
APP in the Neuromuscular Junction for the Development of Sarcopenia and Alzheimer’s Disease

{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. AD Association with Sarcopenia
3. APP Family Proteins in Skeletal Muscles and NMJ
3.1. APP Family Proteins and Their Expressions in Skeletal Muscles and NMJ
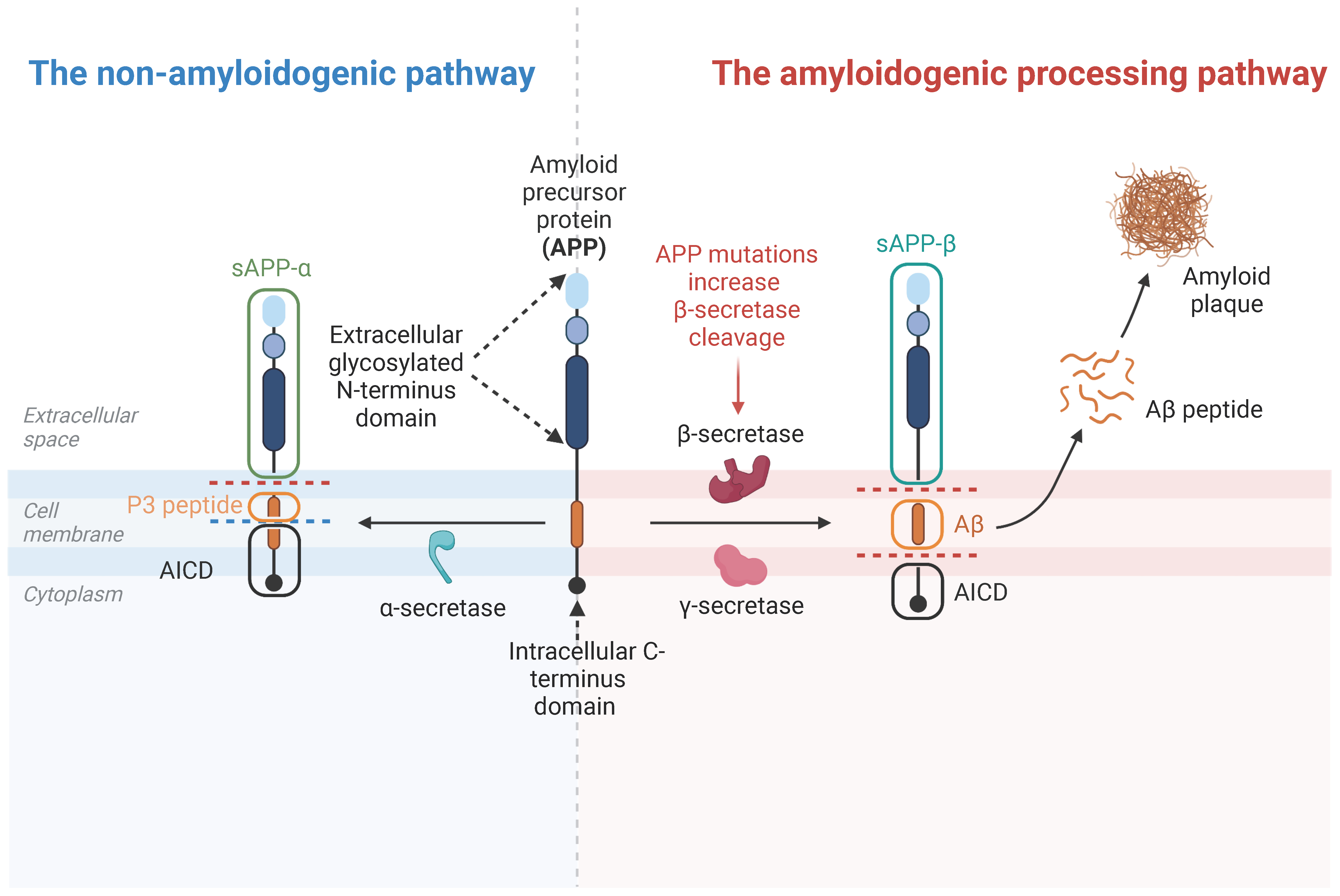
3.2. APP Cleavage Products
3.3. APP Family Proteins’ Functions in NMJ
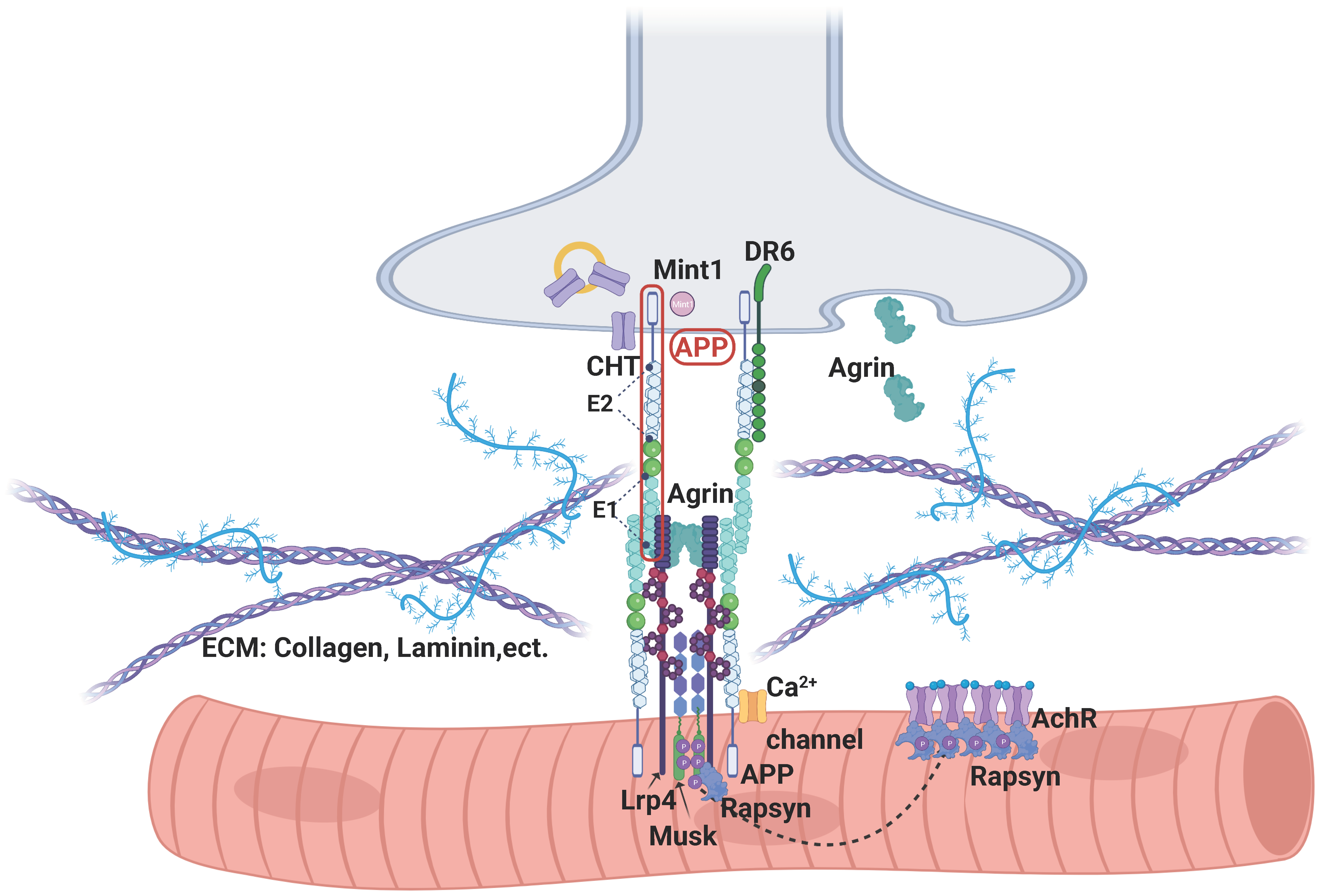
3.4. APP’s Binding Partners in NMJ
4. APP and Aβ in AD and Sarcopenia Development
4.1. APP and Aβ in AD Development
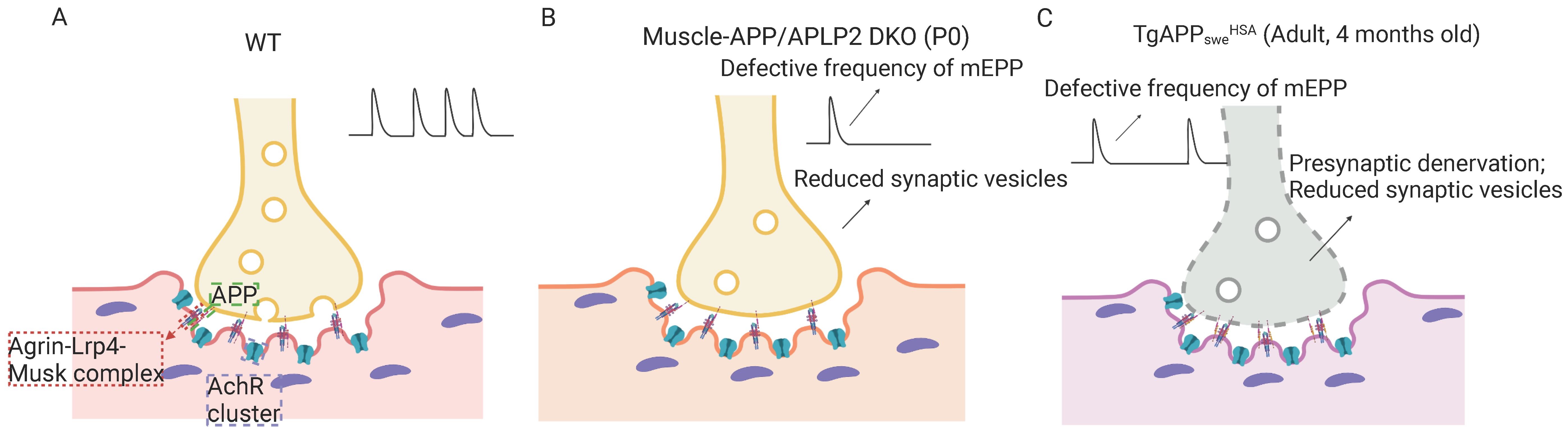
4.2. APP and Aβ in the Development of Sarcopenia-like Phenotype
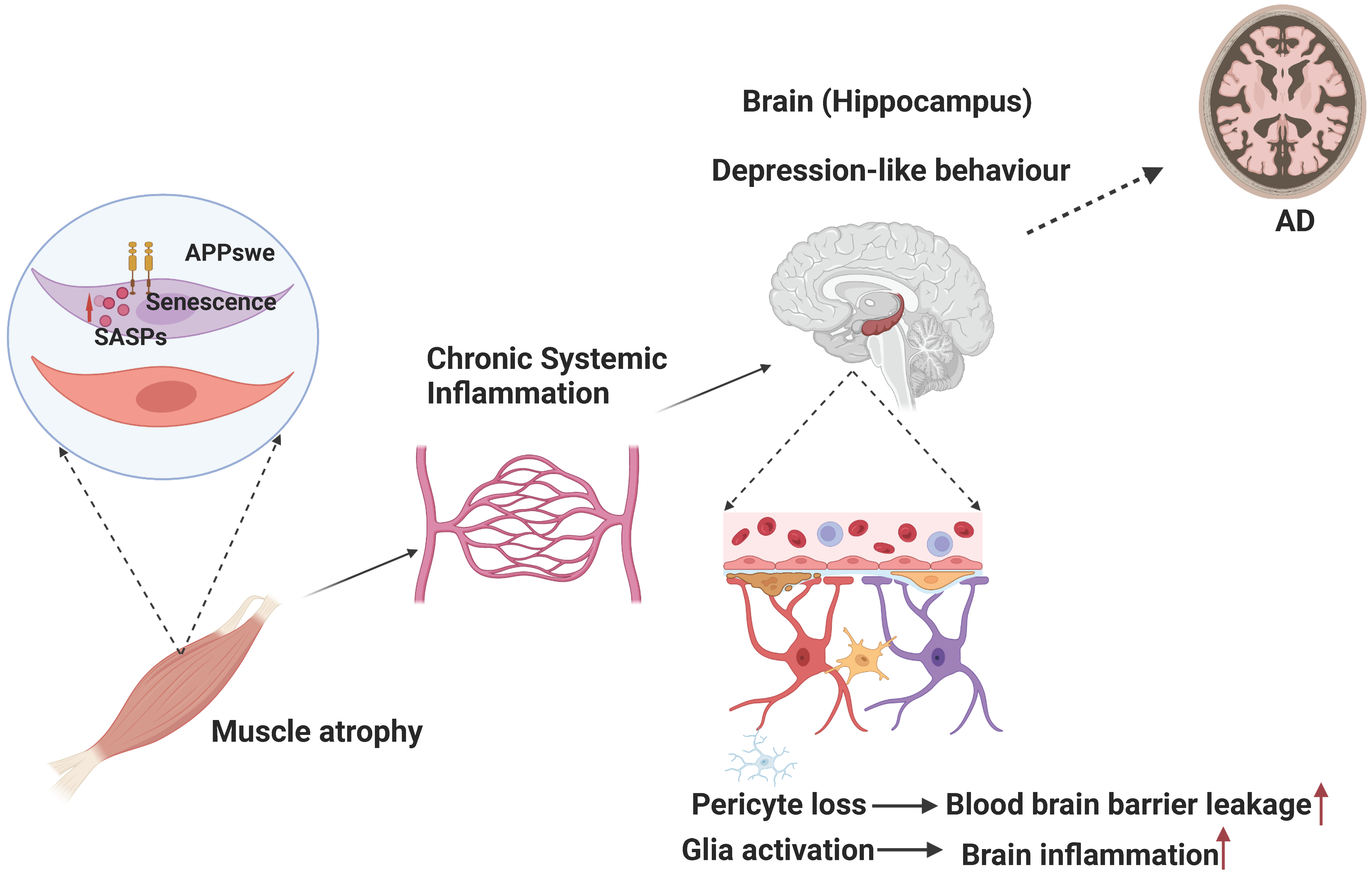
4.3. Muscular Swedish Mutant APP/Aβ’s Contributions to Not Only Sarcopenia-like Deficit but Also AD-Relevant Brain Pathology
5. APP/Aβ’s Contribution to Amyotrophic Lateral Sclerosis (ALS)
6. APP/Aβ’s Contribution to Myopathy
6.1. APP/Aβ’s Contribution to Sporadic Inclusion Body Myositis (sIBM)
6.2. APP/Aβ’s Contribution to GNE Myopathy (GNEM)
6.3. APP/Aβ’s Contribution to Chloroquine-Induced Myopathy
7. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Cruz-Jentoft, A.J.; Baeyens, J.P.; Bauer, J.M.; Boirie, Y.; Cederholm, T.; Landi, F.; Martin, F.C.; Michel, J.P.; Rolland, Y.; Schneider, S.M.; et al. Sarcopenia: European consensus on definition and diagnosis: Report of the European Working Group on Sarcopenia in Older People. Age Ageing 2010, 39, 412–423. [Google Scholar] [CrossRef] [PubMed]
- Do Nascimento, T.G.; Paes-Silva, R.P.; da Luz, M.C.L.; Cabral, P.C.; de Araujo Bezerra, G.K.; Gomes, A.C.B. Phase angle, muscle mass, and functionality in patients with Parkinson’s disease. Neurol. Sci. 2022, 43, 4203–4209. [Google Scholar] [CrossRef] [PubMed]
- Klickovic, U.; Zampedri, L.; Sinclair, C.D.J.; Wastling, S.J.; Trimmel, K.; Howard, R.S.; Malaspina, A.; Sharma, N.; Sidle, K.; Emira, A.; et al. Skeletal muscle MRI differentiates SBMA and ALS and correlates with disease severity. Neurology 2019, 93, e895–e907. [Google Scholar] [CrossRef]
- Bozzi, M.; Sciandra, F. Molecular Mechanisms Underlying Muscle Wasting in Huntington’s Disease. Int. J. Mol. Sci. 2020, 21, 8314. [Google Scholar] [CrossRef] [PubMed]
- Sugimoto, T.; Ono, R.; Murata, S.; Saji, N.; Matsui, Y.; Niida, S.; Toba, K.; Sakurai, T. Prevalence and associated factors of sarcopenia in elderly subjects with amnestic mild cognitive impairment or Alzheimer disease. Curr. Alzheimer Res. 2016, 13, 718–726. [Google Scholar] [CrossRef]
- DeTure, M.A.; Dickson, D.W. The neuropathological diagnosis of Alzheimer’s disease. Mol. Neurodegener. 2019, 14, 32. [Google Scholar] [CrossRef]
- 2023 Alzheimer’s disease facts and figures. Alzheimers Dement. J. Alzheimers Assoc. 2023, 19, 1598–1695. [CrossRef]
- Wang, J.; Gu, B.J.; Masters, C.L.; Wang, Y.-J. A systemic view of Alzheimer disease—Insights from amyloid-β metabolism beyond the brain. Nat. Rev. Neurol. 2017, 13, 612–623. [Google Scholar] [CrossRef]
- Cruz-Jentoft, A.J.; Sayer, A.A. Sarcopenia. Lancet 2019, 393, 2636–2646. [Google Scholar] [CrossRef]
- Cruz-Jentoft, A.J.; Bahat, G.; Bauer, J.; Boirie, Y.; Bruyère, O.; Cederholm, T.; Cooper, C.; Landi, F.; Rolland, Y.; Sayer, A.A.; et al. Sarcopenia: Revised European consensus on definition and diagnosis. Age Ageing 2019, 48, 16–31. [Google Scholar] [CrossRef]
- Ebadi, M.; Bhanji, R.A.; Mazurak, V.C.; Montano-Loza, A.J. Sarcopenia in cirrhosis: From pathogenesis to interventions. J. Gastroenterol. 2019, 54, 845–859. [Google Scholar] [CrossRef]
- Brisendine, M.H.; Drake, J.C. Early-stage Alzheimer’s disease: Are skeletal muscle and exercise the key? J. Appl. Physiol. 2023, 134, 515–520. [Google Scholar] [CrossRef] [PubMed]
- Chang, K.V.; Hsu, T.H.; Wu, W.T.; Huang, K.C.; Han, D.S. Association Between Sarcopenia and Cognitive Impairment: A Systematic Review and Meta-Analysis. J. Am. Med. Dir. Assoc. 2016, 17, 1164.e7–1164.e15. [Google Scholar] [CrossRef] [PubMed]
- Peng, T.C.; Chen, W.L.; Wu, L.W.; Chang, Y.W.; Kao, T.W. Sarcopenia and cognitive impairment: A systematic review and meta-analysis. Clin. Nutr. 2020, 39, 2695–2701. [Google Scholar] [CrossRef] [PubMed]
- Takagi, D.; Hirano, H.; Watanabe, Y.; Edahiro, A.; Ohara, Y.; Yoshida, H.; Kim, H.; Murakami, K.; Hironaka, S. Relationship between skeletal muscle mass and swallowing function in patients with Alzheimer’s disease. Geriatr. Gerontol. Int. 2017, 17, 402–409. [Google Scholar] [CrossRef]
- Kim, J.; Choi, K.H.; Cho, S.G.; Kang, S.R.; Yoo, S.W.; Kwon, S.Y.; Min, J.J.; Bom, H.S.; Song, H.C. Association of muscle and visceral adipose tissues with the probability of Alzheimer’s disease in healthy subjects. Sci. Rep. 2019, 9, 949. [Google Scholar] [CrossRef]
- Boyle, P.A.; Buchman, A.S.; Wilson, R.S.; Leurgans, S.E.; Bennett, D.A. Association of muscle strength with the risk of Alzheimer disease and the rate of cognitive decline in community-dwelling older persons. Arch. Neurol. 2009, 66, 1339–1344. [Google Scholar] [CrossRef]
- Meysami, S.; Raji, C.A.; Glatt, R.M.; Popa, E.S.; Ganapathi, A.S.; Bookheimer, T.; Slyapich, C.B.; Pierce, K.P.; Richards, C.J.; Lampa, M.G.; et al. Handgrip Strength Is Related to Hippocampal and Lobar Brain Volumes in a Cohort of Cognitively Impaired Older Adults with Confirmed Amyloid Burden. J. Alzheimers Dis. 2023, 91, 999–1006. [Google Scholar] [CrossRef]
- Kuo, K.; Zhang, Y.R.; Chen, S.D.; He, X.Y.; Huang, S.Y.; Wu, B.S.; Deng, Y.T.; Yang, L.; Ou, Y.N.; Guo, Y.; et al. Associations of grip strength, walking pace, and the risk of incident dementia: A prospective cohort study of 340,212 participants. Alzheimers Dement. J. Alzheimers Assoc. 2022, 19, 1415–1427. [Google Scholar] [CrossRef]
- He, P.; Zhou, C.; Ye, Z.; Liu, M.; Zhang, Y.; Wu, Q.; Zhang, Y.; Yang, S.; Xiaoqin, G.; Qin, X. Walking pace, handgrip strength, age, APOE genotypes, and new-onset dementia: The UK Biobank prospective cohort study. Alzheimers Res. Ther. 2023, 15, 9. [Google Scholar] [CrossRef]
- Magalhaes-Gomes, M.P.S.; Motta-Santos, D.; Schetino, L.P.L.; Andrade, J.N.; Bastos, C.P.; Guimaraes, D.A.S.; Vaughan, S.K.; Martinelli, P.M.; Guatimosim, S.; Pereira, G.S.; et al. Fast and slow-twitching muscles are differentially affected by reduced cholinergic transmission in mice deficient for VAChT: A mouse model for congenital myasthenia. Neurochem. Int. 2018, 120, 1–12. [Google Scholar] [CrossRef]
- Migliavacca, E.; Tay, S.K.H.; Patel, H.P.; Sonntag, T.; Civiletto, G.; McFarlane, C.; Forrester, T.; Barton, S.J.; Leow, M.K.; Antoun, E.; et al. Mitochondrial oxidative capacity and NAD+ biosynthesis are reduced in human sarcopenia across ethnicities. Nat. Commun. 2019, 10, 5808. [Google Scholar] [CrossRef]
- Kimura, A.; Sugimoto, T.; Niida, S.; Toba, K.; Sakurai, T. Association Between Appetite and Sarcopenia in Patients With Mild Cognitive Impairment and Early-Stage Alzheimer’s Disease: A Case-Control Study. Front. Nutr. 2018, 5, 128. [Google Scholar] [CrossRef]
- Wrann, C.D.; White, J.P.; Salogiannnis, J.; Laznik-Bogoslavski, D.; Wu, J.; Ma, D.; Lin, J.D.; Greenberg, M.E.; Spiegelman, B.M. Exercise induces hippocampal BDNF through a PGC-1α/FNDC5 pathway. Cell Metab. 2013, 18, 649–659. [Google Scholar] [CrossRef] [PubMed]
- Lourenco, M.V.; Frozza, R.L.; de Freitas, G.B.; Zhang, H.; Kincheski, G.C.; Ribeiro, F.C.; Gonçalves, R.A.; Clarke, J.R.; Beckman, D.; Staniszewski, A.; et al. Exercise-linked FNDC5/irisin rescues synaptic plasticity and memory defects in Alzheimer’s models. Nat. Med. 2019, 25, 165–175. [Google Scholar] [CrossRef] [PubMed]
- Wang, T.; Maltez, M.T.; Lee, H.W.; Ahmad, M.; Wang, H.W.; Leenen, F.H.H. Effect of exercise training on the FNDC5/BDNF pathway in spontaneously hypertensive rats. Physiol. Rep. 2019, 7, e14323. [Google Scholar] [CrossRef]
- Trejo, J.L.; Carro, E.; Torres-Aleman, I. Circulating insulin-like growth factor I mediates exercise-induced increases in the number of new neurons in the adult hippocampus. J. Neurosci. 2001, 21, 1628–1634. [Google Scholar] [CrossRef] [PubMed]
- Carro, E.; Nunez, A.; Busiguina, S.; Torres-Aleman, I. Circulating insulin-like growth factor I mediates effects of exercise on the brain. J. Neurosci. 2000, 20, 2926–2933. [Google Scholar] [CrossRef]
- Buchman, A.S.; Boyle, P.A.; Wilson, R.S.; Beck, T.L.; Kelly, J.F.; Bennett, D.A. Apolipoprotein E e4 allele is associated with more rapid motor decline in older persons. Alzheimer Dis. Assoc. Disord. 2009, 23, 63–69. [Google Scholar] [CrossRef]
- Pan, J.X.; Lee, D.; Sun, D.; Zhao, K.; Xiong, L.; Guo, H.H.; Ren, X.; Chen, P.; Lopez de Boer, R.; Lu, Y.; et al. Muscular Swedish mutant APP-to-Brain axis in the development of Alzheimer’s disease. Cell Death Dis. 2022, 13, 952. [Google Scholar] [CrossRef] [PubMed]
- Jones, G.; Trajanoska, K.; Santanasto, A.J.; Stringa, N.; Kuo, C.L.; Atkins, J.L.; Lewis, J.R.; Duong, T.; Hong, S.; Biggs, M.L.; et al. Genome-wide meta-analysis of muscle weakness identifies 15 susceptibility loci in older men and women. Nat. Commun. 2021, 12, 654. [Google Scholar] [CrossRef] [PubMed]
- Ogawa, Y.; Kaneko, Y.; Sato, T.; Shimizu, S.; Kanetaka, H.; Hanyu, H. Sarcopenia and Muscle Functions at Various Stages of Alzheimer Disease. Front. Neurol. 2018, 9, 710. [Google Scholar] [CrossRef] [PubMed]
- Caldwell, J.H.; Klevanski, M.; Saar, M.; Müller, U.C. Roles of the amyloid precursor protein family in the peripheral nervous system. Mech. Dev. 2013, 130, 433–446. [Google Scholar] [CrossRef] [PubMed]
- Klevanski, M.; Saar, M.; Baumkötter, F.; Weyer, S.W.; Kins, S.; Müller, U.C. Differential role of APP and APLPs for neuromuscular synaptic morphology and function. Mol. Cell. Neurosci. 2014, 61, 201–210. [Google Scholar] [CrossRef]
- Akaaboune, M.; Allinquant, B.; Farza, H.; Roy, K.; Magoul, R.; Fiszman, M.; Festoff, B.W.; Hantaï, D. Developmental regulation of amyloid precursor protein at the neuromuscular junction in mouse skeletal muscle. Mol. Cell. Neurosci. 2000, 15, 355–367. [Google Scholar] [CrossRef] [PubMed]
- Weyer, S.W.; Klevanski, M.; Delekate, A.; Voikar, V.; Aydin, D.; Hick, M.; Filippov, M.; Drost, N.; Schaller, K.L.; Saar, M.; et al. APP and APLP2 are essential at PNS and CNS synapses for transmission, spatial learning and LTP. EMBO J. 2011, 30, 2266–2280. [Google Scholar] [CrossRef] [PubMed]
- Jayaraman, M.; Kannayiram, G.; Rajadas, J. Amyloid toxicity in skeletal myoblasts: Implications for inclusion-body myositis. Arch. Biochem. Biophys. 2008, 474, 15–21. [Google Scholar] [CrossRef]
- Lopez, J.R.; Shtifman, A. Intracellular β-amyloid accumulation leads to age-dependent progression of Ca2+ dysregulation in skeletal muscle. Muscle Nerve 2010, 42, 731–738. [Google Scholar] [CrossRef]
- Shtifman, A.; Ward, C.W.; Laver, D.R.; Bannister, M.L.; Lopez, J.R.; Kitazawa, M.; LaFerla, F.M.; Ikemoto, N.; Querfurth, H.W. Amyloid-β protein impairs Ca2+ release and contractility in skeletal muscle. Neurobiol. Aging 2010, 31, 2080–2090. [Google Scholar] [CrossRef]
- Magara, F.; Müller, U.; Li, Z.W.; Lipp, H.P.; Weissmann, C.; Stagljar, M.; Wolfer, D.P. Genetic background changes the pattern of forebrain commissure defects in transgenic mice underexpressing the beta-amyloid-precursor protein. Proc. Natl. Acad. Sci. USA 1999, 96, 4656–4661. [Google Scholar] [CrossRef]
- Ring, S.; Weyer, S.W.; Kilian, S.B.; Waldron, E.; Pietrzik, C.U.; Filippov, M.A.; Herms, J.; Buchholz, C.; Eckman, C.B.; Korte, M.; et al. The secreted beta-amyloid precursor protein ectodomain APPs alpha is sufficient to rescue the anatomical, behavioral, and electrophysiological abnormalities of APP-deficient mice. J. Neurosci. 2007, 27, 7817–7826. [Google Scholar] [CrossRef] [PubMed]
- Heber, S.; Herms, J.; Gajic, V.; Hainfellner, J.; Aguzzi, A.; Rülicke, T.; von Kretzschmar, H.; von Koch, C.; Sisodia, S.; Tremml, P.; et al. Mice with combined gene knock-outs reveal essential and partially redundant functions of amyloid precursor protein family members. J. Neurosci. 2000, 20, 7951–7963. [Google Scholar] [CrossRef] [PubMed]
- Von Koch, C.S.; Zheng, H.; Chen, H.; Trumbauer, M.; Thinakaran, G.; van der Ploeg, L.H.; Price, D.L.; Sisodia, S.S. Generation of APLP2 KO mice and early postnatal lethality in APLP2/APP double KO mice. Neurobiol. Aging 1997, 18, 661–669. [Google Scholar] [CrossRef] [PubMed]
- Wang, Z.; Wang, B.; Yang, L.; Guo, Q.; Aithmitti, N.; Songyang, Z.; Zheng, H. Presynaptic and postsynaptic interaction of the amyloid precursor protein promotes peripheral and central synaptogenesis. J. Neurosci. 2009, 29, 10788–10801. [Google Scholar] [CrossRef]
- Choi, H.Y.; Liu, Y.; Tennert, C.; Sugiura, Y.; Karakatsani, A.; Kröger, S.; Johnson, E.B.; Hammer, R.E.; Lin, W.; Herz, J. APP interacts with LRP4 and agrin to coordinate the development of the neuromuscular junction in mice. Elife 2013, 2, e00220. [Google Scholar] [CrossRef]
- Cao, R.; Chen, P.; Wang, H.; Jing, H.; Zhang, H.; Xing, G.; Luo, B.; Pan, J.; Yu, Z.; Xiong, W.-C.; et al. Intrafusal-fiber LRP4 for muscle spindle formation and maintenance in adult and aged animals. Nat. Commun. 2023, 14, 744. [Google Scholar] [CrossRef] [PubMed]
- Wang, B.; Yang, L.; Wang, Z.; Zheng, H. Amyolid precursor protein mediates presynaptic localization and activity of the high-affinity choline transporter. Proc. Natl. Acad. Sci. USA 2007, 104, 14140–14145. [Google Scholar] [CrossRef] [PubMed]
- Nikolaev, A.; McLaughlin, T.; O’Leary, D.D.; Tessier-Lavigne, M. APP binds DR6 to trigger axon pruning and neuron death via distinct caspases. Nature 2009, 457, 981–989. [Google Scholar] [CrossRef] [PubMed]
- Brunholz, S.; Sisodia, S.; Lorenzo, A.; Deyts, C.; Kins, S.; Morfini, G. Axonal transport of APP and the spatial regulation of APP cleavage and function in neuronal cells. Exp. Brain Res. 2012, 217, 353–364. [Google Scholar] [CrossRef]
- Szodorai, A.; Kuan, Y.H.; Hunzelmann, S.; Engel, U.; Sakane, A.; Sasaki, T.; Takai, Y.; Kirsch, J.; Müller, U.; Beyreuther, K.; et al. APP anterograde transport requires Rab3A GTPase activity for assembly of the transport vesicle. J. Neurosci. 2009, 29, 14534–14544. [Google Scholar] [CrossRef]
- Groemer, T.W.; Thiel, C.S.; Holt, M.; Riedel, D.; Hua, Y.; Hüve, J.; Wilhelm, B.G.; Klingauf, J. Amyloid precursor protein is trafficked and secreted via synaptic vesicles. PLoS ONE 2011, 6, e18754. [Google Scholar] [CrossRef] [PubMed]
- Südhof, T.C.; Rizo, J. Synaptic vesicle exocytosis. Cold Spring Harb. Perspect. Biol. 2011, 3, a005637. [Google Scholar] [CrossRef] [PubMed]
- De Strooper, B. Proteases and proteolysis in Alzheimer disease: A multifactorial view on the disease process. Physiol. Rev. 2010, 90, 465–494. [Google Scholar] [CrossRef] [PubMed]
- Hardy, J. Alzheimer’s disease: The amyloid cascade hypothesis: An update and reappraisal. J. Alzheimers Dis. JAD 2006, 9, 151–153. [Google Scholar] [CrossRef] [PubMed]
- Hardy, J. Has the amyloid cascade hypothesis for Alzheimer’s disease been proved? Curr. Alzheimer Res. 2006, 3, 71–73. [Google Scholar] [CrossRef]
- Tang, Y.P.; Gershon, E.S. Genetic studies in Alzheimer’s disease. Dialogues Clin. Neurosci. 2003, 5, 17–26. [Google Scholar] [CrossRef]
- Wolfe, C.M.; Fitz, N.F.; Nam, K.N.; Lefterov, I.; Koldamova, R. The Role of APOE and TREM2 in Alzheimer’s Disease-Current Understanding and Perspectives. Int. J. Mol. Sci. 2018, 20, 81. [Google Scholar] [CrossRef]
- Griciuc, A.; Serrano-Pozo, A.; Parrado, A.R.; Lesinski, A.N.; Asselin, C.N.; Mullin, K.; Hooli, B.; Choi, S.H.; Hyman, B.T.; Tanzi, R.E. Alzheimer’s disease risk gene CD33 inhibits microglial uptake of amyloid beta. Neuron 2013, 78, 631–643. [Google Scholar] [CrossRef] [PubMed]
- Malik, M.; Parikh, I.; Vasquez, J.B.; Smith, C.; Tai, L.; Bu, G.; LaDu, M.J.; Fardo, D.W.; Rebeck, G.W.; Estus, S. Genetics ignite focus on microglial inflammation in Alzheimer’s disease. Mol. Neurodegener. 2015, 10, 52. [Google Scholar] [CrossRef] [PubMed]
- Lee, S.H.; Kang, J.; Ho, A.; Watanabe, H.; Bolshakov, V.Y.; Shen, J. APP Family Regulates Neuronal Excitability and Synaptic Plasticity but Not Neuronal Survival. Neuron 2020, 108, 676–690. [Google Scholar] [CrossRef]
- Müller, U.C.; Deller, T.; Korte, M. Not just amyloid: Physiological functions of the amyloid precursor protein family. Nat. Rev. Neurosci. 2017, 18, 281–298. [Google Scholar] [CrossRef] [PubMed]
- Fanutza, T.; Del Prete, D.; Ford, M.J.; Castillo, P.E.; D’Adamio, L. APP and APLP2 interact with the synaptic release machinery and facilitate transmitter release at hippocampal synapses. Elife 2015, 4, e09743. [Google Scholar] [CrossRef] [PubMed]
- Xu, H.; Bhaskaran, S.; Piekarz, K.M.; Ranjit, R.; Bian, J.; Kneis, P.; Ellis, A.; Bhandari, S.; Rice, H.C.; Van Remmen, H. Age Related Changes in Muscle Mass and Force Generation in the Triple Transgenic (3xTgAD) Mouse Model of Alzheimer’s Disease. Front. Aging Neurosci. 2022, 14, 876816. [Google Scholar] [CrossRef] [PubMed]
- Torcinaro, A.; Ricci, V.; Strimpakos, G.; De Santa, F.; Middei, S. Peripheral Nerve Impairment in a Mouse Model of Alzheimer’s Disease. Brain Sci. 2021, 11, 1245. [Google Scholar] [CrossRef] [PubMed]
- Meola, G.; Sansone, V.; Perani, D.; Colleluori, A.; Cappa, S.; Cotelli, M.; Fazio, F.; Thornton, C.A.; Moxley, R.T. Reduced cerebral blood flow and impaired visual-spatial function in proximal myotonic myopathy. Neurology 1999, 53, 1042–1050. [Google Scholar] [CrossRef] [PubMed]
- Peristeri, E.; Aloizou, A.M.; Keramida, P.; Tsouris, Z.; Siokas, V.; Mentis, A.A.; Dardiotis, E. Cognitive Deficits in Myopathies. Int. J. Mol. Sci. 2020, 21, 3795. [Google Scholar] [CrossRef] [PubMed]
- Anderson, J.L.; Head, S.I.; Rae, C.; Morley, J.W. Brain function in Duchenne muscular dystrophy. Brain A J. Neurol. 2002, 125, 4–13. [Google Scholar] [CrossRef]
- Bagdatlioglu, E.; Porcari, P.; Greally, E.; Blamire, A.M.; Straub, V.W. Cognitive impairment appears progressive in the mdx mouse. Neuromuscul. Disord. NMD 2020, 30, 368–388. [Google Scholar] [CrossRef]
- Hayward, G.C.; Caceres, D.; Copeland, E.N.; Baranowski, B.J.; Mohammad, A.; Whitley, K.C.; Fajardo, V.A.; MacPherson, R.E.K. Characterization of Alzheimer’s disease-like neuropathology in Duchenne’s muscular dystrophy using the DBA/2J mdx mouse model. FEBS Open Bio 2022, 12, 154–162. [Google Scholar] [CrossRef]
- Tcw, J.; Goate, A.M. Genetics of β-Amyloid Precursor Protein in Alzheimer’s Disease. Cold Spring Harb. Perspect. Med. 2017, 7, a024539. [Google Scholar] [CrossRef]
- Mullan, M.; Crawford, F.; Axelman, K.; Houlden, H.; Lilius, L.; Winblad, B.; Lannfelt, L. A pathogenic mutation for probable Alzheimer’s disease in the APP gene at the N-terminus of beta-amyloid. Nat. Genet. 1992, 1, 345–347. [Google Scholar] [CrossRef] [PubMed]
- Haass, C.; Lemere, C.A.; Capell, A.; Citron, M.; Seubert, P.; Schenk, D.; Lannfelt, L.; Selkoe, D.J. The Swedish mutation causes early-onset Alzheimer’s disease by beta-secretase cleavage within the secretory pathway. Nat. Med. 1995, 1, 1291–1296. [Google Scholar] [CrossRef] [PubMed]
- Kuo, Y.M.; Kokjohn, T.A.; Watson, M.D.; Woods, A.S.; Cotter, R.J.; Sue, L.I.; Kalback, W.M.; Emmerling, M.R.; Beach, T.G.; Roher, A.E. Elevated abeta42 in skeletal muscle of Alzheimer disease patients suggests peripheral alterations of AbetaPP metabolism. Am. J. Pathol. 2000, 156, 797–805. [Google Scholar] [CrossRef]
- Schuh, R.A.; Jackson, K.C.; Schlappal, A.E.; Spangenburg, E.E.; Ward, C.W.; Park, J.H.; Dugger, N.; Shi, G.L.; Fishman, P.S. Mitochondrial oxygen consumption deficits in skeletal muscle isolated from an Alzheimer’s disease-relevant murine model. BMC Neurosci. 2014, 15, 24. [Google Scholar] [CrossRef] [PubMed]
- Askanas, V.; McFerrin, J.; Baque, S.; Alvarez, R.B.; Sarkozi, E.; Engel, W.K. Transfer of beta-amyloid precursor protein gene using adenovirus vector causes mitochondrial abnormalities in cultured normal human muscle. Proc. Natl. Acad. Sci. USA 1996, 93, 1314–1319. [Google Scholar] [CrossRef] [PubMed]
- Boncompagni, S.; Moussa, C.E.; Levy, E.; Pezone, M.J.; Lopez, J.R.; Protasi, F.; Shtifman, A. Mitochondrial dysfunction in skeletal muscle of amyloid precursor protein-overexpressing mice. J. Biol. Chem. 2012, 287, 20534–20544. [Google Scholar] [CrossRef]
- Lin, Y.-S.; Lin, F.-Y.; Hsiao, Y.-H. Myostatin Is Associated With Cognitive Decline in an Animal Model of Alzheimer’s Disease. Mol. Neurobiol. 2019, 56, 1984–1991. [Google Scholar] [CrossRef]
- Kriss, A.; Jenkins, T. Muscle MRI in motor neuron diseases: A systematic review. Amyotroph. Lateral Scler. Front. Degener. 2022, 23, 161–175. [Google Scholar] [CrossRef]
- Calingasan, N.Y.; Chen, J.; Kiaei, M.; Beal, M.F. Beta-amyloid 42 accumulation in the lumbar spinal cord motor neurons of amyotrophic lateral sclerosis patients. Neurobiol. Dis. 2005, 19, 340–347. [Google Scholar] [CrossRef]
- Rabinovich-Toidman, P.; Rabinovich-Nikitin, I.; Ezra, A.; Barbiro, B.; Fogel, H.; Slutsky, I.; Solomon, B. Mutant SOD1 Increases APP Expression and Phosphorylation in Cellular and Animal Models of ALS. PLoS ONE 2015, 10, e0143420. [Google Scholar] [CrossRef]
- Koistinen, H.; Prinjha, R.; Soden, P.; Harper, A.; Banner, S.J.; Pradat, P.F.; Loeffler, J.P.; Dingwall, C. Elevated levels of amyloid precursor protein in muscle of patients with amyotrophic lateral sclerosis and a mouse model of the disease. Muscle Nerve 2006, 34, 444–450. [Google Scholar] [CrossRef]
- Sasaki, S.; Iwata, M. Immunoreactivity of beta-amyloid precursor protein in amyotrophic lateral sclerosis. Acta Neuropathol. 1999, 97, 463–468. [Google Scholar] [CrossRef] [PubMed]
- Bryson, J.B.; Hobbs, C.; Parsons, M.J.; Bosch, K.D.; Pandraud, A.; Walsh, F.S.; Doherty, P.; Greensmith, L. Amyloid precursor protein (APP) contributes to pathology in the SOD1(G93A) mouse model of amyotrophic lateral sclerosis. Hum. Mol. Genet. 2012, 21, 3871–3882. [Google Scholar] [CrossRef] [PubMed]
- Truong, P.H.; Crouch, P.J.; Hilton, J.B.W.; McLean, C.A.; Cappai, R.; Ciccotosto, G.D. Sex-dependent effects of amyloid precursor-like protein 2 in the SOD1-G37R transgenic mouse model of MND. Cell. Mol. Life Sci. CMLS 2021, 78, 6605–6630. [Google Scholar] [CrossRef] [PubMed]
- Rabinovich-Toidman, P.; Becker, M.; Barbiro, B.; Solomon, B. Inhibition of amyloid precursor protein beta-secretase cleavage site affects survival and motor functions of amyotrophic lateral sclerosis transgenic mice. Neurodegener. Dis. 2012, 10, 30–33. [Google Scholar] [CrossRef]
- Li, K.; Pu, C.; Huang, X.; Liu, J.; Mao, Y.; Lu, X. Proteomic study of sporadic inclusion body myositis. Proteome Sci. 2014, 12, 45. [Google Scholar] [CrossRef]
- Sugarman, M.C.; Yamasaki, T.R.; Oddo, S.; Echegoyen, J.C.; Murphy, M.P.; Golde, T.E.; Jannatipour, M.; Leissring, M.A.; LaFerla, F.M. Inclusion body myositis-like phenotype induced by transgenic overexpression of beta APP in skeletal muscle. Proc. Natl. Acad. Sci. USA 2002, 99, 6334–6339. [Google Scholar] [CrossRef]
- Sugarman, M.C.; Kitazawa, M.; Baker, M.; Caiozzo, V.J.; Querfurth, H.W.; LaFerla, F.M. Pathogenic accumulation of APP in fast twitch muscle of IBM patients and a transgenic model. Neurobiol. Aging 2006, 27, 423–432. [Google Scholar] [CrossRef]
- Lünemann, J.D.; Schmidt, J.; Schmid, D.; Barthel, K.; Wrede, A.; Dalakas, M.C.; Münz, C. Beta-amyloid is a substrate of autophagy in sporadic inclusion body myositis. Ann. Neurol. 2007, 61, 476–483. [Google Scholar] [CrossRef]
- Nalini, A.; Gayathri, N.; Dawn, R. Distal myopathy with rimmed vacuoles: Report on clinical characteristics in 23 cases. Neurol. India 2010, 58, 235–241. [Google Scholar] [CrossRef]
- Huizing, M.; Krasnewich, D.M. Hereditary inclusion body myopathy: A decade of progress. Biochim. Biophys. Acta 2009, 1792, 881–887. [Google Scholar] [CrossRef] [PubMed]
- Fischer, C.; Kleinschnitz, K.; Wrede, A.; Muth, I.; Kruse, N.; Nishino, I.; Schmidt, J. Cell stress molecules in the skeletal muscle of GNE myopathy. BMC Neurol. 2013, 13, 24. [Google Scholar] [CrossRef] [PubMed]
- Kizuka, Y.; Kitazume, S.; Taniguchi, N. N-glycan and Alzheimer’s disease. Biochim. Biophys. Acta. Gen. Subj. 2017, 1861, 2447–2454. [Google Scholar] [CrossRef] [PubMed]
- Nakagawa, K.; Kitazume, S.; Oka, R.; Maruyama, K.; Saido, T.C.; Sato, Y.; Endo, T.; Hashimoto, Y. Sialylation enhances the secretion of neurotoxic amyloid-beta peptides. J. Neurochem. 2006, 96, 924–933. [Google Scholar] [CrossRef] [PubMed]
- Chun, Y.S.; Park, Y.; Oh, H.G.; Kim, T.W.; Yang, H.O.; Park, M.K.; Chung, S. O-GlcNAcylation promotes non-amyloidogenic processing of amyloid-β protein precursor via inhibition of endocytosis from the plasma membrane. J. Alzheimers Dis. 2015, 44, 261–275. [Google Scholar] [CrossRef]
- Tsuzuki, K.; Fukatsu, R.; Takamaru, Y.; Kimura, K.; Abe, M.; Shima, K.; Fujii, N.; Takahata, N. Immunohistochemical evidence for amyloid beta in rat soleus muscle in chloroquine-induced myopathy. Neurosci. Lett. 1994, 182, 151–154. [Google Scholar] [CrossRef] [PubMed]
- Tsuzuki, K.; Fukatsu, R.; Takamaru, Y.; Yoshida, T.; Hayashi, Y.; Yamaguchi, H.; Fujii, N.; Takahata, N. Amyloid beta protein in rat soleus muscle in chloroquine-induced myopathy using end-specific antibodies for A beta 40 and A beta 42: Immunohistochemical evidence for amyloid beta protein. Neurosci. Lett. 1995, 202, 77–80. [Google Scholar] [CrossRef] [PubMed]




Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Wu, M.-Y.; Zou, W.-J.; Lee, D.; Mei, L.; Xiong, W.-C. APP in the Neuromuscular Junction for the Development of Sarcopenia and Alzheimer’s Disease. Int. J. Mol. Sci. 2023, 24, 7809. https://doi.org/10.3390/ijms24097809
Wu M-Y, Zou W-J, Lee D, Mei L, Xiong W-C. APP in the Neuromuscular Junction for the Development of Sarcopenia and Alzheimer’s Disease. International Journal of Molecular Sciences. 2023; 24(9):7809. https://doi.org/10.3390/ijms24097809
Chicago/Turabian StyleWu, Min-Yi, Wen-Jun Zou, Daehoon Lee, Lin Mei, and Wen-Cheng Xiong. 2023. "APP in the Neuromuscular Junction for the Development of Sarcopenia and Alzheimer’s Disease" International Journal of Molecular Sciences 24, no. 9: 7809. https://doi.org/10.3390/ijms24097809
APA StyleWu, M.-Y., Zou, W.-J., Lee, D., Mei, L., & Xiong, W.-C. (2023). APP in the Neuromuscular Junction for the Development of Sarcopenia and Alzheimer’s Disease. International Journal of Molecular Sciences, 24(9), 7809. https://doi.org/10.3390/ijms24097809