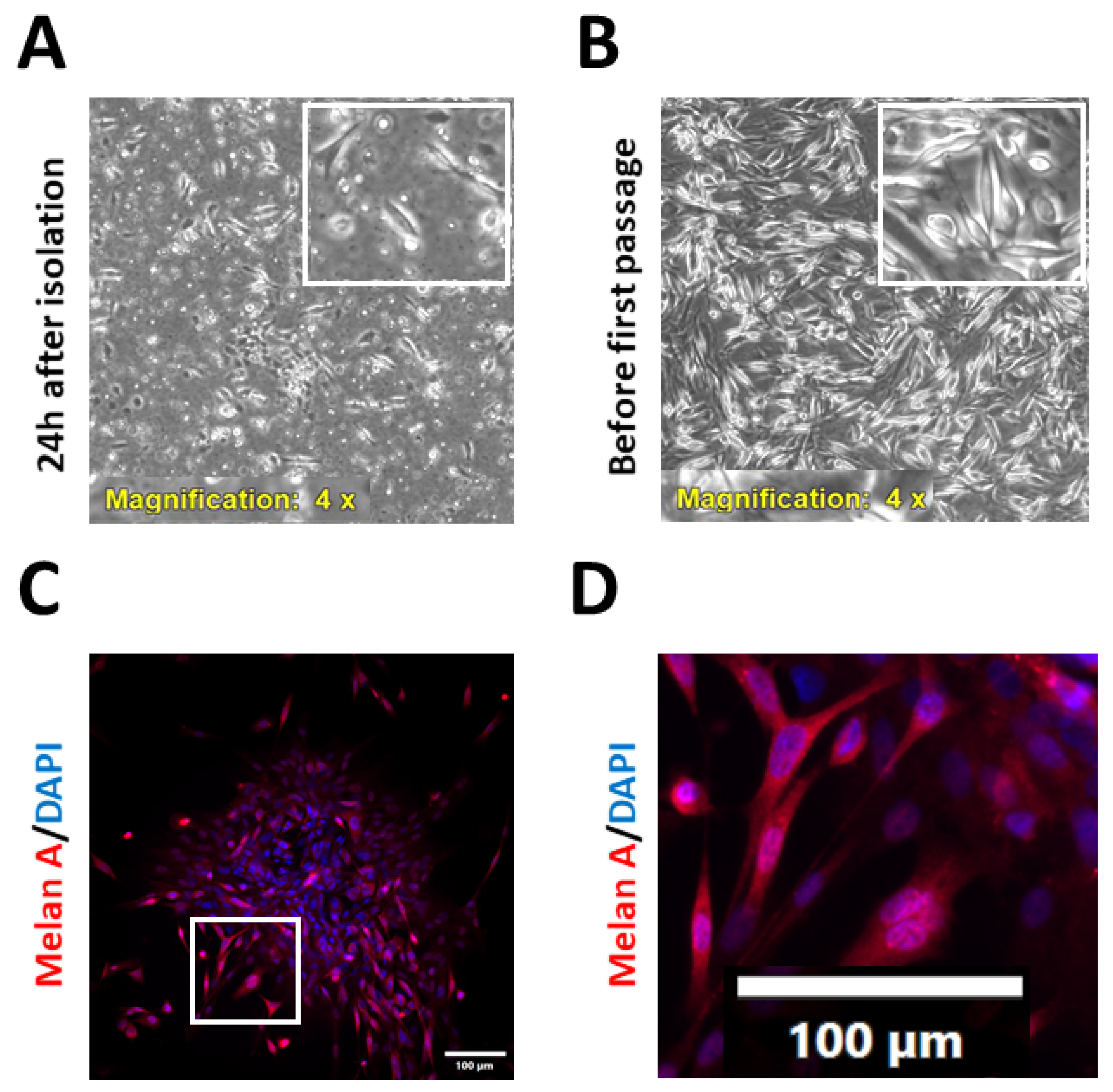
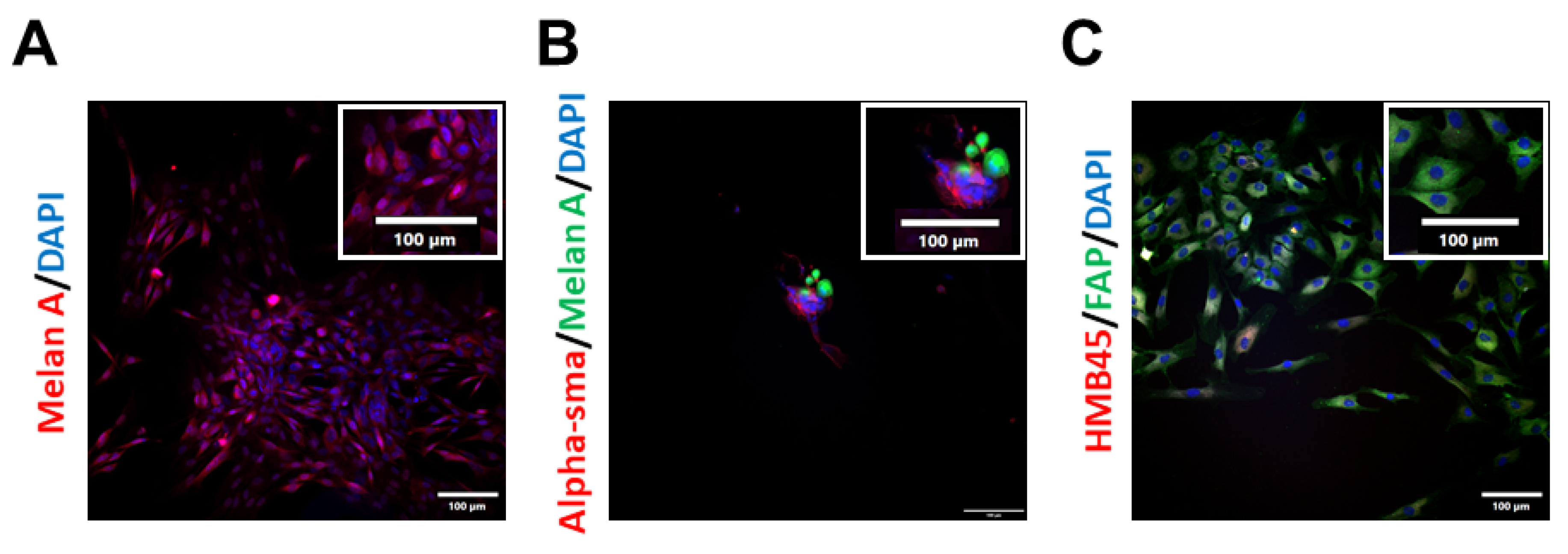
2.1. Isolation and Characterisation of Patient-Derived Melanoma Cell Cultures
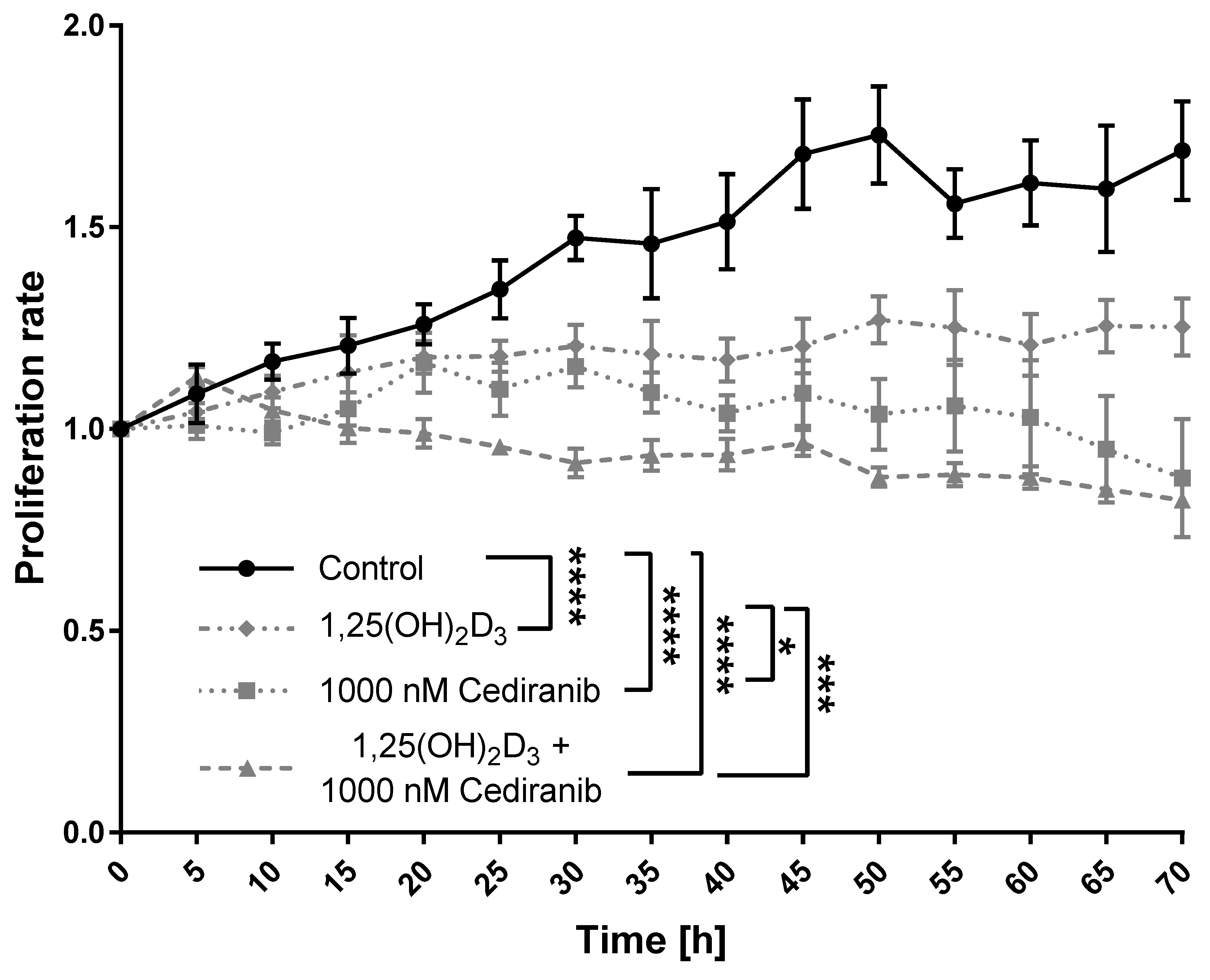
2.2. 1,25(OH)2D3 and Cediranib Compromise the Growth of Patient-Derived Melanoma Cells
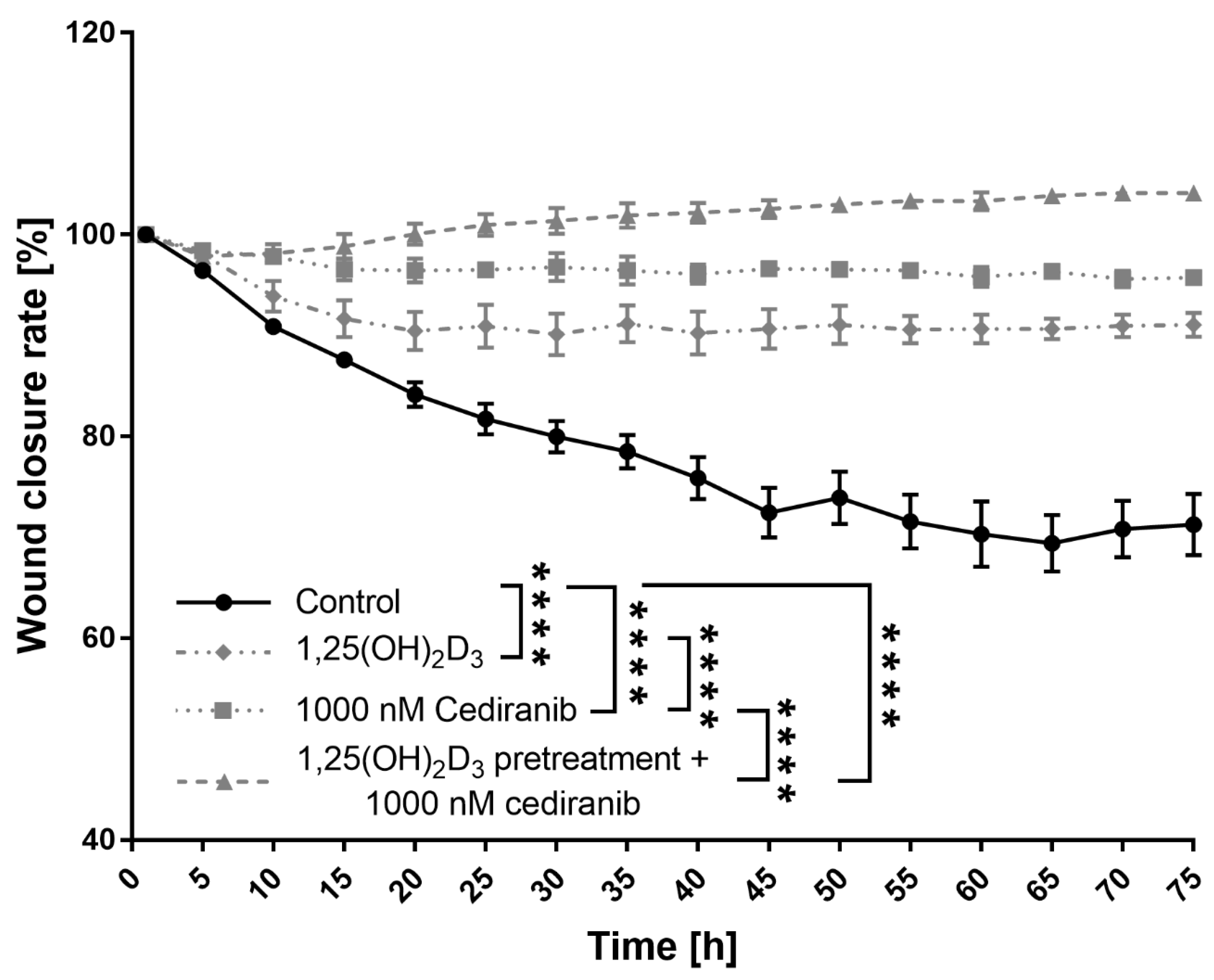
2.3. 1,25(OH)2D3 Preconditioning Significantly Decreases the Mobility of the Patient-Derived Melanoma Cells Treated with Cediranib
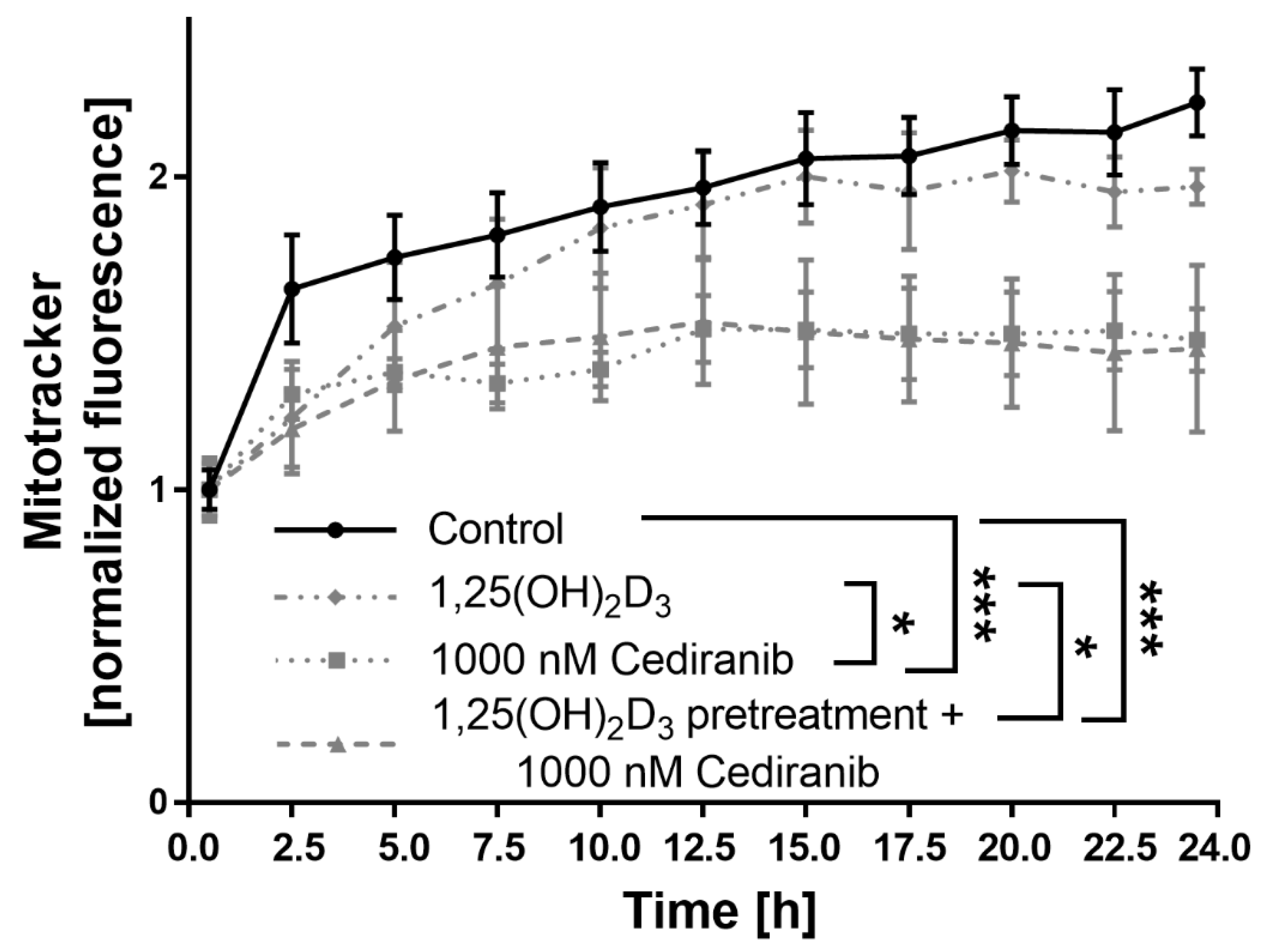
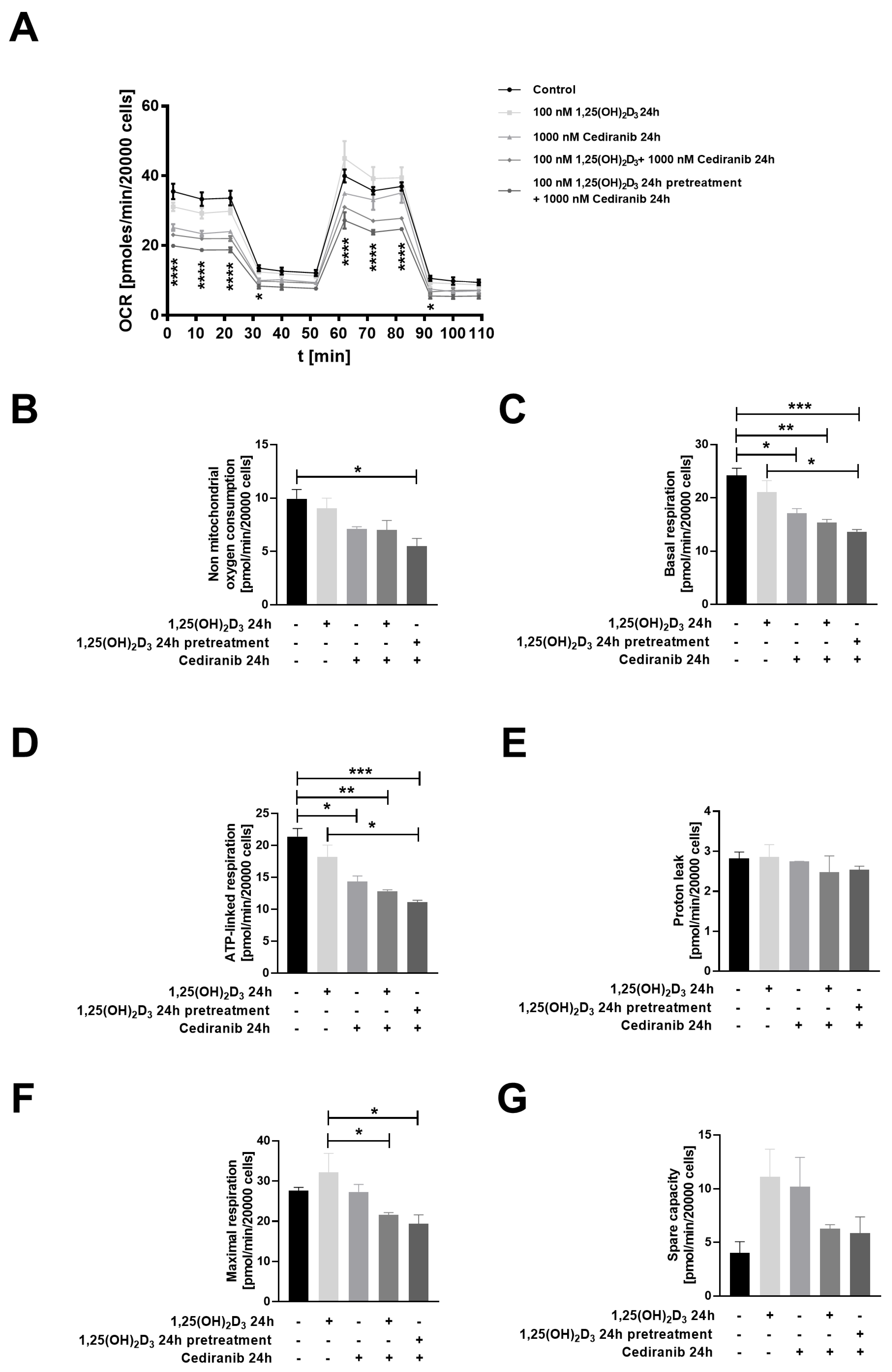
2.4. Cediranib Decreases Mitochondrial Membrane Potential in Patient-Derived Melanoma Cells
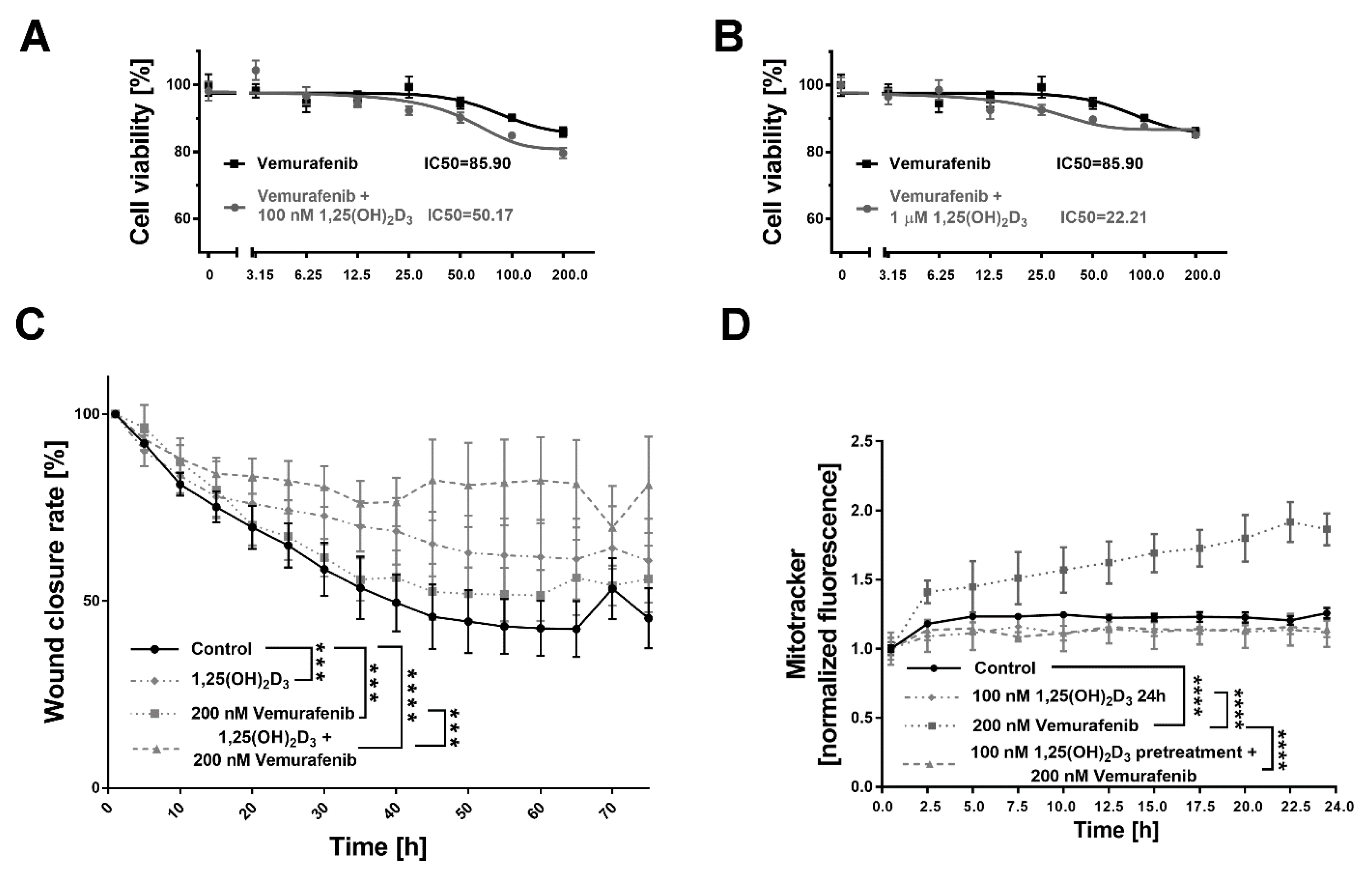
2.5. 1,25(OH)2D3 Significantly Decreases Viability and Mobility of BRAF+ Patient-Derived Cells Treated with Vemurafenib, and the Effect Is Mitochondria-Independent
3. Discussion
4. Materials and Methods
4.1. Chemicals
4.2. Isolation of Melanoma Patient-Derived Cells and Their Cultivation
4.3. Immunofluorescence Staining
4.4. SRB Viability Assay
4.5. Proliferation Rate Assay
4.6. Wound Closure Rate
4.7. Mitochondrial Membrane Potential
4.8. Measurement of Melanoma Bioenergetics
4.9. Statistical Analysis
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Schadendorf, D.; van Akkooi, A.C.J.; Berking, C.; Griewank, K.G.; Gutzmer, R.; Hauschild, A.; Stang, A.; Roesch, A.; Ugurel, S. Melanoma. Lancet 2018, 392, 971–984. [Google Scholar] [CrossRef] [PubMed]
- Switzer, B.; Puzanov, I.; Skitzki, J.J.; Hamad, L.; Ernstoff, M.S. Managing Metastatic Melanoma in 2022: A Clinical Review. JCO Oncol. Pract. 2022, 18, 335–351. [Google Scholar] [CrossRef] [PubMed]
- Dhanyamraju, P.K.; Patel, T.N. Melanoma therapeutics: A literature review. J. Biomed. Res. 2022, 36, 77. [Google Scholar] [CrossRef] [PubMed]
- Eftekhari, A.; Ahmadian, E.; Salatin, S.; Sharifi, S.; Dizaj, S.M.; Khalilov, R.; Hasanzadeh, M. Current analytical approaches in diagnosis of melanoma. TrAC Trends Anal. Chem. 2019, 116, 122–135. [Google Scholar] [CrossRef]
- Eddy, K.; Shah, R.; Chen, S. Decoding Melanoma Development and Progression: Identification of Therapeutic Vulnerabilities. Front. Oncol. 2021, 10. [Google Scholar] [CrossRef] [PubMed]
- Siegel, R.L.; Miller, K.D.; Fuchs, H.E.; Jemal, A. Cancer statistics. CA Cancer J. Clin. 2021, 71, 7–33. [Google Scholar] [CrossRef]
- Siegel, R.L.; Miller, K.D.; Fuchs, H.E.; Jemal, A. Cancer statistics. CA Cancer J. Clin. 2022, 72, 7–33. [Google Scholar] [CrossRef]
- Siegel, R.L.; Miller, K.D.; Wagle, N.S.; Jemal, A. Cancer statistics, 2023. CA A Cancer J. Clin. 2023, 73, 17–48. [Google Scholar] [CrossRef]
- Domingues, B.; Lopes, J.M.; Soares, P.; Populo, H. Melanoma treatment in review. ImmunoTargets Ther. 2018, ume 7, 35–49. [Google Scholar] [CrossRef]
- Patton, E.E.; Mueller, K.L.; Adams, D.J.; Anandasabapathy, N.; Aplin, A.E.; Bertolotto, C.; Bosenberg, M.; Ceol, C.J.; Burd, C.E.; Chi, P.; et al. Melanoma models for the next generation of therapies. Cancer Cell 2021, 39, 610–631. [Google Scholar] [CrossRef]
- Lorusso, P.M.; Schalper, K.; Sosman, J. Targeted therapy and immunotherapy: Emerging biomarkers in metastatic melanoma. Pigment. Cell Melanoma Res. 2020, 33, 390–402. [Google Scholar] [CrossRef] [PubMed]
- Imbert, C.; Montfort, A.; Fraisse, M.; Marcheteau, E.; Gilhodes, J.; Martin, E.; Bertrand, F.; Marcellin, M.; Burlet-Schiltz, O.; Peredo, A.G.; et al. Resistance of melanoma to immune checkpoint inhibitors is overcome by targeting the sphingosine kinase-1. Nat. Commun. 2020, 11, 437. [Google Scholar] [CrossRef]
- Chen, J.; Tang, Z.; Slominski, A.T.; Li, W.; Zmijewski, M.; Liu, Y.; Chen, J. Vitamin D and its analogs as anticancer and anti-inflammatory agents. Eur. J. Med. Chem. 2020, 207, 112738. [Google Scholar] [CrossRef] [PubMed]
- Slominski, A.T.; A Brożyna, A.; A Zmijewski, M.; Jóźwicki, W.; Jetten, A.M.; Mason, R.S.; Tuckey, R.C.; A Elmets, C. Vitamin D signaling and melanoma: Role of vitamin D and its receptors in melanoma progression and management. Lab. Investig. 2017, 97, 706–724. [Google Scholar] [CrossRef]
- Piotrowska, A.; Wierzbicka, J.; Żmijewski, M.A. Vitamin D in the skin physiology and pathology. Acta Biochim. Pol. 2016, 63, 17–29. [Google Scholar] [CrossRef] [PubMed]
- Wierzbicka, J.; Piotrowska, A.; A Żmijewski, M. The renaissance of vitamin D. Acta Biochim. Pol. 2014, 61, 679–686. [Google Scholar] [CrossRef]
- Piotrowska, A.; Beserra, F.P.; Wierzbicka, J.M.; Nowak, J.I.; Żmijewski, M.A. Vitamin D Enhances Anticancer Properties of Cediranib, a VEGFR Inhibitor, by Modulation of VEGFR2 Expression in Melanoma Cells. Front. Oncol. 2021, 11. [Google Scholar] [CrossRef]
- Muñoz, A.; Grant, W.B. Vitamin D and Cancer: An Historical Overview of the Epidemiology and Mechanisms. Nutrients 2022, 14, 1448. [Google Scholar] [CrossRef]
- Fathi, N.; Ahmadian, E.; Shahi, S.; Roshangar, L.; Khan, H.; Kouhsoltani, M.; Dizaj, S.M.; Sharifi, S. Role of vitamin D and vitamin D receptor (VDR) in oral cancer. Biomed. Pharmacother. 2018, 109, 391–401. [Google Scholar] [CrossRef]
- Ferrer-Mayorga, G.; Gómez-López, G.; Barbáchano, A.; Fernández-Barral, A.; Peña, C.; Pisano, D.G.; Cantero, R.; Rojo, F.; Muñoz, A.; Larriba, M.J. Vitamin D receptor expression and associated gene signature in tumour stromal fibroblasts predict clinical outcome in colorectal cancer. Gut 2016, 66, 1449–1462. [Google Scholar] [CrossRef]
- Han, J.; Guo, X.; Yu, X.; Liu, S.; Cui, X.; Zhang, B.; Liang, H. 25-Hydroxyvitamin D and Total Cancer Incidence and Mortality: A Meta-Analysis of Prospective Cohort Studies. Nutrients 2019, 11, 2295. [Google Scholar] [CrossRef]
- Manson, J.E.; Cook, N.R.; Lee, I.M.; Christen, W.; Bassuk, S.S.; Mora, S.; Gibson, H.; Gordon, D.; Copeland, T.; D’Agostino, D.; et al. Vitamin D Supplements and Prevention of Cancer and Cardiovascular Disease. N. Engl. J. Med. 2019, 380, 33–44. [Google Scholar] [CrossRef]
- Grant, W.B.; Boucher, B.J. Why Secondary Analyses in Vitamin D Clinical Trials Are Important and How to Improve Vitamin D Clinical Trial Outcome Analyses-A Comment on "Extra-Skeletal Effects of Vitamin D, Nutrients 2019, 11, 1460". Nutrients 2019, 11, 2182. [Google Scholar] [CrossRef]
- Chandler, P.D.; Chen, W.Y.; Ajala, O.N.; Hazra, A.; Cook, N.; Bubes, V.; Lee, I.-M.; Giovannucci, E.L.; Willett, W.; Buring, J.E.; et al. Effect of Vitamin D3 Supplements on Development of Advanced Cancer: A Secondary Analysis of the VITAL Randomized Clinical Trial. JAMA Netw. Open 2020, 3, e2025850. [Google Scholar] [CrossRef] [PubMed]
- Lappe, J.M.; Travers-Gustafson, D.; Davies, K.M.; Recker, R.R.; Heaney, R.P. Vitamin D and calcium supplementation reduces cancer risk: Results of a randomized trial. Am. J. Clin. Nutr. 2007, 85, 1586–1591. [Google Scholar] [CrossRef]
- Holick, M.F. Cancer, sunlight and vitamin D. J. Clin. Transl. Endocrinol. 2014, 1, 179–186. [Google Scholar] [CrossRef]
- Keum, N.; Lee, D.H.; Greenwood, D.C.; Manson, J.E.; Giovannucci, E. Vitamin D supplementation and total cancer incidence and mortality: A meta-analysis of randomized controlled trials. Ann. Oncol. 2019, 30, 733–743. [Google Scholar] [CrossRef] [PubMed]
- Wyatt, C.; Lucas, R.; Hurst, C.; Kimlin, M. Vitamin D Deficiency at Melanoma Diagnosis Is Associated with Higher Breslow Thickness. PLoS ONE 2015, 10, e0126394. [Google Scholar] [CrossRef] [PubMed]
- De Smedt, J.; Van Kelst, S.; Boecxstaens, V.; Stas, M.; Bogaerts, K.; Vanderschueren, D.; Aura, C.; Vandenberghe, K.; Lambrechts, D.; Wolter, P.; et al. Vitamin D supplementation in cutaneous malignant melanoma outcome (ViDMe): A randomized controlled trial. BMC Cancer 2017, 17, 1–10. [Google Scholar] [CrossRef]
- Piotrowska, A.; Wierzbicka, J.; Rybarczyk, A.; Tuckey, R.C.; Slominski, A.T.; Zmijewski, M.A. Vitamin D and its low calcemic analogs modulate the anticancer properties of cisplatin and dacarbazine in the human melanoma A375 cell line. Int. J. Oncol. 2019, 54, 1481–1495. [Google Scholar] [CrossRef]
- Wasiewicz, T.; Piotrowska, A.; Wierzbicka, J.; Slominski, A.T.; Zmijewski, M.A. Antiproliferative Activity of Non-Calcemic Vitamin D Analogs on Human Melanoma Lines in Relation to VDR and PDIA3 Receptors. Int. J. Mol. Sci. 2018, 19, 2583. [Google Scholar] [CrossRef] [PubMed]
- Slominski, A.T.; Brozyna, A.; Jozwicki, W.; Tuckey, R.C. Vitamin D as an adjuvant in melanoma therapy. Melanoma Manag. 2015, 2, 1–4. [Google Scholar] [CrossRef]
- Ma, Y.; Yu, W.-D.; Trump, D.L.; Johnson, C.S. 1,25D3 Enhances antitumor activity of gemcitabine and cisplatin in human bladder cancer models. Cancer 2010, 116, 3294–3303. [Google Scholar] [CrossRef] [PubMed]
- Rassnick, K.M.; Muindi, J.R.; Johnson, C.S.; Balkman, C.E.; Ramnath, N.; Yu, W.-D.; Engler, K.L.; Page, R.L.; Trump, D.L. In vitro and in vivo evaluation of combined calcitriol and cisplatin in dogs with spontaneously occurring tumors. Cancer Chemother. Pharmacol. 2008, 62, 881–891. [Google Scholar] [CrossRef]
- Podgorska, E.; Drzal, A.; Matuszak, Z.; Swakon, J.; Slominski, A.; Elas, M.; Urbanska, K. Calcitriol and Calcidiol Can Sensitize Melanoma Cells to Low–LET Proton Beam Irradiation. Int. J. Mol. Sci. 2018, 19, 2236. [Google Scholar] [CrossRef]
- Banerjee, S.S.; Harris, M. Morphological and immunophenotypic variations in malignant melanoma. Histopathology 2000, 36, 387–402. [Google Scholar] [CrossRef]
- Saleem, A.; Narala, S.; Raghavan, S.S. Immunohistochemistry in melanocytic lesions: Updates with a practical review for pathologists. Semin. Diagn. Pathol. 2022, 39, 239–247. [Google Scholar] [CrossRef] [PubMed]
- Kuźbicki, Ł.; Brożyna, A.A. The detectability of intraepidermal melanocytes—A narrative review of immunohistochemical studies. J. Cutan. Pathol. 2022, 49, 1074–1089. [Google Scholar] [CrossRef]
- Ordóñez, N.G. Value of melanocytic-associated immunohistochemical markers in the diagnosis of malignant melanoma: A review and update. Hum. Pathol. 2013, 45, 191–205. [Google Scholar] [CrossRef]
- Chapman, S.W.K.; Metzger, N.; Grest, P.; Feige, K.; Von Rechenberg, B.; Auer, J.A.; Hottiger, M.O. Isolation, establishment, and characterization of ex vivo equine melanoma cell cultures. Vitr. Cell. Dev. Biol. Anim. 2008, 45, 152–162. [Google Scholar] [CrossRef]
- Sobiepanek, A.; Kowalska, P.D.; Soszyńska, M.; Kobiela, T.; Ścieżynska, A. A short guide on the selection of melanocytes andmelanoma cells’ isolation procedures for cancer research. Adv. Biomed. Res. Cancer Prev. Treat. 2020, 6, 67–78. [Google Scholar]
- Sobiepanek, A.; Kowalska, P.D.; Soszyńska, M.; Ścieżyńska, A. Implementation of Geneticin in the in vitro cell culture and in vivo studies. Adv. Biomed. Res. COVID Med. Humanit. 2020, 6, 79–87. [Google Scholar]
- Wedge, S.R.; Kendrew, J.; Hennequin, L.F.; Valentine, P.J.; Barry, S.T.; Brave, S.R.; Smith, N.R.; James, N.H.; Dukes, M.; Curwen, J.O.; et al. AZD2171: A Highly Potent, Orally Bioavailable, Vascular Endothelial Growth Factor Receptor-2 Tyrosine Kinase Inhibitor for the Treatment of Cancer. Cancer Res. 2005, 65, 4389–4400. [Google Scholar] [CrossRef]
- Stuelten, C.; Parent, C.A.; Montell, D.J. Cell motility in cancer invasion and metastasis: Insights from simple model organisms. Nat. Rev. Cancer 2018, 18, 296–312. [Google Scholar] [CrossRef] [PubMed]
- Di Renzo, M.F.; Corso, S. Patient-Derived Cancer Models. Cancers 2020, 12, 3779. [Google Scholar] [CrossRef]
- Kondo, T. Current status and future outlook for patient-derived cancer models from a rare cancer research perspective. Cancer Sci. 2020, 112, 953–961. [Google Scholar] [CrossRef]
- Bhadury, J.; Einarsdottir, B.O.; Podraza, A.; Bagge, R.O.; Stierner, U.; Ny, L.; López, M.D.; Nilsson, J.A. Hypoxia-regulated gene expression explains differences between melanoma cell line-derived xenografts and patient-derived xenografts. Oncotarget 2016, 7, 23801–23811. [Google Scholar] [CrossRef]
- Gazdar, A.F.; Kurvari, V.; Virmani, A.; Gollahon, L.; Sakaguchi, M.; Westerfield, M.; Kodagoda, D.; Stasny, V.; Cunningham, H.T.; Wistuba, I.I.; et al. Characterization of paired tumor and non-tumor cell lines established from patients with breast cancer. Int. J. Cancer 1998, 78, 766–774. [Google Scholar] [CrossRef]
- Cailleau, R.; Olivé, M.; Cruciger, Q.V.J. Long-term human breast carcinoma cell lines of metastatic origin: Preliminary characterization. Vitr. Cell. Dev. Biol. Plant 1978, 14, 911–915. [Google Scholar] [CrossRef] [PubMed]
- McCallum, H.M.; Lowther, G.W. Long-term culture of primary breast cancer in defined medium. Breast Cancer Res. Treat. 1996, 39, 247–259. [Google Scholar] [CrossRef] [PubMed]
- Xiao, M.; Rebecca, V.W.; Herlyn, M. A Melanoma Patient-Derived Xenograft Model. J. Vis. Exp. 2019, e59508. [Google Scholar] [CrossRef]
- Sztiller-Sikorska, M.; Hartman, M.L.; Talar, B.; Jakubowska, J.; Zalesna, I.; Czyz, M. Phenotypic diversity of patient-derived melanoma populations in stem cell medium. Lab. Investig. 2015, 95, 672–683. [Google Scholar] [CrossRef]
- Neal, J.T.; Li, X.; Zhu, J.; Giangarra, V.; Grzeskowiak, C.L.; Ju, J.; Liu, I.H.; Chiou, S.-H.; Salahudeen, A.A.; Smith, A.R.; et al. Organoid Modeling of the Tumor Immune Microenvironment. Cell 2018, 175, 1972–1988. [Google Scholar] [CrossRef]
- Tsao, A.S.; Moon, J.; Wistuba, I.I.; Vogelzang, N.J.; Kalemkerian, G.P.; Redman, M.W.; Gandara, D.R.; Kelly, K. Phase I Trial of Cediranib in Combination with Cisplatin and Pemetrexed in Chemonaive Patients with Unresectable Malignant Pleural Mesothelioma (SWOG S0905). J. Thorac. Oncol. 2017, 12, 1299–1308. [Google Scholar] [CrossRef] [PubMed]
- Kaplan, A.R.; Gueble, S.E.; Liu, Y.; Oeck, S.; Kim, H.; Yun, Z.; Glazer, P.M. Cediranib suppresses homology-directed DNA repair through down-regulation of BRCA1/2 and RAD51. Sci. Transl. Med. 2019, 11, eaav4508. [Google Scholar] [CrossRef] [PubMed]
- Hielscher, A.; Gerecht, S. Hypoxia and free radicals: Role in tumor progression and the use of engineering-based platforms to address these relationships. Free. Radic. Biol. Med. 2015, 79, 281–291. [Google Scholar] [CrossRef] [PubMed]
- Kuo, C.-L.; Babuharisankar, A.P.; Lin, Y.-C.; Lien, H.-W.; Lo, Y.K.; Chou, H.-Y.; Tangeda, V.; Cheng, L.-C.; Cheng, A.N.; Lee, A.Y.-L. Mitochondrial oxidative stress in the tumor microenvironment and cancer immunoescape: Foe or friend? J. Biomed. Sci. 2022, 29, 1–25. [Google Scholar] [CrossRef]
- Ricca, C.; Aillon, A.; Bergandi, L.; Alotto, D.; Castagnoli, C.; Silvagno, F. Vitamin D Receptor Is Necessary for Mitochondrial Function and Cell Health. Int. J. Mol. Sci. 2018, 19, 1672. [Google Scholar] [CrossRef] [PubMed]
- Olszewska, A.M.; Sieradzan, A.K.; Bednarczyk, P.; Szewczyk, A.; Żmijewski, M.A. Mitochondrial potassium channels: A novel calcitriol target. Cell. Mol. Biol. Lett. 2022, 27, 1–20. [Google Scholar] [CrossRef]
- McWhirter, E.; Quirt, I.; Gajewski, T.; Pond, G.; Wang, L.; Hui, J.; Oza, A. A phase II study of cediranib, an oral VEGF inhibitor, in previously untreated patients with metastatic or recurrent malignant melanoma. Investig. New Drugs 2016, 34, 231–235. [Google Scholar] [CrossRef]
- Jeon, S.-M.; Shin, E.-A. Exploring vitamin D metabolism and function in cancer. Exp. Mol. Med. 2018, 50, 1–14. [Google Scholar] [CrossRef] [PubMed]
- Koul, P.A.; Ahmad, S.H.; Ahmad, F.; Jan, R.A.; Shah, S.; Khan, U.H. Vitamin D Toxicity in Adults: A Case Series from an Area with Endemic Hypovitaminosis D. Oman Med. J. 2011, 26, 201–204. [Google Scholar] [CrossRef] [PubMed]
- Marcinowska-Suchowierska, E.; Kupisz-Urbańska, M.; Łukaszkiewicz, J.; Płudowski, P.; Jones, G. Vitamin D Toxicity–A Clinical Perspective. Front. Endocrinol. 2018, 9, 550. [Google Scholar] [CrossRef]
- Holick, M.F.; Binkley, N.C.; Bischoff-Ferrari, H.A.; Gordon, C.M.; Hanley, D.A.; Heaney, R.P.; Murad, M.H.; Weaver, C.M. Evaluation, Treatment, and Prevention of Vitamin D Deficiency: An Endocrine Society Clinical Practice Guideline. Med. J. Clin. Endocrinol. Metab. 2011, 96, 1911–1930. [Google Scholar] [CrossRef] [PubMed]
- Dudenkov, D.V.; Yawn, B.P.; Oberhelman, S.S.; Fischer, P.R.; Singh, R.J.; Cha, S.S.; Maxson, J.A.; Quigg, S.M.; Thacher, T. Changing Incidence of Serum 25-Hydroxyvitamin D Values Above 50 ng/mL: A 10-Year Population-Based Study. Mayo Clin. Proc. 2015, 90, 577–586. [Google Scholar] [CrossRef]
- Płudowski, P.; Kos-Kudła, B.; Walczak, M.; Fal, A.; Zozulińska-Ziółkiewicz, D.; Sieroszewski, P.; Peregud-Pogorzelski, J.; Lauterbach, R.; Targowski, T.; Lewiński, A.; et al. Guidelines for Preventing and Treating Vitamin D Deficiency: A 2023 Update in Poland. Nutrients 2023, 15, 695. [Google Scholar] [CrossRef]
- Ng, K.; Nimeiri, H.S.; McCleary, N.J.; Abrams, T.A.; Yurgelun, M.B.; Cleary, J.M.; Rubinson, D.A.; Schrag, D.; Miksad, R.; Bullock, A.J.; et al. Effect of High-Dose vs Standard-Dose Vitamin D3 Supplementation on Progression-Free Survival Among Patients with Advanced or Metastatic Colorectal Cancer: The SUNSHINE Randomized Clinical Trial. JAMA 2019, 321, 1370–1379. [Google Scholar] [CrossRef]
- Slominski, A.T.; Brożyna, A.A.; Skobowiat, C.; Zmijewski, M.A.; Kim, T.-K.; Janjetovic, Z.; Oak, A.S.; Jozwicki, W.; Jetten, A.M.; Mason, R.S.; et al. On the role of classical and novel forms of vitamin D in melanoma progression and management. J. Steroid Biochem. Mol. Biol. 2018, 177, 159–170. [Google Scholar] [CrossRef]
- Timerman, D.; McEnery-Stonelake, M.; Joyce, C.J.; Nambudiri, V.E.; Hodi, F.S.; Claus, E.B.; Ibrahim, N.; Lin, J.Y. Vitamin D deficiency is associated with a worse prognosis in metastatic melanoma. Oncotarget 2016, 8, 6873–6882. [Google Scholar] [CrossRef]
- Johansson, H.; Spadola, G.; Tosti, G.; Mandalà, M.; Minisini, A.M.; Queirolo, P.; Aristarco, V.; Baldini, F.; Cocorocchio, E.; Albertazzi, E.; et al. Vitamin D Supplementation and Disease-Free Survival in Stage II Melanoma: A Randomized Placebo Controlled Trial. Nutrients 2021, 13, 1931. [Google Scholar] [CrossRef]
- Kanwar, R.; Rathee, J.; Salunke, D.B.; Mehta, S.K. Green Nanotechnology-Driven Drug Delivery Assemblies. ACS Omega 2019, 4, 8804–8815. [Google Scholar] [CrossRef]
- Davis, L.E.; Shalin, S.C.; Tackett, A.J. Current state of melanoma diagnosis and treatment. Cancer Biol. Ther. 2019, 20, 1366–1379. [Google Scholar] [CrossRef] [PubMed]
- Trotta, A.P.; Gelles, J.D.; Serasinghe, M.N.; Loi, P.; Arbiser, J.L.; Chipuk, J.E. Disruption of mitochondrial electron transport chain function potentiates the pro-apoptotic effects of MAPK inhibition. J. Biol. Chem. 2017, 292, 11727–11739. [Google Scholar] [CrossRef]
- Davies, H.; Bignell, G.R.; Cox, C.; Stephens, P.; Edkins, S.; Clegg, S.; Teague, J.; Woffendin, H.; Garnett, M.J.; Bottomley, W.; et al. Mutations of the BRAF gene in human cancer. Nature 2002, 417, 949–954. [Google Scholar] [CrossRef]
- Fearfield, L.; Nobbs, J.; Petruckevitch, A.; Harland, C. Severe vitamin D deficiency associated with BRAF-mutated melanoma. Br. J. Dermatol. 2019, 181, 1343. [Google Scholar] [CrossRef]
- Segaoula, Z.; Primot, A.; Lepretre, F.; Hedan, B.; Bouchaert, E.; Minier, K.; Marescaux, L.; Serres, F.; Galiègue-Zouitina, S.; André, C.; et al. Isolation and characterization of two canine melanoma cell lines: New models for comparative oncology. BMC Cancer 2018, 18, 1–16. [Google Scholar] [CrossRef]
- Einarsdottir, B.O.; Bagge, R.O.; Bhadury, J.; Jespersen, H.; Mattsson, J.; Nilsson, L.M.; Truvé, K.; López, M.D.; Naredi, P.; Nilsson, O.; et al. Melanoma patient-derived xenografts accurately model the disease and develop fast enough to guide treatment decisions. Oncotarget 2014, 5, 9609–9618. [Google Scholar] [CrossRef]
- Kovacs, D.; Migliano, E.; Muscardin, L.; Silipo, V.; Catricalà, C.; Picardo, M.; Bellei, B. The role of WNT/β-catenin signaling pathway in melanoma epithelial-to-mesenchymal-like switching: Evidences from patients-derived cell lines. Oncotarget 2016, 7, 43295–43314. [Google Scholar] [CrossRef]
- Piotrowska, A.; Wierzbicka, J.; Ślebioda, T.; Woźniak, M.; Tuckey, R.C.; Slominski, A.T.; Żmijewski, M.A. Vitamin D derivatives enhance cytotoxic effects of H2O2 or cisplatin on human keratinocytes. Steroids 2016, 110, 49–61. [Google Scholar] [CrossRef] [PubMed]
- Franczak, M.; Kutryb-Zajac, B.; El Hassouni, B.; Giovannetti, E.; Granchi, C.; Minutolo, F.; Smolenski, R.T.; Peters, G.J. The effect of lactate dehydrogenase-A inhibition on intracellular nucleotides and mitochondrial respiration in pancreatic cancer cells. Nucleosides Nucleotides Nucleic Acids 2022, 41, 1375–1385. [Google Scholar] [CrossRef] [PubMed]







Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Piotrowska, A.; Zaucha, R.; Król, O.; Żmijewski, M.A. Vitamin D Modulates the Response of Patient-Derived Metastatic Melanoma Cells to Anticancer Drugs. Int. J. Mol. Sci. 2023, 24, 8037. https://doi.org/10.3390/ijms24098037
Piotrowska A, Zaucha R, Król O, Żmijewski MA. Vitamin D Modulates the Response of Patient-Derived Metastatic Melanoma Cells to Anticancer Drugs. International Journal of Molecular Sciences. 2023; 24(9):8037. https://doi.org/10.3390/ijms24098037
Chicago/Turabian StylePiotrowska, Anna, Renata Zaucha, Oliwia Król, and Michał Aleksander Żmijewski. 2023. "Vitamin D Modulates the Response of Patient-Derived Metastatic Melanoma Cells to Anticancer Drugs" International Journal of Molecular Sciences 24, no. 9: 8037. https://doi.org/10.3390/ijms24098037
APA StylePiotrowska, A., Zaucha, R., Król, O., & Żmijewski, M. A. (2023). Vitamin D Modulates the Response of Patient-Derived Metastatic Melanoma Cells to Anticancer Drugs. International Journal of Molecular Sciences, 24(9), 8037. https://doi.org/10.3390/ijms24098037