

The Epithelial Sodium Channel—An Underestimated Drug Target


Abstract
1. Introduction
2. Phylogeny
3. Tissues in Which ENaC Is Expressed
4. Structure and Stoichiometry
5. Structural and Biochemical Regulation of ENaC Function
5.1. Regulation of ENaC Activity by Proteases

5.2. Sequence Motifs Involved in Regulation of ENaC
5.2.1. Selectivity Filter
5.2.2. Amiloride-Binding Site
5.2.3. N-Glycosylation
5.2.4. Cysteine-Rich Domains
5.2.5. Na(+)-Binding Acidic Cleft
5.2.6. HG-Motif
5.2.7. N-Terminal Ubiquitination Motif
5.2.8. PY Motif
5.2.9. Cys-Palmitoylation
5.2.10. Acetylation
5.2.11. Phosphorylation
5.2.12. PIP2 and PIP3 Binding Sites
5.3. Regulation of ENaC via Several Other Proteins
6. Variation in Human SCNN1A, SCNN1B and SCNN1G
6.1. Gain-of-Function: Liddle Syndrome
6.2. Gain-of-Function: Cystic Fibrosis
6.3. Gain-of-Function: Bronchiectasis
6.4. Loss-of-Function Mutations in ENaC: PHA-1B
7. ENaC and Disease Conditions
7.1. Role of ENaC in Salt-Sensitive and Resistant Hypertension
7.1.1. Role of ENaC in HTN with Comorbidities
7.1.2. ENaC Expression in Blood Cells of Hypertensive Patients
7.1.3. Effect of ENaC on Vascular Response in Hypertensive Conditions
7.1.4. Shear Force Sensing of ENaC
7.1.5. Effect of ENaC on Inflammatory Responses in Hypertensive Conditions
7.1.6. Putative Targets and ENaC-Modulating Compounds for Treatment of Cardiovascular Disease
- Amiloride binding site (ENaC inhibitor amiloride and derivatives)
- Extra-renal ENaC including vascular and immune cells
- De-ubiquitination enzyme activating agents
- Intestinal mineralocorticoid receptor
- Inwardly rectifying K(+) (Kir) channels
- Inhibition of ROS activation (NOX subunit-specific inhibitors, tempol, quercetin, allicin)
- Synthetic serine protease inhibitors (camostat)
Amiloride Binding Site—ENaC Inhibitors
Extra-Renal ENaC including Vascular and Immune Cells
De-Ubiquitination Enzyme Activating Agents
Intestinal Mineralocorticoid Receptor
Inwardly Rectifying K(+) (Kir) Channels
Inhibition of ROS Activation (NOX Subunit-Specific Inhibitors, Tempol)
Synthetic Serine Protease Inhibitors
7.2. Role of ENaC in Cystic Fibrosis
7.2.1. Pathology and Therapeutic Approach
- ENaC inhibitors (BI 1265162, GS-9411, AZD5634)
- SPLUNC1 mimetics (SPX-101)
- Epigenetic technology
- ENaC genes (antisense oligonucleotides, siRNAs)
- Proteolytic cleavage (CAPs inhibitor QUB-TL1)
- Inhibition of myeloperoxidase-derived oxidants (AZM198)
7.2.2. Compounds Targeting ENaC (ENaC Inhibitors and SPLUNC1 Mimetics)
7.2.3. Epigenetic Targeting
7.2.4. Targeting ENaC Genes (Antisense Oligonucleotides, siRNAs)
7.2.5. Proteolytic Cleavage (CAPs Inhibitors)
7.2.6. Targeting Myeloperoxidase-Derived Oxidants
7.3. Non-Cardiogenic Respiratory Failure
7.4. Pseudohypoaldosteronism Type 1 (PHA-1B)
7.5. Inflammatory Diseases
Arthropathies, Nephritis
7.6. Concluding Remarks
- Autosomal recessive pseudohypoaldosteronism type 1:
- PHA1B, OMIM:264350 (https://www.omim.org/entry/264350) accessed on 27 March 2023
- Global Variome shared LOVD database [153]:
- https://databases.lovd.nl/shared/genes/SCNN1A accessed on 27 March 2023
- https://databases.lovd.nl/shared/genes/SCNN1B accessed on 27 March 2023
- https://databases.lovd.nl/shared/genes/SCNN1G accessed on 27 March 2023
- Liddle Syndrome (LS):
- LIDLS, OMIM:177200 (https://www.omim.org/entry/177200) accessed on 27 March 2023
- Cystic fibrosis (Mucoviscidosis):
- CF, OMIM:219700 https://www.omim.org/entry/219700 accessed on 27 March 2023
- Bronchiectasis (Cystic fibrosis-like syndrome):
- BESC1, OMIM:211400 https://www.omim.org/entry/211400 accessed on 27 March 2023
- BESC2, OMIM:613021 https://www.omim.org/entry/613021 accessed on 27 March 2023
- BESC3, OMIM:613071 https://www.omim.org/entry/613071 accessed on 27 March 2023
- Clinical trials:
- https://clinicaltrials.gov/amiloride accessed on 27 March 2023
- https://clinicaltrials.gov/ENaC accessed on 27 March 2023
- https://clinicaltrials.gov/setanaxib, https://clinicaltrials.gov/GKT137831 accessed on 27 March 2023
- https://clinicaltrials.gov/solnatide, https://clinicaltrials.gov/AP301 accessed on 27 March 2023
- https://eudract.ema.europa.eu/ENaC accessed on 27 March 2023
- https://eudract.ema.europa.eu/solnatide accessed on 27 March 2023
- https://eudract.ema.europa.eu/AP301 accessed on 27 March 2023
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
References
- Canessa, C.M.; Horisberger, J.-D.; Rossier, B.C. Epithelial Sodium Channel Related to Proteins Involved in Neurodegeneration. Nature 1993, 361, 467–470. [Google Scholar] [CrossRef] [PubMed]
- Lingueglia, E.; Voilley, N.; Waldmann, R.; Lazdunski, M.; Barbry, P. Expression Cloning of an Epithelial Amiloride-Sensitive Na+ Channel: A New Channel Type with Homologies to Caenorhabditis Elegans Degenerins. FEBS Lett. 1993, 318, 95–99. [Google Scholar] [CrossRef] [PubMed]
- Kellenberger, S.; Schild, L. Epithelial Sodium Channel/Degenerin Family of Ion Channels: A Variety of Functions for a Shared Structure. Physiol. Rev. 2002, 82, 735–767. [Google Scholar] [CrossRef] [PubMed]
- Rossier, B.C.; Baker, M.E.; Studer, R.A. Epithelial Sodium Transport and Its Control by Aldosterone: The Story of Our Internal Environment Revisited. Physiol. Rev. 2015, 95, 297–340. [Google Scholar] [CrossRef] [PubMed]
- Kashlan, O.B.; Kleyman, T.R. ENaC Structure and Function in the Wake of a Resolved Structure of a Family Member. Am. J. Physiol.-Ren. Physiol. 2011, 301, F684–F696. [Google Scholar] [CrossRef]
- Boscardin, E.; Alijevic, O.; Hummler, E.; Frateschi, S.; Kellenberger, S. The Function and Regulation of Acid-sensing Ion Channels (ASICs) and the Epithelial Na+ Channel (ENaC): IUPHAR Review 19. Br. J. Pharmacol. 2016, 173, 2671–2701. [Google Scholar] [CrossRef]
- Kleyman, T.R.; Kashlan, O.B.; Hughey, R.P. Epithelial Na+ Channel Regulation by Extracellular and Intracellular Factors. Annu. Rev. Physiol. 2018, 80, 263–281. [Google Scholar] [CrossRef]
- Arnadottir, J.; O’Hagan, R.; Chen, Y.; Goodman, M.B.; Chalfie, M. The DEG/ENaC Protein MEC-10 Regulates the Transduction Channel Complex in Caenorhabditis Elegans Touch Receptor Neurons. J. Neurosci. 2011, 31, 12695–12704. [Google Scholar] [CrossRef]
- Lingueglia, E.; Deval, E.; Lazdunski, M. FMRFamide-Gated Sodium Channel and ASIC Channels: A New Class of Ionotropic Receptors for FMRFamide and Related Peptides. Peptides 2006, 27, 1138–1152. [Google Scholar] [CrossRef]
- Wiemuth, D.; Assmann, M.; Gründer, S. The Bile Acid-Sensitive Ion Channel (BASIC), the Ignored Cousin of ASICs and ENaC. Channels 2014, 8, 29–34. [Google Scholar] [CrossRef]
- Adams, C.M.; Anderson, M.G.; Motto, D.G.; Price, M.P.; Johnson, W.A.; Welsh, M.J. Ripped Pocket and Pickpocket, Novel Drosophila DEG/ENaC Subunits Expressed in Early Development and in Mechanosensory Neurons. J. Cell Biol. 1998, 140, 143–152. [Google Scholar] [CrossRef] [PubMed]
- Gettings, S.M.; Maxeiner, S.; Tzika, M.; Cobain, M.R.D.; Ruf, I.; Benseler, F.; Brose, N.; Krasteva-Christ, G.; Vande Velde, G.; Schönberger, M.; et al. Two Functional Epithelial Sodium Channel Isoforms Are Present in Rodents despite Pronounced Evolutionary Pseudogenization and Exon Fusion. Mol. Biol. Evol. 2021, 38, 5704–5725. [Google Scholar] [CrossRef] [PubMed]
- Rotin, D.; Staub, O. Function and Regulation of the Epithelial Na+ Channel ENaC. In Comprehensive Physiology; Terjung, R., Ed.; Wiley: Hoboken, NJ, USA, 2021; pp. 2017–2045. ISBN 978-0-470-65071-4. [Google Scholar]
- Nemeth, Z.; Ryan, M.J.; Granger, J.P.; Drummond, H.A. Expression of Exogenous Epithelial Sodium Channel Beta Subunit in the Mouse Middle Cerebral Artery Increases Pressure-Induced Constriction. Am. J. Hypertens. 2021, 34, 1227–1235. [Google Scholar] [CrossRef] [PubMed]
- Enuka, Y.; Hanukoglu, I.; Edelheit, O.; Vaknine, H.; Hanukoglu, A. Epithelial Sodium Channels (ENaC) Are Uniformly Distributed on Motile Cilia in the Oviduct and the Respiratory Airways. Histochem. Cell Biol. 2012, 137, 339–353. [Google Scholar] [CrossRef] [PubMed]
- Pearce, D.; Manis, A.D.; Nesterov, V.; Korbmacher, C. Regulation of Distal Tubule Sodium Transport: Mechanisms and Roles in Homeostasis and Pathophysiology. Pflüg. Arch.—Eur. J. Physiol. 2022, 474, 869–884. [Google Scholar] [CrossRef] [PubMed]
- Chandrashekar, J.; Kuhn, C.; Oka, Y.; Yarmolinsky, D.A.; Hummler, E.; Ryba, N.J.P.; Zuker, C.S. The Cells and Peripheral Representation of Sodium Taste in Mice. Nature 2010, 464, 297–301. [Google Scholar] [CrossRef]
- Eaton, D.C.; Helms, M.N.; Koval, M.; Bao, H.F.; Jain, L. The Contribution of Epithelial Sodium Channels to Alveolar Function in Health and Disease. Annu. Rev. Physiol. 2009, 71, 403–423. [Google Scholar] [CrossRef]
- Guo, D.; Liang, S.; Wang, S.; Tang, C.; Yao, B.; Wan, W.; Zhang, H.; Jiang, H.; Ahmed, A.; Zhang, Z.; et al. Role of Epithelial Sodium Channels (ENaCs) in Endothelial Function. J. Cell Sci. 2015, 129, 290–297. [Google Scholar] [CrossRef]
- Paudel, P.; McDonald, F.J.; Fronius, M. The δ Subunit of Epithelial Sodium Channel in Humans-a Potential Player in Vascular Physiology. Am. J. Physiol. Heart Circ. Physiol. 2021, 320, H487–H493. [Google Scholar] [CrossRef]
- Paudel, P.; van Hout, I.; Bunton, R.W.; Parry, D.J.; Coffey, S.; McDonald, F.J.; Fronius, M. Epithelial Sodium Channel δ Subunit Is Expressed in Human Arteries and Has Potential Association with Hypertension. Hypertension 2022, 79, 1385–1394. [Google Scholar] [CrossRef]
- Zhang, J.; Yuan, H.; Chen, S.; Zhang, Z. Detrimental or Beneficial: Role of Endothelial ENaC in Vascular Function. J. Cell. Physiol. 2022, 237, 29–48. [Google Scholar] [CrossRef] [PubMed]
- Mutchler, S.M.; Kleyman, T.R. New Insights Regarding Epithelial Na+ Channel Regulation and Its Role in the Kidney, Immune System and Vasculature. Curr. Opin. Nephrol. Hypertens. 2019, 28, 113–119. [Google Scholar] [CrossRef] [PubMed]
- Kusche-Vihrog, K.; Tarjus, A.; Fels, J.; Jaisser, F. The Epithelial Na+ Channel: A New Player in the Vasculature. Curr. Opin. Nephrol. Hypertens. 2014, 23, 143–148. [Google Scholar] [CrossRef] [PubMed]
- Tarjus, A.; Maase, M.; Jeggle, P.; Martinez-Martinez, E.; Fassot, C.; Loufrani, L.; Henrion, D.; Hansen, P.B.L.; Kusche-Vihrog, K.; Jaisser, F. The Endothelial αENaC Contributes to Vascular Endothelial Function in Vivo. PLoS ONE 2017, 12, e0185319. [Google Scholar] [CrossRef]
- Sharma, K.; Haque, M.; Guidry, R.; Ueta, Y.; Teruyama, R. Effect of Dietary Salt Intake on Epithelial Na+ Channels (ENaC) in Vasopressin Magnocellular Neurosecretory Neurons in the Rat Supraoptic Nucleus: Effect of Dietary Salt Intake on ENaC in Vasopressin Neurons. J. Physiol. 2017, 595, 5857–5874. [Google Scholar] [CrossRef]
- Sudarikova, A.V.; Tsaplina, O.A.; Chubinskiy-Nadezhdin, V.I.; Morachevskaya, E.A.; Negulyaev, Y.A. Amiloride-Insensitive Sodium Channels Are Directly Regulated by Actin Cytoskeleton Dynamics in Human Lymphoma Cells. Biochem. Biophys. Res. Commun. 2015, 461, 54–58. [Google Scholar] [CrossRef]
- Cerecedo, D.; Martínez-Vieyra, I.; Sosa-Peinado, A.; Cornejo-Garrido, J.; Ordaz-Pichardo, C.; Benítez-Cardoza, C. Alterations in Plasma Membrane Promote Overexpression and Increase of Sodium Influx through Epithelial Sodium Channel in Hypertensive Platelets. Biochim. Biophys. Acta BBA—Biomembr. 2016, 1858, 1891–1903. [Google Scholar] [CrossRef]
- Reus-Chavarría, E.; Martínez-Vieyra, I.; Salinas-Nolasco, C.; Chávez-Piña, A.E.; Méndez-Méndez, J.V.; López-Villegas, E.O.; Sosa-Peinado, A.; Cerecedo, D. Enhanced Expression of the Epithelial Sodium Channel in Neutrophils from Hypertensive Patients. Biochim. Biophys. Acta BBA—Biomembr. 2019, 1861, 387–402. [Google Scholar] [CrossRef]
- Wang, R.-Y.; Yang, S.-H.; Xu, W.-H. Role of Epithelium Sodium Channel in Bone Formation. Chin. Med. J. 2016, 129, 594–600. [Google Scholar] [CrossRef]
- Cui, Y.; Sun, K.; Xiao, Y.; Li, X.; Mo, S.; Yuan, Y.; Wang, P.; Yang, L.; Zhang, R.; Zhu, X. High-Salt Diet Accelerates Bone Loss Accompanied by Activation of Ion Channels Related to Kidney and Bone Tissue in Ovariectomized Rats. Ecotoxicol. Environ. Saf. 2022, 244, 114024. [Google Scholar] [CrossRef]
- Salker, M.S.; Hosseinzadeh, Z.; Alowayed, N.; Zeng, N.; Umbach, A.T.; Webster, Z.; Singh, Y.; Brosens, J.J.; Lang, F. LEFTYA Activates the Epithelial Na+ Channel (ENaC) in Endometrial Cells via Serum and Glucocorticoid Inducible Kinase SGK1. Cell. Physiol. Biochem. 2016, 39, 1295–1306. [Google Scholar] [CrossRef] [PubMed]
- Sharma, S.; Hanukoglu, A.; Hanukoglu, I. Localization of Epithelial Sodium Channel (ENaC) and CFTR in the Germinal Epithelium of the Testis, Sertoli Cells, and Spermatozoa. J. Mol. Histol. 2018, 49, 195–208. [Google Scholar] [CrossRef] [PubMed]
- Sharma, S.; Hanukoglu, I. Mapping the Sites of Localization of Epithelial Sodium Channel (ENaC) and CFTR in Segments of the Mammalian Epididymis. J. Mol. Histol. 2019, 50, 141–154. [Google Scholar] [CrossRef]
- Mutchler, S.M.; Shi, S.; Whelan, S.C.M.; Kleyman, T.R. Validation of Commercially Available Antibodies Directed against Subunits of the Epithelial Na+ Channel. Physiol. Rep. 2023, 11, e15554. [Google Scholar] [CrossRef]
- Jasti, J.; Furukawa, H.; Gonzales, E.B.; Gouaux, E. Structure of Acid-Sensing Ion Channel 1 at 1.9 Å Resolution and Low PH. Nature 2007, 449, 316–323. [Google Scholar] [CrossRef]
- Kashlan, O.B.; Adelman, J.L.; Okumura, S.; Blobner, B.M.; Zuzek, Z.; Hughey, R.P.; Kleyman, T.R.; Grabe, M. Constraint-Based, Homology Model of the Extracellular Domain of the Epithelial Na+ Channel α Subunit Reveals a Mechanism of Channel Activation by Proteases. J. Biol. Chem. 2011, 286, 649–660. [Google Scholar] [CrossRef]
- Noreng, S.; Bharadwaj, A.; Posert, R.; Yoshioka, C.; Baconguis, I. Structure of the Human Epithelial Sodium Channel by Cryo-Electron Microscopy. eLife 2018, 7, e39340. [Google Scholar] [CrossRef]
- Noreng, S.; Posert, R.; Bharadwaj, A.; Houser, A.; Baconguis, I. Molecular Principles of Assembly, Activation, and Inhibition in Epithelial Sodium Channel. eLife 2020, 9, e59038. [Google Scholar] [CrossRef]
- Waldmann, R.; Champigny, G.; Bassilana, F.; Voilley, N.; Lazdunski, M. Molecular Cloning and Functional Expression of a Novel Amiloride-Sensitive Na+ Channel. J. Biol. Chem. 1995, 270, 27411–27414. [Google Scholar] [CrossRef]
- Ji, H.-L.; Zhao, R.-Z.; Chen, Z.-X.; Shetty, S.; Idell, S.; Matalon, S. δ ENaC: A Novel Divergent Amiloride-Inhibitable Sodium Channel. Am. J. Physiol.-Lung Cell. Mol. Physiol. 2012, 303, L1013–L1026. [Google Scholar] [CrossRef]
- Krueger, B.; Schlötzer-Schrehardt, U.; Haerteis, S.; Zenkel, M.; Chankiewitz, V.E.; Amann, K.U.; Kruse, F.E.; Korbmacher, C. Four Subunits (αβγδ) of the Epithelial Sodium Channel (ENaC) Are Expressed in the Human Eye in Various Locations. Investig. Opthalmol. Vis. Sci. 2012, 53, 596. [Google Scholar] [CrossRef] [PubMed]
- Firsov, D.; Gautschi, I.; Merillat, A.M.; Rossier, B.C.; Schild, L. The Heterotetrameric Architecture of the Epithelial Sodium Channel (ENaC). EMBO J. 1998, 17, 344–352. [Google Scholar] [CrossRef] [PubMed]
- Kosari, F.; Sheng, S.; Li, J.; Mak, D.-O.D.; Foskett, J.K.; Kleyman, T.R. Subunit Stoichiometry of the Epithelial Sodium Channel. J. Biol. Chem. 1998, 273, 13469–13474. [Google Scholar] [CrossRef] [PubMed]
- Anantharam, A.; Palmer, L.G. Determination of Epithelial Na+ Channel Subunit Stoichiometry from Single-Channel Conductances. J. Gen. Physiol. 2007, 130, 55–70. [Google Scholar] [CrossRef] [PubMed]
- Trac, P.T.; Thai, T.L.; Linck, V.; Zou, L.; Greenlee, M.; Yue, Q.; Al-Khalili, O.; Alli, A.A.; Eaton, A.F.; Eaton, D.C. Alveolar Nonselective Channels Are ASIC1a/α-ENaC Channels and Contribute to AFC. Am. J. Physiol.-Lung Cell. Mol. Physiol. 2017, 312, L797–L811. [Google Scholar] [CrossRef] [PubMed]
- Shabbir, W.; Scherbaum-Hazemi, P.; Tzotzos, S.; Fischer, B.; Fischer, H.; Pietschmann, H.; Lucas, R.; Lemmens-Gruber, R. Mechanism of Action of Novel Lung Edema Therapeutic AP301 by Activation of the Epithelial Sodium Channel. Mol. Pharmacol. 2013, 84, 899–910. [Google Scholar] [CrossRef] [PubMed]
- Kleyman, T.R.; Eaton, D.C. Regulating ENaC’s Gate. Am. J. Physiol.-Cell Physiol. 2020, 318, C150–C162. [Google Scholar] [CrossRef]
- Santesso, M.R.; Oliveira, F.A.; Tokuhara, C.K.; Oliveira, G.S.N.; Levy, F.M.; Antonio, L.S.; Buzalaf, M.A.R.; Oliveira, R.C. Fluoride Effects on Cell Viability and ENaC Expression in Kidney Epithelial Cells. Toxicol. Mech. Methods 2021, 31, 566–571. [Google Scholar] [CrossRef]
- Knoepp, F.; Ashley, Z.; Barth, D.; Baldin, J.-P.; Jennings, M.; Kazantseva, M.; Saw, E.L.; Katare, R.; Alvarez de la Rosa, D.; Weissmann, N.; et al. Shear Force Sensing of Epithelial Na+ Channel (ENaC) Relies on N-Glycosylated Asparagines in the Palm and Knuckle Domains of αENaC. Proc. Natl. Acad. Sci. USA 2020, 117, 717–726. [Google Scholar] [CrossRef]
- Buck, T.M.; Brodsky, J.L. Epithelial Sodium Channel Biogenesis and Quality Control in the Early Secretory Pathway. Curr. Opin. Nephrol. Hypertens. 2018, 27, 364–372. [Google Scholar] [CrossRef]
- Ishigami, T.; Kino, T.; Minegishi, S.; Araki, N.; Umemura, M.; Ushio, H.; Saigoh, S.; Sugiyama, M. Regulators of Epithelial Sodium Channels in Aldosterone-Sensitive Distal Nephrons (ASDN): Critical Roles of Nedd4L/Nedd4-2 and Salt-Sensitive Hypertension. Int. J. Mol. Sci. 2020, 21, 3871. [Google Scholar] [CrossRef] [PubMed]
- Pitzer, A.L.; Van Beusecum, J.P.; Kleyman, T.R.; Kirabo, A. ENaC in Salt-Sensitive Hypertension: Kidney and Beyond. Curr. Hypertens. Rep. 2020, 22, 69. [Google Scholar] [CrossRef] [PubMed]
- Hanssens, L.S.; Duchateau, J.; Casimir, G.J. CFTR Protein: Not Just a Chloride Channel? Cells 2021, 10, 2844. [Google Scholar] [CrossRef]
- Mutchler, S.M.; Kirabo, A.; Kleyman, T.R. Epithelial Sodium Channel and Salt-Sensitive Hypertension. Hypertension 2021, 77, 759–767. [Google Scholar] [CrossRef]
- Ertuglu, L.A.; Kirabo, A. Dendritic Cell Epithelial Sodium Channel in Inflammation, Salt-Sensitive Hypertension, and Kidney Damage. Kidney360 2022, 3, 1620–1629. [Google Scholar] [CrossRef] [PubMed]
- Vallet, V.; Chraibi, A.; Gaeggeler, H.-P.; Horisberger, J.-D.; Rossier, B.C. An Epithelial Serine Protease Activates the Amiloride-Sensitive Sodium Channel. Nature 1997, 389, 607–610. [Google Scholar] [CrossRef]
- Sheng, S.; Carattino, M.D.; Bruns, J.B.; Hughey, R.P.; Kleyman, T.R. Furin Cleavage Activates the Epithelial Na+ Channel by Relieving Na+ Self-Inhibition. Am. J. Physiol.-Ren. Physiol. 2006, 290, F1488–F1496. [Google Scholar] [CrossRef]
- Kashlan, O.B.; Kleyman, T.R. Epithelial Na+ Channel Regulation by Cytoplasmic and Extracellular Factors. Exp. Cell Res. 2012, 318, 1011–1019. [Google Scholar] [CrossRef]
- Carattino, M.D.; Sheng, S.; Bruns, J.B.; Pilewski, J.M.; Hughey, R.P.; Kleyman, T.R. The Epithelial Na+ Channel Is Inhibited by a Peptide Derived from Proteolytic Processing of Its α Subunit. J. Biol. Chem. 2006, 281, 18901–18907. [Google Scholar] [CrossRef]
- Bruns, J.B.; Carattino, M.D.; Sheng, S.; Maarouf, A.B.; Weisz, O.A.; Pilewski, J.M.; Hughey, R.P.; Kleyman, T.R. Epithelial Na+ Channels Are Fully Activated by Furin- and Prostasin-Dependent Release of an Inhibitory Peptide from the γ-Subunit. J. Biol. Chem. 2007, 282, 6153–6160. [Google Scholar] [CrossRef]
- Hughey, R.P.; Bruns, J.B.; Kinlough, C.L.; Harkleroad, K.L.; Tong, Q.; Carattino, M.D.; Johnson, J.P.; Stockand, J.D.; Kleyman, T.R. Epithelial Sodium Channels Are Activated by Furin-Dependent Proteolysis. J. Biol. Chem. 2004, 279, 18111–18114. [Google Scholar] [CrossRef] [PubMed]
- Carattino, M.D.; Hughey, R.P.; Kleyman, T.R. Proteolytic Processing of the Epithelial Sodium Channel γ Subunit Has a Dominant Role in Channel Activation. J. Biol. Chem. 2008, 283, 25290–25295. [Google Scholar] [CrossRef] [PubMed]
- Passero, C.J.; Carattino, M.D.; Kashlan, O.B.; Myerburg, M.M.; Hughey, R.P.; Kleyman, T.R. Defining an Inhibitory Domain in the Gamma Subunit of the Epithelial Sodium Channel. Am. J. Physiol.-Ren. Physiol. 2010, 299, F854–F861. [Google Scholar] [CrossRef] [PubMed]
- Zachar, R.; Mikkelsen, M.K.; Skjødt, K.; Marcussen, N.; Zamani, R.; Jensen, B.L.; Svenningsen, P. The Epithelial Na+ Channel α- and γ-Subunits Are Cleaved at Predicted Furin-Cleavage Sites, Glycosylated and Membrane Associated in Human Kidney. Pflüg. Arch.—Eur. J. Physiol. 2019, 471, 1383–1396. [Google Scholar] [CrossRef]
- Anand, D.; Hummler, E.; Rickman, O.J. ENaC Activation by Proteases. Acta Physiol. 2022, 235, e13811. [Google Scholar] [CrossRef]
- Carattino, M.D.; Passero, C.J.; Steren, C.A.; Maarouf, A.B.; Pilewski, J.M.; Myerburg, M.M.; Hughey, R.P.; Kleyman, T.R. Defining an Inhibitory Domain in the α-Subunit of the Epithelial Sodium Channel. Am. J. Physiol.-Ren. Physiol. 2008, 294, F47–F52. [Google Scholar] [CrossRef]
- Sure, F.; Bertog, M.; Afonso, S.; Diakov, A.; Rinke, R.; Madej, M.G.; Wittmann, S.; Gramberg, T.; Korbmacher, C.; Ilyaskin, A.V. Transmembrane Serine Protease 2 (TMPRSS2) Proteolytically Activates the Epithelial Sodium Channel (ENaC) by Cleaving the Channel’s γ-Subunit. J. Biol. Chem. 2022, 298, 102004. [Google Scholar] [CrossRef]
- Artunc, F.; Bohnert, B.N.; Schneider, J.C.; Staudner, T.; Sure, F.; Ilyaskin, A.V.; Wörn, M.; Essigke, D.; Janessa, A.; Nielsen, N.V.; et al. Proteolytic Activation of the Epithelial Sodium Channel (ENaC) by Factor VII Activating Protease (FSAP) and Its Relevance for Sodium Retention in Nephrotic Mice. Pflüg. Arch.—Eur. J. Physiol. 2022, 474, 217–229. [Google Scholar] [CrossRef]
- Anand, P.; Puranik, A.; Aravamudan, M.; Venkatakrishnan, A.; Soundararajan, V. SARS-CoV-2 Strategically Mimics Proteolytic Activation of Human ENaC. eLife 2020, 9, e58603. [Google Scholar] [CrossRef]
- Gentzsch, M.; Rossier, B.C. A Pathophysiological Model for COVID-19: Critical Importance of Transepithelial Sodium Transport upon Airway Infection. Function 2020, 1, zqaa024. [Google Scholar] [CrossRef]
- Haerteis, S.; Krappitz, A.; Krappitz, M.; Murphy, J.E.; Bertog, M.; Krueger, B.; Nacken, R.; Chung, H.; Hollenberg, M.D.; Knecht, W.; et al. Proteolytic Activation of the Human Epithelial Sodium Channel by Trypsin IV and Trypsin I Involves Distinct Cleavage Sites. J. Biol. Chem. 2014, 289, 19067–19078. [Google Scholar] [CrossRef] [PubMed]
- Kota, P.; García-Caballero, A.; Dang, H.; Gentzsch, M.; Stutts, M.J.; Dokholyan, N.V. Energetic and Structural Basis for Activation of the Epithelial Sodium Channel by Matriptase. Biochemistry 2012, 51, 3460–3469. [Google Scholar] [CrossRef] [PubMed]
- Althaus, M.; Lawong, R.Y. Proteolytic ENaC Activation in Health and Disease—A Complicated Puzzle. Pflüg. Arch.—Eur. J. Physiol. 2022, 474, 177–179. [Google Scholar] [CrossRef]
- Schild, L.; Schneeberger, E.; Gautschi, I.; Firsov, D. Identification of Amino Acid Residues in the α, β, and γ Subunits of the Epithelial Sodium Channel (ENaC) Involved in Amiloride Block and Ion Permeation. J. Gen. Physiol. 1997, 109, 15–26. [Google Scholar] [CrossRef]
- Mernea, M.; Ulăreanu, R.Ș.; Cucu, D.; Al-Saedi, J.H.; Pop, C.-E.; Fendrihan, S.; Anghelescu, G.D.C.; Mihăilescu, D.F. Epithelial Sodium Channel Inhibition by Amiloride Addressed with THz Spectroscopy and Molecular Modeling. Molecules 2022, 27, 3271. [Google Scholar] [CrossRef]
- Canessa, C.M.; Merillat, A.M.; Rossier, B.C. Membrane Topology of the Epithelial Sodium Channel in Intact Cells. Am. J. Physiol.-Cell Physiol. 1994, 267, C1682–C1690. [Google Scholar] [CrossRef]
- Snyder, P.M.; McDonald, F.J.; Stokes, J.B.; Welsh, M.J. Membrane Topology of the Amiloride-Sensitive Epithelial Sodium Channel. J. Biol. Chem. 1994, 269, 24379–24383. [Google Scholar] [CrossRef]
- Kashlan, O.B.; Kinlough, C.L.; Myerburg, M.M.; Shi, S.; Chen, J.; Blobner, B.M.; Buck, T.M.; Brodsky, J.L.; Hughey, R.P.; Kleyman, T.R. N-Linked Glycans Are Required on Epithelial Na+ Channel Subunits for Maturation and Surface Expression. Am. J. Physiol.-Ren. Physiol. 2018, 314, F483–F492. [Google Scholar] [CrossRef]
- Barth, D.; Knoepp, F.; Fronius, M. Enhanced Shear Force Responsiveness of Epithelial Na+ Channel’s (ENaC) δ Subunit Following the Insertion of N-Glycosylation Motifs Relies on the Extracellular Matrix. Int. J. Mol. Sci. 2021, 22, 2500. [Google Scholar] [CrossRef]
- Firsov, D.; Robert-Nicoud, M.; Gruender, S.; Schild, L.; Rossier, B.C. Mutational Analysis of Cysteine-Rich Domains of the Epithelium Sodium Channel (ENaC). J. Biol. Chem. 1999, 274, 2743–2749. [Google Scholar] [CrossRef]
- Sheng, S.; Maarouf, A.B.; Bruns, J.B.; Hughey, R.P.; Kleyman, T.R. Functional Role of Extracellular Loop Cysteine Residues of the Epithelial Na+ Channel in Na+ Self-Inhibition. J. Biol. Chem. 2007, 282, 20180–20190. [Google Scholar] [CrossRef] [PubMed]
- Blobner, B.M.; Wang, X.-P.; Kashlan, O.B. Conserved Cysteines in the Finger Domain of the Epithelial Na+ Channel α and γ Subunits Are Proximal to the Dynamic Finger–Thumb Domain Interface. J. Biol. Chem. 2018, 293, 4928–4939. [Google Scholar] [CrossRef] [PubMed]
- Salih, M.; Gautschi, I.; van Bemmelen, M.X.; Di Benedetto, M.; Brooks, A.S.; Lugtenberg, D.; Schild, L.; Hoorn, E.J. A Missense Mutation in the Extracellular Domain of αENaC Causes Liddle Syndrome. J. Am. Soc. Nephrol. 2017, 28, 3291–3299. [Google Scholar] [CrossRef] [PubMed]
- Kashlan, O.B.; Blobner, B.M.; Zuzek, Z.; Tolino, M.; Kleyman, T.R. Na+ Inhibits the Epithelial Na+ Channel by Binding to a Site in an Extracellular Acidic Cleft. J. Biol. Chem. 2015, 290, 568–576. [Google Scholar] [CrossRef] [PubMed]
- Winarski, K.L.; Sheng, N.; Chen, J.; Kleyman, T.R.; Sheng, S. Extracellular Allosteric Regulatory Subdomain within the γ Subunit of the Epithelial Na+ Channel. J. Biol. Chem. 2010, 285, 26088–26096. [Google Scholar] [CrossRef]
- Shi, S.; Ghosh, D.D.; Okumura, S.; Carattino, M.D.; Kashlan, O.B.; Sheng, S.; Kleyman, T.R. Base of the Thumb Domain Modulates Epithelial Sodium Channel Gating. J. Biol. Chem. 2011, 286, 14753–14761. [Google Scholar] [CrossRef]
- Shi, S.; Blobner, B.M.; Kashlan, O.B.; Kleyman, T.R. Extracellular Finger Domain Modulates the Response of the Epithelial Sodium Channel to Shear Stress. J. Biol. Chem. 2012, 287, 15439–15444. [Google Scholar] [CrossRef]
- Sheng, S.; Bruns, J.B.; Kleyman, T.R. Extracellular Histidine Residues Crucial for Na+ Self-Inhibition of Epithelial Na+ Channels. J. Biol. Chem. 2004, 279, 9743–9749. [Google Scholar] [CrossRef]
- Shi, S.; Kleyman, T.R. Gamma Subunit Second Transmembrane Domain Contributes to Epithelial Sodium Channel Gating and Amiloride Block. Am. J. Physiol.-Ren. Physiol. 2013, 305, F1585–F1592. [Google Scholar] [CrossRef]
- Maarouf, A.B.; Sheng, N.; Chen, J.; Winarski, K.L.; Okumura, S.; Carattino, M.D.; Boyd, C.R.; Kleyman, T.R.; Sheng, S. Novel Determinants of Epithelial Sodium Channel Gating within Extracellular Thumb Domains. J. Biol. Chem. 2009, 284, 7756–7765. [Google Scholar] [CrossRef]
- Gründer, S.; Firsov, D.; Chang, S.S.; Jaeger, N.F.; Gautschi, I.; Schild, L.; Lifton, R.P.; Rossier, B.C. A Mutation Causing Pseudohypoaldosteronism Type 1 Identifies a Conserved Glycine That Is Involved in the Gating of the Epithelial Sodium Channel. EMBO J. 1997, 16, 899–907. [Google Scholar] [CrossRef] [PubMed]
- Gründer, S.; Fowler Jaeger, N.; Gautschi, I.; Schild, L.; Rossier, B.C. Identification of a Highly Conserved Sequence at the N-Terminus of the Epithelial Na+ Channel α Subunit Involved in Gating. Pflüg. Arch.—Eur. J. Physiol. 1999, 438, 709–715. [Google Scholar] [CrossRef] [PubMed]
- Yoder, N.; Gouaux, E. The His-Gly Motif of Acid-Sensing Ion Channels Resides in a Reentrant ‘Loop’ Implicated in Gating and Ion Selectivity. eLife 2020, 9, e56527. [Google Scholar] [CrossRef] [PubMed]
- Staub, O. Regulation of Stability and Function of the Epithelial Na+ Channel (ENaC) by Ubiquitination. EMBO J. 1997, 16, 6325–6336. [Google Scholar] [CrossRef]
- Frindt, G.; Bertog, M.; Korbmacher, C.; Palmer, L.G. Ubiquitination of Renal ENaC Subunits In Vivo. Am. J. Physiol.-Ren. Physiol. 2020, 318, F1113–F1121. [Google Scholar] [CrossRef]
- Ilyaskin, A.V.; Korbmacher, C.; Diakov, A. Inhibition of the Epithelial Sodium Channel (ENaC) by Connexin 30 Involves Stimulation of Clathrin-Mediated Endocytosis. J. Biol. Chem. 2021, 296, 100404. [Google Scholar] [CrossRef]
- Wang, H.; Traub, L.M.; Weixel, K.M.; Hawryluk, M.J.; Shah, N.; Edinger, R.S.; Perry, C.J.; Kester, L.; Butterworth, M.B.; Peters, K.W.; et al. Clathrin-Mediated Endocytosis of the Epithelial Sodium Channel. J. Biol. Chem. 2006, 281, 14129–14135. [Google Scholar] [CrossRef]
- Abriel, H.; Loffing, J.; Rebhun, J.F.; Pratt, J.H.; Schild, L.; Horisberger, J.-D.; Rotin, D.; Staub, O. Defective Regulation of the Epithelial Na+ Channel by Nedd4 in Liddle’s Syndrome. J. Clin. Investig. 1999, 103, 667–673. [Google Scholar] [CrossRef]
- Schild, L.; Lu, Y.; Gautschi, I.; Schneeberger, E.; Lifton, R.P.; Rossier, B.C. Identification of a PY Motif in the Epithelial Na Channel Subunits as a Target Sequence for Mutations Causing Channel Activation Found in Liddle Syndrome. EMBO J. 1996, 15, 2381–2387. [Google Scholar] [CrossRef]
- Staub, O.; Dho, S.; Henry, P.; Correa, J.; Ishikawa, T.; McGlade, J.; Rotin, D. WW Domains of Nedd4 Bind to the Proline-Rich PY Motifs in the Epithelial Na+ Channel Deleted in Liddle’s Syndrome. EMBO J. 1996, 15, 2371–2380. [Google Scholar] [CrossRef]
- Zhou, R.; Patel, S.V.; Snyder, P.M. Nedd4-2 Catalyzes Ubiquitination and Degradation of Cell Surface ENaC. J. Biol. Chem. 2007, 282, 20207–20212. [Google Scholar] [CrossRef] [PubMed]
- Snyder, P.M.; Price, M.P.; McDonald, F.J.; Adams, C.M.; Volk, K.A.; Zeiher, B.G.; Stokes, J.B.; Welsh, M.J. Mechanism by which Liddle’s syndrome mutations increase activity of a human epithelial Na+ channel. Cell 1995, 83, 969–978. [Google Scholar] [CrossRef] [PubMed]
- Staruschenko, A.; Pochynyuk, O.; Stockand, J.D. Regulation of Epithelial Na+ Channel Activity by Conserved Serine/Threonine Switches within Sorting Signals. J. Biol. Chem. 2005, 280, 39161–39167. [Google Scholar] [CrossRef] [PubMed]
- Mueller, G.M.; Maarouf, A.B.; Kinlough, C.L.; Sheng, N.; Kashlan, O.B.; Okumura, S.; Luthy, S.; Kleyman, T.R.; Hughey, R.P. Cys Palmitoylation of the β Subunit Modulates Gating of the Epithelial Sodium Channel. J. Biol. Chem. 2010, 285, 30453–30462. [Google Scholar] [CrossRef]
- Mueller, G.M.; Yan, W.; Copelovitch, L.; Jarman, S.; Wang, Z.; Kinlough, C.L.; Tolino, M.A.; Hughey, R.P.; Kleyman, T.R.; Rubenstein, R.C. Multiple Residues in the Distal C Terminus of the α-Subunit Have Roles in Modulating Human Epithelial Sodium Channel Activity. Am. J. Physiol.-Ren. Physiol. 2012, 303, F220–F228. [Google Scholar] [CrossRef][Green Version]
- Mukherjee, A.; Mueller, G.M.; Kinlough, C.L.; Sheng, N.; Wang, Z.; Mustafa, S.A.; Kashlan, O.B.; Kleyman, T.R.; Hughey, R.P. Cysteine Palmitoylation of the γ Subunit Has a Dominant Role in Modulating Activity of the Epithelial Sodium Channel. J. Biol. Chem. 2014, 289, 14351–14359. [Google Scholar] [CrossRef]
- Butler, P.L.; Staruschenko, A.; Snyder, P.M. Acetylation Stimulates the Epithelial Sodium Channel by Reducing Its Ubiquitination and Degradation. J. Biol. Chem. 2015, 290, 12497–12503. [Google Scholar] [CrossRef]
- Dinudom, A.; Fotia, A.B.; Lefkowitz, R.J.; Young, J.A.; Kumar, S.; Cook, D.I. The Kinase Grk2 Regulates Nedd4/Nedd4-2-Dependent Control of Epithelial Na+ Channels. Proc. Natl. Acad. Sci. USA 2004, 101, 11886–11890. [Google Scholar] [CrossRef]
- Diakov, A.; Nesterov, V.; Dahlmann, A.; Korbmacher, C. Two Adjacent Phosphorylation Sites in the C-Terminus of the Channel’s α-Subunit Have Opposing Effects on Epithelial Sodium Channel (ENaC) Activity. Pflüg. Arch.—Eur. J. Physiol. 2022, 474, 681–697. [Google Scholar] [CrossRef]
- Abd El-Aziz, T.M.; Soares, A.G.; Mironova, E.; Boiko, N.; Kaur, A.; Archer, C.R.; Stockand, J.D.; Berman, J.M. Mechanisms and Consequences of Casein Kinase II and Ankyrin-3 Regulation of the Epithelial Na+ Channel. Sci. Rep. 2021, 11, 14600. [Google Scholar] [CrossRef]
- Ho, P.-Y.; Li, H.; Cheng, L.; Bhalla, V.; Fenton, R.A.; Hallows, K.R. AMPK Phosphorylation of the β1 Pix Exchange Factor Regulates the Assembly and Function of an ENaC Inhibitory Complex in Kidney Epithelial Cells. Am. J. Physiol.-Ren. Physiol. 2019, 317, F1513–F1525. [Google Scholar] [CrossRef] [PubMed]
- Archer, C.R.; Enslow, B.T.; Carver, C.M.; Stockand, J.D. Phosphatidylinositol 4,5-Bisphosphate Directly Interacts with the β and γ Subunits of the Sodium Channel ENaC. J. Biol. Chem. 2020, 295, 7958–7969. [Google Scholar] [CrossRef] [PubMed]
- Song, C.; Yue, Q.; Moseley, A.; Al-Khalili, O.; Wynne, B.M.; Ma, H.; Wang, L.; Eaton, D.C. Myristoylated Alanine-Rich C Kinase Substrate-like Protein-1 Regulates Epithelial Sodium Channel Activity in Renal Distal Convoluted Tubule Cells. Am. J. Physiol.-Cell Physiol. 2020, 319, C589–C604. [Google Scholar] [CrossRef] [PubMed]
- Yue, Q.; Al-Khalili, O.; Moseley, A.; Yoshigi, M.; Wynne, B.M.; Ma, H.; Eaton, D.C. PIP2 Interacts Electrostatically with MARCKS-like Protein-1 and ENaC in Renal Epithelial Cells. Biology 2022, 11, 1694. [Google Scholar] [CrossRef] [PubMed]
- Helms, M.N.; Liu, L.; Liang, Y.-Y.; Al-Khalili, O.; Vandewalle, A.; Saxena, S.; Eaton, D.C.; Ma, H.-P. Phosphatidylinositol 3,4,5-Trisphosphate Mediates Aldosterone Stimulation of Epithelial Sodium Channel (ENaC) and Interacts with γ-ENaC. J. Biol. Chem. 2005, 280, 40885–40891. [Google Scholar] [CrossRef] [PubMed]
- Pochynyuk, O.; Tong, Q.; Staruschenko, A.; Ma, H.-P.; Stockand, J.D. Regulation of the Epithelial Na+ Channel (ENaC) by Phosphatidylinositides. Am. J. Physiol.-Ren. Physiol. 2006, 290, F949–F957. [Google Scholar] [CrossRef]
- Alli, A.A.; Bao, H.-F.; Alli, A.A.; Aldrugh, Y.; Song, J.Z.; Ma, H.-P.; Yu, L.; Al-Khalili, O.; Eaton, D.C. Phosphatidylinositol Phosphate-Dependent Regulation of Xenopus ENaC by MARCKS Protein. Am. J. Physiol.-Ren. Physiol. 2012, 303, F800–F811. [Google Scholar] [CrossRef]
- Xu, C.; Yang, G.; Fu, Z.; Chen, Y.; Xie, S.; Wang, F.; Yang, T. Na+-Retaining Action of COX-2 (Cyclooxygenase-2)/EP1 Pathway in the Collecting Duct via Activation of Intrarenal Renin-Angiotensin-Aldosterone System and Epithelial Sodium Channel. Hypertension 2022, 79, 1190–1202. [Google Scholar] [CrossRef]
- Vendrov, A.E.; Stevenson, M.D.; Lozhkin, A.; Hayami, T.; Holland, N.A.; Yang, X.; Moss, N.; Pan, H.; Wickline, S.A.; Stockand, J.D.; et al. Renal NOXA1/NOX1 Signaling Regulates Epithelial Sodium Channel and Sodium Retention in Angiotensin II-Induced Hypertension. Antioxid. Redox Signal. 2022, 36, 550–566. [Google Scholar] [CrossRef]
- Shi, S.; Montalbetti, N.; Wang, X.; Rush, B.M.; Marciszyn, A.L.; Baty, C.J.; Tan, R.J.; Carattino, M.D.; Kleyman, T.R. Paraoxonase 3 Functions as a Chaperone to Decrease Functional Expression of the Epithelial Sodium Channel. J. Biol. Chem. 2020, 295, 4950–4962. [Google Scholar] [CrossRef]
- Sherry, S.T. DbSNP: The NCBI Database of Genetic Variation. Nucleic Acids Res. 2001, 29, 308–311. [Google Scholar] [CrossRef] [PubMed]
- Chen, J.; Kleyman, T.R.; Sheng, S. Gain-of-Function Variant of the Human Epithelial Sodium Channel. Am. J. Physiol. Renal Physiol. 2013, 304, F207-213. [Google Scholar] [CrossRef] [PubMed]
- Samaha, F.F.; Rubenstein, R.C.; Yan, W.; Ramkumar, M.; Levy, D.I.; Ahn, Y.J.; Sheng, S.; Kleyman, T.R. Functional Polymorphism in the Carboxyl Terminus of the α-Subunit of the Human Epithelial Sodium Channel. J. Biol. Chem. 2004, 279, 23900–23907. [Google Scholar] [CrossRef]
- Azad, A.K.; Rauh, R.; Vermeulen, F.; Jaspers, M.; Korbmacher, J.; Boissier, B.; Bassinet, L.; Fichou, Y.; des Georges, M.; Stanke, F.; et al. Mutations in the Amiloride-Sensitive Epithelial Sodium Channel in Patients with Cystic Fibrosis-like Disease. Hum. Mutat. 2009, 30, 1093–1103. [Google Scholar] [CrossRef]
- Huber, R.; Krueger, B.; Diakov, A.; Korbmacher, J.; Haerteis, S.; Einsiedel, J.; Gmeiner, P.; Azad, A.; Cuppens, H.; Cassiman, J.-J.; et al. Functional Characterization of a Partial Loss-of-Function Mutation of the Epithelial Sodium Channel (ENaC) Associated with Atypical Cystic Fibrosis. Cell. Physiol. Biochem. 2010, 25, 145–158. [Google Scholar] [CrossRef]
- Rauh, R.; Diakov, A.; Tzschoppe, A.; Korbmacher, J.; Azad, A.K.; Cuppens, H.; Cassiman, J.-J.; Dötsch, J.; Sticht, H.; Korbmacher, C. A Mutation of the Epithelial Sodium Channel Associated with Atypical Cystic Fibrosis Increases Channel Open Probability and Reduces Na+ Self Inhibition: ENaC Gain-of-Function Mutation Associated with Atypical CF. J. Physiol. 2010, 588, 1211–1225. [Google Scholar] [CrossRef]
- Ray, E.C.; Chen, J.; Kelly, T.N.; He, J.; Hamm, L.L.; Gu, D.; Shimmin, L.C.; Hixson, J.E.; Rao, D.C.; Sheng, S.; et al. Human Epithelial Na+ Channel Missense Variants Identified in the GenSalt Study Alter Channel Activity. Am. J. Physiol.-Ren. Physiol. 2016, 311, F908–F914. [Google Scholar] [CrossRef]
- Shimkets, R.A.; Warnock, D.G.; Bositis, C.M.; Nelson-Williams, C.; Hansson, J.H.; Schambelan, M.; Gill, J.R.; Ulick, S.; Milora, R.V.; Findling, J.W.; et al. Liddle’s Syndrome: Heritable Human Hypertension Caused by Mutations in the β Subunit of the Epithelial Sodium Channel. Cell 1994, 79, 407–414. [Google Scholar] [CrossRef]
- Schild, L.; Canessa, C.M.; Shimkets, R.A.; Gautschi, I.; Lifton, R.P.; Rossier, B.C. A Mutation in the Epithelial Sodium Channel Causing Liddle Disease Increases Channel Activity in the Xenopus Laevis Oocyte Expression System. Proc. Natl. Acad. Sci. USA 1995, 92, 5699–5703. [Google Scholar] [CrossRef]
- Firsov, D.; Schild, L.; Gautschi, I.; Mérillat, A.-M.; Schneeberger, E.; Rossier, B.C. Cell Surface Expression of the Epithelial Na Channel and a Mutant Causing Liddle Syndrome: A Quantitative Approach. Proc. Natl. Acad. Sci. USA 1996, 93, 15370–15375. [Google Scholar] [CrossRef]
- Rotin, D. Regulation of the Epithelial Sodium Channel (ENaC) by Accessory Proteins. Curr. Opin. Nephrol. Hypertens. 2000, 9, 529–534. [Google Scholar] [CrossRef] [PubMed]
- Staub, O.; Abriel, H.; Plant, P.; Ishikawa, T.; Kanelis, V.; Saleki, R.; Horisberger, J.-D.; Schild, L.; Rotin, D. Regulation of the Epithelial Na+ Channel by Nedd4 and Ubiquitination. Kidney Int. 2000, 57, 809–815. [Google Scholar] [CrossRef]
- Debonneville, C. Phosphorylation of Nedd4-2 by Sgk1 Regulates Epithelial Na+ Channel Cell Surface Expression. EMBO J. 2001, 20, 7052–7059. [Google Scholar] [CrossRef] [PubMed]
- Chang, S.S.; Grunder, S.; Hanukoglu, A.; Rösler, A.; Mathew, P.M.; Hanukoglu, I.; Schild, L.; Lu, Y.; Shimkets, R.A.; Nelson-Williams, C.; et al. Mutations in Subunits of the Epithelial Sodium Channel Cause Salt Wasting with Hyperkalaemic Acidosis, Pseudohypoaldosteronism Type 1. Nat. Genet. 1996, 12, 248–253. [Google Scholar] [CrossRef] [PubMed]
- Boiko, N.; Kucher, V.; Stockand, J.D. Pseudohypoaldosteronism Type 1 and Liddle’s Syndrome Mutations That Affect the Single-Channel Properties of the Epithelial Na+ Channel. Physiol. Rep. 2015, 3, e12600. [Google Scholar] [CrossRef]
- Ramsey, B.W.; Banks-Schlegel, S.; Accurso, F.J.; Boucher, R.C.; Cutting, G.R.; Engelhardt, J.F.; Guggino, W.B.; Karp, C.L.; Knowles, M.R.; Kolls, J.K.; et al. Future Directions in Early Cystic Fibrosis Lung Disease Research: An NHLBI Workshop Report. Am. J. Respir. Crit. Care Med. 2012, 185, 887–892. [Google Scholar] [CrossRef]
- Stanke, F.; Becker, T.; Ismer, H.S.; Dunsche, I.; Hedtfeld, S.; Kontsendorn, J.; Dittrich, A.-M.; Tümmler, B. Consistent Assignment of Risk and Benign Allele at Rs2303153 in the CF Modifier Gene SCNN1B in Three Independent F508del-CFTR Homozygous Patient Populations. Genes 2021, 12, 1554. [Google Scholar] [CrossRef]
- Flume, P.A.; Chalmers, J.D.; Olivier, K.N. Advances in Bronchiectasis: Endotyping, Genetics, Microbiome, and Disease Heterogeneity. Lancet 2018, 392, 880–890. [Google Scholar] [CrossRef]
- Sheridan, M.B.; Fong, P.; Groman, J.D.; Conrad, C.; Flume, P.; Diaz, R.; Harris, C.; Knowles, M.; Cutting, G.R. Mutations in the Beta-Subunit of the Epithelial Na+ Channel in Patients with a Cystic Fibrosis-like Syndrome. Hum. Mol. Genet. 2005, 14, 3493–3498. [Google Scholar] [CrossRef]
- Fajac, I.; Viel, M.; Sublemontier, S.; Hubert, D.; Bienvenu, T. Could a Defective Epithelial Sodium Channel Lead to Bronchiectasis. Respir. Res. 2008, 9, 46. [Google Scholar] [CrossRef]
- Casals, T.; De-Gracia, J.; Gallego, M.; Dorca, J.; Rodríguez-Sanchón, B.; Ramos, M.; Giménez, J.; Cisteró-Bahima, A.; Olveira, C.; Estivill, X. Bronchiectasis in Adult Patients: An Expression of Heterozygosity for CFTR Gene Mutations?: Heterozygosity for CFTR Mutations in Bronchiectasis Adult Patients. Clin. Genet. 2004, 65, 490–495. [Google Scholar] [CrossRef] [PubMed]
- Mutesa, L.; Azad, A.K.; Verhaeghe, C.; Segers, K.; Vanbellinghen, J.-F.; Ngendahayo, L.; Rusingiza, E.K.; Mutwa, P.R.; Rulisa, S.; Koulischer, L.; et al. Genetic Analysis of Rwandan Patients With Cystic Fibrosis-Like Symptoms. Chest 2009, 135, 1233–1242. [Google Scholar] [CrossRef] [PubMed]
- Geller, D.S.; Rodriguez-Soriano, J.; Boado, A.V.; Schifter, S.; Bayer, M.; Chang, S.S.; Lifton, R.P. Mutations in the Mineralocorticoid Receptor Gene Cause Autosomal Dominant Pseudohypoaldosteronism Type I. Nat. Genet. 1998, 19, 279–281. [Google Scholar] [CrossRef] [PubMed]
- Serra, G.; Antona, V.; D’Alessandro, M.M.; Maggio, M.C.; Verde, V.; Corsello, G. Novel SCNN1A Gene Splicing-Site Mutation Causing Autosomal Recessive Pseudohypoaldosteronism Type 1 (PHA1) in Two Italian Patients Belonging to the Same Small Town. Ital. J. Pediatr. 2021, 47, 138. [Google Scholar] [CrossRef] [PubMed]
- Liu, Z.; Wang, X.; Zhang, Z.; Yang, Z.; Wang, J.; Wang, Y. Case Report: A Novel Compound Heterozygote Mutation of the SCNN1B Gene Identified in a Chinese Familial Pseudohypoaldosteronism Disease Type I With Persistent Hyperkalemia. Front. Pediatr. 2022, 10, 831284. [Google Scholar] [CrossRef]
- Bandhakavi, M.; Wanaguru, A.; Ayuk, L.; Kirk, J.M.; Barrett, T.G.; Kershaw, M.; Högler, W.; Shaw, N.J. Clinical Characteristics and Treatment Requirements of Children with Autosomal Recessive Pseudohypoaldosteronism. Eur. J. Endocrinol. 2021, 184, K15–K20. [Google Scholar] [CrossRef]
- Bockenhauer, D.; Kleta, R. Tubulopathy Meets Sherlock Holmes: Biochemical Fingerprinting of Disorders of Altered Kidney Tubular Salt Handling. Pediatr. Nephrol. 2021, 36, 2553–2561. [Google Scholar] [CrossRef]
- Babar, G.S.; Tariq, M. Challenges of Diagnosing Pseudohypoaldosteronism (PHA) in an Infant. Case Rep. Endocrinol. 2022, 2022, 9921003. [Google Scholar] [CrossRef]
- McVadon, D.H.; Costello, J.M.; Twombley, K.E.; Zyblewski, S.C. A Late Diagnosis of Pseudohypoaldosteronism Type I in an Infant with Hypoplastic Left Heart Syndrome Presenting with Failure to Thrive. Cardiol. Young 2022, 32, 491–493. [Google Scholar] [CrossRef]
- Abdalla, A.; Alhassan, M.A.; Tawfeeg, R.; Sanad, A.; Tawamie, H.; Abdullah, M. Systemic Pseudohypoaldosteronism-1 with Episodic Dyslipidemia in a Sudanese Child. Endocrinol. Diabetes Metab. Case Rep. 2021, 2021, 21-0010. [Google Scholar] [CrossRef]
- Mora-Lopez, F.; Bernal-Quiros, M.; Lechuga-Sancho, A.M.; Lechuga-Campoy, J.L.; Hernandez-Trujillo, N.; Nieto, A. Novel Mutation in the Epithelial Sodium Channel Causing Type I Pseudohypoaldosteronism in a Patient Misdiagnosed with Cystic Fibrosis. Eur. J. Pediatr. 2012, 171, 997–1000. [Google Scholar] [CrossRef] [PubMed]
- Fokkema, I.F.A.C.; Taschner, P.E.M.; Schaafsma, G.C.P.; Celli, J.; Laros, J.F.J.; den Dunnen, J.T. LOVD v.2.0: The next Generation in Gene Variant Databases. Hum. Mutat. 2011, 32, 557–563. [Google Scholar] [CrossRef] [PubMed]
- Canessa, C.M.; Schild, L.; Buell, G.; Thorens, B.; Gautschi, I.; Horisberger, J.-D.; Rossier, B.C. Amiloride-Sensitive Epithelial Na+ Channel Is Made of Three Homologous Subunits. Nature 1994, 367, 463–467. [Google Scholar] [CrossRef] [PubMed]
- Blobner, B.M.; Kirabo, A.; Kashlan, O.B.; Sheng, S.; Arnett, D.K.; Becker, L.C.; Boerwinkle, E.; Carlson, J.C.; Gao, Y.; Gibbs, R.A.; et al. Rare Variants in Genes Encoding Subunits of the Epithelial Na+ Channel Are Associated with Blood Pressure and Kidney Function. Hypertension 2022, 79, 2573–2582. [Google Scholar] [CrossRef]
- Zilbermint, M.; Hannah-Shmouni, F.; Stratakis, C. Genetics of Hypertension in African Americans and Others of African Descent. Int. J. Mol. Sci. 2019, 20, 1081. [Google Scholar] [CrossRef]
- Liu, F.; Yang, X.; Mo, X.; Huang, J.; Chen, J.; Kelly, T.N.; Hixson, J.E.; Rao, D.C.; Gu, C.C.; Shimmin, L.C.; et al. Associations of Epithelial Sodium Channel Genes with Blood Pressure: The GenSalt Study. J. Hum. Hypertens. 2015, 29, 224–228. [Google Scholar] [CrossRef]
- Gu, X.; Gu, D.; He, J.; Rao, D.C.; Hixson, J.E.; Chen, J.; Li, J.; Huang, J.; Wu, X.; Rice, T.K.; et al. Resequencing Epithelial Sodium Channel Genes Identifies Rare Variants Associated With Blood Pressure Salt-Sensitivity: The GenSalt Study. Am. J. Hypertens. 2018, 31, 205–211. [Google Scholar] [CrossRef]
- Mareš, Š.; Filipovský, J.; Vlková, K.; Pešta, M.; Černá, V.; Hrabák, J.; Mlíková Seidlerová, J.; Mayer, O. A Novel Nonsense Mutation in the β-Subunit of the Epithelial Sodium Channel Causing Liddle Syndrome. Blood Press. 2021, 30, 291–299. [Google Scholar] [CrossRef]
- Enslow, B.T.; Stockand, J.D.; Berman, J.M. Liddle’s Syndrome Mechanisms, Diagnosis and Management. Integr. Blood Press. Control 2019, 12, 13–22. [Google Scholar] [CrossRef]
- Capdevila, J.H.; Pidkovka, N.; Mei, S.; Gong, Y.; Falck, J.R.; Imig, J.D.; Harris, R.C.; Wang, W. The Cyp2c44 Epoxygenase Regulates Epithelial Sodium Channel Activity and the Blood Pressure Responses to Increased Dietary Salt. J. Biol. Chem. 2014, 289, 4377–4386. [Google Scholar] [CrossRef]
- Wall, S.M. Regulation of Blood Pressure and Salt Balance By Pendrin-Positive Intercalated Cells: Donald Seldin Lecture 2020. Hypertension 2022, 79, 706–716. [Google Scholar] [CrossRef] [PubMed]
- Pech, V.; Wall, S.M.; Nanami, M.; Bao, H.-F.; Kim, Y.H.; Lazo-Fernandez, Y.; Yue, Q.; Pham, T.D.; Eaton, D.C.; Verlander, J.W. Pendrin Gene Ablation Alters ENaC Subcellular Distribution and Open Probability. Am. J. Physiol.-Ren. Physiol. 2015, 309, F154–F163. [Google Scholar] [CrossRef] [PubMed]
- Pham, T.D.; Elengickal, A.J.; Verlander, J.W.; Al-Qusairi, L.; Chen, C.; Abood, D.C.; King, S.A.; Loffing, J.; Welling, P.A.; Wall, S.M. Pendrin-Null Mice Develop Severe Hypokalemia Following Dietary Na+ and K+ Restriction: Role of ENaC. Am. J. Physiol.-Ren. Physiol. 2022, 322, F486–F497. [Google Scholar] [CrossRef] [PubMed]
- Andersen, H.; Hansen, M.H.; Buhl, K.B.; Stæhr, M.; Friis, U.G.; Enggaard, C.; Supramaniyam, S.; Lund, I.K.; Svenningsen, P.; Hansen, P.B.L.; et al. Plasminogen Deficiency and Amiloride Mitigate Angiotensin II–Induced Hypertension in Type 1 Diabetic Mice Suggesting Effects through the Epithelial Sodium Channel. J. Am. Heart Assoc. 2020, 9, e016387. [Google Scholar] [CrossRef] [PubMed]
- Hinrichs, G.R.; Jensen, B.L.; Svenningsen, P. Mechanisms of Sodium Retention in Nephrotic Syndrome. Curr. Opin. Nephrol. Hypertens. 2020, 29, 207–212. [Google Scholar] [CrossRef]
- Ray, E.C.; Pitzer, A.; Lam, T.; Jordahl, A.; Patel, R.; Ao, M.; Marciszyn, A.; Winfrey, A.; Barak, Y.; Sheng, S.; et al. Salt Sensitivity of Volume and Blood Pressure in a Mouse with Globally Reduced ENaC γ-Subunit Expression. Am. J. Physiol.-Ren. Physiol. 2021, 321, F705–F714. [Google Scholar] [CrossRef]
- Xu, W.; Huang, Y.; Li, L.; Sun, Z.; Shen, Y.; Xing, J.; Li, M.; Su, D.; Liang, X. Hyperuricemia Induces Hypertension through Activation of Renal Epithelial Sodium Channel (ENaC). Metabolism 2016, 65, 73–83. [Google Scholar] [CrossRef]
- Quadri, S.S.; Culver, S.; Ramkumar, N.; Kohan, D.E.; Siragy, H.M. (Pro)Renin Receptor Mediates Obesity-Induced Antinatriuresis and Elevated Blood Pressure via Upregulation of the Renal Epithelial Sodium Channel. PLoS ONE 2018, 13, e0202419. [Google Scholar] [CrossRef]
- Nielsen, M.R.; Frederiksen-Møller, B.; Zachar, R.; Jørgensen, J.S.; Hansen, M.R.; Ydegaard, R.; Svenningsen, P.; Buhl, K.; Jensen, B.L. Urine Exosomes from Healthy and Hypertensive Pregnancies Display Elevated Level of α-Subunit and Cleaved α- and γ-Subunits of the Epithelial Sodium Channel—ENaC. Pflüg. Arch.—Eur. J. Physiol. 2017, 469, 1107–1119. [Google Scholar] [CrossRef]
- García-Rubio, D.; Martínez-Vieyra, I.; de la Mora, M.B.; Fuentes-García, M.A.; Cerecedo, D. Clinical Application of Epithelial Sodium Channel (ENaC) as a Biomarker for Arterial Hypertension. Biosensors 2022, 12, 806. [Google Scholar] [CrossRef]
- Jia, G.; Habibi, J.; Aroor, A.R.; Hill, M.A.; Yang, Y.; Whaley-Connell, A.; Jaisser, F.; Sowers, J.R. Epithelial Sodium Channel in Aldosterone-Induced Endothelium Stiffness and Aortic Dysfunction. Hypertension 2018, 72, 731–738. [Google Scholar] [CrossRef] [PubMed]
- Nakamura, T.; Kurihara, I.; Kobayashi, S.; Yokota, K.; Murai-Takeda, A.; Mitsuishi, Y.; Morisaki, M.; Kohata, N.; Oshima, Y.; Minami, Y.; et al. Intestinal Mineralocorticoid Receptor Contributes to Epithelial Sodium Channel–Mediated Intestinal Sodium Absorption and Blood Pressure Regulation. J. Am. Heart Assoc. 2018, 7, e008259. [Google Scholar] [CrossRef] [PubMed]
- Jia, G.; Sowers, J.R. Hypertension in Diabetes: An Update of Basic Mechanisms and Clinical Disease. Hypertension 2021, 78, 1197–1205. [Google Scholar] [CrossRef]
- Yang, X.; Niu, N.; Liang, C.; Wu, M.-M.; Tang, L.-L.; Wang, Q.-S.; Lou, J.; Song, B.-L.; Zheng, W.-W.; Ma, H.-P.; et al. Stimulation of Epithelial Sodium Channels in Endothelial Cells by Bone Morphogenetic Protein-4 Contributes to Salt-Sensitive Hypertension in Rats. Oxid. Med. Cell. Longev. 2020, 2020, 3921897. [Google Scholar] [CrossRef] [PubMed]
- Ashley, Z.; Mugloo, S.; McDonald, F.J.; Fronius, M. Epithelial Na+ Channel Differentially Contributes to Shear Stress-Mediated Vascular Responsiveness in Carotid and Mesenteric Arteries from Mice. Am. J. Physiol.-Heart Circ. Physiol. 2018, 314, H1022–H1032. [Google Scholar] [CrossRef] [PubMed]
- Althaus, M.; Bogdan, R.; Clauss, W.G.; Fronius, M. Mechano-sensitivity of Epithelial Sodium Channels (ENaCs): Laminar Shear Stress Increases Ion Channel Open Probability. FASEB J. 2007, 21, 2389–2399. [Google Scholar] [CrossRef]
- Coste, B.; Mathur, J.; Schmidt, M.; Earley, T.J.; Ranade, S.; Petrus, M.J.; Dubin, A.E.; Patapoutian, A. Piezo1 and Piezo2 are essential components of distinct mechanically activated cation channels. Science 2010, 330, 55–60. [Google Scholar] [CrossRef]
- Yang, H.; Tenorio Lopes, L.; Barioni, N.O.; Roeske, J.; Incognito, A.V.; Baker, J.; Raj, S.R.; Wilson, R.J.A. The Molecular Makeup of Peripheral and Central Baroreceptors: Stretching a Role for Transient Receptor Potential (TRP), Epithelial Sodium Channel (ENaC), Acid Sensing Ion Channel (ASIC), and Piezo Channels. Cardiovasc. Res. 2022, 118, 3052–3070. [Google Scholar] [CrossRef]
- Fronius, M. Epithelial Na+ Channel and the Glycocalyx: A Sweet and Salty Relationship for Arterial Shear Stress Sensing. Curr. Opin. Nephrol. Hypertens. 2022, 31, 142–150. [Google Scholar] [CrossRef]
- Pérez, F.R.; Venegas, F.; González, M.; Andrés, S.; Vallejos, C.; Riquelme, G.; Sierralta, J.; Michea, L. Endothelial Epithelial Sodium Channel Inhibition Activates Endothelial Nitric Oxide Synthase via Phosphoinositide 3-Kinase/Akt in Small-Diameter Mesenteric Arteries. Hypertension 2009, 53, 1000–1007. [Google Scholar] [CrossRef]
- Tarjus, A.; González-Rivas, C.; Amador-Martínez, I.; Bonnard, B.; López-Marure, R.; Jaisser, F.; Barrera-Chimal, J. The Absence of Endothelial Sodium Channel α (AENaC) Reduces Renal Ischemia/Reperfusion Injury. Int. J. Mol. Sci. 2019, 20, 3132. [Google Scholar] [CrossRef] [PubMed]
- Ydegaard, R.; Andersen, H.; Oxlund, C.S.; Jacobsen, I.A.; Hansen, P.B.L.; Jürgensen, J.F.; Peluso, A.A.; Vanhoutte, P.M.; Staehr, M.; Svenningsen, P.; et al. The Acute Blood Pressure-Lowering Effect of Amiloride Is Independent of Endothelial ENaC and ENOS in Humans and Mice: XXXX. Acta Physiol. 2019, 225, e13189. [Google Scholar] [CrossRef] [PubMed]
- Solak, Y.; Afsar, B.; Vaziri, N.D.; Aslan, G.; Yalcin, C.E.; Covic, A.; Kanbay, M. Hypertension as an Autoimmune and Inflammatory Disease. Hypertens. Res. 2016, 39, 567–573. [Google Scholar] [CrossRef] [PubMed]
- Veiras, L.C.; Bernstein, E.A.; Cao, D.; Okwan-Duodu, D.; Khan, Z.; Gibb, D.R.; Roach, A.; Skelton, R.; Williams, R.M.; Bernstein, K.E.; et al. Tubular IL-1β Induces Salt Sensitivity in Diabetes by Activating Renal Macrophages. Circ. Res. 2022, 131, 59–73. [Google Scholar] [CrossRef]
- Barbaro, N.R.; Foss, J.D.; Kryshtal, D.O.; Tsyba, N.; Kumaresan, S.; Xiao, L.; Mernaugh, R.L.; Itani, H.A.; Loperena, R.; Chen, W.; et al. Dendritic Cell Amiloride-Sensitive Channels Mediate Sodium-Induced Inflammation and Hypertension. Cell Rep. 2017, 21, 1009–1020. [Google Scholar] [CrossRef]
- Sahinoz, M.; Elijovich, F.; Ertuglu, L.A.; Ishimwe, J.; Pitzer, A.; Saleem, M.; Mwesigwa, N.; Kleyman, T.R.; Laffer, C.L.; Kirabo, A. Salt Sensitivity of Blood Pressure in Blacks and Women: A Role of Inflammation, Oxidative Stress, and Epithelial Na+ Channel. Antioxid. Redox Signal. 2021, 35, 1477–1493. [Google Scholar] [CrossRef]
- Spence, J.D. Controlling Resistant Hypertension. Stroke Vasc. Neurol. 2018, 3, 69–75. [Google Scholar] [CrossRef]
- Hebert, P.R.; Coffey, C.S.; Byrne, D.W.; Scott, T.A.; Fagard, R.H.; Rottman, J.N.; Murray, K.T.; Oates, J.A. Treatment of Elderly Hypertensive Patients with Epithelial Sodium Channel Inhibitors Combined with a Thiazide Diuretic Reduces Coronary Mortality and Sudden Cardiac Death. J. Am. Soc. Hypertens. 2008, 2, 355–365. [Google Scholar] [CrossRef][Green Version]
- Elias, S.O.; Sofola, O.A.; Jaja, S.I. Epithelial Sodium Channel Blockade and New β-ENaC Polymorphisms among Normotensive and Hypertensive Adult Nigerians. Clin. Exp. Hypertens. 2019, 41, 144–151. [Google Scholar] [CrossRef]
- Witte, J.; Lampe, J.; Koenen, A.; Urbaneck, I.; Steinbach, A.; Rettig, R.; Grisk, O. The Role of Distal Tubule and Collecting Duct Sodium Reabsorption in Sunitinib-Induced Hypertension. J. Hypertens. 2018, 36, 892–903. [Google Scholar] [CrossRef]
- Isaeva, E.; Bohovyk, R.; Fedoriuk, M.; Shalygin, A.; Klemens, C.A.; Zietara, A.; Levchenko, V.; Denton, J.S.; Staruschenko, A.; Palygin, O. Crosstalk between Epithelial Sodium Channels (ENaC) and Basolateral Potassium Channels (K ir 4.1/K ir 5.1) in the Cortical Collecting Duct. Br. J. Pharmacol. 2022, 179, 2953–2968. [Google Scholar] [CrossRef] [PubMed]
- Zheng, H.; Xu, N.; Zhang, Z.; Wang, F.; Xiao, J.; Ji, X. Setanaxib (GKT137831) Ameliorates Doxorubicin-Induced Cardiotoxicity by Inhibiting the NOX1/NOX4/Reactive Oxygen Species/MAPK Pathway. Front. Pharmacol. 2022, 13, 823975. [Google Scholar] [CrossRef] [PubMed]
- Owada, S.; Endo, H.; Okada, C.; Yoshida, K.; Shida, Y.; Tatemichi, M. Setanaxib as a Potent Hypoxia-Specific Therapeutic Agent Against Liver Cancer. Anticancer Res. 2020, 40, 5071–5079. [Google Scholar] [CrossRef] [PubMed]
- Demircan, M.B.; Mgbecheta, P.C.; Kresinsky, A.; Schnoeder, T.M.; Schröder, K.; Heidel, F.H.; Böhmer, F.D. Combined Activity of the Redox-Modulating Compound Setanaxib (GKT137831) with Cytotoxic Agents in the Killing of Acute Myeloid Leukemia Cells. Antioxidants 2022, 11, 513. [Google Scholar] [CrossRef]
- Jones, D.; Carbone, M.; Invernizzi, P.; Little, N.; Nevens, F.; Swain, M.G.; Wiesel, P.; Levy, C. Impact of Setanaxib on Quality of Life Outcomes in Primary Biliary Cholangitis in a Phase 2 Randomized Controlled Trial. Hepatol. Commun. 2023, 7, e0057. [Google Scholar] [CrossRef]
- Wilcox, C.S.; Pearlman, A. Chemistry and Antihypertensive Effects of Tempol and Other Nitroxides. Pharmacol. Rev. 2008, 60, 418–469. [Google Scholar] [CrossRef]
- Chacko, K.M.; Nouri, M.-Z.; Schramm, W.C.; Malik, Z.; Liu, L.P.; Denslow, N.D.; Alli, A.A. Tempol Alters Urinary Extracellular Vesicle Lipid Content and Release While Reducing Blood Pressure during the Development of Salt-Sensitive Hypertension. Biomolecules 2021, 11, 1804. [Google Scholar] [CrossRef]
- Marunaka, Y.; Marunaka, R.; Sun, H.; Yamamoto, T.; Kanamura, N.; Inui, T.; Taruno, A. Actions of Quercetin, a Polyphenol, on Blood Pressure. Molecules 2017, 22, 209. [Google Scholar] [CrossRef]
- Krumm, P.; Giraldez, T.; de la Rosa, D.A.; Clauss, W.G.; Fronius, M.; Althaus, M. Thiol-Reactive Compounds from Garlic Inhibit the Epithelial Sodium Channel (ENaC). Bioorg. Med. Chem. 2012, 20, 3979–3984. [Google Scholar] [CrossRef]
- Lee, Y.-J.; Jang, Y.-N.; Han, Y.-M.; Kim, H.-M.; Seo, H.S. 6-Gingerol Normalizes the Expression of Biomarkers Related to Hypertension via PPAR δ in HUVECs, HEK293, and Differentiated 3T3-L1 Cells. PPAR Res. 2018, 2018, 6485064. [Google Scholar] [CrossRef]
- Sies, H.; Jones, D.P. Reactive Oxygen Species (ROS) as Pleiotropic Physiological Signalling Agents. Nat. Rev. Mol. Cell Biol. 2020, 21, 363–383. [Google Scholar] [CrossRef] [PubMed]
- Kitamura, K.; Tomita, K. Proteolytic Activation of the Epithelial Sodium Channel and Therapeutic Application of a Serine Protease Inhibitor for the Treatment of Salt-Sensitive Hypertension. Clin. Exp. Nephrol. 2012, 16, 44–48. [Google Scholar] [CrossRef] [PubMed]
- Rooj, A.K.; Cormet-Boyaka, E.; Clark, E.B.; Qadri, Y.J.; Lee, W.; Boddu, R.; Agarwal, A.; Tambi, R.; Uddin, M.; Parpura, V.; et al. Association of Cystic Fibrosis Transmembrane Conductance Regulator with Epithelial Sodium Channel Subunits Carrying Liddle’s Syndrome Mutations. Am. J. Physiol.-Lung Cell. Mol. Physiol. 2021, 321, L308–L320. [Google Scholar] [CrossRef] [PubMed]
- Saber, A.; Nakka, S.S.; Hussain, R.; Hugosson, S. Staphylococcus Aureus in Chronic Rhinosinusitis: The Effect on the Epithelial Chloride Channel (Cystic Fibrosis Transmembrane Conductance Regulator, CFTR) and the Epithelial Sodium Channel (ENaC) Physiology. Acta Otolaryngol. 2019, 139, 652–658. [Google Scholar] [CrossRef] [PubMed]
- Scambler, T.; Jarosz-Griffiths, H.H.; Lara-Reyna, S.; Pathak, S.; Wong, C.; Holbrook, J.; Martinon, F.; Savic, S.; Peckham, D.; McDermott, M.F. ENaC-Mediated Sodium Influx Exacerbates NLRP3-Dependent Inflammation in Cystic Fibrosis. eLife 2019, 8, e49248. [Google Scholar] [CrossRef]
- Grant, G.J.; Liou, T.G.; Paine, R.; Helms, M.N. High-Mobility Group Box-1 Increases Epithelial Sodium Channel Activity and Inflammation via the Receptor for Advanced Glycation End Products. Am. J. Physiol.-Cell Physiol. 2020, 318, C570–C580. [Google Scholar] [CrossRef]
- Haq, I.J.; Gray, M.A.; Garnett, J.P.; Ward, C.; Brodlie, M. Airway Surface Liquid Homeostasis in Cystic Fibrosis: Pathophysiology and Therapeutic Targets. Thorax 2016, 71, 284–287. [Google Scholar] [CrossRef]
- Sasaki, S.; Guo, S. Nucleic Acid Therapies for Cystic Fibrosis. Nucleic Acid Ther. 2018, 28, 1–9. [Google Scholar] [CrossRef]
- Shei, R.-J.; Peabody, J.E.; Kaza, N.; Rowe, S.M. The Epithelial Sodium Channel (ENaC) as a Therapeutic Target for Cystic Fibrosis. Curr. Opin. Pharmacol. 2018, 43, 152–165. [Google Scholar] [CrossRef]
- Gentzsch, M.; Mall, M.A. Ion Channel Modulators in Cystic Fibrosis. Chest 2018, 154, 383–393. [Google Scholar] [CrossRef]
- Moore, P.J.; Tarran, R. The Epithelial Sodium Channel (ENaC) as a Therapeutic Target for Cystic Fibrosis Lung Disease. Expert Opin. Ther. Targets 2018, 22, 687–701. [Google Scholar] [CrossRef] [PubMed]
- Martin, S.L.; Saint-Criq, V.; Hwang, T.-C.; Csanády, L. Ion Channels as Targets to Treat Cystic Fibrosis Lung Disease. J. Cyst. Fibros. 2018, 17, S22–S27. [Google Scholar] [CrossRef] [PubMed]
- Mall, M.A.; Mayer-Hamblett, N.; Rowe, S.M. Cystic Fibrosis: Emergence of Highly Effective Targeted Therapeutics and Potential Clinical Implications. Am. J. Respir. Crit. Care Med. 2020, 201, 1193–1208. [Google Scholar] [CrossRef] [PubMed]
- Laselva, O.; Guerra, L.; Castellani, S.; Favia, M.; Di Gioia, S.; Conese, M. Small-Molecule Drugs for Cystic Fibrosis: Where Are We Now? Pulm. Pharmacol. Ther. 2022, 72, 102098. [Google Scholar] [CrossRef] [PubMed]
- De Boeck, K. Cystic fibrosis in the year 2020: A disease with a new face. Acta Paediatr. 2020, 109, 893–899. [Google Scholar] [CrossRef]
- Bierlaagh, M.C.; Muilwijk, D.; Beekman, J.M.; van der Ent, C.K. A new era for people with cystic fibrosis. Eur. J. Pediatr. 2021, 180, 2731–2739. [Google Scholar] [CrossRef]
- O’Riordan, T.G.; Donn, K.H.; Hodsman, P.; Ansede, J.H.; Newcomb, T.; Lewis, S.A.; Flitter, W.D.; White, V.S.; Johnson, M.R.; Montgomery, A.B.; et al. Acute Hyperkalemia Associated with Inhalation of a Potent ENaC Antagonist: Phase 1 Trial of GS-9411. J. Aerosol Med. Pulm. Drug Deliv. 2014, 27, 200–208. [Google Scholar] [CrossRef]
- Goss, C.H.; Fajac, I.; Jain, R.; Seibold, W.; Gupta, A.; Hsu, M.-C.; Sutharsan, S.; Davies, J.C.; Mall, M.A. Efficacy and Safety of Inhaled ENaC Inhibitor BI 1265162 in Patients with Cystic Fibrosis: BALANCE-CF 1, a Randomised, Phase II Study. Eur. Respir. J. 2022, 59, 2100746. [Google Scholar] [CrossRef]
- Coote, K.J.; Paisley, D.; Czarnecki, S.; Tweed, M.; Watson, H.; Young, A.; Sugar, R.; Vyas, M.; Smith, N.J.; Baettig, U.; et al. NVP-QBE170: An Inhaled Blocker of the Epithelial Sodium Channel with a Reduced Potential to Induce Hyperkalaemia: NVP-QBE170: A Novel Inhaled ENaC Blocker. Br. J. Pharmacol. 2015, 172, 2814–2826. [Google Scholar] [CrossRef]
- Åstrand, A.; Libby, E.F.; Shei, R.-J.; Lever, J.E.P.; Kaza, N.; Adewale, A.T.; Boitet, E.; Edwards, L.; Hemmerling, M.; Root, J.; et al. Preclinical Evaluation of the Epithelial Sodium Channel Inhibitor AZD5634 and Implications on Human Translation. Am. J. Physiol.-Lung Cell. Mol. Physiol. 2022, 323, L536–L547. [Google Scholar] [CrossRef]
- Kristensson, C.; Åstrand, A.; Donaldson, S.; Goldwater, R.; Abdulai, R.; Patel, N.; Gardiner, P.; Tehler, U.; Mercier, A.-K.; Olsson, M.; et al. AZD5634, an Inhaled ENaC Inhibitor, in Healthy Subjects and Patients with Cystic Fibrosis. J. Cyst. Fibros. 2022, 21, 684–690. [Google Scholar] [CrossRef] [PubMed]
- Giorgetti, M.; Klymiuk, N.; Bähr, A.; Hemmerling, M.; Jinton, L.; Tarran, R.; Malmgren, A.; Åstrand, A.; Hansson, G.C.; Ermund, A. New Generation ENaC Inhibitors Detach Cystic Fibrosis Airway Mucus Bundles via Sodium/Hydrogen Exchanger Inhibition. Eur. J. Pharmacol. 2021, 904, 174123. [Google Scholar] [CrossRef] [PubMed]
- Terryah, S.T.; Fellner, R.C.; Ahmad, S.; Moore, P.J.; Reidel, B.; Sesma, J.I.; Kim, C.S.; Garland, A.L.; Scott, D.W.; Sabater, J.R.; et al. Evaluation of a SPLUNC1-Derived Peptide for the Treatment of Cystic Fibrosis Lung Disease. Am. J. Physiol.-Lung Cell. Mol. Physiol. 2018, 314, L192–L205. [Google Scholar] [CrossRef] [PubMed]
- Kim, C.S.; Ahmad, S.; Wu, T.; Walton, W.G.; Redinbo, M.R.; Tarran, R. SPLUNC1 Is an Allosteric Modulator of the Epithelial Sodium Channel. FASEB J. 2018, 32, 2478–2491. [Google Scholar] [CrossRef] [PubMed]
- Scott, D.W.; Walker, M.P.; Sesma, J.; Wu, B.; Stuhlmiller, T.J.; Sabater, J.R.; Abraham, W.M.; Crowder, T.M.; Christensen, D.J.; Tarran, R. SPX-101 Is a Novel Epithelial Sodium Channel–Targeted Therapeutic for Cystic Fibrosis That Restores Mucus Transport. Am. J. Respir. Crit. Care Med. 2017, 196, 734–744. [Google Scholar] [CrossRef]
- Sesma, J.I.; Wu, B.; Stuhlmiller, T.J.; Scott, D.W. SPX-101 Is Stable in and Retains Function after Exposure to Cystic Fibrosis Sputum. J. Cyst. Fibros. 2019, 18, 244–250. [Google Scholar] [CrossRef]
- Walker, M.P.; Cowlen, M.; Christensen, D.; Miyamoto, M.; Barley, P.; Crowder, T. Nonclinical Safety Assessment of SPX-101, a Novel Peptide Promoter of Epithelial Sodium Channel Internalization for the Treatment of Cystic Fibrosis. Inhal. Toxicol. 2017, 29, 356–365. [Google Scholar] [CrossRef]
- Couroux, P.; Farias, P.; Rizvi, L.; Griffin, K.; Hudson, C.; Crowder, T.; Tarran, R.; Tullis, E. First Clinical Trials of Novel ENaC Targeting Therapy, SPX-101, in Healthy Volunteers and Adults with Cystic Fibrosis. Pulm. Pharmacol. Ther. 2019, 58, 101819. [Google Scholar] [CrossRef]
- Fujikawa, H.; Kawakami, T.; Nakashima, R.; Nasu, A.; Kamei, S.; Nohara, H.; Eto, Y.; Ueno-Shuto, K.; Takeo, T.; Nakagata, N.; et al. Azithromycin Inhibits Constitutive Airway Epithelial Sodium Channel Activation in Vitro and Modulates Downstream Pathogenesis in Vivo. Biol. Pharm. Bull. 2020, 43, 725–730. [Google Scholar] [CrossRef]
- Gróf, I.; Bocsik, A.; Harazin, A.; Santa-Maria, A.R.; Vizsnyiczai, G.; Barna, L.; Kiss, L.; Fűr, G.; Rakonczay, Z.; Ambrus, R.; et al. The Effect of Sodium Bicarbonate, a Beneficial Adjuvant Molecule in Cystic Fibrosis, on Bronchial Epithelial Cells Expressing a Wild-Type or Mutant CFTR Channel. Int. J. Mol. Sci. 2020, 21, 4024. [Google Scholar] [CrossRef]
- Pierandrei, S.; Truglio, G.; Ceci, F.; Del Porto, P.; Bruno, S.M.; Castellani, S.; Conese, M.; Ascenzioni, F.; Lucarelli, M. DNA Methylation Patterns Correlate with the Expression of SCNN1A, SCNN1B, and SCNN1G (Epithelial Sodium Channel, ENaC) Genes. Int. J. Mol. Sci. 2021, 22, 3754. [Google Scholar] [CrossRef] [PubMed]
- Hey, J.; Paulsen, M.; Toth, R.; Weichenhan, D.; Butz, S.; Schatterny, J.; Liebers, R.; Lutsik, P.; Plass, C.; Mall, M.A. Epigenetic Reprogramming of Airway Macrophages Promotes Polarization and Inflammation in Muco-Obstructive Lung Disease. Nat. Commun. 2021, 12, 6520. [Google Scholar] [CrossRef] [PubMed]
- Blaconà, G.; Raso, R.; Castellani, S.; Pierandrei, S.; Del Porto, P.; Ferraguti, G.; Ascenzioni, F.; Conese, M.; Lucarelli, M. Downregulation of Epithelial Sodium Channel (ENaC) Activity in Cystic Fibrosis Cells by Epigenetic Targeting. Cell. Mol. Life Sci. 2022, 79, 257. [Google Scholar] [CrossRef] [PubMed]
- Boyd, C.A.; Guo, S.; Huang, L.; Kerem, B.; Oren, Y.S.; Walker, A.J.; Hart, S.L. New Approaches to Genetic Therapies for Cystic Fibrosis. J. Cyst. Fibros. 2020, 19, S54–S59. [Google Scholar] [CrossRef] [PubMed]
- Almughem, F.A.; Aldossary, A.M.; Tawfik, E.A.; Alomary, M.N.; Alharbi, W.S.; Alshahrani, M.Y.; Alshehri, A.A. Cystic Fibrosis: Overview of the Current Development Trends and Innovative Therapeutic Strategies. Pharmaceutics 2020, 12, 616. [Google Scholar] [CrossRef]
- Zhao, C.; Crosby, J.; Lv, T.; Bai, D.; Monia, B.P.; Guo, S. Antisense Oligonucleotide Targeting of MRNAs Encoding ENaC Subunits α, β, and γ Improves Cystic Fibrosis-like Disease in Mice. J. Cyst. Fibros. 2019, 18, 334–341. [Google Scholar] [CrossRef]
- Crosby, J.R.; Zhao, C.; Jiang, C.; Bai, D.; Katz, M.; Greenlee, S.; Kawabe, H.; McCaleb, M.; Rotin, D.; Guo, S.; et al. Inhaled ENaC Antisense Oligonucleotide Ameliorates Cystic Fibrosis-like Lung Disease in Mice. J. Cyst. Fibros. 2017, 16, 671–680. [Google Scholar] [CrossRef]
- Tagalakis, A.D.; Munye, M.M.; Ivanova, R.; Chen, H.; Smith, C.M.; Aldossary, A.M.; Rosa, L.Z.; Moulding, D.; Barnes, J.L.; Kafetzis, K.N.; et al. Effective Silencing of ENaC by SiRNA Delivered with Epithelial-Targeted Nanocomplexes in Human Cystic Fibrosis Cells and in Mouse Lung. Thorax 2018, 73, 847–856. [Google Scholar] [CrossRef]
- Reihill, J.A.; Walker, B.; Hamilton, R.A.; Ferguson, T.E.G.; Elborn, J.S.; Stutts, M.J.; Harvey, B.J.; Saint-Criq, V.; Hendrick, S.M.; Martin, S.L. Inhibition of Protease–Epithelial Sodium Channel Signaling Improves Mucociliary Function in Cystic Fibrosis Airways. Am. J. Respir. Crit. Care Med. 2016, 194, 701–710. [Google Scholar] [CrossRef]
- Dickerhof, N.; Huang, J.; Min, E.; Michaëlsson, E.; Lindstedt, E.-L.; Pearson, J.F.; Kettle, A.J.; Day, B.J. Myeloperoxidase Inhibition Decreases Morbidity and Oxidative Stress in Mice with Cystic Fibrosis-like Lung Inflammation. Free Radic. Biol. Med. 2020, 152, 91–99. [Google Scholar] [CrossRef]
- Lucas, R.; Magez, S.; De Leys, R.; Fransen, L.; Scheerlinck, J.-P.; Rampelberg, M.; Sablon, E.; De Baetselier, P. Mapping the Lectin-Like Activity of Tumor Necrosis Factor. Science 1994, 263, 814–817. [Google Scholar] [CrossRef] [PubMed]
- Hazemi, P.; Tzotzos, S.J.; Fischer, B.; Andavan, G.S.B.; Fischer, H.; Pietschmann, H.; Lucas, R.; Lemmens-Gruber, R. Essential Structural Features of TNF-α Lectin-like Domain Derived Peptides for Activation of Amiloride-Sensitive Sodium Current in A549 Cells. J. Med. Chem. 2010, 53, 8021–8029. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Shabbir, W.; Tzotzos, S.; Bedak, M.; Aufy, M.; Willam, A.; Kraihammer, M.; Holzner, A.; Czikora, I.; Scherbaum-Hazemi, P.; Fischer, H.; et al. Glycosylation-Dependent Activation of Epithelial Sodium Channel by Solnatide. Biochem. Pharmacol. 2015, 98, 740–753. [Google Scholar] [CrossRef] [PubMed]
- Lazrak, A.; Matalon, S. CAMP-Induced Changes of Apical Membrane Potentials of Confluent H441 Monolayers. Am. J. Physiol. Lung Cell. Mol. Physiol. 2003, 285, L443–L450. [Google Scholar] [CrossRef] [PubMed]
- Czikora, I.; Alli, A.; Bao, H.-F.; Kaftan, D.; Sridhar, S.; Apell, H.-J.; Gorshkov, B.; White, R.; Zimmermann, A.; Wendel, A.; et al. A Novel Tumor Necrosis Factor–Mediated Mechanism of Direct Epithelial Sodium Channel Activation. Am. J. Respir. Crit. Care Med. 2014, 190, 522–532. [Google Scholar] [CrossRef] [PubMed]
- Lazrak, A.; Samanta, A.; Matalon, S. Biophysical Properties and Molecular Characterization of Amiloride-Sensitive Sodium Channels in A549 Cells. Am. J. Physiol. Lung Cell. Mol. Physiol. 2000, 278, L848–L857. [Google Scholar] [CrossRef]
- Tzotzos, S.; Fischer, B.; Fischer, H.; Pietschmann, H.; Lucas, R.; Dupré, G.; Lemmens-Gruber, R.; Hazemi, P.; Prymaka, V.; Shabbir, W. AP301, a Synthetic Peptide Mimicking the Lectin-like Domain of TNF, Enhances Amiloride-Sensitive Na+ Current in Primary Dog, Pig and Rat Alveolar Type II Cells. Pulm. Pharmacol. Ther. 2013, 26, 356–363. [Google Scholar] [CrossRef]
- Braun, C.; Hamacher, J.; Morel, D.R.; Wendel, A.; Lucas, R. Dichotomal Role of TNF in Experimental Pulmonary Edema Reabsorption. J. Immunol. 2005, 175, 3402–3408. [Google Scholar] [CrossRef]
- Vadász, I.; Schermuly, R.T.; Ghofrani, H.A.; Rummel, S.; Wehner, S.; Mühldorfer, I.; Schäfer, K.P.; Seeger, W.; Morty, R.E.; Grimminger, F.; et al. The Lectin-like Domain of Tumor Necrosis Factor-Alpha Improves Alveolar Fluid Balance in Injured Isolated Rabbit Lungs. Crit. Care Med. 2008, 36, 1543–1550. [Google Scholar] [CrossRef]
- Hamacher, J.; Stammberger, U.; Roux, J.; Kumar, S.; Yang, G.; Xiong, C.; Schmid, R.A.; Fakin, R.M.; Chakraborty, T.; Hossain, H.M.D.; et al. The Lectin-like Domain of Tumor Necrosis Factor Improves Lung Function after Rat Lung Transplantation—Potential Role for a Reduction in Reactive Oxygen Species Generation. Crit. Care Med. 2010, 38, 871–878. [Google Scholar] [CrossRef]
- Lucas, R.; Czikora, I.; Sridhar, S.; Zemskov, E.; Gorshkov, B.; Siddaramappa, U.; Oseghale, A.; Lawson, J.; Verin, A.; Rick, F.G.; et al. Mini-Review: Novel Therapeutic Strategies to Blunt Actions of Pneumolysin in the Lungs. Toxins 2013, 5, 1244–1260. [Google Scholar] [CrossRef] [PubMed]
- Xiong, C.; Yang, G.; Kumar, S.; Aggarwal, S.; Leustik, M.; Snead, C.; Hamacher, J.; Fischer, B.; Umapathy, N.S.; Hossain, H.; et al. The Lectin-like Domain of TNF Protects from Listeriolysin-Induced Hyperpermeability in Human Pulmonary Microvascular Endothelial Cells—A Crucial Role for Protein Kinase C-α Inhibition. Vascul. Pharmacol. 2010, 52, 207–213. [Google Scholar] [CrossRef] [PubMed]
- Lucas, R.; Yue, Q.; Alli, A.; Duke, B.J.; Al-Khalili, O.; Thai, T.L.; Hamacher, J.; Sridhar, S.; Lebedyeva, I.; Su, H.; et al. The Lectin-like Domain of TNF Increases ENaC Open Probability through a Novel Site at the Interface between the Second Transmembrane and C-Terminal Domains of the α-Subunit. J. Biol. Chem. 2016, 291, 23440–23451. [Google Scholar] [CrossRef]
- Fukuda, N.; Jayr, C.; Lazrak, A.; Wang, Y.; Lucas, R.; Matalon, S.; Matthay, M.A. Mechanisms of TNF-Alpha Stimulation of Amiloride-Sensitive Sodium Transport across Alveolar Epithelium. Am. J. Physiol. Lung Cell. Mol. Physiol. 2001, 280, L1258–L1265. [Google Scholar] [CrossRef] [PubMed]
- Martin-Malpartida, P.; Arrastia-Casado, S.; Farrera-Sinfreu, J.; Lucas, R.; Fischer, H.; Fischer, B.; Eaton, D.C.; Tzotzos, S.; Macias, M.J. Conformational Ensemble of the TNF-Derived Peptide Solnatide in Solution. Comput. Struct. Biotechnol. J. 2022, 20, 2082–2090. [Google Scholar] [CrossRef] [PubMed]
- Yang, G.; Pillich, H.; White, R.; Czikora, I.; Pochic, I.; Yue, Q.; Hudel, M.; Gorshkov, B.; Verin, A.; Sridhar, S.; et al. Listeriolysin O Causes ENaC Dysfunction in Human Airway Epithelial Cells. Toxins 2018, 10, 79. [Google Scholar] [CrossRef]
- Czikora, I.; Alli, A.A.; Sridhar, S.; Matthay, M.A.; Pillich, H.; Hudel, M.; Berisha, B.; Gorshkov, B.; Romero, M.J.; Gonzales, J.; et al. Epithelial Sodium Channel-α Mediates the Protective Effect of the TNF-Derived TIP Peptide in Pneumolysin-Induced Endothelial Barrier Dysfunction. Front. Immunol. 2017, 8, 842. [Google Scholar] [CrossRef]
- Schwameis, R.; Eder, S.; Pietschmann, H.; Fischer, B.; Mascher, H.; Tzotzos, S.; Fischer, H.; Lucas, R.; Zeitlinger, M.; Hermann, R. A FIM Study to Assess Safety and Exposure of Inhaled Single Doses of AP301-A Specific ENaC Channel Activator for the Treatment of Acute Lung Injury: The Journal of Clinical Pharmacology. J. Clin. Pharmacol. 2014, 54, 341–350. [Google Scholar] [CrossRef]
- Krenn, K.; Lucas, R.; Croizé, A.; Boehme, S.; Klein, K.U.; Hermann, R.; Markstaller, K.; Ullrich, R. Inhaled AP301 for Treatment of Pulmonary Edema in Mechanically Ventilated Patients with Acute Respiratory Distress Syndrome: A Phase IIa Randomized Placebo-Controlled Trial. Crit. Care 2017, 21, 194. [Google Scholar] [CrossRef]
- Schmid, B.; Kredel, M.; Ullrich, R.; Krenn, K.; Lucas, R.; Markstaller, K.; Fischer, B.; Kranke, P.; Meybohm, P.; Zwißler, B.; et al. Safety and Preliminary Efficacy of Sequential Multiple Ascending Doses of Solnatide to Treat Pulmonary Permeability Edema in Patients with Moderate-to-Severe ARDS—A Randomized, Placebo-Controlled, Double-Blind Trial. Trials 2021, 22, 643. [Google Scholar] [CrossRef]
- Schmid, B.; Kranke, P.; Lucas, R.; Meybohm, P.; Zwissler, B.; Frank, S. Safety and Preliminary Efficacy of Sequential Multiple Ascending Doses of Solnatide to Treat Pulmonary Permeability Edema in Patients with Moderate to Severe ARDS in a Randomized, Placebo-Controlled, Double-Blind Trial: Preliminary Evaluation of Safety and Feasibility in Light of the COVID-19 Pandemic. Trials 2022, 23, 252. [Google Scholar] [CrossRef] [PubMed]
- Artigas, A.; Camprubí-Rimblas, M.; Tantinyà, N.; Bringué, J.; Guillamat-Prats, R.; Matthay, M.A. Inhalation Therapies in Acute Respiratory Distress Syndrome. Ann. Transl. Med. 2017, 5, 293. [Google Scholar] [CrossRef] [PubMed]
- Horie, S.; McNicholas, B.; Rezoagli, E.; Pham, T.; Curley, G.; McAuley, D.; O’Kane, C.; Nichol, A.; dos Santos, C.; Rocco, P.R.M.; et al. Emerging Pharmacological Therapies for ARDS: COVID-19 and Beyond. Intensive Care Med. 2020, 46, 2265–2283. [Google Scholar] [CrossRef] [PubMed]
- Tzotzos, S.J.; Fischer, B.; Fischer, H.; Zeitlinger, M. Incidence of ARDS and Outcomes in Hospitalized Patients with COVID-19: A Global Literature Survey. Crit. Care 2020, 24, 516. [Google Scholar] [CrossRef]
- Cattel, F.; Giordano, S.; Bertiond, C.; Lupia, T.; Corcione, S.; Scaldaferri, M.; Angelone, L.; De Rosa, F.G. Use of Exogenous Pulmonary Surfactant in Acute Respiratory Distress Syndrome (ARDS): Role in SARS-CoV-2-Related Lung Injury. Respir. Physiol. Neurobiol. 2021, 288, 103645. [Google Scholar] [CrossRef]
- Aigner, C.; Slama, A.; Barta, M.; Mitterbauer, A.; Lang, G.; Taghavi, S.; Matilla, J.; Ullrich, R.; Krenn, K.; Jaksch, P.; et al. Treatment of Primary Graft Dysfunction after Lung Transplantation with Orally Inhaled AP301: A Prospective, Randomized Pilot Study. J. Heart Lung Transplant. 2018, 37, 225–231. [Google Scholar] [CrossRef]
- Zhou, Q.; Wang, D.; Liu, Y.; Yang, X.; Lucas, R.; Fischer, B. Solnatide Demonstrates Profound Therapeutic Activity in a Rat Model of Pulmonary Edema Induced by Acute Hypobaric Hypoxia and Exercise. Chest 2017, 151, 658–667. [Google Scholar] [CrossRef]
- Dirlewanger, M.; Huser, D.; Zennaro, M.-C.; Girardin, E.; Schild, L.; Schwitzgebel, V.M. A Homozygous Missense Mutation in SCNN1A Is Responsible for a Transient Neonatal Form of Pseudohypoaldosteronism Type 1. Am. J. Physiol.-Endocrinol. Metab. 2011, 301, E467–E473. [Google Scholar] [CrossRef][Green Version]
- Riepe, F.G. Pseudohypoaldosteronism. In Endocrine Development; Maghnie, M., Loche, S., Cappa, M., Ghizzoni, L., Lorini, R., Eds.; Karger: Basel, Switzerland, 2013; Volume 24, pp. 86–95. ISBN 978-3-318-02267-4. [Google Scholar]
- Güran, T.; Değirmenci, S.; Bulut, İ.K.; Say, A.; Riepe, F.G.; Güran, Ö. Critical Points in the Management of Pseudohypoaldosteronism Type 1. J. Clin. Res. Pediatr. Endocrinol. 2011, 3, 98–100. [Google Scholar] [CrossRef]
- Kerem, E.; Bistritzer, T.; Hanukoglu, A.; Hofmann, T.; Zhou, Z.; Bennett, W.; MacLaughlin, E.; Barker, P.; Nash, M.; Quittell, L.; et al. Pulmonary Epithelial Sodium-Channel Dysfunction and Excess Airway Liquid in Pseudohypoaldosteronism. N. Engl. J. Med. 1999, 341, 156–162. [Google Scholar] [CrossRef]
- Welzel, M.; Akin, L.; Büscher, A.; Güran, T.; Hauffa, B.P.; Högler, W.; Leonards, J.; Karges, B.; Kentrup, H.; Kirel, B.; et al. Five Novel Mutations in the SCNN1A Gene Causing Autosomal Recessive Pseudohypoaldosteronism Type 1. Eur. J. Endocrinol. 2013, 168, 707–715. [Google Scholar] [CrossRef] [PubMed]
- Cayir, A.; Demirelli, Y.; Yildiz, D.; Kahveci, H.; Yarali, O.; Kurnaz, E.; Vuralli, D.; Demirbilek, H. Systemic Pseudohypoaldosteronism Type 1 Due to 3 Novel Mutations in SCNN1Aand SCNN1BGenes. Horm. Res. Paediatr. 2019, 91, 175–185. [Google Scholar] [CrossRef] [PubMed]
- Gopal-Kothandapani, J.S.; Doshi, A.B.; Smith, K.; Christian, M.; Mushtaq, T.; Banerjee, I.; Padidela, R.; Ramakrishnan, R.; Owen, C.; Cheetham, T.; et al. Phenotypic Diversity and Correlation with the Genotypes of Pseudohypoaldosteronism Type 1. J. Pediatr. Endocrinol. Metab. JPEM 2019, 32, 959–967. [Google Scholar] [CrossRef] [PubMed]
- Bonny, O.; Knoers, N.; Monnens, L.; Rossier, B.C. A Novel Mutation of the Epithelial Na+ Channel Causes Type 1 Pseudohypoaldosteronism. Pediatr. Nephrol. Berl. Ger. 2002, 17, 804–808. [Google Scholar] [CrossRef] [PubMed]
- Kala Ahluwalia, G.; Dasouki, M.; Lennon, A. Phenotypic Variation of Autosomal Recessive Pseudohypoaldosteronism Type I: A Case in Point. Clin. Case Rep. 2014, 2, 326–330. [Google Scholar] [CrossRef] [PubMed]
- Ekinci, Z.; Aytac, M.B.; Cheong, H.I. A Case of SCNN1A Splicing Mutation Presenting as Mild Systemic Pseudohypoaldosteronism Type 1. J. Pediatr. Endocrinol. Metab. 2013, 26, 1197–1200. [Google Scholar] [CrossRef] [PubMed]
- Schaedel, C.; Marthinsen, L.; Kristoffersson, A.C.; Kornfält, R.; Nilsson, K.O.; Orlenius, B.; Holmberg, L. Lung Symptoms in Pseudohypoaldosteronism Type 1 Are Associated with Deficiency of the Alpha-Subunit of the Epithelial Sodium Channel. J. Pediatr. 1999, 135, 739–745. [Google Scholar] [CrossRef]
- Huneif, M.A.; Alhazmy, Z.H.; Shoomi, A.M.; Alghofely, M.A.; Heena, H.; Mushiba, A.M.; AlSaheel, A. A Novel SCNN1A Variation in a Patient with Autosomal-Recessive Pseudohypoaldosteronism Type 1. J. Clin. Res. Pediatr. Endocrinol. 2022, 14, 244–250. [Google Scholar] [CrossRef]
- Wang, J.; Yu, T.; Yin, L.; Li, J.; Yu, L.; Shen, Y.; Yu, Y.; Shen, Y.; Fu, Q. Novel Mutations in the SCNN1A Gene Causing Pseudohypoaldosteronism Type 1. PLoS ONE 2013, 8, e65676. [Google Scholar] [CrossRef]
- Silva, N.; Costa, M.; Silva, A.; Sá, C.; Martins, S.; Antunes, A.; Marques, O.; Castedo, S.; Pereira, A. A Case of Systemic Pseudohypoaldosteronism with a Novel Mutation in the SCNN1A Gene. Endocrinol. Nutr. Organo Soc. Esp. Endocrinol. Nutr. 2013, 60, 33–36. [Google Scholar] [CrossRef]
- Alzahrani, A.S.; Alswailem, M.; Abbas, B.B.; Qasem, E.; Alsagheir, A.; Al Shidhani, A.; Al Sinani, A.; Al Badi, M.; Al-Maqbali, A.; Al Shawi, M.; et al. A Unique Genotype of Pseudohypoaldosteronism Type 1b in a Highly Consanguineous Population. J. Endocr. Soc. 2021, 5, bvab095. [Google Scholar] [CrossRef] [PubMed]
- Edelheit, O.; Hanukoglu, I.; Gizewska, M.; Kandemir, N.; Tenenbaum-Rakover, Y.; Yurdakök, M.; Zajaczek, S.; Hanukoglu, A. Novel Mutations in Epithelial Sodium Channel (ENaC) Subunit Genes and Phenotypic Expression of Multisystem Pseudohypoaldosteronism. Clin. Endocrinol. 2005, 62, 547–553. [Google Scholar] [CrossRef] [PubMed]
- Saxena, A.; Hanukoglu, I.; Saxena, D.; Thompson, R.J.; Gardiner, R.M.; Hanukoglu, A. Novel Mutations Responsible for Autosomal Recessive Multisystem Pseudohypoaldosteronism and Sequence Variants in Epithelial Sodium Channel Alpha-, Beta-, and Gamma-Subunit Genes. J. Clin. Endocrinol. Metab. 2002, 87, 3344–3350. [Google Scholar] [CrossRef] [PubMed]
- Turan, I.; Kotan, L.D.; Tastan, M.; Gurbuz, F.; Topaloglu, A.K.; Yuksel, B. Molecular Genetic Studies in a Case Series of Isolated Hypoaldosteronism Due to Biosynthesis Defects or Aldosterone Resistance. Clin. Endocrinol. 2018, 88, 799–805. [Google Scholar] [CrossRef] [PubMed]
- Huppmann, S.; Lankes, E.; Schnabel, D.; Bührer, C. Unimpaired Postnatal Respiratory Adaptation in a Preterm Human Infant with a Homozygous ENaC-α Unit Loss-of-Function Mutation. J. Perinatol. Off. J. Calif. Perinat. Assoc. 2011, 31, 802–803. [Google Scholar] [CrossRef] [PubMed]
- Riepe, F.G.; van Bemmelen, M.X.P.; Cachat, F.; Plendl, H.; Gautschi, I.; Krone, N.; Holterhus, P.-M.; Theintz, G.; Schild, L. Revealing a Subclinical Salt-Losing Phenotype in Heterozygous Carriers of the Novel S562P Mutation in the Alpha Subunit of the Epithelial Sodium Channel. Clin. Endocrinol. 2009, 70, 252–258. [Google Scholar] [CrossRef] [PubMed]
- Joshi, K.; Verma, P.K.; Barman, M. Systemic Pseudohypoaldosteronism Type 1 Due to a Novel Mutation in SCNN1B Gene: A Case Report. EJIFCC 2022, 33, 268–273. [Google Scholar]
- Belot, A.; Ranchin, B.; Fichtner, C.; Pujo, L.; Rossier, B.C.; Liutkus, A.; Morlat, C.; Nicolino, M.; Zennaro, M.C.; Cochat, P. Pseudohypoaldosteronisms, Report on a 10-Patient Series. Nephrol. Dial. Transplant. Off. Publ. Eur. Dial. Transpl. Assoc.-Eur. Ren. Assoc. 2008, 23, 1636–1641. [Google Scholar] [CrossRef]
- Edelheit, O.; Hanukoglu, I.; Shriki, Y.; Tfilin, M.; Dascal, N.; Gillis, D.; Hanukoglu, A. Truncated Beta Epithelial Sodium Channel (ENaC) Subunits Responsible for Multi-System Pseudohypoaldosteronism Support Partial Activity of ENaC. J. Steroid Biochem. Mol. Biol. 2010, 119, 84–88. [Google Scholar] [CrossRef]
- Pugh, C.P. Pseudohypoaldosteronism Type 1: The Presentation and Management of a Neonate With a Novel Mutation of the SCNN1B Gene Found in Two Hispanic Siblings. Cureus 2022, 14, e23918. [Google Scholar] [CrossRef]
- Karacan Küçükali, G.; Çetinkaya, S.; Tunç, G.; Oğuz, M.M.; Çelik, N.; Akkaş, K.Y.; Şenel, S.; Güleray Lafcı, N.; Savaş Erdeve, Ş. Clinical Management in Systemic Type Pseudohypoaldosteronism Due to SCNN1B Variant and Literature Review. J. Clin. Res. Pediatr. Endocrinol. 2021, 13, 446–451. [Google Scholar] [CrossRef] [PubMed]
- Seyhanli, M.; Ilhan, O.; Gumus, E.; Bor, M.; Karaca, M. Pseudohypoaldosteronism Type 1 Newborn Patient with a Novel Mutation in SCNN1B. J. Pediatr. Intensive Care 2020, 9, 145–148. [Google Scholar] [CrossRef] [PubMed]
- Dogan, C.S.; Erdem, D.; Mesut, P.; Merve, A.; Sema, A.; Iffet, B.; Afig, B. A Novel Splice Site Mutation of the Beta Subunit Gene of Epithelial Sodium Channel (ENaC) in One Turkish Patient with a Systemic Form of Pseudohypoaldosteronism Type 1. J. Pediatr. Endocrinol. Metab. 2012, 25, 1035–1039. [Google Scholar] [CrossRef] [PubMed]
- Nobel, Y.R.; Lodish, M.B.; Raygada, M.; Rivero, J.D.; Faucz, F.R.; Abraham, S.B.; Lyssikatos, C.; Belyavskaya, E.; Stratakis, C.A.; Zilbermint, M. Pseudohypoaldosteronism Type 1 Due to Novel Variants of SCNN1B Gene. Endocrinol. Diabetes Metab. Case Rep. 2016, 2016, 150104. [Google Scholar] [CrossRef]
- Thomas, C.P.; Zhou, J.; Liu, K.Z.; Mick, V.E.; MacLaughlin, E.; Knowles, M. Systemic Pseudohypoaldosteronism from Deletion of the Promoter Region of the Human Beta Epithelial Na(+) Channel Subunit. Am. J. Respir. Cell Mol. Biol. 2002, 27, 314–319. [Google Scholar] [CrossRef]
- Yin, L.-P.; Zhu, H.; Zhu, R.-Y.; Huang, L. A Novel SCNN1G Mutation in a PHA I Infant Patient Correlates with Nephropathy. Biochem. Biophys. Res. Commun. 2019, 519, 415–421. [Google Scholar] [CrossRef]
- Strautnieks, S.S.; Thompson, R.J.; Gardiner, R.M.; Chung, E. A Novel Splice-Site Mutation in the Gamma Subunit of the Epithelial Sodium Channel Gene in Three Pseudohypoaldosteronism Type 1 Families. Nat. Genet. 1996, 13, 248–250. [Google Scholar] [CrossRef]
- Adachi, M.; Tachibana, K.; Asakura, Y.; Abe, S.; Nakae, J.; Tajima, T.; Fujieda, K. Compound Heterozygous Mutations in the Gamma Subunit Gene of ENaC (1627delG and 1570-1G-->A) in One Sporadic Japanese Patient with a Systemic Form of Pseudohypoaldosteronism Type 1. J. Clin. Endocrinol. Metab. 2001, 86, 9–12. [Google Scholar] [CrossRef][Green Version]
- Pettersen, E.F.; Goddard, T.D.; Huang, C.C.; Couch, G.S.; Greenblatt, D.M.; Meng, E.C.; Ferrin, T.E. UCSF Chimera—A visualization system for exploratory research and analysis. J. Comput. Chem. 2004, 25, 1605–1612. [Google Scholar] [CrossRef]
- Willam, A.; Aufy, M.; Tzotzos, S.; Evanzin, H.; Chytracek, S.; Geppert, S.; Fischer, B.; Fischer, H.; Pietschmann, H.; Czikora, I.; et al. Restoration of Epithelial Sodium Channel Function by Synthetic Peptides in Pseudohypoaldosteronism Type 1B Mutants. Front. Pharmacol. 2017, 8, 85. [Google Scholar] [CrossRef]
- Willam, A.; Aufy, M.; Tzotzos, S.; El-Malazi, D.; Poser, F.; Wagner, A.; Unterköfler, B.; Gurmani, D.; Martan, D.; Iqbal, S.M.; et al. TNF Lectin-Like Domain Restores Epithelial Sodium Channel Function in Frameshift Mutants Associated with Pseudohypoaldosteronism Type 1B. Front. Immunol. 2017, 8, 601. [Google Scholar] [CrossRef] [PubMed]
- Kim, M.; Lee, S.-W.; Kim, J.; Shin, Y.; Chang, F.; Kim, J.M.; Cong, X.; Yu, G.-Y.; Park, K. LPS-Induced Epithelial Barrier Disruption via Hyperactivation of CACC and ENaC. Am. J. Physiol.-Cell Physiol. 2021, 320, C448–C461. [Google Scholar] [CrossRef] [PubMed]
- Lucas, R.; Hadizamani, Y.; Enkhbaatar, P.; Csanyi, G.; Caldwell, R.W.; Hundsberger, H.; Sridhar, S.; Lever, A.A.; Hudel, M.; Ash, D.; et al. Dichotomous Role of Tumor Necrosis Factor in Pulmonary Barrier Function and Alveolar Fluid Clearance. Front. Physiol. 2022, 12, 793251. [Google Scholar] [CrossRef] [PubMed]
- Madaio, M.P.; Czikora, I.; Kvirkvelia, N.; McMenamin, M.; Yue, Q.; Liu, T.; Toque, H.A.; Sridhar, S.; Covington, K.; Alaisami, R.; et al. The TNF-Derived TIP Peptide Activates the Epithelial Sodium Channel and Ameliorates Experimental Nephrotoxic Serum Nephritis. Kidney Int. 2019, 95, 1359–1372. [Google Scholar] [CrossRef] [PubMed]
- Pinto, A.C.M.D.; Nunes, R.d.M.; Nogueira, I.A.; Fischer, B.; Lucas, R.; Girão-Carmona, V.C.C.; de Oliveira, V.L.S.; Amaral, F.A.; Schett, G.; Rocha, F.A.C. Potent Anti-Inflammatory Activity of the Lectin-like Domain of TNF in Joints. Front. Immunol. 2022, 13, 1049368. [Google Scholar] [CrossRef]





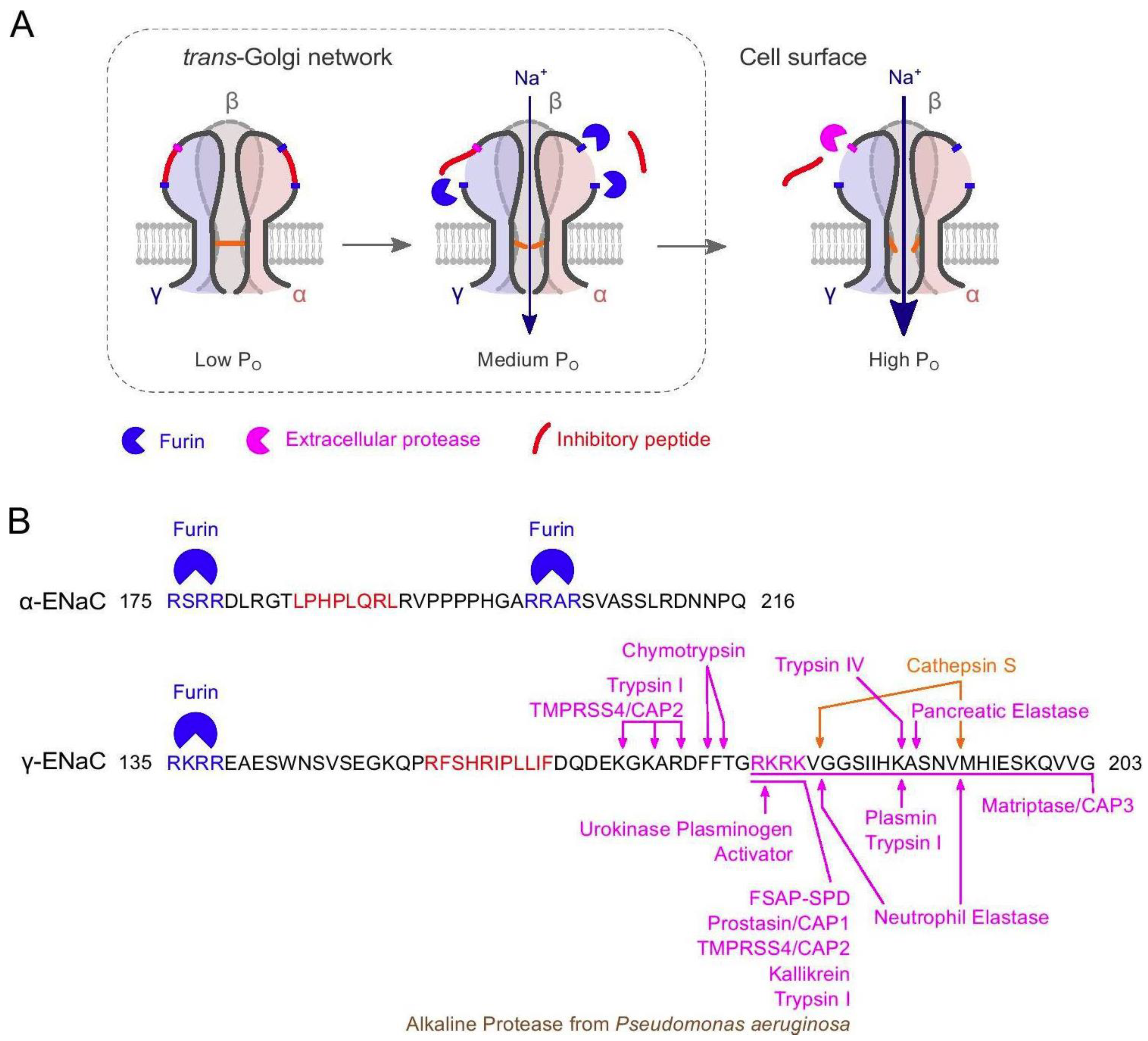{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Type of Mutation * | Gene | ||
|---|---|---|---|
| SCNN1A | SCNN1B | SCNN1G | |
| Missense | 13 | 2 | 1 |
| Nonsense (stop) | 6 | 3 | 1 |
| Frameshift | 14 | 10 | 2 |
| In-frame deletion | 1 | 1 | |
| Deletion, insertion or substitution of nucleotides in intron or splice junction | 7 | 5 | 2 |
| Large upstream deletion in non-coding region | 1 | ||
| TOTAL per gene | 41 | 21 | 7 |
| Study Title/Study Number | Intervention | Trial Phase/ Recruitment Status | Disease/ Condition |
|---|---|---|---|
| Data Analysis for Drug Repurposing for Effective Alzheimer’s Medicines (DREAM)-Amiloride vs Triamterene NCT05125237 | Amiloride Triamterene | Retrospective 2021–2023 Active, Not recruiting | Hypertension |
| The effect of Amiloride and Spironolactone in Patients With Hypertension (hass) NCT01388088 | Spironolactone Amiloride placebo | Phase 4 2010–2012 completed | Hypertension |
| Bioequivalence Study Between GSK3542503 Hydrochlorothiazide + Amiloride Hydrochloride 50 mg: 5 mg Tablets and Reference Product in Healthy Adult Participants Under Fasting Conditions NCT03031496 | GSK3542503 (HCTZ 50 mg/ Amiloride HCl 5 mg tablets) Moduretic (HCTZ 50 mg/ Amiloride HCl 5 mg tablets) | Phase 1 2017 completed | Hypertension |
| Comparison of Spironolactone and Amiloride on Home Blood Pressure in Resistant Hypertension NCT04331691 | Spironolactone amiloride | Phase 4 2020–2024 recruiting | Resistant hypertension |
| Diuretics and Angiotensin-Receptor Blocker Agents in Patients With Stage I Hypertension NCT00971165 | Losartan Chlorthalidone plus amiloride | Phase 3 2010–2014 completed | Hypertension Cardiovascular Disease |
| Amiloride for Resistant Hypertension NCT02122731 | Amiloride added to triple antihypertensive therapy | Phase 4 2010–2012 completed | Hypertension Type 2 Diabetes Microalbuminuria |
| Epithelial Sodium Channel (ENaC) as a Novel Mechanism for Hypertension and Volume Expansion in Type 2 Diabetes NCT01804777 | Amiloride hydrochlorothiazide | Early phase 1 2013–2014 terminated | Proteinuria Hypertension Type II Diabetes |
| Amiloride in Nephrotic Syndrome (AMILOR) NCT05079789 | amiloride 5 mg furosemide 40 mg | Phase 3 2020–2023 recruiting | Nephrotic Syndrome Edema Na(+) Retention |
| Safety and Efficacy of Furosemide 40 mg + Amiloride Hydrochloride 10 mg to Reduct Edema NCT01210365 | fixed combination of furosemide and amiloride (10 mg) furosemide (40 mg) | Phase 3 2011–2013 terminated | Congestive heart failure Oedema |
| Treatment of Vascular Stiffness in ADPKD NCT05228574 | NaCl Placebo Amiloride 5 mg | Phase 4 2022–2024 recruiting | Autosomal Dominant Polycystic Kidney |
| ENaC Blockade and Arterial Stiffness NCT03837626 | Placebo-Cap | Phase 2 2019–2024 recruiting | overweight and obesity |
| Amiloride Pill | Phase 3 2019–2024 recruiting | Insulin resistance |
| Study Title/ Study Number | Intervention | Trial Phase/ Recruitment Status | Disease/ Condition |
|---|---|---|---|
| Study to Evaluate the Pharmacokinetics and Drug-Drug Interactions of Setanaxib in Healthy Adult Male and Female Subjects NCT04327089 | Setanaxib | Phase 1 2020–2022 completed | Pharmacokinetics |
| A Trial of Setanaxib in Patients With Primary Biliary Cholangitis (PBC) and Liver Stiffness NCT05014672 EudraCT Nr. 2021-001810-13 | Placebo Setanaxib (GKT137831) | Phase 2 2021–2023 recruiting Phase 3 | Primary Biliary Choleangitis Liver Stiffness |
| A Study of Setanaxib Co-Administered With Pembrolizumab in Patients With Recurrent or Metastatic Squamous Cell Carcinoma of the Head and Neck (SCCHN) NCT05323656 | Setanaxib Pembrolizumab Placebo | Phase 2 2022–2023 recruiting | Squamous Cell Carcinoma of Head and Neck |
| Study to Assess Safety & Efficacy of GKT137831 in Patients With Primary Biliary Cholangitis Receiving Ursodiol. NCT03226067 | Placebo Setanaxib (GKT137831) | Phase 2 2017–2022 completed | Primary Biliary Cirrhosis |
| Test Compound | Study Title/Study Number | Intervention | Trial Phase/ Recruitment Status | Disease/ Condition |
|---|---|---|---|---|
| ENaC inhibitors | ||||
| VX-371 | Clearing Lungs with ENaC Inhibition in Primary Ciliary Dyskinesia (CLEAN-PCD) NCT02871778 | VX-371 Hypertonic Saline Placebo VX-371 + HS Ivacaftor | Phase 2 2016–2021 completed | Primary Ciliary Dyskinesia |
| VX-371 | Safety and Efficacy of VX-371 Solution for Inhalation With and Without Oral Ivacaftor in Subjects With Primary Ciliary Dyskinesia EudraCT2015-004917-26 | VX-371 (inhalation) Ivacaftor (oral) Placebo | Phase 2a 2016–2018 completed | Primary Ciliary Dyskinesia |
| P-1037 | Clearing Lungs With Epithelial Sodium Channel (ENaC) Inhibition in Cystic Fibrosis (CF) (CLEAN-CF) NCT02343445 | P-1037 Hypertonic Saline Saline | phase 2 2015–2021 completed | Cystic fibrosis |
| ETD001 | A First in Human Study to Evaluate the Safety, Tolerability and Pharmacokinetics of Single and Multiple Ascending Doses of Inhaled ETD001 NCT04926701 | inhaled, single dose, and multiple once/twice daily ETD001 Placebo | Phase 1 2021–2022 completed | Cystic fibrosis |
| GS-9411 | A Phase 1 Trial to Assess the Safety, Tolerability, and Pharmacokinetics of GS 9411 in Subjects With Cystic Fibrosis (CF) NCT01025713 | GS-9411 Placebo | Phase 1 2009–2010 withdrawn | Cystic fibrosis Mucociliary Clearance |
| GS-9411 | Trial to Assess the Safety, Tolerability and Pharmacokinetics of GS-9411 in Healthy Male Volunteers NCT00800579 | GS-9411 Placebo | Phase 1 2008–2009 completed | Cystic fibrosis |
| GS-9411 | A Trial to Assess the Safety, Tolerability, and Pharmacokinetics of GS-9411 in Healthy Male Volunteers NCT00951522 | GS-9411 Placebo | Phase 1 2009 | Cystic fibrosis Mucociliary Clearance Airway Hydration |
| AZD5634 | To Assess the Safety, Tolerability and Pharmacokinetics of AZD5634 Following Inhaled and Intravenous (IV)Dose Administration NCT02679729 | AZD5634 for inhalation AZD5634 for infusion Placebo | Phase 1 2016 completed | Cystic fibrosis |
| AZD5634 | A Study to Assess the Effect of AZD5634 on Mucociliary Clearance, Safety, Tolerability and Pharmacokinetic Parameters in Patients With Cystic Fibrosis NCT02950805 | Placebo AZD5634 | Phase 1 2017–2018 completed | Pulmonary/Respiratory Diseases |
| BI 1265162 | A 4-week Study to Test Different Doses of BI 1265162 in Adolescents and Adults With Cystic Fibrosis Using the Respimat® Inhaler NCT04059094 | BI 1265162 Placebo | Phase 2 2020 Terminated | Cystic fibrosis |
| Analogues of SPLUNC1 | ||||
| SPX-101 | A Safety Study of SPX-101 Inhalation Solution in Subjects With Cystic Fibrosis NCT03056989 | SPX-101 | Phase 1 2017 completed | Cystic fibrosis |
| SPX-101 | An Efficacy and Safety Study of SPX-101 Inhalation Solution in Subjects With Cystic Fibrosis NCT03229252 | Placebo SPX-101 | Phase 2 2017–2019 completed | Cystic fibrosis |
| Antisense oligonucleotides | ||||
| IONIS-ENaCRx | A Phase 1/2a Study to Assess the Safety, Tolerability, Pharmacokinetics and Pharmacodynamics of Single and Multiple Doses of IONIS-ENaCRx in Healthy Volunteers and Patients With Cystic Fibrosis NCT 03647228 | IONIS-ENaCRx Placebo Ascending single and multiple doses inhaled or nebulized | Phase 1 2018–2020 Completed | Healthy Subjects Cystic fibrosis |
| ION-827359 Antisense Inhibitor of ENaC (ENaCCRx) | A Double-Blind, Placebo-Controlled, Phase 2a Study to Assess the Safety, Tolerability, and Efficacy of ION-827359 in Patients with Mild to Moderate COPD with Chronic Bronchitis EudraCT2020-000210-15 NCT04441788 | Placebo ION-827359 Oral inhalation of single-dose once every week up to 13 weeks | Phase 2a 2020–2021 terminated | COPD with Chronic Bronchitis |
| Short interfering α-ENaC RNA | ||||
| ARO-ENaC | Study of ARO-ENaC in Healthy Volunteers and in Patients With Cystic Fibrosis NCT04375514 | ARO-ENaC Placebo | Phase 1 2020–2022 terminated | Cystic Fibrosis Pulmonary |
| Study Title/ Number | Intervention | Trial Phase/ Recruitment Status | Disease/ Condition |
|---|---|---|---|
| Study in Intensive Care Patients to Investigate the Clinical Effect of Repetitive Orally Inhaled Doses of AP301 on Alveolar Liquid Clearance in Acute Lung Injury NCT01627613 EudraCT Nr. 2012-001863-64 | AP301 Saline Solution | Phase 2 2012–2019 completed | Acute Lung Injury (ALI) |
| Study in Intensive Care Patients Regarding the Effect of Inhaled AP301 After Primary Graft Dysfunction After Lung Transplantation NCT02095626 EudraCT Nr. 2013-000716-21 | AP301 Saline Solution | Phase 2 2014–2019 completed | Primary Graft Dysfunction |
| Safety and Preliminary Efficacy of Sequential Multiple Ascending Doses of Solnatide to Treat Pulmonary Permeability Oedema in Patients With Moderate-to-severe ARDS NCT03567577 | Solnatide 25 mg powder (solution for inhalation) 0.9% Saline Solution | Phase 2 2018–2022 recruiting | ARDS |
| COVID-19: Efficacy of solnatide to treat pulmonary permeability oedema in SARS-CoV-2 positive patients with moderate-to-severe ARDS—a pilot-trial. EudraCT Nr. 2020-001244-26 | Solnatide Powder for nebuliser suspension (inhalation) Placebo | Phase 2 2020–2021 Prematurely ended | Oedema in SARS-Co-2 positive patients with ARDS |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Lemmens-Gruber, R.; Tzotzos, S. The Epithelial Sodium Channel—An Underestimated Drug Target. Int. J. Mol. Sci. 2023, 24, 7775. https://doi.org/10.3390/ijms24097775
Lemmens-Gruber R, Tzotzos S. The Epithelial Sodium Channel—An Underestimated Drug Target. International Journal of Molecular Sciences. 2023; 24(9):7775. https://doi.org/10.3390/ijms24097775
Chicago/Turabian StyleLemmens-Gruber, Rosa, and Susan Tzotzos. 2023. "The Epithelial Sodium Channel—An Underestimated Drug Target" International Journal of Molecular Sciences 24, no. 9: 7775. https://doi.org/10.3390/ijms24097775
APA StyleLemmens-Gruber, R., & Tzotzos, S. (2023). The Epithelial Sodium Channel—An Underestimated Drug Target. International Journal of Molecular Sciences, 24(9), 7775. https://doi.org/10.3390/ijms24097775