
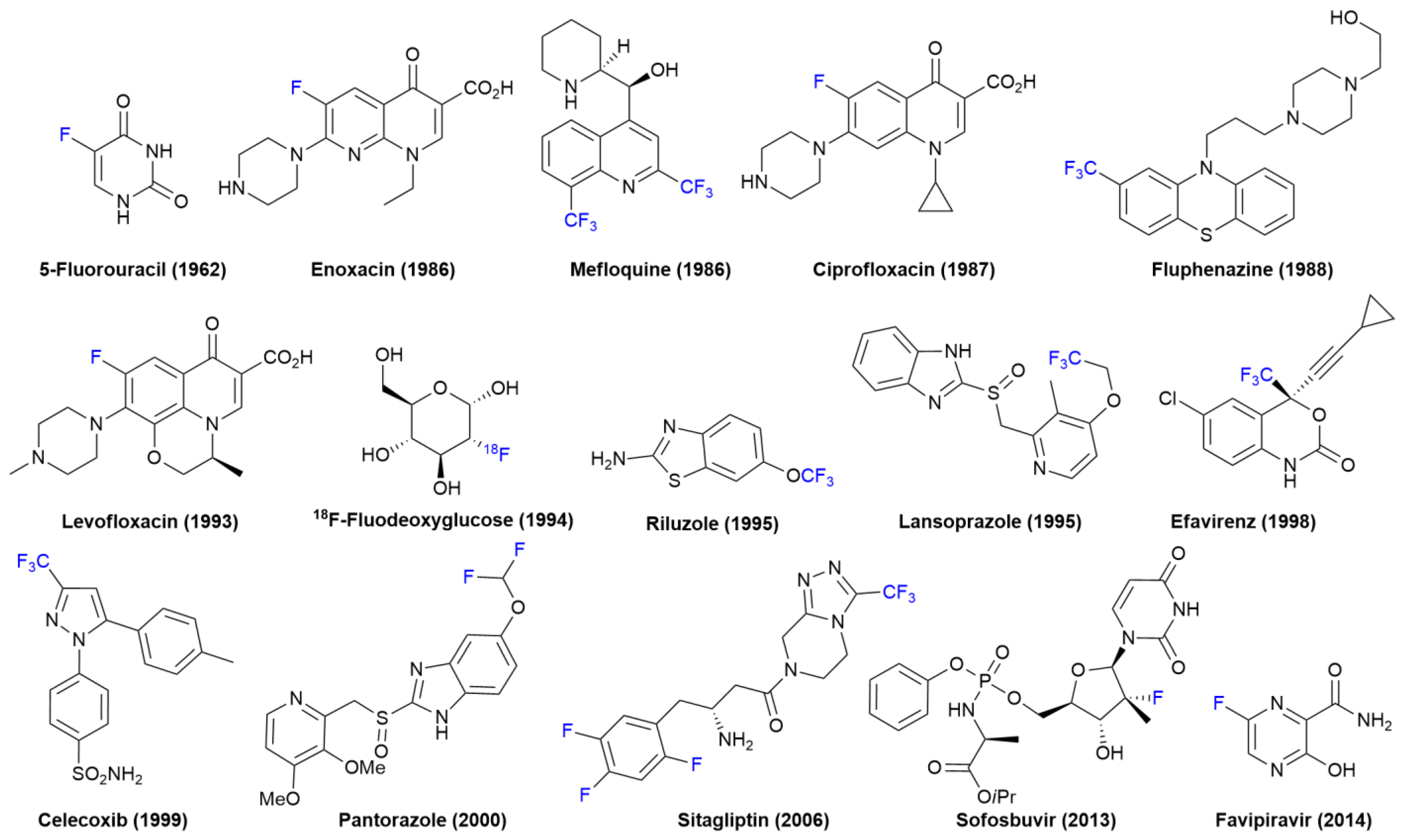
FDA-Approved Fluorinated Heterocyclic Drugs from 2016 to 2022

 ,
,  , ,
, ,  ,
, 
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
Introduction of Fluorine Atoms in Organic Molecules
2. FDA-Approved Drugs in 2022
2.1. Lenacapavir
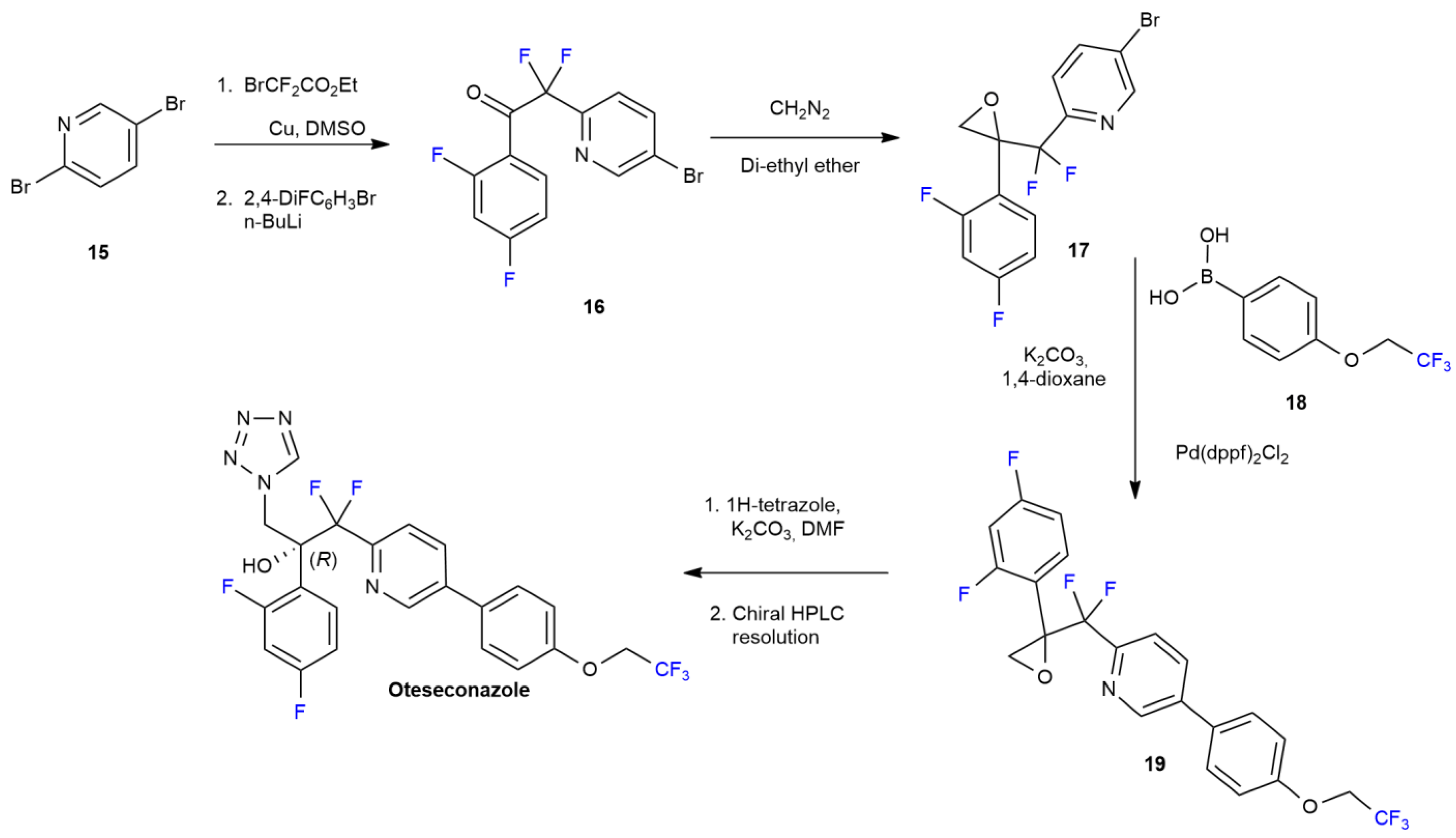
2.2. Oteseconazole
3. FDA-Approved Drugs in 2021
3.1. Atogepant

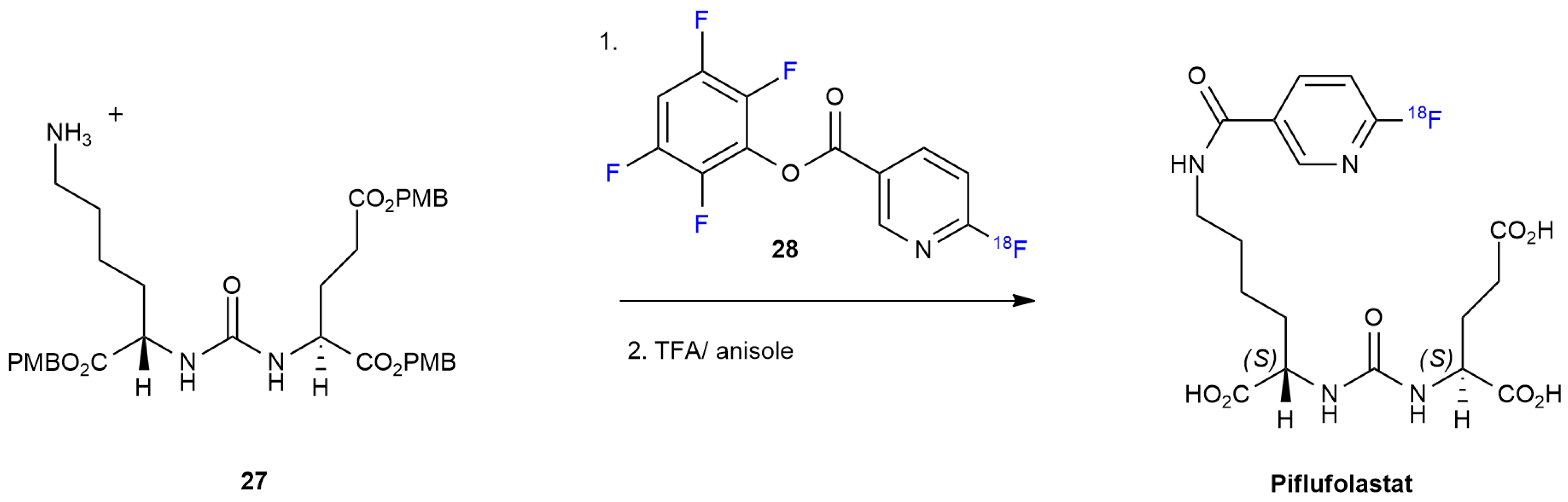
3.2. Piflufolastat F 18

3.3. Sotorasib

3.4. Umbralisib

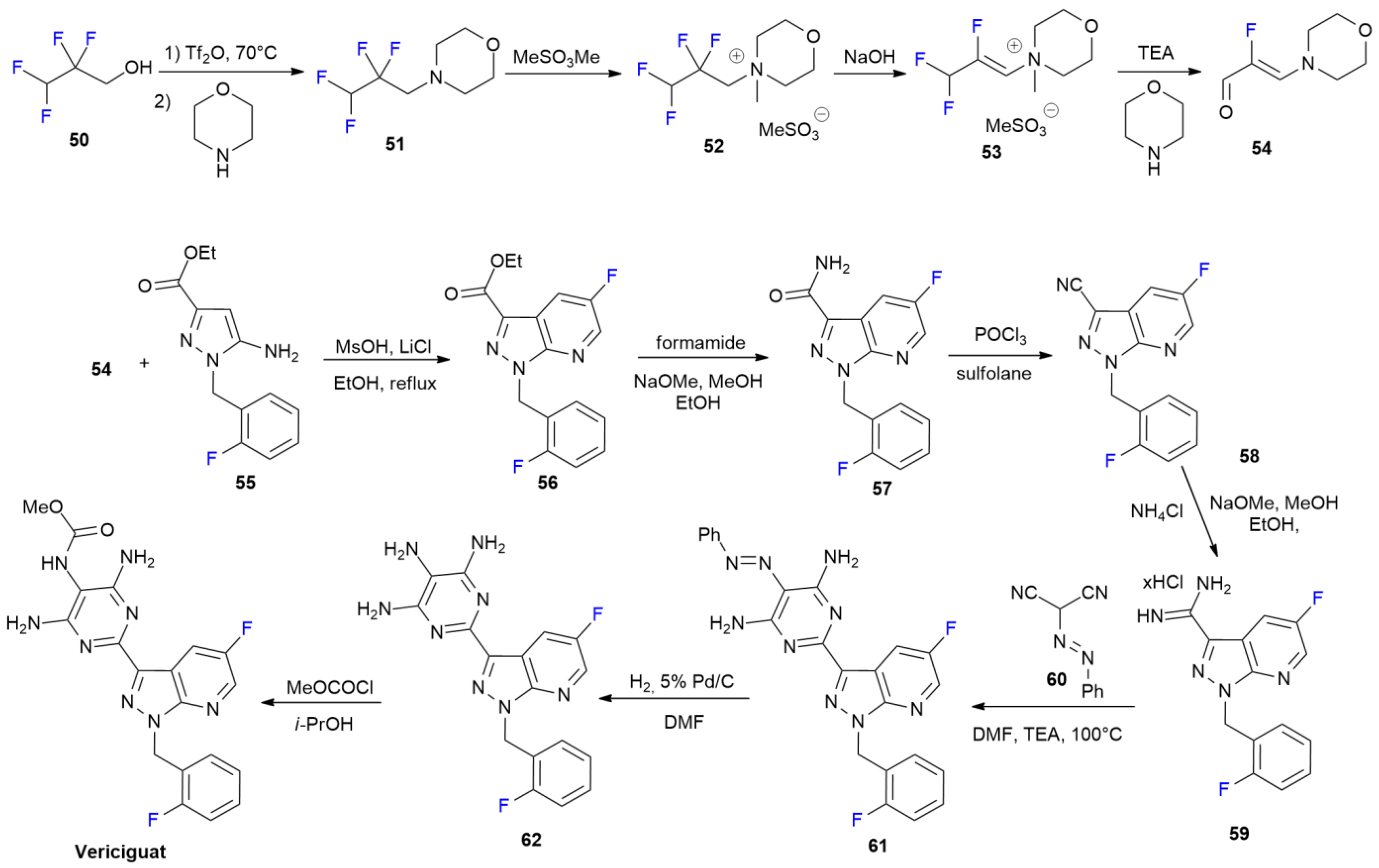
3.5. Vericiguat

4. FDA-Approved Drugs in 2020
4.1. Berotralstat

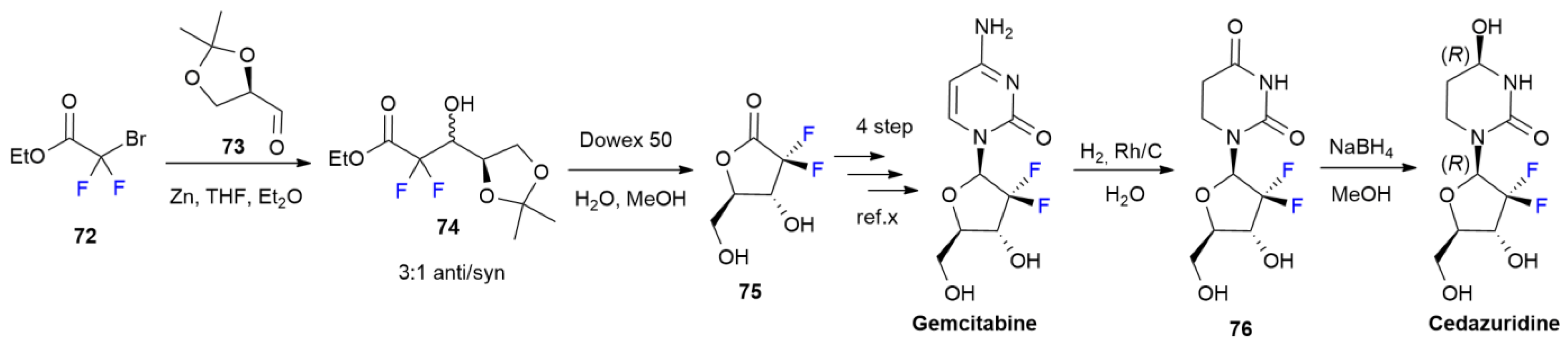
4.2. Cedazuridine

4.3. Pralsetinib

4.4. Selumetinib

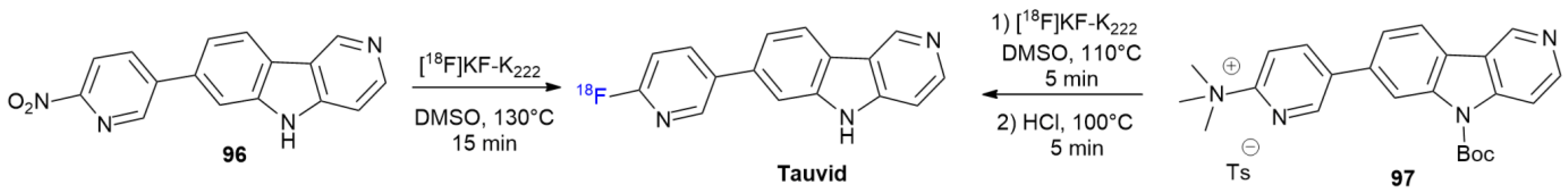
4.5. Tauvid

5. FDA-Approved Drugs in 2019
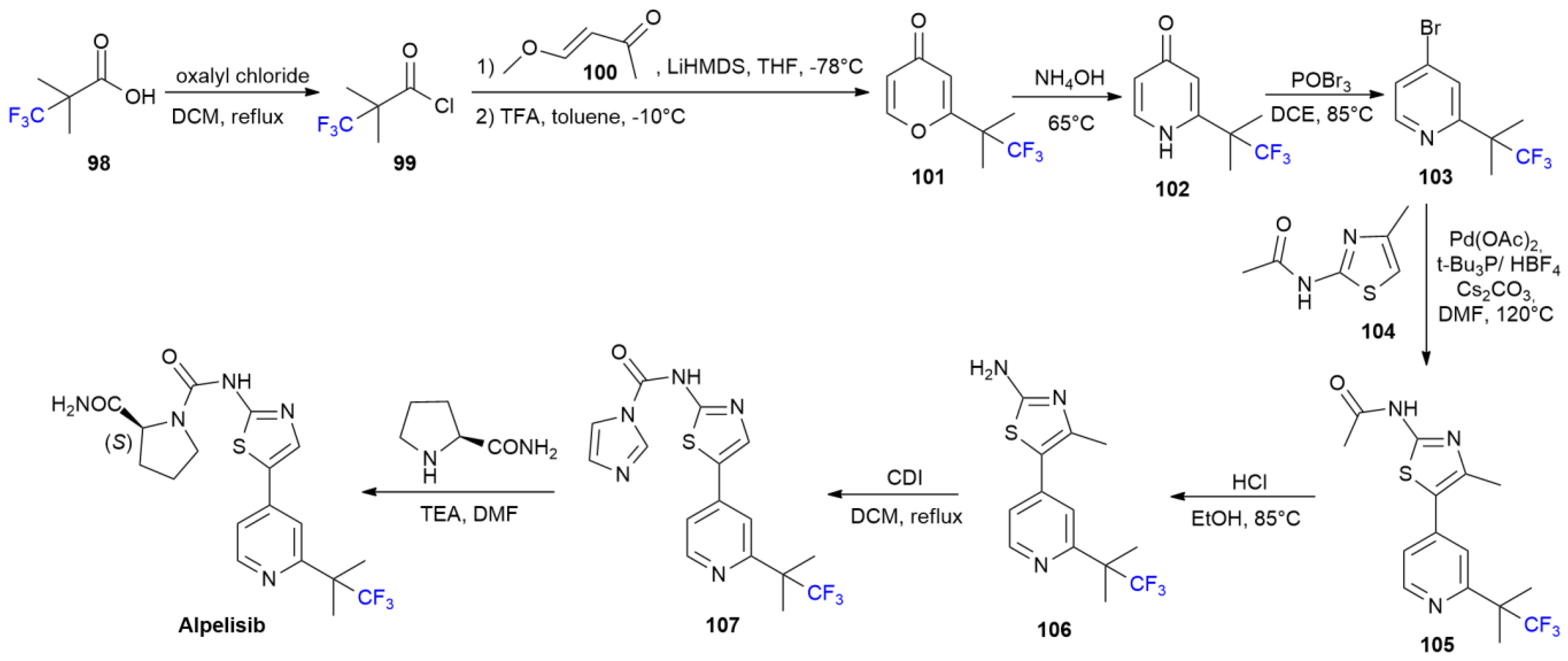
5.1. Alpelisib

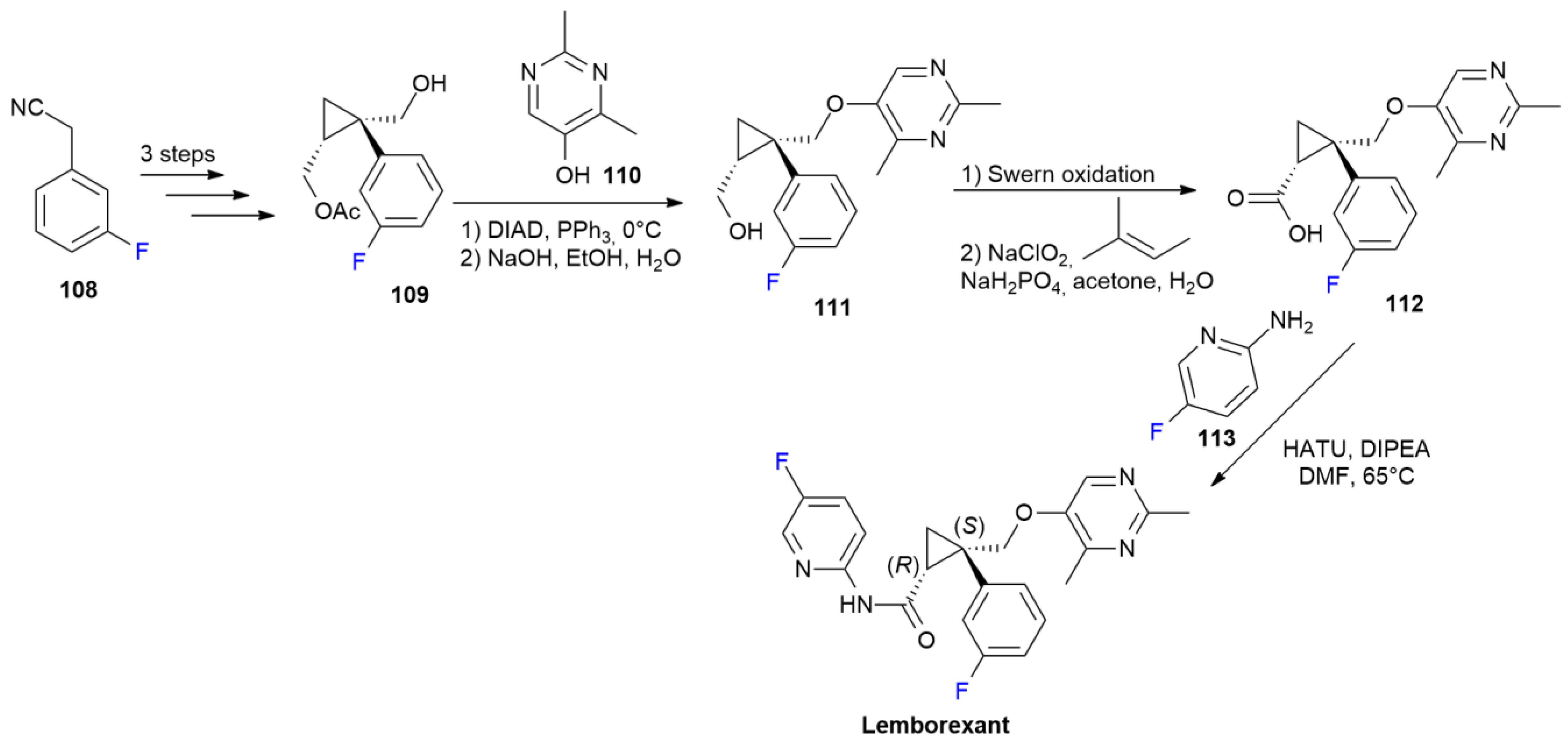
5.2. Lemborexant

5.3. Pexidartinib

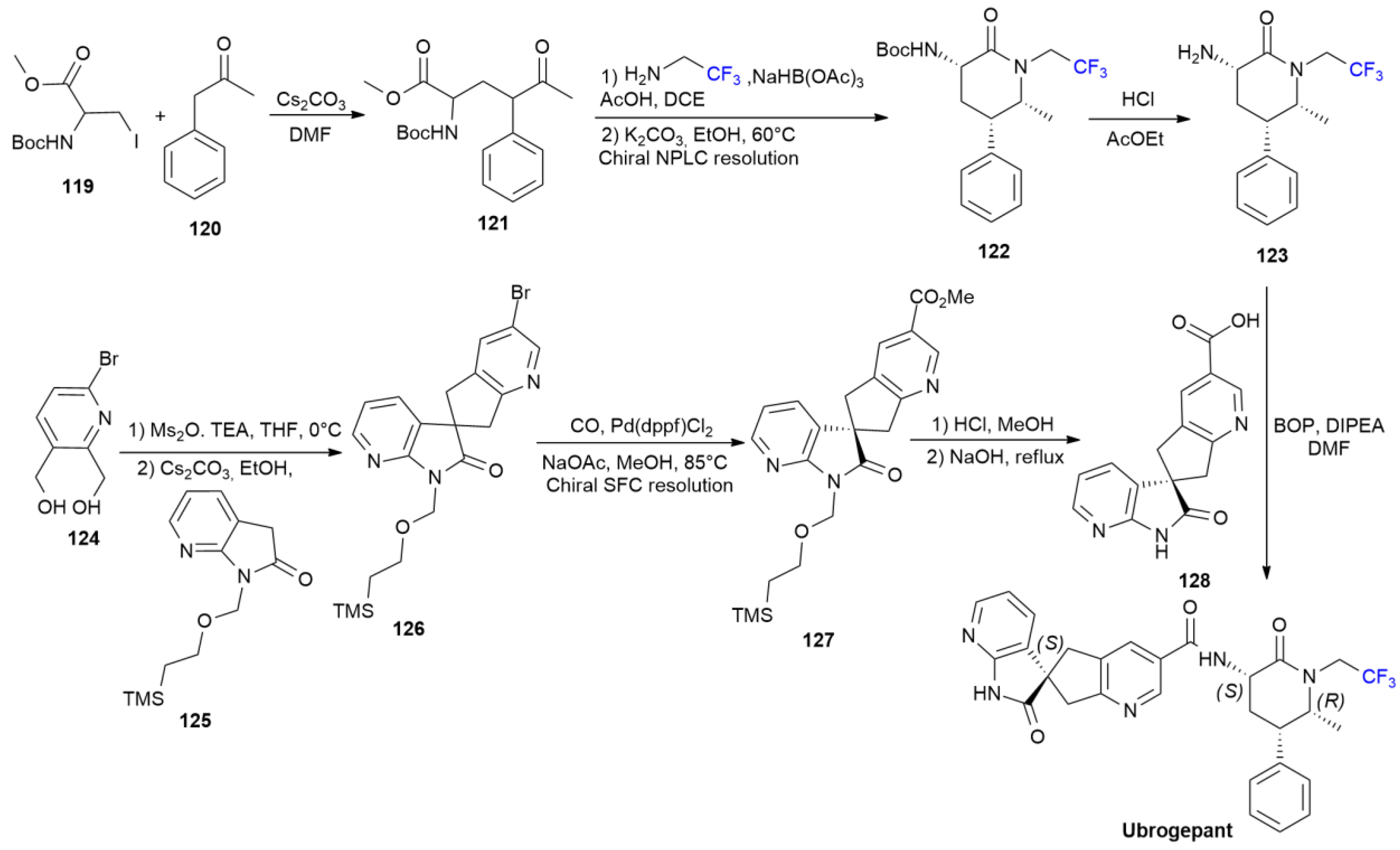
5.4. Ubrogepant

6. FDA-Approved Drugs in 2018
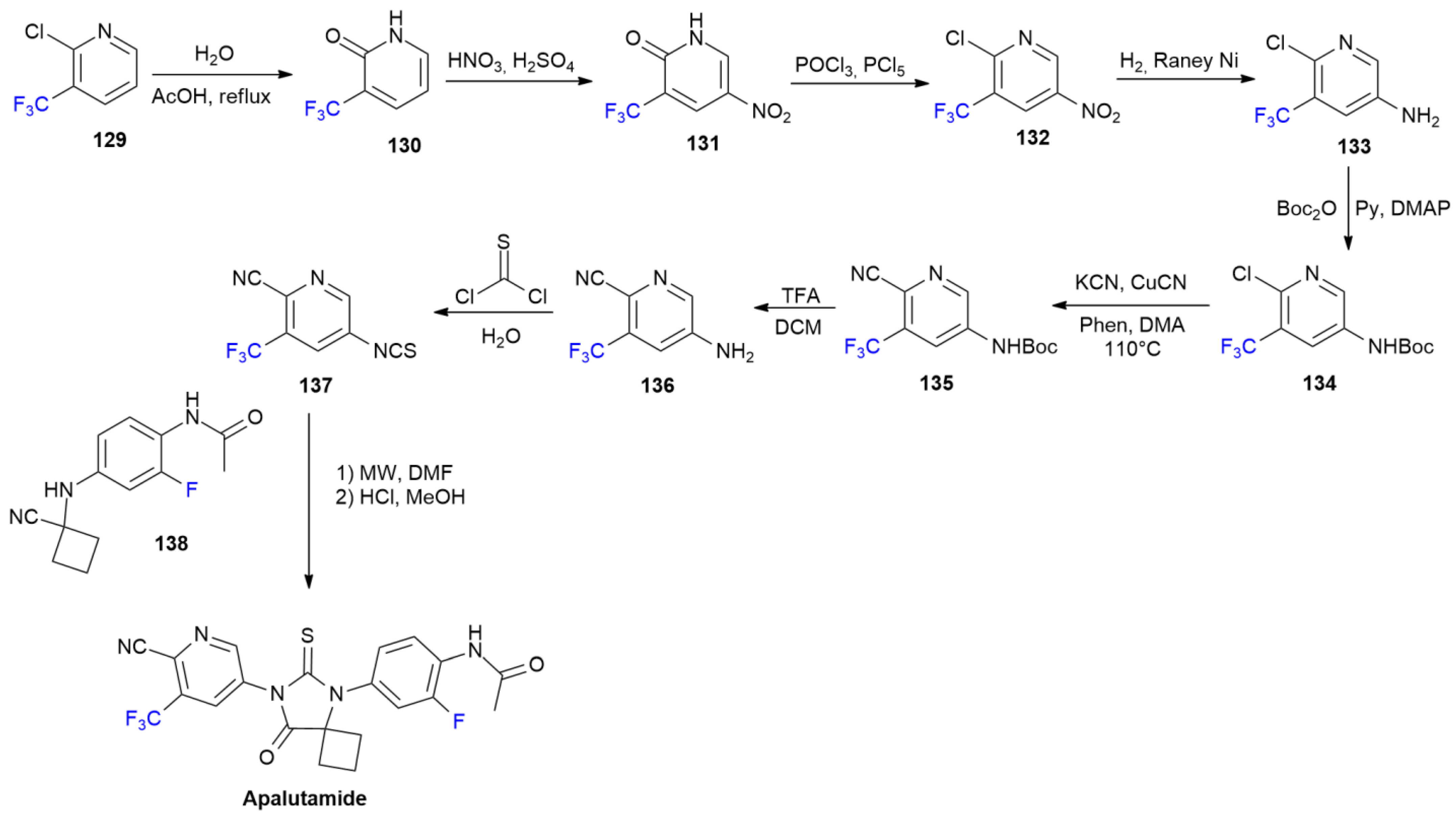
6.1. Apalutamide

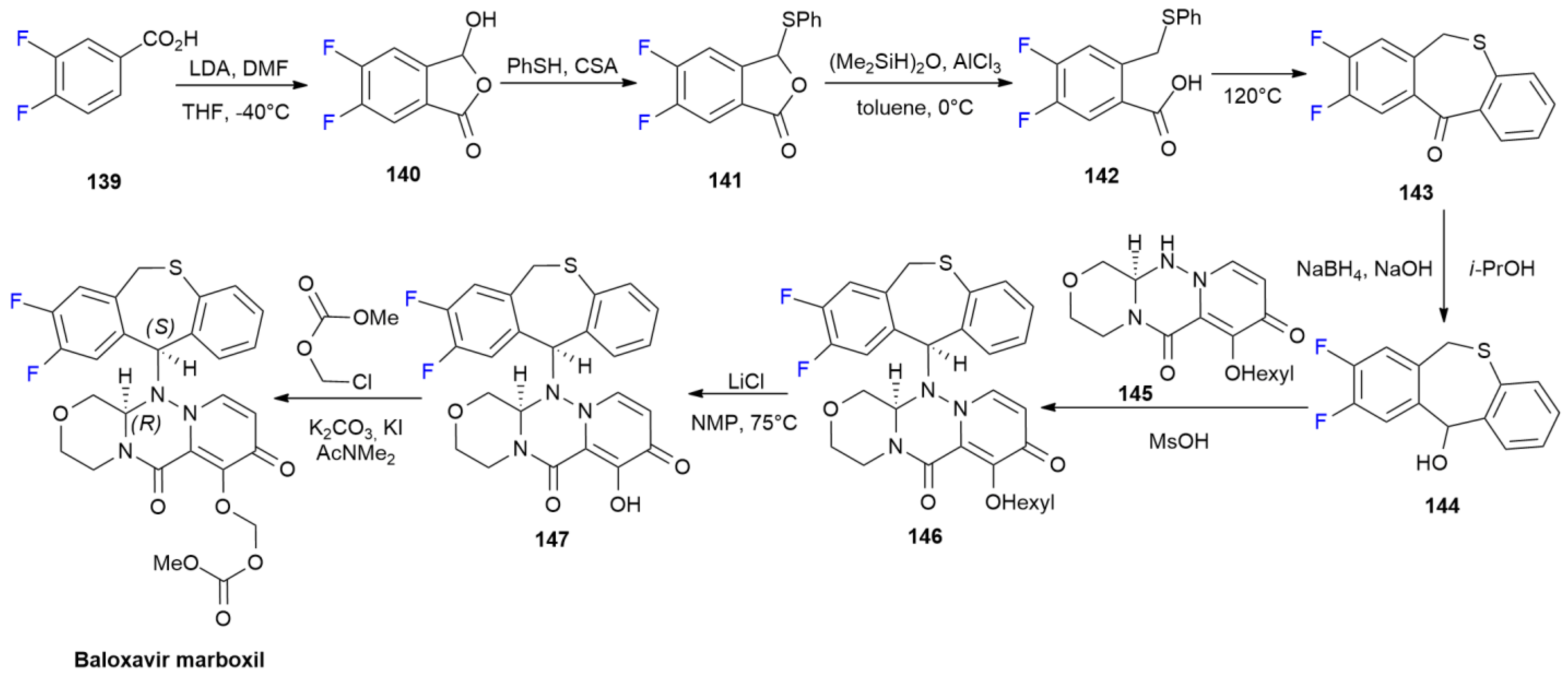
6.2. Baloxavir Marboxil

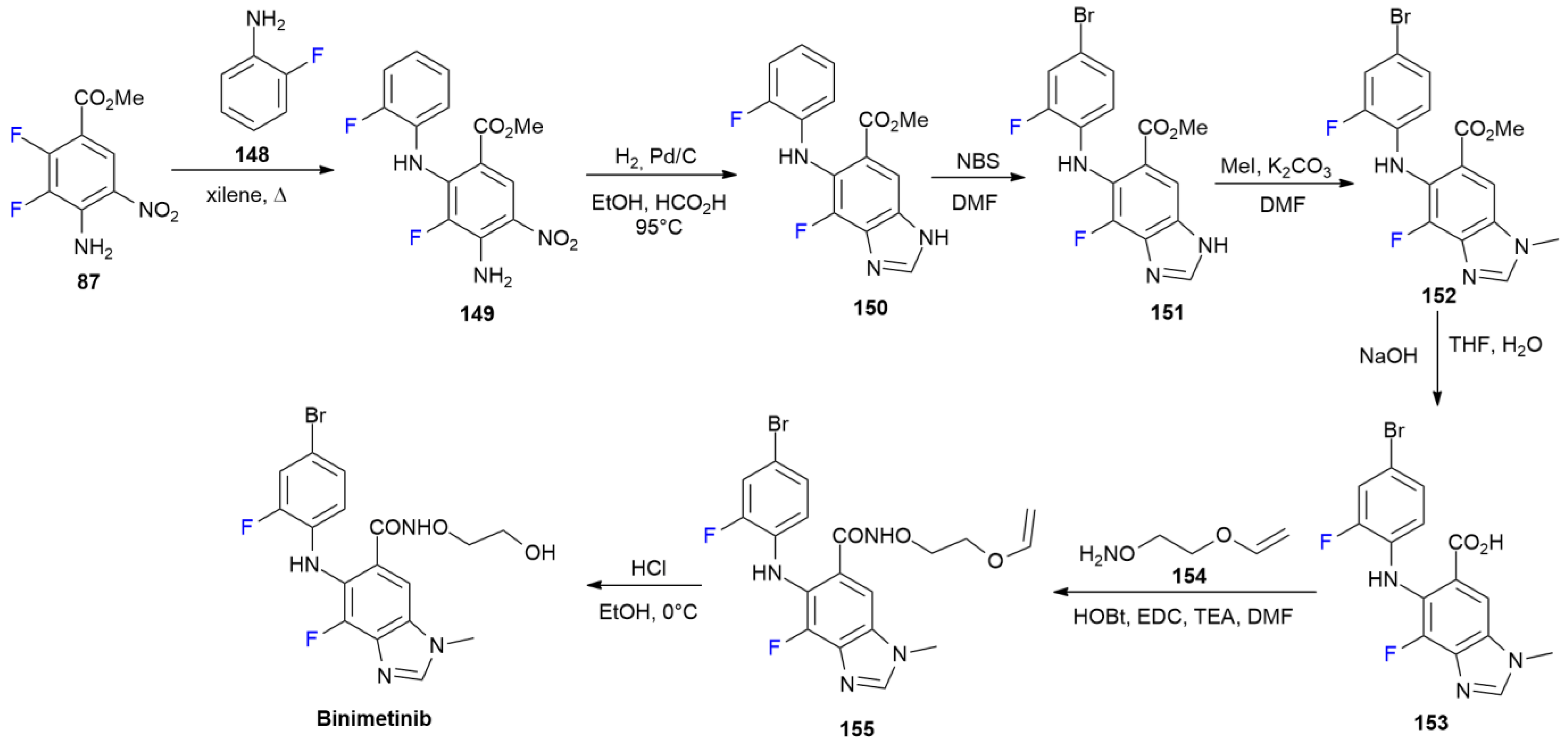
6.3. Binimetinib

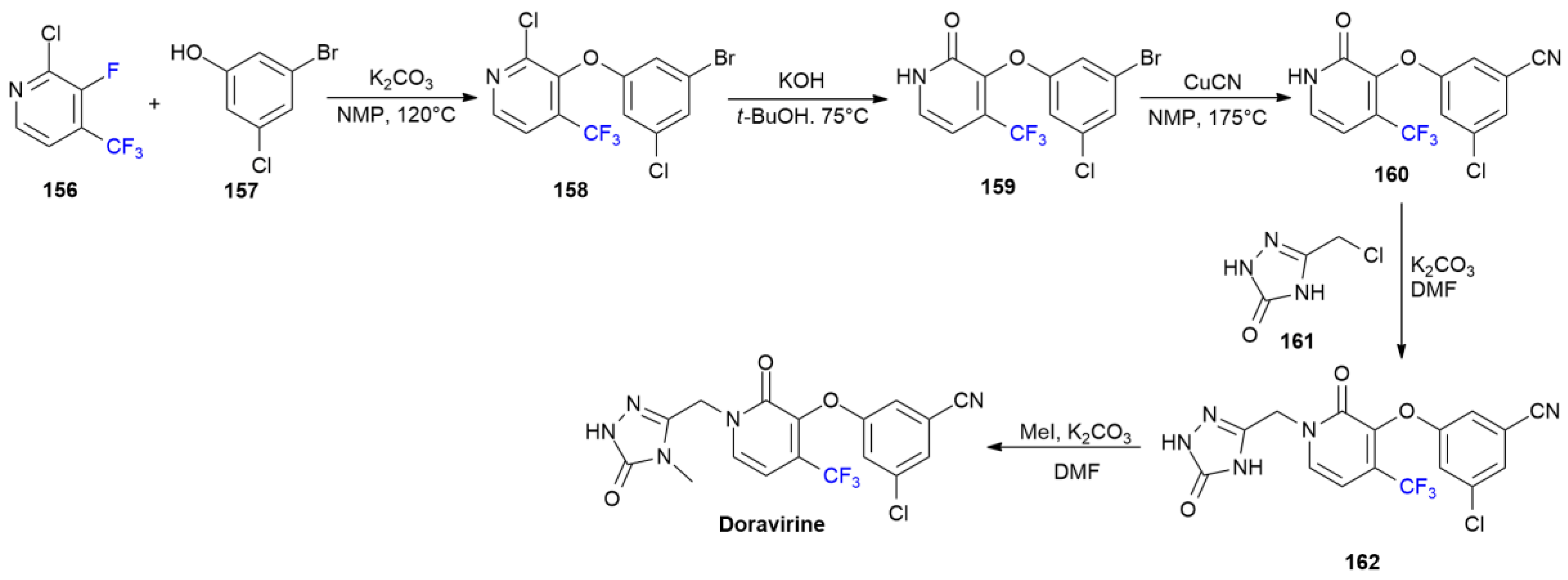
6.4. Doravirine

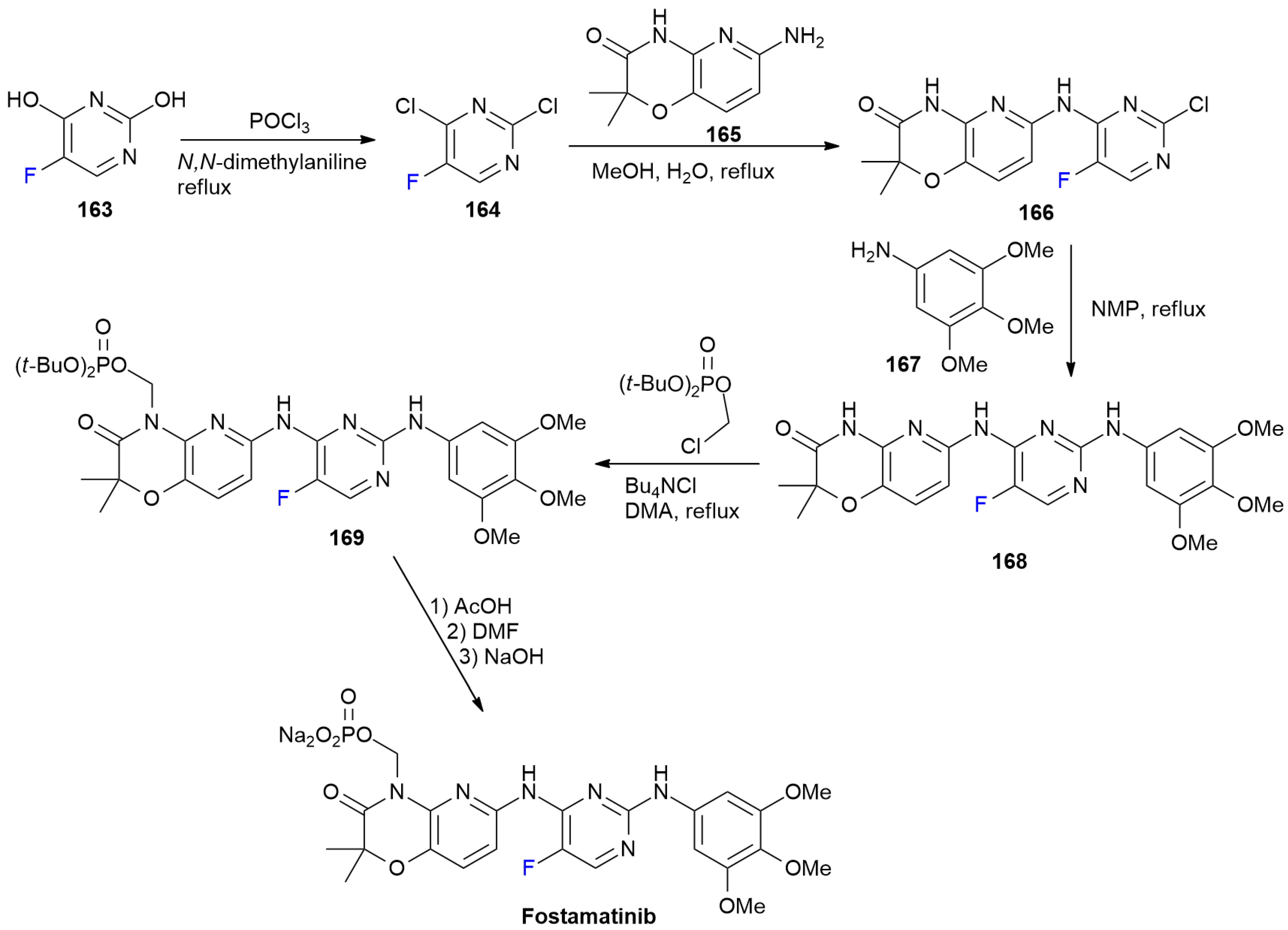
6.5. Fostamatinib

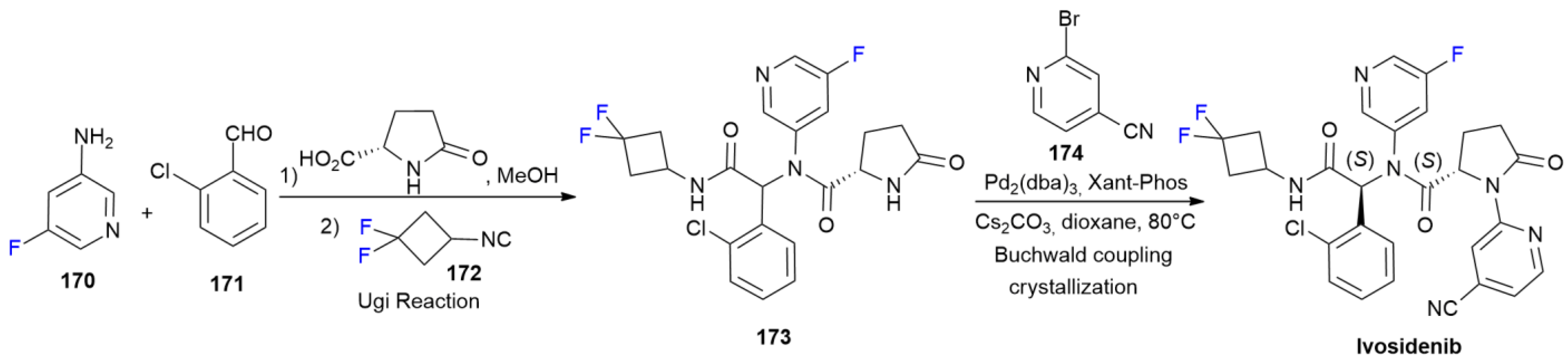
6.6. Ivosidenib

6.7. Talazoparib

6.8. Tezacaftor

7. FDA-Approved Drugs in 2017
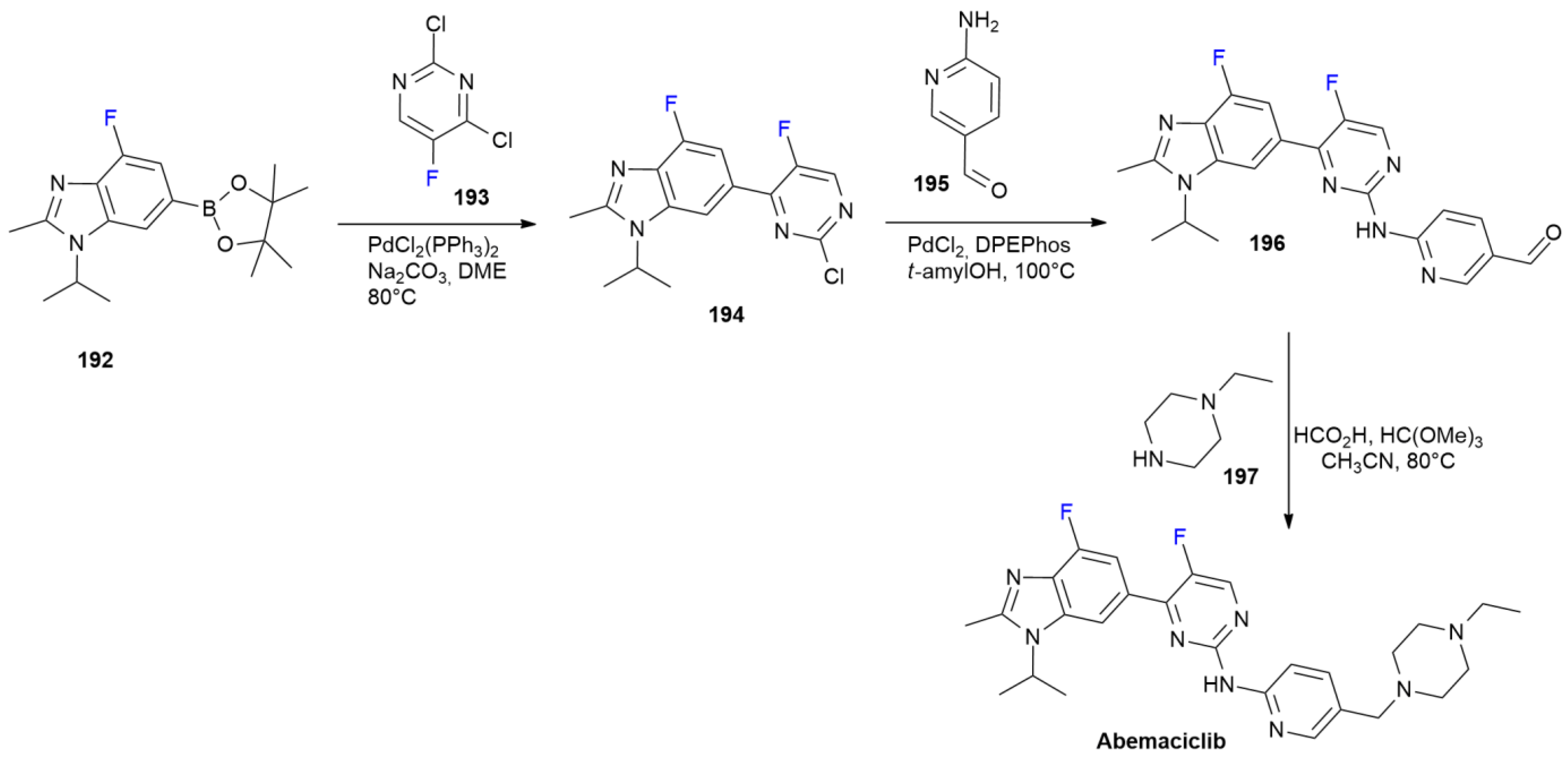
7.1. Abemaciclib

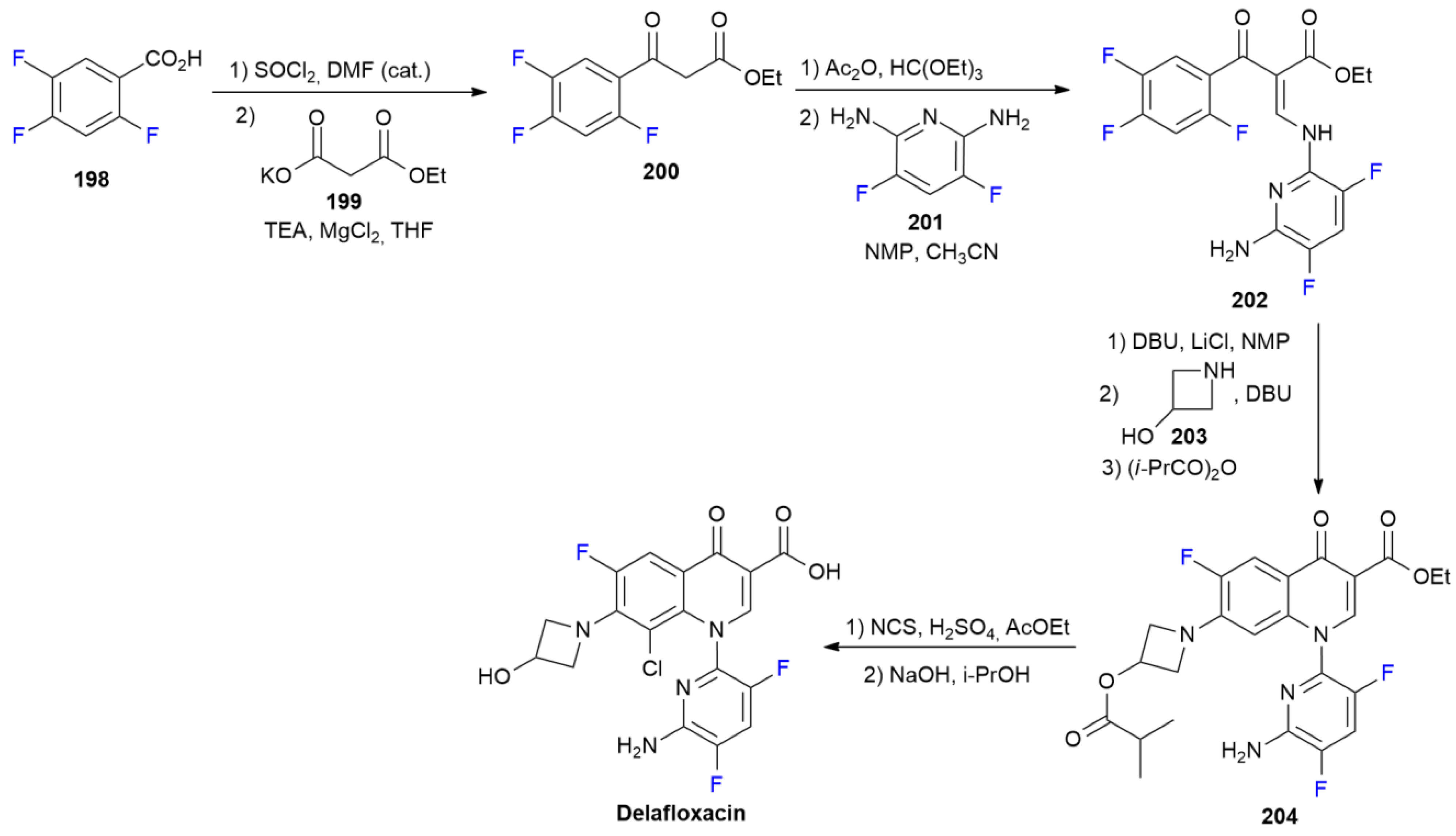
7.2. Delafloxacin

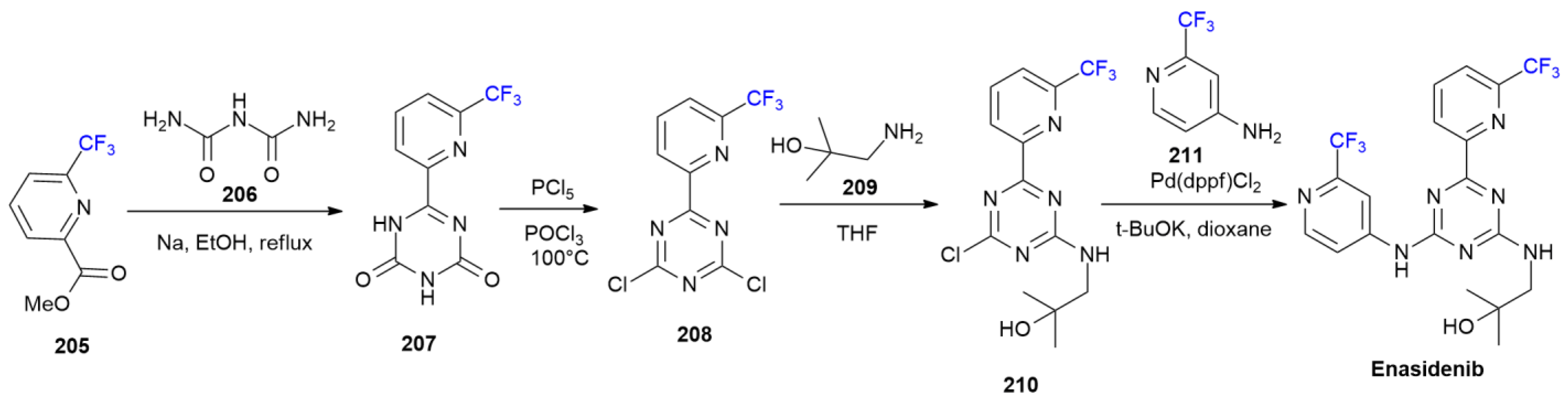
7.3. Enasidenib

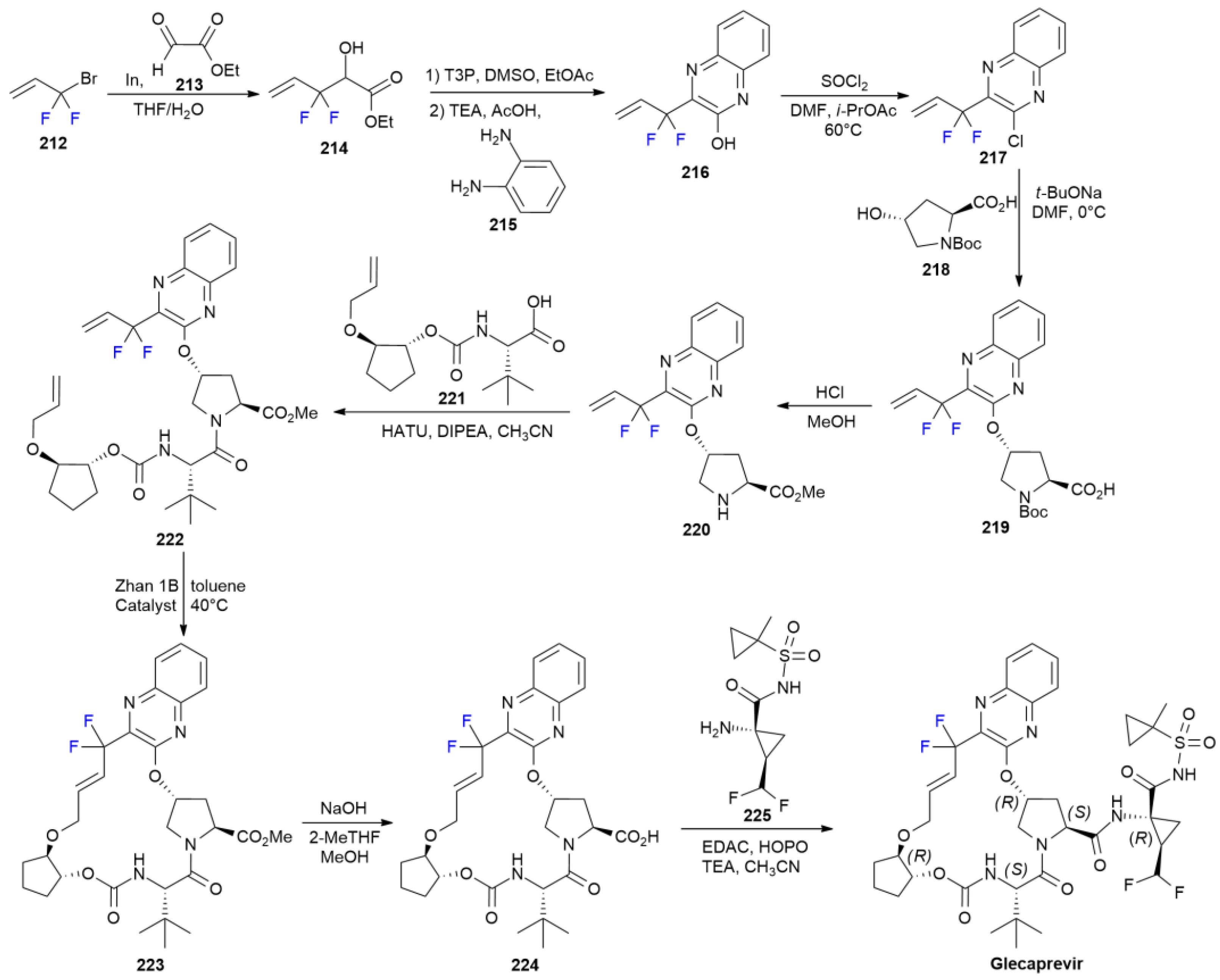
7.4. Glecaprevir

7.5. Letermovir

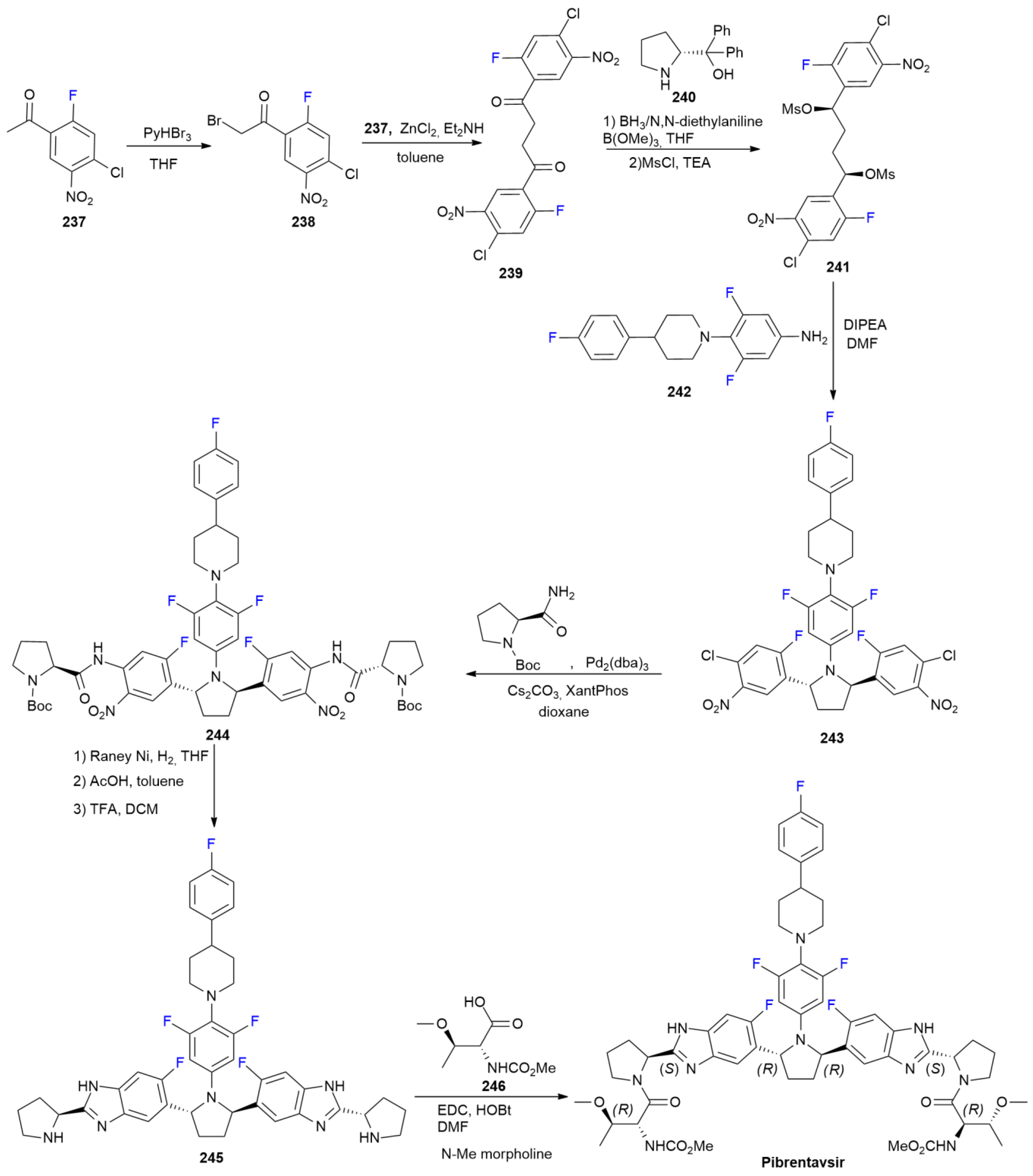
7.6. Pibrentasvir

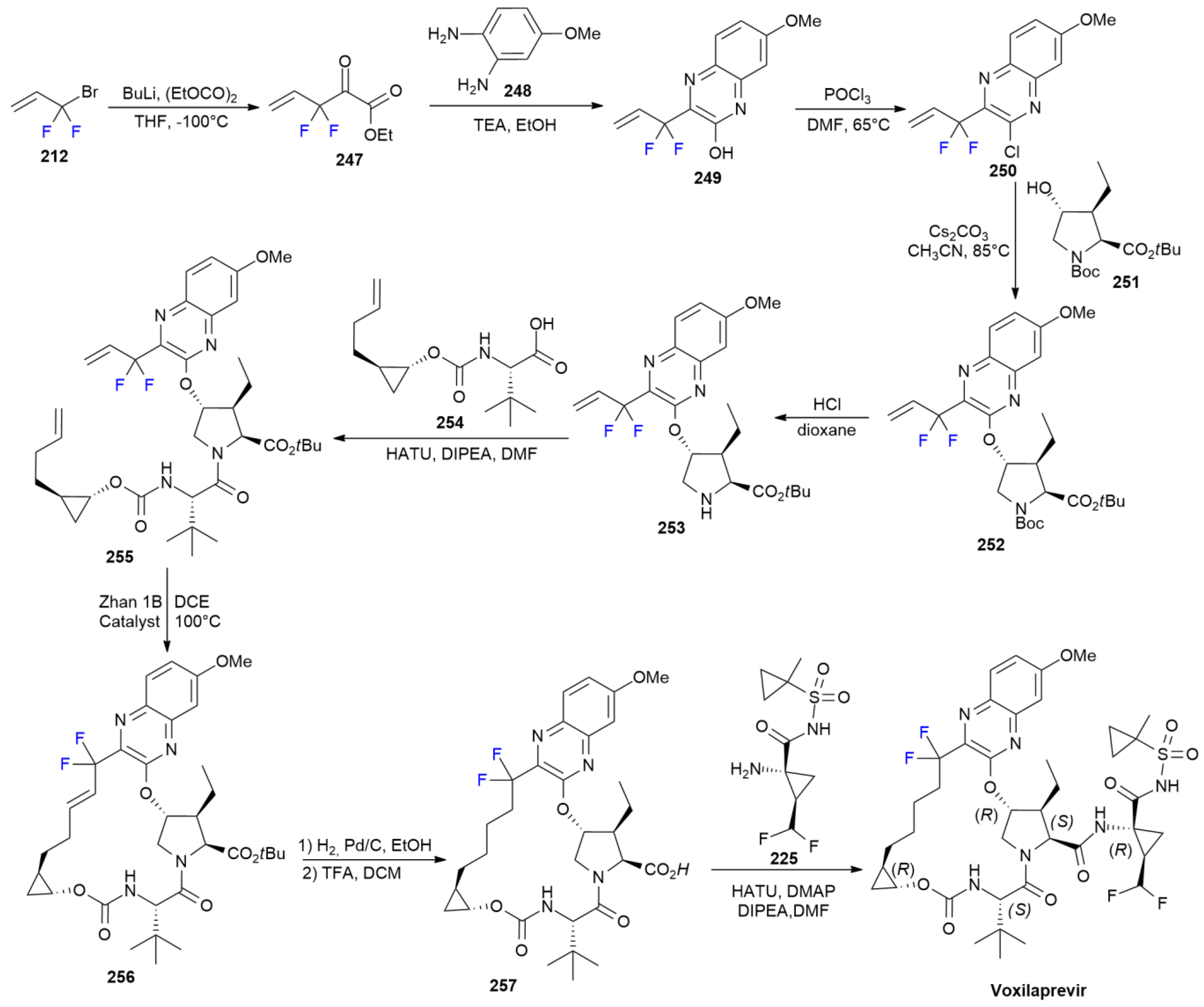
7.7. Voxilaprevir

8. FDA-Approved Drugs in 2016
8.1. Rucaparib

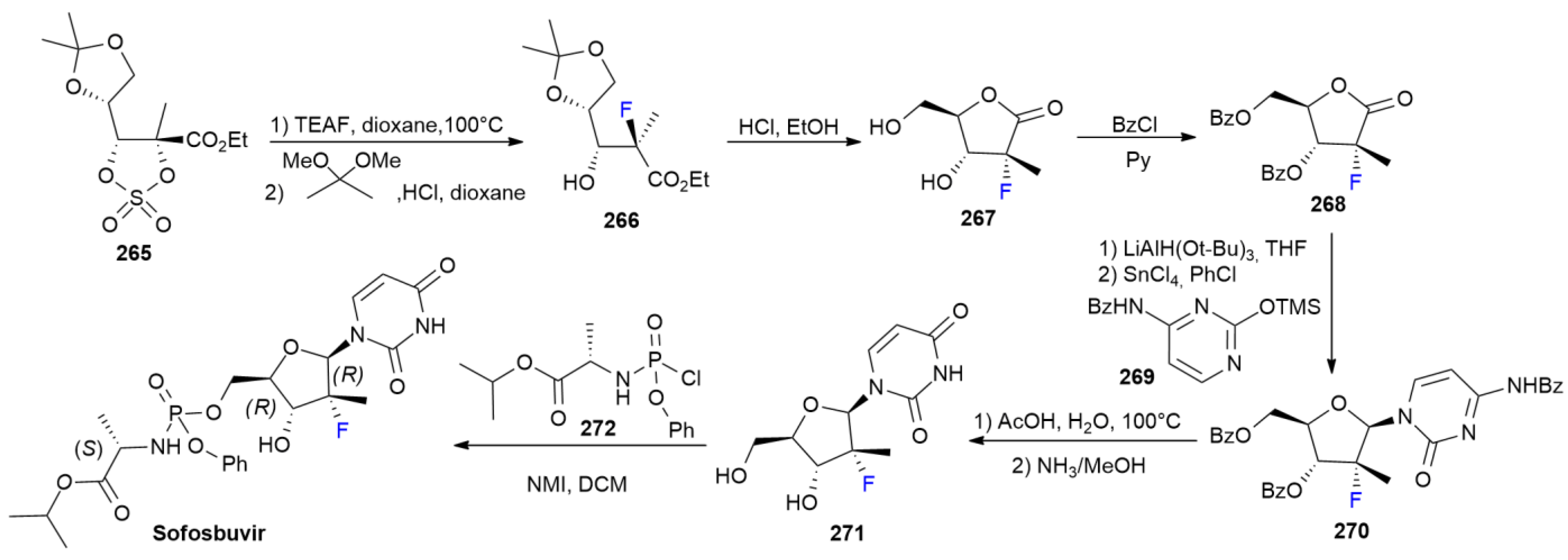
8.2. Sofosbuvir

9. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Kerru, N.; Gummidi, L.; Maddila, S.; Gangu, K.K.; Jonnalagadda, S.B. A Review on Recent Advances in Nitrogen-Containing Molecules and Their Biological Applications. Molecules 2020, 25, 1909. [Google Scholar] [CrossRef]
- Martorana, A.; Giacalone, V.; Bonsignore, R.; Pace, A.; Gentile, C.; Pibiri, I.; Buscemi, S.; Lauria, A.; Piccionello, P.A. Heterocyclic Scaffolds for the Treatment of Alzheimer’s Disease. Curr. Pharm. Des. 2016, 22, 3971–3995. [Google Scholar] [CrossRef]
- Pathania, S.; Narang, R.K.; Rawal, R.K. Role of sulphur-heterocycles in medicinal chemistry: An update. Eur. J. Med. Chem. 2019, 180, 486–508. [Google Scholar] [CrossRef]
- Wetzel, C.; Lonneman, M.; Wu, C. Polypharmacological drug actions of recently FDA approved antibiotics. Eur. J. Med. Chem. 2021, 209, 112931. [Google Scholar] [CrossRef] [PubMed]
- Heravi, M.M.; Zadsirjan, V. Prescribed drugs containing nitrogen heterocycles: An overview. RSC Adv. 2020, 10, 44247–44311. [Google Scholar] [CrossRef]
- Inoue, M.; Sumii, Y.; Shibata, N. Contribution of Organofluorine Compounds to Pharmaceuticals. ACS Omega 2020, 5, 10633–10640. [Google Scholar] [CrossRef] [PubMed]
- Upadhyay, C.; Chaudhary, M.; De Oliveira, R.N.; Borbas, A.; Kempaiah, P.S.; Rathi, B. Fluorinated scaffolds for antimalarial drug discovery. Expert Opin. Drug Discov. 2020, 15, 705–718. [Google Scholar] [CrossRef]
- O’Hagan, D. Fluorine in health care: Organofluorine containing blockbuster drugs. J. Fluor. Chem. 2010, 131, 1071–1081. [Google Scholar] [CrossRef]
- Fried, J.; Sabo, E.F. 9α-Fluoro Derivatives of Cortisone and Hydrocortisone. J. Am. Chem. Soc. 1954, 76, 1455–1456. [Google Scholar] [CrossRef]
- Purser, S.; Moore, P.R.; Swallow, S.; Gouverneur, V. Fluorine in medicinal chemistry. Chem. Soc. Rev. 2008, 37, 320–330. [Google Scholar] [CrossRef] [PubMed]
- Meanwell, N.A. Fluorine and Fluorinated Motifs in the Design and Application of Bioisosteres for Drug Design. J. Med. Chem. 2018, 61, 5822–5880. [Google Scholar] [CrossRef] [PubMed]
- Johnson, B.M.; Shu, Y.Z.; Zhuo, X.; Meanwell, N.A. Metabolic and Pharmaceutical Aspects of Fluorinated Compounds. J. Med. Chem. 2020, 63, 6315–6386. [Google Scholar] [CrossRef]
- Morgenthaler, M.; Schweizer, E.; Hoffmann-Röder, A.; Benini, F.; Martin, R.E.; Jaeschke, G.; Wagner, B.; Fischer, H.; Bendels, S.; Zimmerli, D.; et al. Predicting and Tuning Physicochemical Properties in Lead Optimization: Amine Basicities. ChemMedChem 2007, 2, 1100–1115. [Google Scholar] [CrossRef] [PubMed]
- Rowley, M.; Hallett, D.J.; Goodacre, S.; Moyes, C.; Crawforth, J.; Sparey, T.J.; Patel, S.; Marwood, R.; Patel, S.; Thomas, S.; et al. 3-(4-Fluoropiperidin-3-yl)-2-phenylindoles as High Affinity, Selective, and Orally Bioavailable h5-HT2A Receptor Antagonists. J. Med. Chem. 2001, 44, 1603–1614. [Google Scholar] [CrossRef] [PubMed]
- Shah, P.; Westwell, A.D. The role of fluorine in medicinal chemistry. J. Enzym. Inhib. Med. Chem. 2007, 22, 527–540. [Google Scholar] [CrossRef]
- Mei, H.; Han, J.; Fustero, S.; Medio-Simon, M.; Sedgwick, D.M.; Santi, C.; Ruzziconi, R.; Soloshonok, V.A. Fluorine-Containing Drugs Approved by the FDA in 2018. Chem. A Eur. J. 2019, 25, 11797–11819. [Google Scholar] [CrossRef] [PubMed]
- Knight, J.C.; Edwards, P.G.; Paisey, S.J. Fluorinated contrast agents for magnetic resonance imaging; a review of recent developments. RSC Adv. 2011, 1, 1415–1425. [Google Scholar] [CrossRef]
- Van der Born, D.; Pees, A.; Poot, A.J.; Orru, R.V.A.; Windhorst, A.D.; Vugts, D.J. Fluorine-18 labelled building blocks for PET tracer synthesis. Chem. Soc. Rev. 2017, 46, 4709–4773. [Google Scholar] [CrossRef]
- Das, P.; Delost, M.D.; Qureshi, M.H.; Smith, D.T.; Njardarson, J.T. A Survey of the Structures of US FDA Approved Combination Drugs. J. Med. Chem. 2019, 62, 4265–4311. [Google Scholar] [CrossRef]
- Ivasyshyn, V.; Smit, H.; Chiechi, R.C. Synthesis of a Hominal Bis(difluoromethyl) Fragment. ACS Omega 2019, 4, 14140–14150. [Google Scholar] [CrossRef]
- Liang, T.; Neumann, C.N.; Ritter, T. Introduction of Fluorine and Fluorine-Containing Functional Groups. Angew. Chem. Int. Ed. 2013, 52, 8214–8264. [Google Scholar] [CrossRef]
- López, S.E.; Salazar, J. Trifluoroacetic acid: Uses and recent applications in organic synthesis. J. Fluor. Chem. 2013, 156, 73–100. [Google Scholar] [CrossRef]
- Shibata, N.; Matsnev, A.; Cahard, D. Shelf-stable electrophilic trifluoromethylating reagents: A brief historical perspective. Beilstein J. Org. Chem. 2010, 6, 65. [Google Scholar] [CrossRef]
- Eisenberger, P.; Gischig, S.; Togni, A. Novel 10-I-3 Hypervalent Iodine-Based Compounds for Electrophilic Trifluoromethylation. Chem. A Eur. J. 2006, 12, 2579–2586. [Google Scholar] [CrossRef]
- Magnier, E.; Blazejewski, J.-C.; Tordeux, M.; Wakselman, C. Straightforward One-Pot Synthesis of Trifluoromethyl Sulfonium Salts. Angew. Chem. Int. Ed. 2006, 45, 1279–1282. [Google Scholar] [CrossRef] [PubMed]
- FDA. Novel Drug Approvals for 2022; FDA: Silver Spring, MD, USA, 2022.
- De la Torre, B.G.; Albericio, F. The Pharmaceutical Industry in 2022: An Analysis of FDA Drug Approvals from the Perspective of Molecules. Molecules 2023, 28, 1038. [Google Scholar] [CrossRef] [PubMed]
- Paik, J. Lenacapavir: First Approval. Drugs 2022, 82, 1499–1504. [Google Scholar] [CrossRef]
- Segal-Maurer, S.; DeJesus, E.; Stellbrink, H.-J.; Castagna, A.; Richmond, G.J.; Sinclair, G.I.; Siripassorn, K.; Ruane, P.J.; Berhe, M.; Wang, H.; et al. Capsid Inhibition with Lenacapavir in Multidrug-Resistant HIV-1 Infection. N. Engl. J. Med. 2022, 386, 1793–1803. [Google Scholar] [CrossRef]
- Graupe, M.; Henry, S.J.; Link, J.O.; Rowe, C.W.; Saito, R.D.; Schroeder, S.; Stefanidis, D.; Tse, W.C.; Zhang, J.R. Therapeutic Compounds Useful for the Prophylactic or Treatment of an HIV Virus Infection. Patent WO 2018035359, 22 February 2018. [Google Scholar]
- Hoy, S.M. Oteseconazole: First Approval. Drugs 2022, 82, 1017–1023. [Google Scholar] [CrossRef]
- Sobel, J.D.; Nyirjesy, P. Oteseconazole: An advance in treatment of recurrent vulvovaginal candidiasis. Future Microbiol. 2021, 16, 1453–1461. [Google Scholar] [CrossRef] [PubMed]
- Hoekstra, W.J.; Schotzinger, R.J.; Rafferty, S.W. Metalloenzyme Inhibitor Compounds. Patent WO 2011/133875, 27 October 2011. [Google Scholar]
- Benedetto Tiz, D.; Bagnoli, L.; Rosati, O.; Marini, F.; Sancineto, L.; Santi, C. New Halogen-Containing Drugs Approved by FDA in 2021: An Overview on Their Syntheses and Pharmaceutical Use. Molecules 2022, 27, 1643. [Google Scholar] [CrossRef] [PubMed]
- Deeks, E.D. Atogepant: First Approval. Drugs 2022, 82, 65–70. [Google Scholar] [CrossRef]
- Hay, D.L.; Walker, C.S.; Harris, P.W.R. Atogepant (Qulipta®) for migraine prevention. Trends Pharmacol. Sci. 2022, 43, 701–702. [Google Scholar] [CrossRef] [PubMed]
- Belyk, K.M.; Cleator, E.; Kuo, S.; Maligres, P.E.; Xiang, B.; Yasuda, N.; Yin, J. Process for Making CGRP Receptor Antagonists. Patent WO 2013/138418, 19 September 2013. [Google Scholar]
- Wood, M.R.; Bell, I.M.; Gallicchio, S.N.; Selnick, H.G.; Stump, C.A.; Zartman, C.B. Substituted Spirocyclic CGRP Receptor Antagonists. Patent WO 2007133491, 22 December 2007. [Google Scholar]
- Keam, S.J. Piflufolastat F 18: Diagnostic First Approval. Mol. Diagn. Ther. 2021, 25, 647–656. [Google Scholar] [CrossRef]
- Chen, Y.; Pullambhatla, M.; Foss, C.A.; Byun, Y.; Nimmagadda, S.; Senthamizhchelvan, S.; Sgouros, G.; Mease, R.C.; Pomper, M.G. 2-(3-{1-Carboxy-5-[(6-[18F]fluoro-pyridine-3-carbonyl)-amino]-pentyl}-ureido)-pentanedioic acid, [18F]DCFPyL, a PSMA-based PET imaging agent for prostate cancer. Clin. Cancer Res. Off. J. Am. Assoc. Cancer Res. 2011, 17, 7645–7653. [Google Scholar] [CrossRef] [PubMed]
- Olberg, D.E.; Arukwe, J.M.; Grace, D.; Hjelstuen, O.K.; Solbakken, M.; Kindberg, G.M.; Cuthbertson, A. One Step Radiosynthesis of 6-[18F]Fluoronicotinic Acid 2,3,5,6-Tetrafluorophenyl Ester ([18F]F-Py-TFP): A New Prosthetic Group for Efficient Labeling of Biomolecules with Fluorine-18. J. Med. Chem. 2010, 53, 1732–1740. [Google Scholar] [CrossRef] [PubMed]
- Canon, J.; Rex, K.; Saiki, A.Y.; Mohr, C.; Cooke, K.; Bagal, D.; Gaida, K.; Holt, T.; Knutson, C.G.; Koppada, N.; et al. The clinical KRAS(G12C) inhibitor AMG 510 drives anti-tumour immunity. Nature 2019, 575, 217–223. [Google Scholar] [CrossRef]
- Lanman, B.A.; Allen, J.R.; Allen, J.G.; Amegadzie, A.K.; Ashton, K.S.; Booker, S.K.; Chen, J.J.; Chen, N.; Frohn, M.J.; Goodman, G.; et al. Discovery of a Covalent Inhibitor of KRASG12C (AMG 510) for the Treatment of Solid Tumors. J. Med. Chem. 2020, 63, 52–65. [Google Scholar] [CrossRef]
- Lanman, B.A.; Chen, J.; Reed Anthony, B.; Cee Victor, J.; Liu, L.; Kopecky, D.J.; Lopez, P.; Wurz, R.P.; Nguyen, T.T.; Booker, S.; et al. Kras G12C Inhibitors and Method of Using the Same. Patent US 2018/0334454, 22 December 2018. [Google Scholar]
- Zhang, L.; Griffin, D.J.; Beaver, M.G.; Blue, L.E.; Borths, C.J.; Brown, D.B.; Caille, S.; Chen, Y.; Cherney, A.H.; Cochran, B.M.; et al. Development of a Commercial Manufacturing Process for Sotorasib, a First-in-Class KRASG12C Inhibitor. Org. Process Res. Dev. 2022, 26, 3115–3125. [Google Scholar] [CrossRef]
- Lunning, M.; Vose, J.; Nastoupil, L.; Fowler, N.; Burger, J.A.; Wierda, W.G.; Schreeder, M.T.; Siddiqi, T.; Flowers, C.R.; Cohen, J.B.; et al. Ublituximab and umbralisib in relapsed/refractory B-cell non-Hodgkin lymphoma and chronic lymphocytic leukemia. Blood 2019, 134, 1811–1820. [Google Scholar] [CrossRef]
- Weiss, M.; Miskin, H.; Sportelli, P.S.; Vakkalanka, K.V.S. Combination of Anti-CD20 Antibody and PI3 Kinase Selective Inhibitor. Patent WO 2014071125, 8 May 2014. [Google Scholar]
- Follmann, M.; Ackerstaff, J.; Redlich, G.; Wunder, F.; Lang, D.; Kern, A.; Fey, P.; Griebenow, N.; Kroh, W.; Becker-Pelster, E.-M.; et al. Discovery of the Soluble Guanylate Cyclase Stimulator Vericiguat (BAY 1021189) for the Treatment of Chronic Heart Failure. J. Med. Chem. 2017, 60, 5146–5161. [Google Scholar] [CrossRef] [PubMed]
- Yuan, S.; Luo, Y.Q.; Zuo, J.H.; Liu, H.; Li, F.; Yu, B. New drug approvals for 2020: Synthesis and clinical applications. Eur. J. Med. Chem. 2021, 215, 113284. [Google Scholar] [CrossRef]
- Ohsawa, I.; Honda, D.; Suzuki, Y.; Fukuda, T.; Kohga, K.; Morita, E.; Moriwaki, S.; Ishikawa, O.; Sasaki, Y.; Tago, M.; et al. Oral berotralstat for the prophylaxis of hereditary angioedema attacks in patients in Japan: A phase 3 randomized trial. Allergy 2021, 76, 1789–1799. [Google Scholar] [CrossRef] [PubMed]
- Yahya El-Kattan, Y.S.B. Crystalline Salts of a Plasma Kallikrein Inhibitor. Patent US 202010662160, 7 May 2020. [Google Scholar]
- Dhillon, S. Decitabine/Cedazuridine: First Approval. Drugs 2020, 80, 1373–1378. [Google Scholar] [CrossRef] [PubMed]
- Ferraris, D.; Duvall, B.; Delahanty, G.; Mistry, B.; Alt, J.; Rojas, C.; Rowbottom, C.; Sanders, K.; Schuck, E.; Huang, K.-C.; et al. Design, Synthesis, and Pharmacological Evaluation of Fluorinated Tetrahydrouridine Derivatives as Inhibitors of Cytidine Deaminase. J. Med. Chem. 2014, 57, 2582–2588. [Google Scholar] [CrossRef]
- Brown, K.; Dixey, M.; Weymouth-Wilson, A.; Linclau, B. The synthesis of gemcitabine. Carbohydr. Res. 2014, 387, 59–73. [Google Scholar] [CrossRef] [PubMed]
- Markham, A. Pralsetinib: First Approval. Drugs 2020, 80, 1865–1870. [Google Scholar] [CrossRef] [PubMed]
- Subbiah, V.; Shen, T.; Terzyan, S.S.; Liu, X.; Hu, X.; Patel, K.P.; Hu, M.; Cabanillas, M.; Behrang, A.; Meric-Bernstam, F.; et al. Structural basis of acquired resistance to selpercatinib and pralsetinib mediated by non-gatekeeper RET mutations. Ann. Oncol. Off. J. Eur. Soc. Med. Oncol. 2021, 32, 261–268. [Google Scholar] [CrossRef]
- Brubaker, J.D.; Kim, J.L.; Wilson, K.J.; Wilson, D.; Di Pietro, L.V. Inhibitors of Ret. Patent US 20170121312, 4 May 2017. [Google Scholar]
- Markham, A.; Keam, S.J. Selumetinib: First Approval. Drugs 2020, 80, 931–937. [Google Scholar] [CrossRef]
- Wallace, E.M.; Lyssikatos, J.P.; Hurley, B.T.; Marlow, A.L. N3 Alkylated Benzimiadazole Derivates Ad MEK Inhibitors. Patent WO 2003077914, 25 September 2003. [Google Scholar]
- Mu, L.; Jie, C.V.M.L.; Treyer, V.; Schibli, R. Tauvid™: The First FDA-Approved PET Tracer for Imaging Tau Pathology in Alzheimer’s Disease. Pharmaceuticals 2021, 14, 110. [Google Scholar] [CrossRef]
- Xia, C.F.; Arteaga, J.; Chen, G.; Gangadharmath, U.; Gomez, L.F.; Kasi, D.; Lam, C.; Liang, Q.; Liu, C.; Mocharla, V.P.; et al. [(18)F]T807, a novel tau positron emission tomography imaging agent for Alzheimer’s disease. Alzheimer’s Dement. J. Alzheimer’s Assoc. 2013, 9, 666–676. [Google Scholar] [CrossRef] [PubMed]
- Attardo, G.; Lister-James, J.; Xiong, H.; Lim, N. Compounds and Their Use for Preparation of Tau Imaging Agents and Tau Imaging Formulations. Patent WO 2015047902, 2 April 2015. [Google Scholar]
- Patridge, E.; Gareiss, P.; Kinch, M.S.; Hoyer, D. An analysis of FDA-approved drugs: Natural products and their derivatives. Drug Discov. Today 2016, 21, 204–207. [Google Scholar] [CrossRef] [PubMed]
- Yuan, S.; Yu, B.; Liu, H.M. New drug approvals for 2019: Synthesis and clinical applications. Eur. J. Med. Chem. 2020, 205, 112667. [Google Scholar] [CrossRef] [PubMed]
- Mei, H.; Remete, A.M.; Zou, Y.; Moriwaki, H.; Fustero, S.; Kiss, L.; Soloshonok, V.A.; Han, J. Fluorine-containing drugs approved by the FDA in 2019. Chin. Chem. Lett. 2020, 31, 2401–2413. [Google Scholar] [CrossRef]
- Markham, A. Alpelisib: First Global Approval. Drugs 2019, 79, 1249–1253. [Google Scholar] [CrossRef] [PubMed]
- André, F.; Ciruelos, E.; Rubovszky, G.; Campone, M.; Loibl, S.; Rugo, H.S.; Iwata, H.; Conte, P.; Mayer, I.A.; Kaufman, B.; et al. Alpelisib for PIK3CA-Mutated, Hormone Receptor-Positive Advanced Breast Cancer. N. Engl. J. Med. 2019, 380, 1929–1940. [Google Scholar] [CrossRef] [PubMed]
- Furet, P.; Guagnano, V.; Fairhurst, R.A.; Imbach-Weese, P.; Bruce, I.; Knapp, M.; Fritsch, C.; Blasco, F.; Blanz, J.; Aichholz, R.; et al. Discovery of NVP-BYL719 a potent and selective phosphatidylinositol-3 kinase alpha inhibitor selected for clinical evaluation. Bioorg. Med. Chem. Lett. 2013, 23, 3741–3748. [Google Scholar] [CrossRef] [PubMed]
- Caravatti, G.; Fairhutst, R.A.; Guagnano, V.; Imbach, P.; Furet, P. Thiazole Derivate Used as PI 3 KINASE Inhibitors. Patent WO 2009080694, 2 July 2009. [Google Scholar]
- Caravatti, G.; Fairhutst, R.A.; Guagnano, V.; Imbach, P.; Furet, P. Organic Compounds. Patent WO 2010029082, 10 September 2010. [Google Scholar]
- Beuckmann, C.T.; Suzuki, M.; Ueno, T.; Nagaoka, K.; Arai, T.; Higashiyama, H. In Vitro and In Silico Characterization of Lemborexant (E2006), a Novel Dual Orexin Receptor Antagonist. J. Pharmacol. Exp. Ther. 2017, 362, 287–295. [Google Scholar] [CrossRef] [PubMed]
- Beuckmann, C.T.; Ueno, T.; Nakagawa, M.; Suzuki, M.; Akasofu, S. Preclinical in vivo characterization of lemborexant (E2006), a novel dual orexin receptor antagonist for sleep/wake regulation. Sleep 2019, 42, zsz076. [Google Scholar] [CrossRef]
- Yoshida, Y.; Naoe, Y.; Terauchi, T.; Ozaki, F.; Doko, T.; Takemura, A.; Tanaka, T.; Sorimachi, K.; Beuckmann, C.T.; Suzuki, M.; et al. Discovery of (1R,2S)-2-{[(2,4-Dimethylpyrimidin-5-yl)oxy]methyl}-2-(3-fluorophenyl)-N-(5-fluoropyridin-2-yl)cyclopropanecarboxamide (E2006): A Potent and Efficacious Oral Orexin Receptor Antagonist. J. Med. Chem. 2015, 58, 4648–4664. [Google Scholar] [CrossRef]
- Lamb, Y.N. Pexidartinib: First Approval. Drugs 2019, 79, 1805–1812. [Google Scholar] [CrossRef]
- Ibrahim, P.N.; Jin, M.; Matsuura, S. Synthesis of 1H-Pyrrolo[2,3-B]pyridin Derivates That Modulate Kinases. Patent WO 2016179412, 10 November 2016. [Google Scholar]
- Dodick, D.W.; Lipton, R.B.; Ailani, J.; Lu, K.; Finnegan, M.; Trugman, J.M.; Szegedi, A. Ubrogepant for the Treatment of Migraine. N. Engl. J. Med. 2019, 381, 2230–2241. [Google Scholar] [CrossRef] [PubMed]
- Bell, I.M.; Fraley, M.E.; Gallicchio, S.N.; Ginnetti, A.; Mitchell, H.; Paone, D.V.; Wang, S.D.D.C.; Zartman, C.B.; Stevenson, H.E. Piperidone Carbozamide CGRP Receptor Antagonists. Patent WO 2012064910, 18 May 2012. [Google Scholar]
- Flick, A.C.; Leverett, C.A.; Ding, H.X.; McInturff, E. Synthetic Approaches to New Drugs Approved during 2018. J. Med. Chem. 2020, 63, 10652–10704. [Google Scholar] [CrossRef]
- Al-Salama, Z.T. Apalutamide: First Global Approval. Drugs 2018, 78, 699–705. [Google Scholar] [CrossRef] [PubMed]
- Smith, M.R.; Saad, F.; Chowdhury, S.; Oudard, S.; Hadaschik, B.A.; Graff, J.N.; Olmos, D.; Mainwaring, P.N.; Lee, J.Y.; Uemura, H.; et al. Apalutamide Treatment and Metastasis-free Survival in Prostate Cancer. N. Engl. J. Med. 2018, 378, 1408–1418. [Google Scholar] [CrossRef] [PubMed]
- Clegg, N.J.; Wongvipat, J.; Joseph, J.D.; Tran, C.; Ouk, S.; Dilhas, A.; Chen, Y.; Grillot, K.; Bischoff, E.D.; Cai, L.; et al. ARN-509: A novel antiandrogen for prostate cancer treatment. Cancer Res. 2012, 72, 1494–1503. [Google Scholar] [CrossRef]
- Jung, M.; Sawyers, E.C.; Ouk, S.L.; Tran, C.; Wongvipat, J. Androgen Receptor Modulator for the Treatment of Prostate Cancer and Androgen Receptor-Associated Disease. Patent WO 2007126765, 8 November 2007. [Google Scholar]
- Hughes, D.L. Review of Synthetic Routes and Crystalline Forms of the Antiandrogen Oncology Drugs Enzalutamide, Apalutamide, and Darolutamide. Org. Process Res. Dev. 2020, 24, 347–362. [Google Scholar] [CrossRef]
- Heo, Y.A. Baloxavir: First Global Approval. Drugs 2018, 78, 693–697. [Google Scholar] [CrossRef]
- Noshi, T.; Kitano, M.; Taniguchi, K.; Yamamoto, A.; Omoto, S.; Baba, K.; Hashimoto, T.; Ishida, K.; Kushima, Y.; Hattori, K.; et al. In vitro characterization of baloxavir acid, a first-in-class cap-dependent endonuclease inhibitor of the influenza virus polymerase PA subunit. Antivir. Res. 2018, 160, 109–117. [Google Scholar] [CrossRef]
- Takashita, E.; Morita, H.; Ogawa, R.; Nakamura, K.; Fujisaki, S.; Shirakura, M.; Kuwahara, T.; Kishida, N.; Watanabe, S.; Odagiri, T. Susceptibility of Influenza Viruses to the Novel Cap-Dependent Endonuclease Inhibitor Baloxavir Marboxil. Front. Microbiol. 2018, 9, 3026. [Google Scholar] [CrossRef]
- Shibahara, S.F.; Nobuaki; Toshikatsu, M. Method for Producing Substituted Polycyclic Derivate and Crystal of Same. Patent JP 20176212678, 28 December 2017. [Google Scholar]
- Koelblinger, P.; Dornbierer, J.; Dummer, R. A review of binimetinib for the treatment of mutant cutaneous melanoma. Future Oncol. 2017, 13, 1755–1766. [Google Scholar] [CrossRef]
- Ascierto, P.A.; Schadendorf, D.; Berking, C.; Agarwala, S.S.; van Herpen, C.M.; Queirolo, P.; Blank, C.U.; Hauschild, A.; Beck, J.T.; St-Pierre, A.; et al. MEK162 for patients with advanced melanoma harbouring NRAS or Val600 BRAF mutations: A non-randomised, open-label phase 2 study. Lancet Oncol. 2013, 14, 249–256. [Google Scholar] [CrossRef] [PubMed]
- Chen, J. Synthetic Method of Binimetinib. Patent CN 2016105820124, 3 August 2016. [Google Scholar]
- Colombier, M.A.; Molina, J.M. Doravirine: A review. Curr. Opin. HIV AIDS 2018, 13, 308–314. [Google Scholar] [CrossRef] [PubMed]
- Côté, B.; Burch, J.D.; Asante-Appiah, E.; Bayly, C.; Bédard; Blouin, L.M.; Campeau, L.C.; Cauchon, E.; Chan, M.; Chefson, A.; et al. Discovery of MK-1439, an orally bioavailable non-nucleoside reverse transcriptase inhibitor potent against a wide range of resistant mutant HIV viruses. Bioorganic Med. Chem. Lett. 2014, 24, 917–922. [Google Scholar] [CrossRef]
- Burch, J.; Nguyen, N.; Li, C.S.; St-onge, M.; Gauvreau, D.; Cote, B. Non-Nucleoside Reverse Transcriptase Inhibitors. Patent WO 2011120133, 6 October 2011. [Google Scholar]
- Markham, A. Fostamatinib: First Global Approval. Drugs 2018, 78, 959–963. [Google Scholar] [CrossRef] [PubMed]
- Pine, P.R.; Chang, B.; Schoettler, N.; Banquerigo, M.L.; Wang, S.; Lau, A.; Zhao, F.; Grossbard, E.B.; Payan, D.G.; Brahn, E. Inflammation and bone erosion are suppressed in models of rheumatoid arthritis following treatment with a novel Syk inhibitor. Clin. Immunol. 2007, 124, 244–257. [Google Scholar] [CrossRef]
- Felfer, U.; Giselbrecht, K.-H.; Wolberg, M. Syntesis of N4-(2,2-Dimethyl-4-[(Dihydrogen Phosphonoxy]-3-Oxo-5-Pyrydo [1,4] Oxazin6-yl)-5-Fluoro-N2-(3,4,5,-Trimethoxyphenyl)-2,4-Pyrimidinediamine Disodium Salt. Patent WO 2011002999, 6 January 2011. [Google Scholar]
- Dhillon, S. Ivosidenib: First Global Approval. Drugs 2018, 78, 1509–1516. [Google Scholar] [CrossRef]
- Popovici-Muller, J.; Lemieux, R.M.; Artin, E.; Saunders, J.O.; Salituro, F.G.; Travins, J.; Cianchetta, G.; Cai, Z.; Zhou, D.; Cui, D.; et al. Discovery of AG-120 (Ivosidenib): A First-in-Class Mutant IDH1 Inhibitor for the Treatment of IDH1 Mutant Cancers. ACS Med. Chem. Lett. 2018, 9, 300–305. [Google Scholar] [CrossRef]
- Lemieux, R.M.; Popovici-Muller, J.; Travins, J.; Cai, Z.; Cui, D.; Zhou, D. Therapeutically Active Compounds and Their Methods of Use. Patent WO 2013107291, 25 July 2013. [Google Scholar]
- Hoy, S.M. Talazoparib: First Global Approval. Drugs 2018, 78, 1939–1946. [Google Scholar] [CrossRef]
- Wang, B.; Chu, D.; Feng, Y.; Shen, Y.; Aoyagi-Scharber, M.; Post, L.E. Discovery and Characterization of (8S,9R)-5-Fluoro-8-(4-fluorophenyl)-9-(1-methyl-1H-1,2,4-triazol-5-yl)-2,7,8,9-tetrahydro-3H-pyrido[4,3,2-de]phthalazin-3-one(BMN673,Talazoparib), a Novel, Highly Potent, and Orally Efficacious Poly(ADP-ribose) Polymerase-1/2 Inhibitor, as an Anticancer Agent. J. Med. Chem. 2016, 59, 335–357. [Google Scholar] [CrossRef] [PubMed]
- Xu, Y.Y.; Peter, W.; Michael, X.; Douglas, C. Synthesis of Parp Inhibitor Talazoparib. Patent US 20179708319, 21 December 2017. [Google Scholar]
- Mospan, C.; Mospan, G.; Byland, E.; Whitaker, W.B.; Xiong, L.; Dunlap, J.; Canupp, K. Drug updates and approvals: 2018 in review. Nurse Pract. 2018, 43, 23–32. [Google Scholar] [CrossRef]
- Donaldson, S.H.; Pilewski, J.M.; Griese, M.; Cooke, J.; Viswanathan, L.; Tullis, E.; Davies, J.C.; Lekstrom-Himes, J.A.; Wang, L.T. Tezacaftor/Ivacaftor in Subjects with Cystic Fibrosis and F508del/F508del-CFTR or F508del/G551D-CFTR. Am. J. Respir. Crit. Care Med. 2018, 197, 214–224. [Google Scholar] [CrossRef]
- Hughes, D.L. Patent Review of Synthetic Routes and Crystalline Forms of the CFTR-Modulator Drugs Ivacaftor, Lumacaftor, Tezacaftor, and Elexacaftor. Org. Process Res. Dev. 2019, 23, 2302–2322. [Google Scholar] [CrossRef]
- Tanoury, G.J.; Harrison, C.; Littler, B.J.; Rose, P.J.; Hughes, R.M.; Jung, Y.C.; Siesel, D.A.; Lee, E.C.; Belmont, D.T. Process of Producing Cycloalkylcarboxamido-Indole Compounds. Patent WO 2011133751, 27 October 2011. [Google Scholar]
- De la Torre, B.G.; Albericio, F. The Pharmaceutical Industry in 2017. An Analysis of FDA Drug Approvals from the Perspective of Molecules. Molecules 2018, 23, 533. [Google Scholar] [CrossRef]
- Mullard, A. 2017 FDA drug approvals, Nature reviews. Drug Discov. 2018, 17, 81–85. [Google Scholar] [CrossRef] [PubMed]
- Martin, M.; Garcia-Saenz, J.A. Abemaciclib, a CDK4 and CDK6 inhibitor for the treatment of metastatic breast cancer. Futur. Oncol. 2020, 16, 2763–2778. [Google Scholar] [CrossRef] [PubMed]
- Frederick, M.O.; Kjell, D.P. A synthesis of abemaciclib utilizing a Leuckart–Wallach reaction. Tetrahedron Lett. 2015, 56, 949–951. [Google Scholar] [CrossRef]
- Frederick, M.O.; Pietz, M.A.; Kjell, D.P.; Richey, R.N.; Tharp, G.A.; Touge, T.; Yokoyama, N.; Kida, M.; Matsuo, T. Development of a Leuckart–Wallach Reaction in Flow for the Synthesis of Abemaciclib. Org. Process Res. Dev. 2017, 21, 1447–1451. [Google Scholar] [CrossRef]
- Markham, A. Delafloxacin: First Global Approval. Drugs 2017, 77, 1481–1486. [Google Scholar] [CrossRef] [PubMed]
- Harnett, S.J.; Fraise, A.P.; Andrews, J.M.; Jevons, G.; Brenwald, N.P.; Wise, R. Comparative study of the in vitro activity of a new fluoroquinolone, ABT-492. J. Antimicrob. Chemother. 2004, 53, 783–792. [Google Scholar] [CrossRef]
- Barnes, D.M.; Christesen, A.C.; Engstrom, K.M.; Haight, A.R.; Hsu, M.C.; Lee, E.C.; Peterson, M.J.; Plata, D.J.; Raje, P.S.; Stoner, E.J.; et al. Chlorination at the 8-Position of a Functionalized Quinolone and the Synthesis of Quinolone Antibiotic ABT-492. Org. Process Res. Dev. 2006, 10, 803–807. [Google Scholar] [CrossRef]
- Kim, E.S. Enasidenib: First Global Approval. Drugs 2017, 77, 1705–1711. [Google Scholar] [CrossRef] [PubMed]
- Stein, E.M.; DiNardo, C.D.; Pollyea, D.A.; Fathi, A.T.; Roboz, G.J.; Altman, J.K.; Stone, R.M.; DeAngelo, D.J.; Levine, R.L.; Flinn, I.W.; et al. Enasidenib in mutant IDH2 relapsed or refractory acute myeloid leukemia. Blood 2017, 130, 722–731. [Google Scholar] [CrossRef]
- García-Llinás, X.; Bauzá, A.; Seth, S.K.; Frontera, A. Importance of R–CF3···O Tetrel Bonding Interactions in Biological Systems. J. Phys. Chem. A 2017, 121, 5371–5376. [Google Scholar] [CrossRef] [PubMed]
- Cianchetta, B.D.G.; Popovici-Muller, J.F.; Salituro, G.; Saunders, J.O.; Travins, J.; Yan, S.; Guo, T.; Zhang, L. Therapeutically Active Compounds and Their Methods of Use. Patent WO 2013102431, 7 November 2013. [Google Scholar]
- Lamb, Y.N. Glecaprevir/Pibrentasvir: First Global Approval. Drugs 2017, 77, 1797–1804. [Google Scholar] [CrossRef]
- Lawitz Eric, J.; O’Riordan William, D.; Asatryan, A.; Freilich Bradley, L.; Box Terry, D.; Overcash, J.S.; Lovell, S.; Ng Teresa, I.; Liu, W.; Campbell, A.; et al. Potent Antiviral Activities of the Direct-Acting Antivirals ABT-493 and ABT-530 with Three-Day Monotherapy for Hepatitis C Virus Genotype 1 Infection. Antimicrob. Agents Chemother. 2016, 60, 1546–1555. [Google Scholar] [CrossRef]
- Cink, R.D.; Lukin, K.A.; Bishop, R.D.; Zhao, G.; Pelc, M.J.; Towne, T.B.; Gates, B.D.; Ravn, M.M.; Hill, D.R.; Ding, C.; et al. Development of the Enabling Route for Glecaprevir via Ring-Closing Metathesis. Org. Process Res. Dev. 2020, 24, 183–200. [Google Scholar] [CrossRef]
- Kallemeyn, J.M.; Engstrom, K.M.; Pelc, M.J.; Lukin, K.A.; Morrill, W.H.; Wei, H.; Towne, T.B.; Henle, J.; Nere, N.K.; Welch, D.S.; et al. Development of a Large-Scale Route to Glecaprevir: Synthesis of the Macrocycle via Intramolecular Etherification. Org. Process Res. Dev. 2020, 24, 1373–1392. [Google Scholar] [CrossRef]
- Kim, E.S. Letermovir: First Global Approval. Drugs 2018, 78, 147–152. [Google Scholar] [CrossRef] [PubMed]
- Goldner, T.; Hewlett, G.; Ettischer, N.; Ruebsamen-Schaeff, H.; Zimmermann, H.; Lischka, P. The novel anticytomegalovirus compound AIC246 (Letermovir) inhibits human cytomegalovirus replication through a specific antiviral mechanism that involves the viral terminase. J. Virol. 2011, 85, 10884–10893. [Google Scholar] [CrossRef]
- Humphrey, G.R.; Dalby, S.M.; Andreani, T.; Xiang, B.; Luzung, M.R.; Song, Z.J.; Shevlin, M.; Christensen, M.; Belyk, K.M.; Tschaen, D.M. Asymmetric Synthesis of Letermovir Using a Novel Phase-Transfer-Catalyzed Aza-Michael Reaction. Org. Process Res. Dev. 2016, 20, 1097–1103. [Google Scholar] [CrossRef]
- Chung, C.K.; Liu, Z.; Lexa, K.W.; Andreani, T.; Xu, Y.; Ji, Y.; DiRocco, D.A.; Humphrey, G.R.; Ruck, R.T. Asymmetric Hydrogen Bonding Catalysis for the Synthesis of Dihydroquinazoline-Containing Antiviral, Letermovir. J. Am. Chem. Soc. 2017, 139, 10637–10640. [Google Scholar] [CrossRef]
- Wang, P.S.; Shen, M.L.; Wang, T.C.; Lin, H.C.; Gong, L.Z. Access to Chiral Hydropyrimidines through Palladium-Catalyzed Asymmetric Allylic C-H Amination. Angew. Chem. 2017, 56, 16032–16036. [Google Scholar] [CrossRef] [PubMed]
- Ng Teresa, I.; Krishnan, P.; Pilot-Matias, T.; Kati, W.; Schnell, G.; Beyer, J.; Reisch, T.; Lu, L.; Dekhtyar, T.; Irvin, M.; et al. In Vitro Antiviral Activity and Resistance Profile of the Next-Generation Hepatitis C Virus NS5A Inhibitor Pibrentasvir. Antimicrob. Agents Chemother. 2017, 61, e02558-16. [Google Scholar] [CrossRef] [PubMed]
- Degoey, D.A.; Kati, W.M.; Hutchins, C.W.; Donner, P.L.; Krueger, A.C.; Randolph, J.T.; Motter, C.E.; Nelson, L.T.; Patel, S.V.; Matulenko, M.A.; et al. Anti-Viral Compounds. Patent WO 2012051361, 19 April 2012. [Google Scholar]
- Heo, Y.-A.; Deeks, E.D. Sofosbuvir/Velpatasvir/Voxilaprevir: A Review in Chronic Hepatitis C. Drugs 2018, 78, 577–587. [Google Scholar] [CrossRef]
- Bjornson, K.; Canales, E.; Cotell, J.; Karki, J.K.; Katana, K.A.; Kato, A.D.; Kobayashi, T.; Link, J.O.; Martinez, R.; Phillips, B.; et al. Inhibitors of Hepatitis C Virus. Patent WO 2014008285, 9 January 2014. [Google Scholar]
- Mullard, A. 2016 FDA drug approvals. Nat. Rev. Drug Discov. 2017, 16, 73–76. [Google Scholar] [CrossRef]
- Musella, A.; Bardhi, E.; Marchetti, C.; Vertechy, L.; Santangelo, G.; Sassu, C.; Tomao, F.; Rech, F.; D’Amelio, R.; Monti, M.; et al. Rucaparib: An emerging parp inhibitor for treatment of recurrent ovarian cancer. Cancer Treat. Rev. 2018, 66, 7–14. [Google Scholar] [CrossRef]
- Thomas, H.D.; Calabrese, C.R.; Batey, M.A.; Canan, S.; Hostomsky, Z.; Kyle, S.; Maegley, K.A.; Newell, D.R.; Skalitzky, D.; Wang, L.-Z.; et al. Preclinical selection of a novel poly(ADP-ribose) polymerase inhibitor for clinical trial. Mol. Cancer Ther. 2007, 6, 945–956. [Google Scholar] [CrossRef]
- Webber, S.E.; Canan-Koch, S.S.; Tikhe, J.G.; Thoresen, L.H. Tricyclic Inhibitors of Poly(ADP-Ribose) Polymerases. Patent WO 2000042040, 20 July 2000. [Google Scholar]
- Bonaventura, A.; Montecucco, F. Sofosbuvir/velpatasvir: A promising combination. World J. Hepatol. 2016, 8, 785–789. [Google Scholar] [CrossRef] [PubMed]
- Keating, G.M.; Vaidya, A. Sofosbuvir: First Global Approval. Drugs 2014, 74, 273–282. [Google Scholar] [CrossRef]
- Asselah, T. Sofosbuvir for the treatment of hepatitis C virus. Expert Opin. Pharmacother. 2014, 15, 121–130. [Google Scholar] [CrossRef] [PubMed]
- Sofia, M.J.; Bao, D.; Chang, W.; Du, J.; Nagarathnam, D.; Rachakonda, S.; Reddy, P.G.; Ross, B.S.; Wang, P.; Zhang, H.-R.; et al. Discovery of a β-d-2′-Deoxy-2′-α-fluoro-2′-β-C-methyluridine Nucleotide Prodrug (PSI-7977) for the Treatment of Hepatitis C Virus. J. Med. Chem. 2010, 53, 7202–7218. [Google Scholar] [CrossRef] [PubMed]
- Wang, P.; Chun, B.-K.; Rachakonda, S.; Du, J.; Khan, N.; Shi, J.; Stec, W.; Cleary, D.; Ross, B.S.; Sofia, M.J. An Efficient and Diastereoselective Synthesis of PSI-6130: A Clinically Efficacious Inhibitor of HCV NS5B Polymerase. J. Org. Chem. 2009, 74, 6819–6824. [Google Scholar] [CrossRef] [PubMed]





Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Rizzo, C.; Amata, S.; Pibiri, I.; Pace, A.; Buscemi, S.; Palumbo Piccionello, A. FDA-Approved Fluorinated Heterocyclic Drugs from 2016 to 2022. Int. J. Mol. Sci. 2023, 24, 7728. https://doi.org/10.3390/ijms24097728
Rizzo C, Amata S, Pibiri I, Pace A, Buscemi S, Palumbo Piccionello A. FDA-Approved Fluorinated Heterocyclic Drugs from 2016 to 2022. International Journal of Molecular Sciences. 2023; 24(9):7728. https://doi.org/10.3390/ijms24097728
Chicago/Turabian StyleRizzo, Carla, Sara Amata, Ivana Pibiri, Andrea Pace, Silvestre Buscemi, and Antonio Palumbo Piccionello. 2023. "FDA-Approved Fluorinated Heterocyclic Drugs from 2016 to 2022" International Journal of Molecular Sciences 24, no. 9: 7728. https://doi.org/10.3390/ijms24097728
APA StyleRizzo, C., Amata, S., Pibiri, I., Pace, A., Buscemi, S., & Palumbo Piccionello, A. (2023). FDA-Approved Fluorinated Heterocyclic Drugs from 2016 to 2022. International Journal of Molecular Sciences, 24(9), 7728. https://doi.org/10.3390/ijms24097728