

Uremic Toxin Indoxyl Sulfate Promotes Macrophage-Associated Low-Grade Inflammation and Epithelial Cell Senescence


 ,
,  , and
, and 
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Results
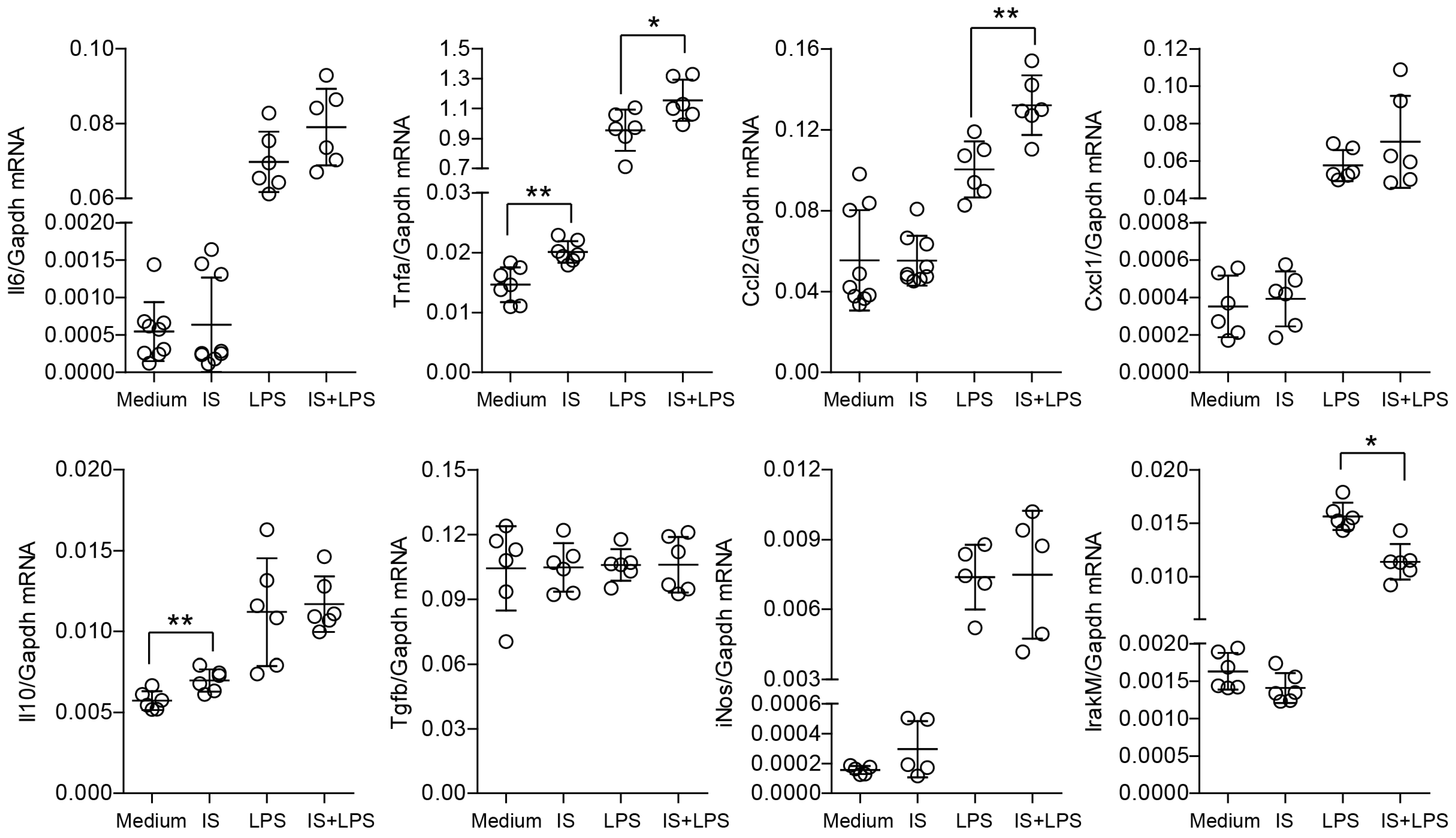
2.1. Indoxyl Sulfate Induces Proinflammatory Transcripts in Primary Macrophages In Vitro and Modulates Their Immune Response and Metabolic Activity upon LPS Stimulation
2.2. Indoxyl Sulfate Does Not Affect the Self-Limiting Nature of NF-kB-Associated Inflammatory Signaling
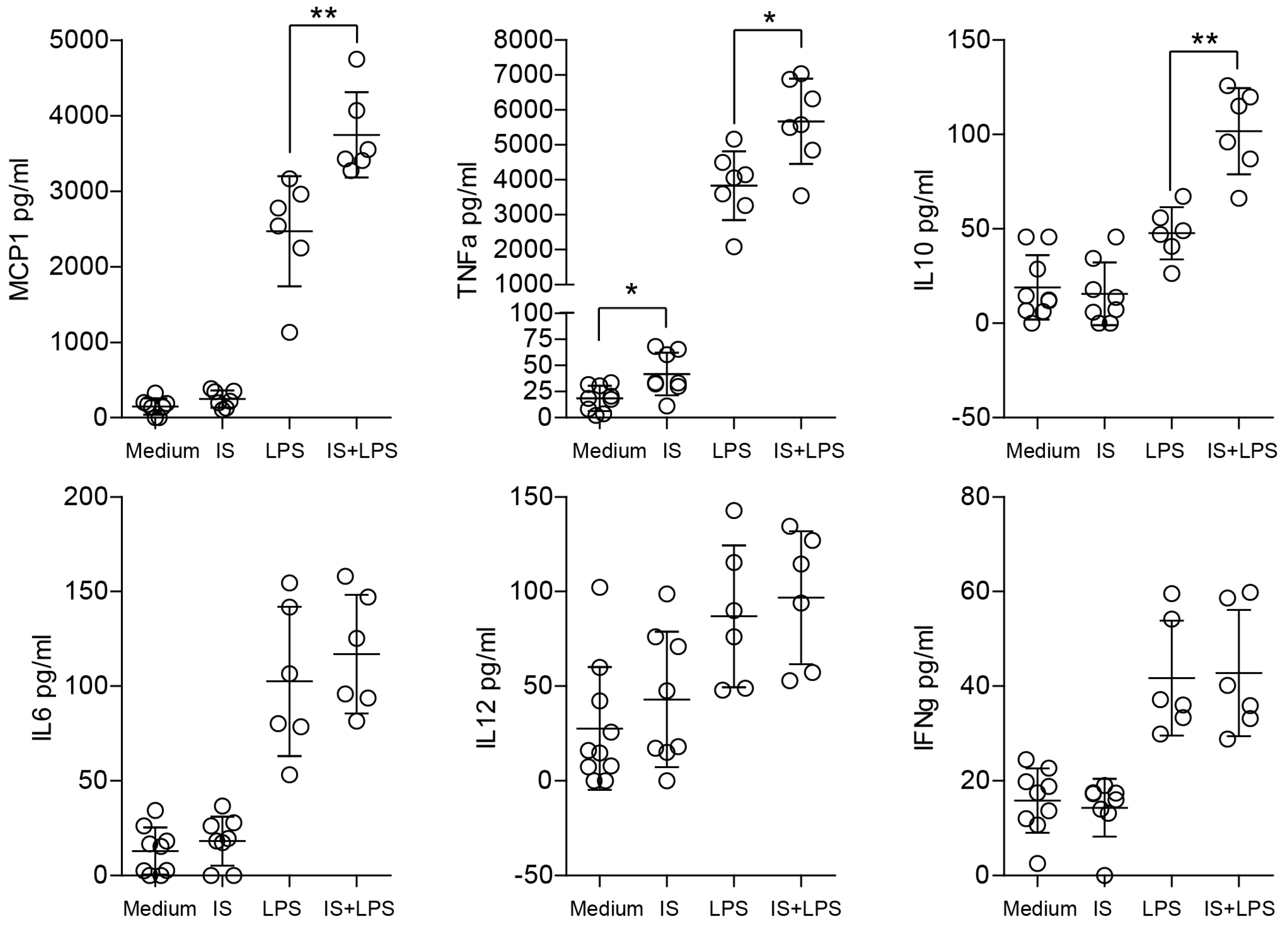
2.3. Indoxyl Sulfate Induces Moderate Inflammation by Enhancing Inflammatory Cytokine Production in Macrophages
2.4. Indoxyl Sulfate Induces ROS Production, Enhances LPS-Induced ROS Release, and Increases Mitochondrial Superoxide Production
2.5. Indoxyl Sulfate Promotes Macrophage Polarization toward a Proinflammatory Phenotype
2.6. Indoxyl Sulfate and LPS Co-Stimulation Induce Ahr Expression
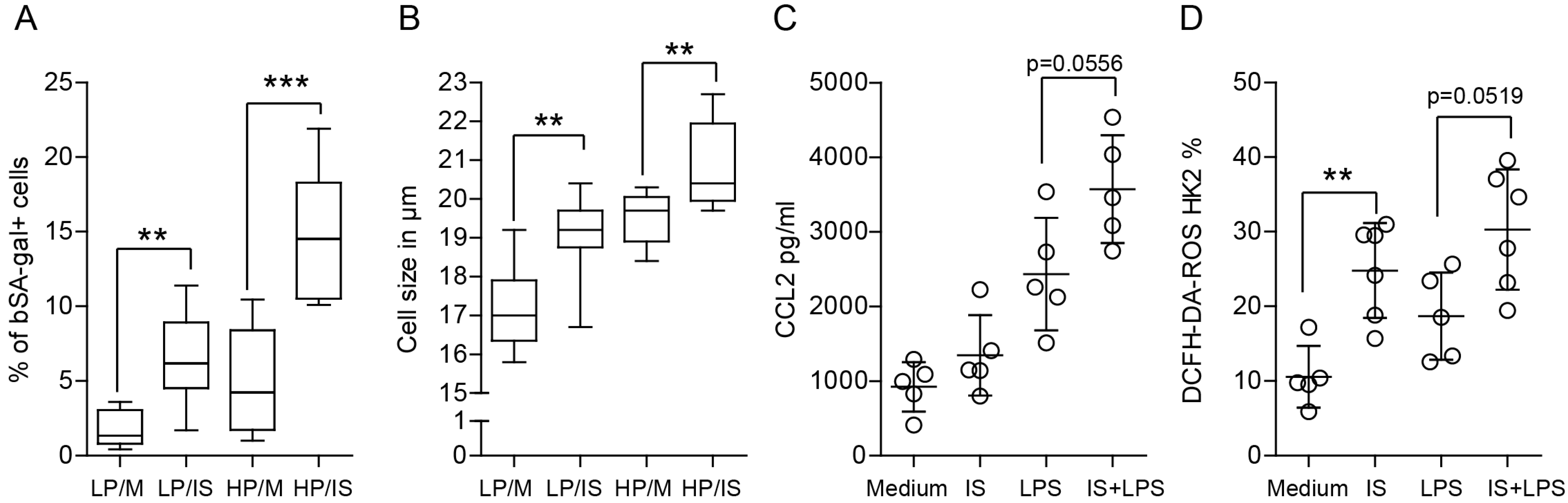
2.7. Indoxyl Sulfate Affects the Proliferation and Metabolic Activity of Renal Tubular Epithelial Cells
2.8. Indoxyl Sulfate Promotes Cellular Senescence by Activating Oxidative Stress
3. Discussion
4. Materials and Methods
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- GBD Chronic Kidney Disease Collaboration (2020). Global, regional, and national burden of chronic kidney disease, 1990–2017: A systematic analysis for the Global Burden of Disease Study. Lancet 2017, 395, 709–733. [Google Scholar]
- Sundström, J.; Bodegard, J. Prevalence, outcomes, and cost of chronic kidney disease in a contemporary population of 2·4 million patients from 11 countries: The CaReMe CKD study. Lancet Reg. Health Eur. 2022, 20, 100438. [Google Scholar] [CrossRef]
- Chi, M.; Tian, Z. The diseased kidney: Aging and senescent immunology. Immun. Ageing 2022, 19, 58. [Google Scholar] [CrossRef]
- Romagnani, P.; Remuzzi, G. Chronic kidney disease. Nat. Rev. 2017, 3, 17088. [Google Scholar] [CrossRef]
- Tejeswini Sen, T.; Kale, A. Promising novel therapeutic targets for kidney disease: Emphasis on kidney-specific proteins. Drug Discov Today 2023, 28, 103466. [Google Scholar] [CrossRef] [PubMed]
- Lichtnekert, J.; Anders, H.-J. Lupus Nephritis: Current Perspectives and Moving Forward. J. Inflamm. Res. 2022, 15, 6533–6552. [Google Scholar] [CrossRef] [PubMed]
- Vanholder, R.; Baurmeister, U. A bench to bedside view of uremic toxins. J. Am. Soc. Nephrol. 2008, 19, 863–870. [Google Scholar] [CrossRef] [PubMed]
- Duranton, F.; Cohen, G. Normal and pathologic concentrations of uremic toxins. J. Am. Soc. Nephrol. 2012, 23, 1258–1270. [Google Scholar] [CrossRef]
- Vanholder, R.; de Smet, R. Review on uremic toxins: Classification, concentration, and interindividual variability. Kidney Int. 2003, 63, 1934–1943. [Google Scholar] [CrossRef]
- Almeras, C.; Argilés, A. The general picture of uremia. Semin. Dial. 2009, 22, 329–333. [Google Scholar] [CrossRef]
- Steiger, S.; Rossaint, J. Secondary Immunodeficiency Related to Kidney Disease (SIDKD)-Definition, Unmet Need, and Mechanisms. J. Am. Soc. Nephrol. 2022, 33, 259–278. [Google Scholar] [CrossRef]
- Falconi, C.A.; Junho, C.V. Uremic Toxins: An Alarming Danger Concerning the Cardiovascular System. Front. Physiol. 2021, 12, 686249. [Google Scholar] [CrossRef]
- Rock, K.L.; Kataoka, H. Uric acid as a danger signal in gout and its comorbidities. Nat. Rev. Rheumatol. 2013, 9, 13–23. [Google Scholar] [CrossRef]
- Desai, J.; Steiger, S. Molecular Pathophysiology of Gout. Trends Mol. Med. 2017, 23, 756–768. [Google Scholar] [CrossRef]
- Sellmayr, M.; Hernandez Petzsche, M.R. Only Hyperuricemia with Crystalluria, but not Asymptomatic Hyperuricemia, Drives Progression of Chronic Kidney Disease. J. Am. Soc. Nephrol. 2020, 31, 2773–2792. [Google Scholar] [CrossRef]
- Kolachalama, V.B.; Shashar, M. Uremic Solute-Aryl Hydrocarbon Receptor-Tissue Factor Axis Associates with Thrombosis after Vascular Injury in Humans. J. Am. Soc. Nephrol. 2018, 29, 1063–1072. [Google Scholar] [CrossRef]
- Zhu, W.; Gregory, J.C. Gut Microbial Metabolite TMAO Enhances Platelet Hyperreactivity and Thrombosis Risk. Cell 2016, 165, 111–124. [Google Scholar] [CrossRef]
- Koppe, L.; Nyam, E. Urea impairs β cell glycolysis and insulin secretion in chronic kidney disease. J. Clin. Investig. 2016, 126, 3598–3612. [Google Scholar] [CrossRef]
- D’Apolito, M.; Du, X. Urea-induced ROS generation causes insulin resistance in mice with chronic renal failure. J. Clin. Investig. 2010, 120, 203–213. [Google Scholar] [CrossRef]
- Koppe, L.; Alix, P.M. p-Cresyl glucuronide is a major metabolite of p-cresol in mouse: In contrast to p-cresyl sulphate, p-cresyl glucuronide fails to promote insulin resistance. Nephrol. Dial. Transplant. 2017, 32, 2000–2009. [Google Scholar] [CrossRef]
- Koppe, L.; Pillon, N.J. p-Cresyl sulfate promotes insulin resistance associated with CKD. J. Am. Soc. Nephrol. 2013, 24, 88–99. [Google Scholar] [CrossRef] [PubMed]
- Leong, S.C.; Sirich, T.L. Indoxyl Sulfate-Review of Toxicity and Therapeutic Strategies. Toxins 2016, 8, 358. [Google Scholar] [CrossRef] [PubMed]
- Vanholder, R.; Schepers, E. The uremic toxicity of indoxyl sulfate and p-cresyl sulfate: A systematic review. J. Am. Soc. Nephrol. 2014, 25, 1897–1907. [Google Scholar] [CrossRef] [PubMed]
- Berg, A.H.; Kumar, S. Indoxyl sulfate in uremia: An old idea with updated concepts. J. Clin. Investig. 2022, 132, e155860. [Google Scholar] [CrossRef]
- Lin, X.; Liang, W. The Accumulation of Gut Microbiome-derived Indoxyl Sulfate and P-Cresyl Sulfate in Patients With End-stage Renal Disease. J. Ren. Nutr. 2022, 32, 578–586. [Google Scholar] [CrossRef]
- Espi, M.; Koppe, L. Chronic Kidney Disease-Associated Immune Dysfunctions: Impact of Protein-Bound Uremic Retention Solutes on Immune Cells. Toxins 2020, 12, 300. [Google Scholar] [CrossRef]
- Kim, H.Y.; Yoo, T.H. Indoxyl sulfate (IS)-mediated immune dysfunction provokes endothelial damage in patients with end-stage renal disease (ESRD). Sci. Rep. 2017, 7, 3057. [Google Scholar] [CrossRef]
- Niwa, T.; Ise, M. Indoxyl sulfate, a circulating uremic toxin, stimulates the progression of glomerular sclerosis. J. Lab. Clin. Med. 1994, 124, 96–104. [Google Scholar]
- Steiger, S.; Kumar, S.V. Immunomodulatory Molecule IRAK-M Balances Macrophage Polarization and Determines Macrophage Responses during Renal Fibrosis. J. Immunol. 2017, 199, 1440–1452. [Google Scholar] [CrossRef]
- Lech, M.; Kantner, C. Interleukin-1 receptor-associated kinase-M suppresses systemic lupus erythematosus. Ann. Rheum. Dis. 2011, 70, 2207–2217. [Google Scholar] [CrossRef]
- Kemmner, S.; Bachmann, Q. STAT1 regulates macrophage number and phenotype and prevents renal fibrosis after ischemia-reperfusion injury. Am. J. Physiol. Ren. Physiol. 2019, 316, F277–F291. [Google Scholar] [CrossRef]
- Ribeiro, A.; Dobosz, E. Macrophage-Specific MCPIP1/Regnase-1 Attenuates Kidney Ischemia-Reperfusion Injury by Shaping the Local Inflammatory Response and Tissue Regeneration. Cells 2022, 11, 397. [Google Scholar] [CrossRef]
- Mulay, S.R.; Lech, M. Editorial: Negative Regulators of Innate Immunity and Their Role in Host Responses to Injury and Infection. Front. Immunol. 2022, 13, 891919. [Google Scholar] [CrossRef]
- Lorenz, G.; Moschovaki-Filippidou, F. IFN Regulatory Factor 4 Controls Post-ischemic Inflammation and Prevents Chronic Kidney Disease. Front. Immunol. 2019, 10, 2162. [Google Scholar] [CrossRef]
- Canton, M.; Sánchez-Rodríguez, R. Reactive Oxygen Species in Macrophages: Sources and Targets. Front. Immunol. 2021, 12, 734229. [Google Scholar] [CrossRef]
- Schmidlin, C.J.; Dodson, M.B. Redox regulation by NRF2 in aging and disease. Free Radic. Biol. Med. 2019, 134, 702–707. [Google Scholar] [CrossRef]
- Murray, P.J. Macrophage Polarization. Annu. Rev. Physiol. 2017, 79, 541–566. [Google Scholar] [CrossRef]
- Baseler, W.A.; Davies, L.C. Autocrine IL-10 functions as a rheostat for M1 macrophage glycolytic commitment by tuning nitric oxide production. Redox Biol. 2016, 10, 12–23. [Google Scholar] [CrossRef]
- Schroeder, J.C.; Dinatale, B.C. The uremic toxin 3-indoxyl sulfate is a potent endogenous agonist for the human aryl hydrocarbon receptor. Biochemistry 2010, 49, 393–400. [Google Scholar] [CrossRef]
- Wu, W.; Bush, K.T. Key Role for the Organic Anion Transporters, OAT1 and OAT3, in the in vivo Handling of Uremic Toxins and Solutes. Sci. Rep. 2017, 7, 4939. [Google Scholar] [CrossRef]
- Muteliefu, G.; Enomoto, A. Indoxyl sulphate induces oxidative stress and the expression of osteoblast-specific proteins in vascular smooth muscle cells. Nephrol. Dial. Transplant. 2009, 24, 2051–2058. [Google Scholar] [CrossRef] [PubMed]
- Sturmlechner, I.; Durik, M. Cellular senescence in renal ageing and disease. Nat. Rev. Nephrol. 2017, 13, 77–89. [Google Scholar] [CrossRef] [PubMed]
- Barreto, F.C.; Barreto, D.V. Serum indoxyl sulfate is associated with vascular disease and mortality in chronic kidney disease patients. Clin. J. Am. Soc. Nephrol. 2009, 4, 1551–1558. [Google Scholar] [CrossRef] [PubMed]
- Ma, Q.; Honarpisheh, M. Soluble Uric Acid Is an Intrinsic Negative Regulator of Monocyte Activation in Monosodium Urate Crystal-Induced Tissue Inflammation. J. Immunol. 2020, 205, 789–800. [Google Scholar] [CrossRef]
- Ma, Q.; Immler, R. Soluble uric acid inhibits β2 integrin-mediated neutrophil recruitment in innate immunity. Blood 2022, 139, 3402–3417. [Google Scholar] [CrossRef]
- Shimizu, H.; Yisireyili, M. Indoxyl sulfate enhances p53-TGF-β1-Smad3 pathway in proximal tubular cells. Am. J. Nephrol. 2013, 37, 97–103. [Google Scholar] [CrossRef]
- Shimizu, H.; Yisireyili, M. Indoxyl sulfate upregulates renal expression of ICAM-1 via production of ROS and activation of NF-κB and p53 in proximal tubular cells. Life Sci. 2013, 92, 143–148. [Google Scholar] [CrossRef]
- Bolati, D.; Shimizu, H. Indoxyl sulfate, a uremic toxin, downregulates renal expression of Nrf2 through activation of NF-κB. BMC Nephrol. 2013, 14, 56. [Google Scholar] [CrossRef]
- Castillo-Rodriguez, E.; Fernandez-Prado, R. Impact of Altered Intestinal Microbiota on Chronic Kidney Disease Progression. Toxins 2018, 10, 300. [Google Scholar] [CrossRef]
- Rapa, S.F.; Prisco, F. Pro-Inflammatory Effects of Indoxyl Sulfate in Mice: Impairment of Intestinal Homeostasis and Immune Response. Int. J. Mol. Sci. 2021, 22, 1135. [Google Scholar] [CrossRef]
- Holle, J.; Querfeld, U. Indoxyl sulfate associates with cardiovascular phenotype in children with chronic kidney disease. Pediatr. Nephrol. 2019, 34, 2571–2582. [Google Scholar] [CrossRef]
- Wang, C.H.; Lai, Y.H. Association between Serum Indoxyl Sulfate Levels and Endothelial Function in Non-Dialysis Chronic Kidney Disease. Toxins 2019, 11, 589. [Google Scholar] [CrossRef]
- Popolo, A.; Autore, G. Oxidative stress in patients with cardiovascular disease and chronic renal failure. Free Radic Res. 2013, 47, 346–356. [Google Scholar] [CrossRef]
- Adesso, S.; Popolo, A. The uremic toxin indoxyl sulphate enhances macrophage response to LPS. PLoS ONE 2013, 8, e76778. [Google Scholar] [CrossRef]
- Dou, L.; Jourde-Chiche, N. The uremic solute indoxyl sulfate induces oxidative stress in endothelial cells. J. Thromb. Haemost. 2007, 5, 1302–1308. [Google Scholar] [CrossRef]
- Yu, M.; Kim, Y.J. Indoxyl sulfate-induced endothelial dysfunction in patients with chronic kidney disease via an induction of oxidative stress. Clin. J. Am. Soc. Nephrol. 2011, 6, 30–39. [Google Scholar] [CrossRef]
- Mozar, A.; Louvet, L. Uremic toxin indoxyl sulfate inhibits human vascular smooth muscle cell proliferation. Ther. Apher. Dial. 2011, 15, 135–139. [Google Scholar] [CrossRef]
- Tsutsumi, S.; Tokunaga, Y. Investigation of the effects of indoxyl sulfate, a uremic toxin, on the intracellular oxidation level and phagocytic activity using an HL-60-differentiated human macrophage cell model. Biosci. Biotechnol. Biochem. 2020, 84, 1023–1029. [Google Scholar] [CrossRef]
- Wadowska, M.; Dobosz, E. MCP-Induced Protein 1 Participates in Macrophage-Dependent Endotoxin Tolerance. J. Immunol. 2022, 209, 1348–1358. [Google Scholar] [CrossRef]
- Hamza, E.; Vallejo-Mudarra, M. Indoxyl sulfate impairs erythropoiesis at BFU-E stage in chronic kidney disease. Cell Signal. 2023, 104, 110583. [Google Scholar] [CrossRef]
- Lorenz, G.; Ribeiro, A. GDF15 Suppresses Lymphoproliferation and Humoral Autoimmunity in a Murine Model of Systemic Lupus Erythematosus. J. Innate Immun. 2022, 14, 673–689. [Google Scholar] [CrossRef] [PubMed]
- Dobosz, E.; Lorenz, G. Murine myeloid cell MCPIP1 suppresses autoimmunity by regulating B-cell expansion and differentiation. Dis. Model. Mech. 2021, 14, dmm047589. [Google Scholar] [CrossRef] [PubMed]
- Moschovaki-Filippidou, F.; Steiger, S. Growth Differentiation Factor 15 Ameliorates Anti-Glomerular Basement Membrane Glomerulonephritis in Mice. Int. J. Mol. Sci. 2020, 21, 6978. [Google Scholar] [CrossRef] [PubMed]
- Hotamisligil, G.S. Inflammation and metabolic disorders. Nature 2006, 444, 860–867. [Google Scholar] [CrossRef]
- Libby, P. Inflammation in atherosclerosis. Nature 2002, 420, 868–874. [Google Scholar] [CrossRef]
- Chen, L.; Chen, R. Mechanisms Linking Inflammation to Insulin Resistance. Int. J. Endocrinol. 2015, 2015, 508409. [Google Scholar] [CrossRef]
- Rahtes, A.; Li, L. Polarization of Low-Grade Inflammatory Monocytes Through TRAM-Mediated Up-Regulation of Keap1 by Super-Low Dose Endotoxin. Front. Immunol. 2020, 11, 1478. [Google Scholar] [CrossRef]
- Quintana, F.J.; Murugaiyan, G. An endogenous aryl hydrocarbon receptor ligand acts on dendritic cells and T cells to suppress experimental autoimmune encephalomyelitis. Proc. Natl. Acad. Sci. USA 2010, 107, 20768–20773. [Google Scholar] [CrossRef]
- Rothhammer, V.; Quintana, F.J. The aryl hydrocarbon receptor: An environmental sensor integrating immune responses in health and disease. Nat. Rev. Immunol. 2019, 19, 184–197. [Google Scholar] [CrossRef]
- Shinde, R.; Hezaveh, K. Apoptotic cell-induced AhR activity is required for immunological tolerance and suppression of systemic lupus erythematosus in mice and humans. Nat. Immunol. 2018, 19, 571–582. [Google Scholar] [CrossRef]
- Tummalapalli, L.; Nadkarni, G.N. Biomarkers for predicting outcomes in chronic kidney disease. Curr. Opin. Nephrol. Hypertens. 2016, 25, 480–486. [Google Scholar] [CrossRef]
- Lin, C.J.; Wu, V. Meta-Analysis of the Associations of p-Cresyl Sulfate (PCS) and Indoxyl Sulfate (IS) with Cardiovascular Events and All-Cause Mortality in Patients with Chronic Renal Failure. PLoS ONE 2015, 10, e0132589. [Google Scholar] [CrossRef]
- Fourdinier, O.; Glorieux, G. Syndecan-1 and Free Indoxyl Sulfate Levels Are Associated with miR-126 in Chronic Kidney Disease. Int. J. Mol. Sci. 2021, 22, 10549. [Google Scholar] [CrossRef]
- Dugan, B.; Conway, J. Inflammaging as a target for healthy ageing. Age Ageing 2023, 52, afac328. [Google Scholar] [CrossRef]
- Andrade, B.; Jara-Gutiérrez, C. The Relationship between Reactive Oxygen Species and the cGAS/STING Signaling Pathway in the Inflammaging Process. Int. J. Mol. Sci. 2022, 23, 15182. [Google Scholar] [CrossRef]
- Huang, W.; Hickson, L.J. Cellular senescence: The good, the bad and the unknown. Nat. Rev. Nephrol. 2022, 18, 611–627. [Google Scholar] [CrossRef]
- Niwa, T.; Shimizu, H. Indoxyl sulfate induces nephrovascular senescence. J. Ren. Nutr. 2012, 22, 102–106. [Google Scholar] [CrossRef]
- Adelibieke, Y.; Shimizu, H. Indoxyl sulfate induces endothelial cell senescence by increasing reactive oxygen species production and p53 activity. J. Ren. Nutr. 2012, 22, 86–89. [Google Scholar] [CrossRef]
- Justus, C.R.; Leffler, N. In vitro cell migration and invasion assays. J. Vis. Exp. 2014, 88, 51046. [Google Scholar]













Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ribeiro, A.; Liu, F.; Srebrzynski, M.; Rother, S.; Adamowicz, K.; Wadowska, M.; Steiger, S.; Anders, H.-J.; Schmaderer, C.; Koziel, J.; et al. Uremic Toxin Indoxyl Sulfate Promotes Macrophage-Associated Low-Grade Inflammation and Epithelial Cell Senescence. Int. J. Mol. Sci. 2023, 24, 8031. https://doi.org/10.3390/ijms24098031
Ribeiro A, Liu F, Srebrzynski M, Rother S, Adamowicz K, Wadowska M, Steiger S, Anders H-J, Schmaderer C, Koziel J, et al. Uremic Toxin Indoxyl Sulfate Promotes Macrophage-Associated Low-Grade Inflammation and Epithelial Cell Senescence. International Journal of Molecular Sciences. 2023; 24(9):8031. https://doi.org/10.3390/ijms24098031
Chicago/Turabian StyleRibeiro, Andrea, Feiyue Liu, Matthias Srebrzynski, Simone Rother, Karina Adamowicz, Marta Wadowska, Stefanie Steiger, Hans-Joachim Anders, Christoph Schmaderer, Joanna Koziel, and et al. 2023. "Uremic Toxin Indoxyl Sulfate Promotes Macrophage-Associated Low-Grade Inflammation and Epithelial Cell Senescence" International Journal of Molecular Sciences 24, no. 9: 8031. https://doi.org/10.3390/ijms24098031
APA StyleRibeiro, A., Liu, F., Srebrzynski, M., Rother, S., Adamowicz, K., Wadowska, M., Steiger, S., Anders, H.-J., Schmaderer, C., Koziel, J., & Lech, M. (2023). Uremic Toxin Indoxyl Sulfate Promotes Macrophage-Associated Low-Grade Inflammation and Epithelial Cell Senescence. International Journal of Molecular Sciences, 24(9), 8031. https://doi.org/10.3390/ijms24098031