

PPARδ Agonist GW501516 Suppresses the TGF-β-Induced Profibrotic Response of Human Bronchial Fibroblasts from Asthmatic Patients



Abstract
1. Introduction
2. Results
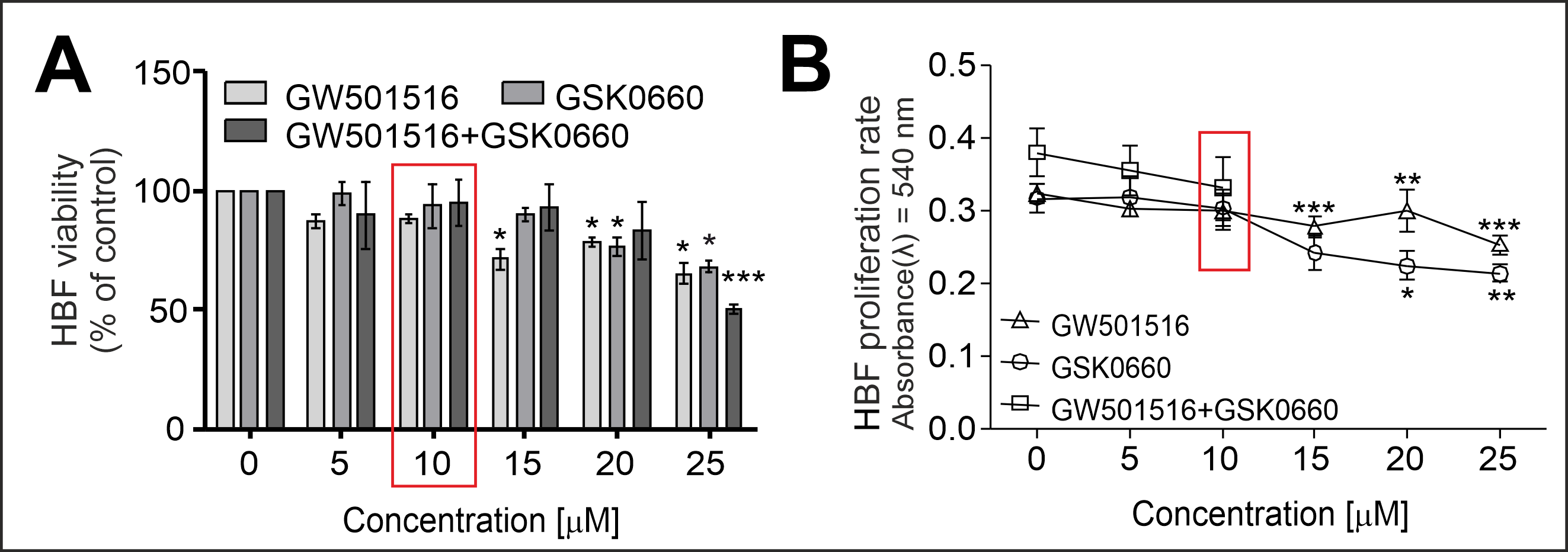
2.1. PPARδ Agonist GW501516 and PPARδ Antagonist GSK0660 Administered Separately or in Combination Inhibit the Proliferation and Viability of HBFs in a Dose-Dependent Manner
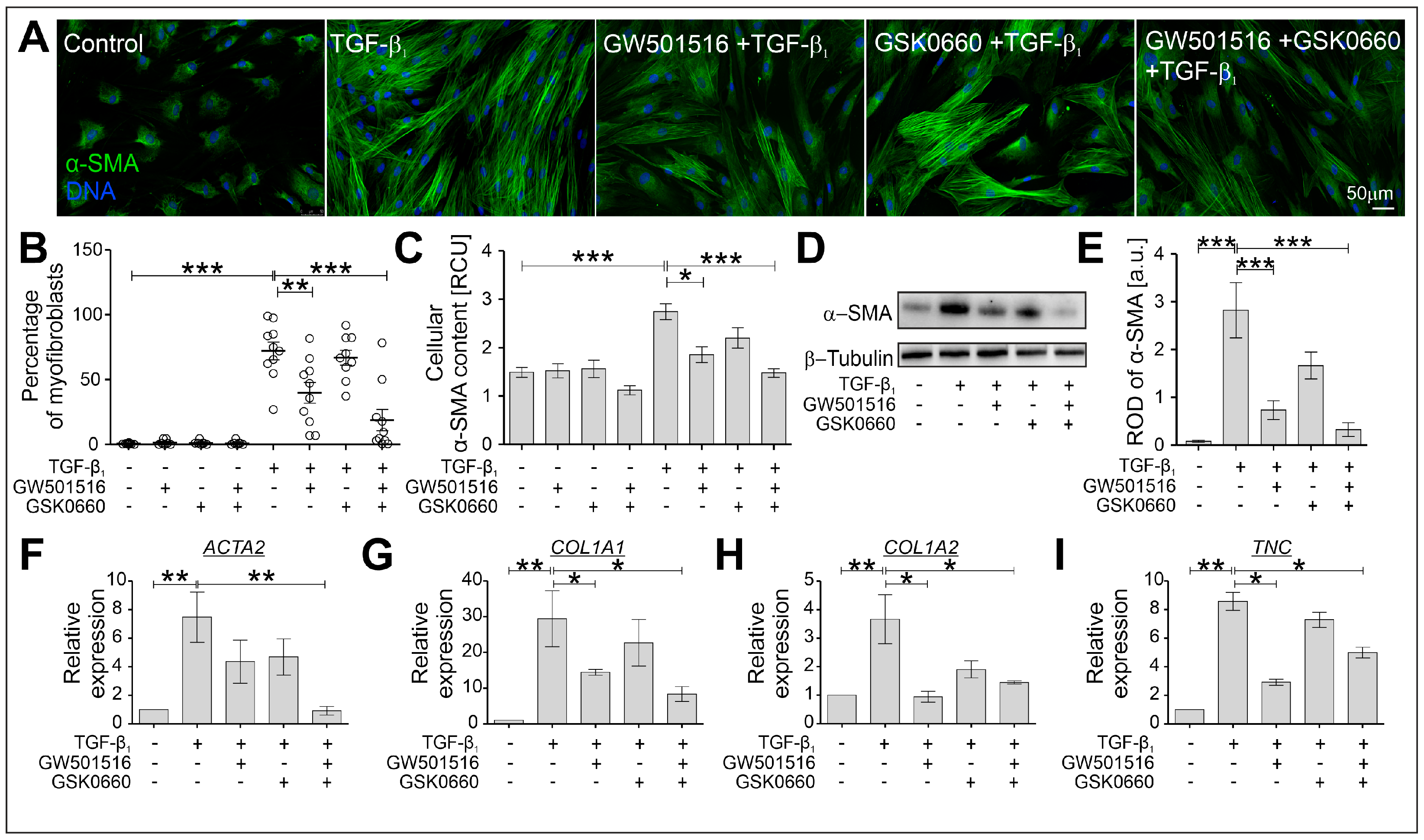
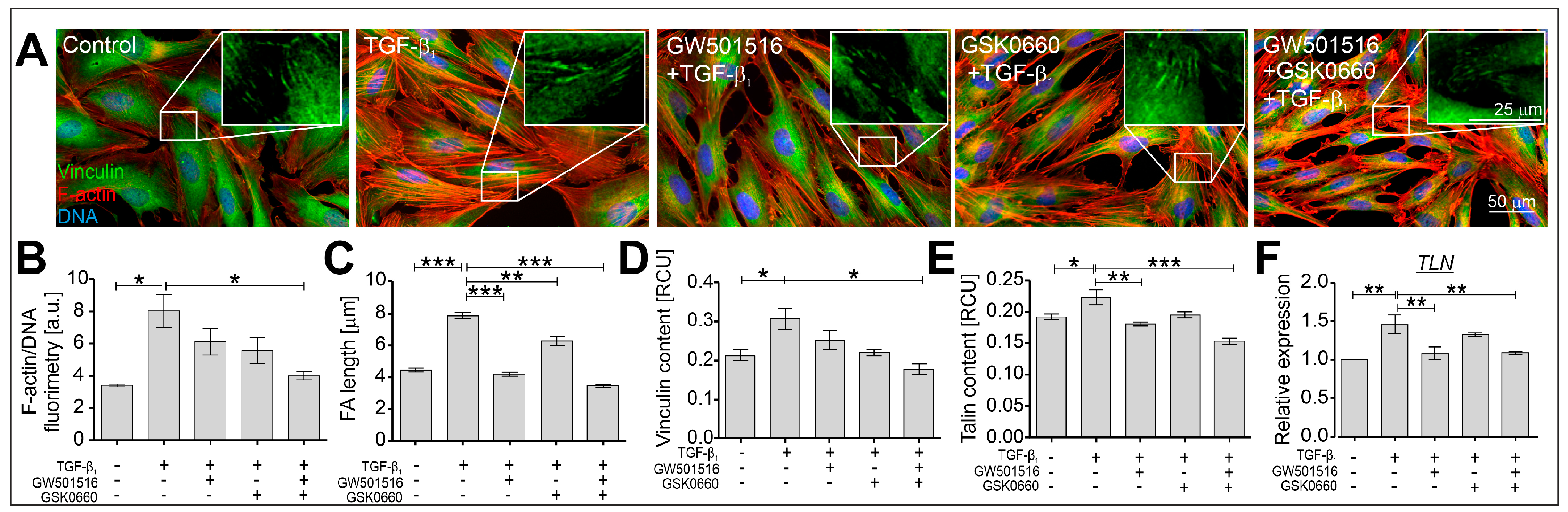
2.2. The TGF-β-Induced Myofibroblastic Transitions of HBFs Are Suppressed after PPARδ Agonist Administration
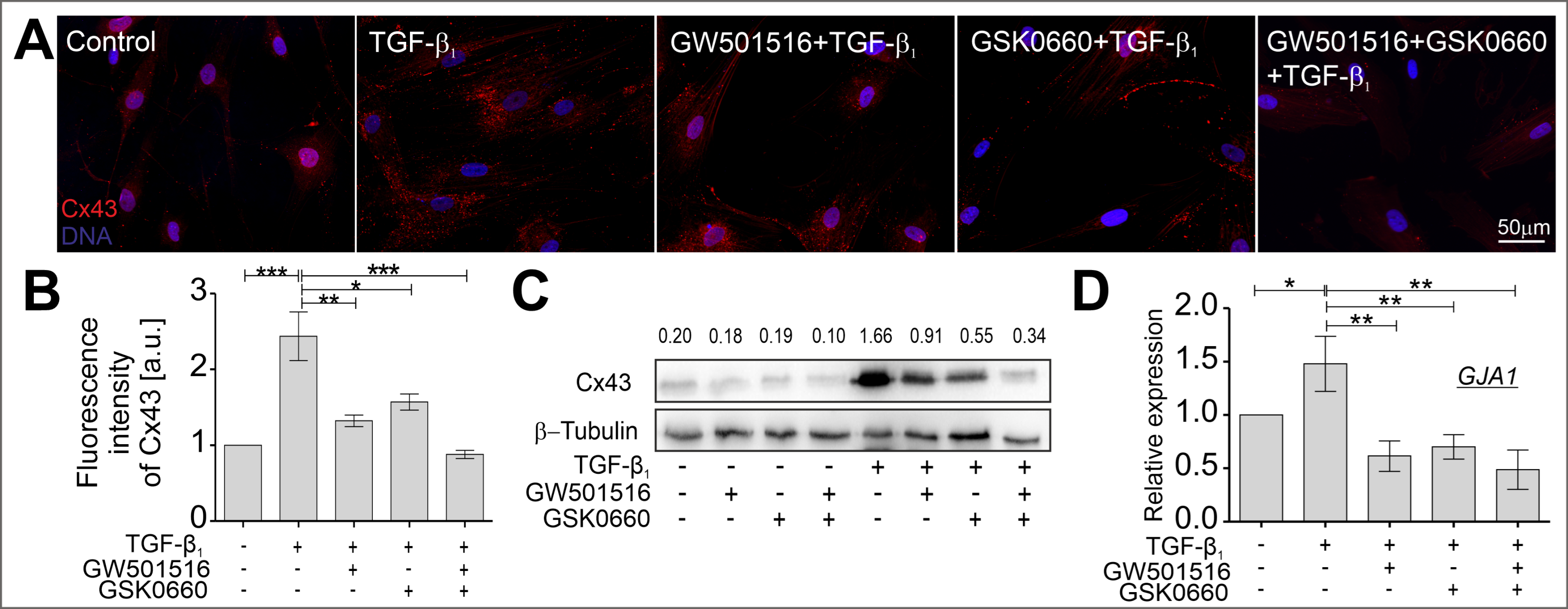
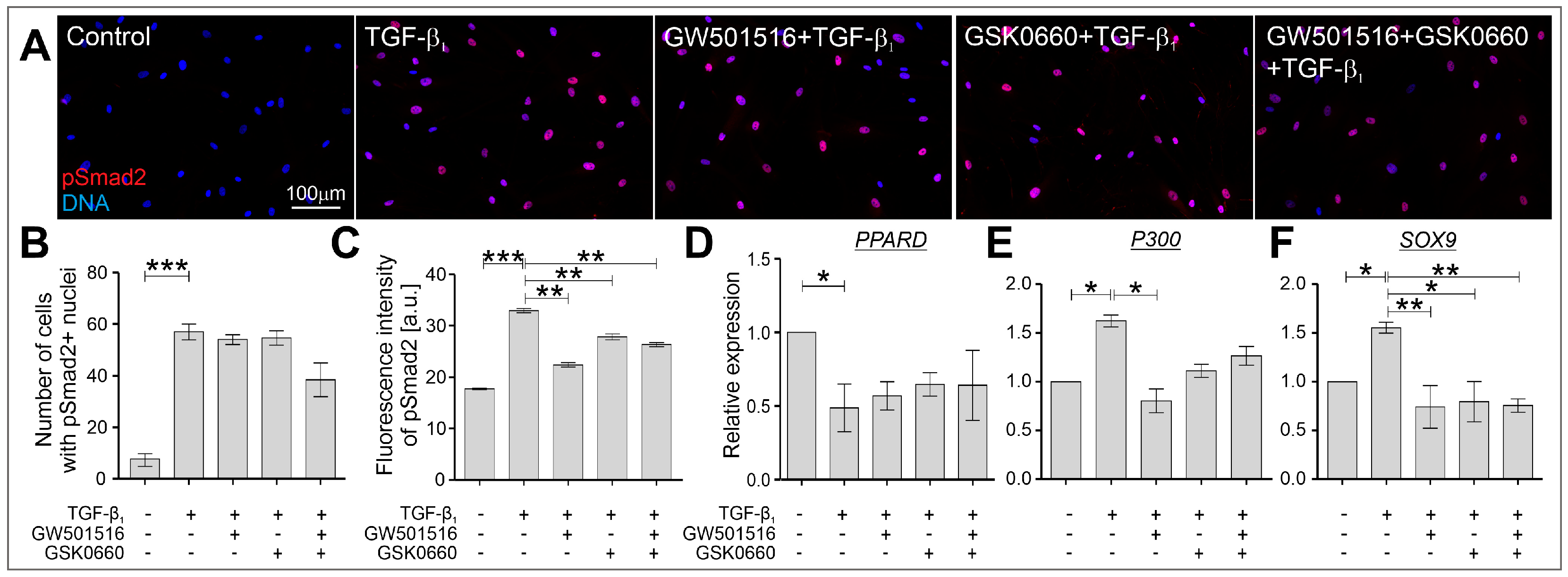
2.3. The TGF-β-Induced Upregulation of Cx43 Is Strongly Suppressed in HBFs Treated with GW501516 and GSK0660 Alone or in Combination via Smad-Dependent Signaling
3. Discussion
4. Materials and Methods
4.1. Cell Culture
4.2. Proliferation and Viability Assay
4.3. Immunofluorescence Staining
4.4. Cell-Based Enzyme-Linked Immunosorbent (In-Cell ELISA) Assay
4.5. Western Blot Analyses
4.6. Real-Time PCR Analyses
4.7. Statistics
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Dharmage, S.C.; Perret, J.L.; Custovic, A. Epidemiology of Asthma in Children and Adults. Front. Pediatr. 2019, 7, 246. [Google Scholar] [CrossRef] [PubMed]
- Serebrisky, D.; Wiznia, A. Pediatric Asthma: A Global Epidemic. Ann. Glob. Health 2019, 85, 6. [Google Scholar] [CrossRef] [PubMed]
- Stern, J.; Pier, J.; Litonjua, A.A. Asthma epidemiology and risk factors. Semin. Immunopathol. 2020, 42, 5–15. [Google Scholar] [CrossRef] [PubMed]
- Contreras, Z.A.; Chen, Z.; Roumeliotaki, T.; Annesi-Maesano, I.; Baïz, N.; von Berg, A.; Bergström, A.; Crozier, S.; Duijts, L.; Ekström, S.; et al. Does early onset asthma increase childhood obesity risk? A pooled analysis of 16 European cohorts. Eur. Respir. J. 2018, 52, 1800504. [Google Scholar] [CrossRef] [PubMed]
- Pascual, R.M.; Peters, S.P. The irreversible component of persistent asthma. J. Allergy Clin. Immunol. 2009, 124, 882–883. [Google Scholar] [CrossRef]
- Manuyakorn, W.; Smart, D.E.; Noto, A.; Bucchieri, F.; Haitchi, H.M.; Holgate, S.T.; Howarth, P.H.; Davies, D.E. Mechanical Strain Causes Adaptive Change in Bronchial Fibroblasts Enhancing Profibrotic and Inflammatory Responses. PLoS ONE 2016, 11, e0153926. [Google Scholar] [CrossRef]
- Veerati, P.C.; Mitchel, J.A.; Reid, A.T.; Knight, D.A.; Bartlett, N.W.; Park, J.-A.; Grainge, C.L. Airway mechanical compression: Its role in asthma pathogenesis and progression. Eur. Respir. Rev. 2020, 29, 190123. [Google Scholar] [CrossRef]
- O’Sullivan, M.J.; Phung, T.-K.N.; Park, J.-A. Bronchoconstriction: A potential missing link in airway remodelling. Open Biol. 2020, 10, 200254. [Google Scholar] [CrossRef]
- Holgate, S.T.; Holloway, J.; Wilson, S.; Bucchieri, F.; Puddicombe, S.; Davies, D.E. Epithelial-mesenchymal communication in the pathogenesis of chronic asthma. Proc. Am. Thorac. Soc. 2004, 1, 93–98. [Google Scholar] [CrossRef]
- Paw, M.; Wnuk, D.; Jakieła, B.; Bochenek, G.; Sładek, K.; Madeja, Z.; Michalik, M. Responsiveness of human bronchial fibroblasts and epithelial cells from asthmatic and non-asthmatic donors to the transforming growth factor-β(1) in epithelial-mesenchymal trophic unit model. BMC Mol. Cell Biol. 2021, 22, 19. [Google Scholar] [CrossRef]
- Michalik, M.; Wójcik-Pszczoła, K.; Paw, M.; Wnuk, D.; Koczurkiewicz, P.; Sanak, M.; Pękala, E.; Madeja, Z. Fibroblast-to-myofibroblast transition in bronchial asthma. Cell. Mol. Life Sci. 2018, 75, 3943–3961. [Google Scholar] [CrossRef]
- Vallée, A.; Lecarpentier, Y. TGF-β in fibrosis by acting as a conductor for contractile properties of myofibroblasts. Cell Biosci. 2019, 9, 98. [Google Scholar] [CrossRef]
- Lecarpentier, Y.; Schussler, O.; Claes, V.; Vallée, A. The Myofibroblast: TGFβ-1, A Conductor which Plays a Key Role in Fibrosis by Regulating the Balance between PPARγ and the Canonical WNT Pathway. Nucl. Recept. Res. 2017, 4, 101299. [Google Scholar] [CrossRef]
- Boser, S.R.; Mauad, T.; de Araújo-Paulino, B.B.; Mitchell, I.; Shrestha, G.; Chiu, A.; Butt, J.; Kelly, M.M.; Caldini, E.; James, A.; et al. Myofibroblasts are increased in the lung parenchyma in asthma. PLoS ONE 2017, 12, e0182378. [Google Scholar] [CrossRef]
- Chen, Y.C.; Tung, K.Y.; Tsai, C.H.; Su, M.W.; Wang, P.C.; Chen, C.H.; Lee, Y.L. Lipid profiles in children with and without asthma: Interaction of asthma and obesity on hyperlipidemia. Diabetes Metab. Syndr. Clin. Res. Rev. 2013, 7, 20–25. [Google Scholar] [CrossRef]
- Yuan, Y.; Ran, N.; Xiong, L.; Wang, G.; Guan, X.; Wang, Z.; Guo, Y.; Pang, Z.; Fang, K.; Lu, J.; et al. Obesity-Related Asthma: Immune Regulation and Potential Targeted Therapies. J. Immunol. Res. 2018, 2018, 1943497. [Google Scholar] [CrossRef]
- Bibi, H.; Shoseyov, D.; Feigenbaum, D.; Genis, M.; Friger, M.; Peled, R.; Sharff, S. The Relationship Between Asthma and Obesity in Children: Is It Real or a Case of Over Diagnosis? J. Asthma 2004, 41, 403–410. [Google Scholar] [CrossRef]
- Mohanan, S.; Tapp, H.; McWilliams, A.; Dulin, M. Obesity and asthma: Pathophysiology and implications for diagnosis and management in primary care. Exp. Biol. Med. 2014, 239, 1531–1540. [Google Scholar] [CrossRef]
- Monga, N.; Sethi, G.S.; Kondepudi, K.K.; Naura, A.S. Lipid mediators and asthma: Scope of therapeutics. Biochem. Pharmacol. 2020, 179, 113925. [Google Scholar] [CrossRef]
- Umetsu, D.T. Mechanisms by which obesity impacts upon asthma. Thorax 2017, 72, 174–177. [Google Scholar] [CrossRef]
- Al-Sawalha, N.A.; Knoll, B.J. Statins in Asthma: A Closer Look into the Pharmacological Mechanism of Action. Pharmacology 2016, 98, 279–283. [Google Scholar] [CrossRef] [PubMed]
- Michalik, M.; Soczek, E.; Kosińska, M.; Rak, M.; Wójcik, K.A.; Lasota, S.; Pierzchalska, M.; Czyz, J.; Madeja, Z. Lovastatin-induced decrease of intracellular cholesterol level attenuates fibroblast-to-myofibroblast transition in bronchial fibroblasts derived from asthmatic patients. Eur. J. Pharmacol. 2013, 704, 23–32. [Google Scholar] [CrossRef] [PubMed]
- Paw, M.; Wnuk, D.; Kadziołka, D.; Sęk, A.; Lasota, S.; Czyż, J.; Madeja, Z.; Michalik, M. Fenofibrate reduces the asthma-related fibroblast-to-myofibroblast transition by TGF-B/Smad2/3 signaling attenuation and connexin 43-dependent phenotype destabilization. Int. J. Mol. Sci. 2018, 19, 2571. [Google Scholar] [CrossRef] [PubMed]
- Kikuchi, R.; Maeda, Y.; Tsuji, T.; Yamaguchi, K.; Abe, S.; Nakamura, H.; Aoshiba, K. Fenofibrate inhibits TGF-β-induced myofibroblast differentiation and activation in human lung fibroblasts in vitro. FEBS Open Bio. 2021, 11, 2340–2349. [Google Scholar] [CrossRef] [PubMed]
- Yang, Q.; Long, Q. PPARδ, a Potential Therapeutic Target for Heart Disease. Nucl. Recept. Res. 2018, 5, 101375. [Google Scholar] [CrossRef]
- Teunissen, B.E.J.; Smeets, P.J.H.; Willemsen, P.H.M.; De Windt, L.J.; Van der Vusse, G.J.; Van Bilsen, M. Activation of PPARdelta inhibits cardiac fibroblast proliferation and the transdifferentiation into myofibroblasts. Cardiovasc. Res. 2007, 75, 519–529. [Google Scholar] [CrossRef]
- Ali, F.Y.; Egan, K.; FitzGerald, G.A.; Desvergne, B.; Wahli, W.; Bishop-Balley, D.; Warner, T.D.; Mitchell, J.A. Role of prostacyclin versus peroxisome proliferator-activated receptor β receptors in prostacyclin sensing by lung fibroblasts. Am. J. Respir. Cell Mol. Biol. 2006, 34, 242–246. [Google Scholar] [CrossRef]
- Sng, M.K.; Chan, J.S.K.; Teo, Z.; Phua, T.; Tan, E.H.P.; Wee, J.W.K.; Koh, N.J.N.; Tan, C.K.; Chen, J.P.; Pal, M.; et al. Selective deletion of PPARβ/δ in fibroblasts causes dermal fibrosis by attenuated LRG1 expression. Cell Discov. 2018, 4, 15. [Google Scholar] [CrossRef]
- Liu, C.; Lim, S.T.; Teo, M.H.Y.; Tan, M.S.Y.; Kulkarni, M.D.; Qiu, B.; Li, A.; Lal, S.; Dos Remedios, C.G.; Tan, N.S.; et al. Collaborative Regulation of LRG1 by TGF-β1 and PPAR-β/δ Modulates Chronic Pressure Overload-Induced Cardiac Fibrosis. Circ. Heart Fail. 2019, 12, e005962. [Google Scholar] [CrossRef]
- Banno, A.; Reddy, A.T.; Lakshmi, S.P.; Reddy, R.C. PPARs: Key Regulators of Airway Inflammation and Potential Therapeutic Targets in Asthma. Nucl. Recept. Res. 2018, 5, 101306. [Google Scholar] [CrossRef]
- Haskova, Z.; Hoang, B.; Luo, G.; Morgan, L.A.; Billin, A.N.; Barone, F.C.; Shearer, B.G.; Barton, M.E.; Kilgore, K.S. Modulation of LPS-induced pulmonary neutrophil infiltration and cytokine production by the selective PPARbeta/delta ligand GW0742. Inflamm. Res. 2008, 57, 314–321. [Google Scholar] [CrossRef]
- Perez Diaz, N.; Lione, L.A.; Hutter, V.; Mackenzie, L.S. Co-Incubation with PPARβ/δ Agonists and Antagonists Modeled Using Computational Chemistry: Effect on LPS Induced Inflammatory Markers in Pulmonary Artery. Int. J. Mol. Sci. 2021, 22, 3158. [Google Scholar] [CrossRef]
- Hack, K.; Reilly, L.; Palmer, C.; Read, K.D.; Norval, S.; Kime, R.; Booth, K.; Foerster, J. Skin-targeted inhibition of PPAR β/δ by selective antagonists to treat PPAR β/δ-mediated psoriasis-like skin disease in vivo. PLoS ONE 2012, 7, e37097. [Google Scholar] [CrossRef]
- Zhang, Z.; Xie, X.; Yao, Q.; Liu, J.; Tian, Y.; Yang, C.; Xiao, L.; Wang, N. PPARδ agonist prevents endothelial dysfunction via induction of dihydrofolate reductase gene and activation of tetrahydrobiopterin salvage pathway. Br. J. Pharmacol. 2019, 176, 2945–2961. [Google Scholar] [CrossRef]
- Coll, T.; Alvarez-Guardia, D.; Barroso, E.; Gómez-Foix, A.M.; Palomer, X.; Laguna, J.C.; Vázquez-Carrera, M. Activation of peroxisome proliferator-activated receptor-{delta} by GW501516 prevents fatty acid-induced nuclear factor-{kappa}B activation and insulin resistance in skeletal muscle cells. Endocrinology 2010, 151, 1560–1569. [Google Scholar] [CrossRef]
- Shearer, B.G.; Steger, D.J.; Way, J.M.; Stanley, T.B.; Lobe, D.C.; Grillot, D.A.; Iannone, M.A.; Lazar, M.A.; Willson, T.M.; Billin, A.N. Identification and characterization of a selective peroxisome proliferator-activated receptor beta/delta (NR1C2) antagonist. Mol. Endocrinol. 2008, 22, 523–529. [Google Scholar] [CrossRef]
- Hall, J.M.; McDonnell, D.P. The molecular mechanisms underlying the proinflammatory actions of thiazolidinediones in human macrophages. Mol. Endocrinol. 2007, 21, 1756–1768. [Google Scholar] [CrossRef]
- Capozzi, M.E.; Savage, S.R.; McCollum, G.W.; Hammer, S.S.; Ramos, C.J.; Yang, R.; Bretz, C.A.; Penn, J.S. The peroxisome proliferator-activated receptor-β/δ antagonist GSK0660 mitigates retinal cell inflammation and leukostasis. Exp. Eye Res. 2020, 190, 107885. [Google Scholar] [CrossRef]
- Paw, M.; Borek, I.; Wnuk, D.; Ryszawy, D.; Piwowarczyk, K.; Kmiotek, K.; Wojcik-Pszczoła, K.A.; Pierzchalska, M.; Madeja, Z.; Sanak, M.; et al. Connexin43 controls the myofibroblastic differentiation of bronchial fibroblasts from patients with asthma. Am. J. Respir. Cell Mol. Biol. 2017, 57, 100–110. [Google Scholar] [CrossRef]
- Paw, M.; Wnuk, D.; Nit, K.; Bobis-Wozowicz, S.; Szychowski, R.; Ślusarczyk, A.; Madeja, Z.; Michalik, M. SB203580-A Potent p38 MAPK Inhibitor Reduces the Profibrotic Bronchial Fibroblasts Transition Associated with Asthma. Int. J. Mol. Sci. 2021, 22, 12790. [Google Scholar] [CrossRef]
- Wnuk, D.; Paw, M.; Ryczek, K.; Bochenek, G.; Sładek, K.; Madeja, Z.; Michalik, M. Enhanced asthma-related fibroblast to myofibroblast transition is the result of profibrotic TGF-β/Smad2/3 pathway intensification and antifibrotic TGF-β/Smad1/5/(8)9 pathway impairment. Sci. Rep. 2020, 10, 16492. [Google Scholar] [CrossRef] [PubMed]
- Michalik, M.; Pierzchalska, M.; Legutko, A.; Ura, M.; Ostaszewska, A.; Soja, J.; Sanak, M. Asthmatic bronchial fibroblasts demonstrate enhanced potential to differentiate into myofibroblasts in culture. Med. Sci. Monit. 2009, 15, 194–201. [Google Scholar]
- Thannickal, V.J.; Lee, D.Y.; White, E.S.; Cui, Z.; Larios, J.M.; Chacon, R.; Horowitz, J.C.; Day, R.M.; Thomas, P.E. Myofibroblast differentiation by transforming growth factor-β1 is dependent on cell adhesion and integrin signaling via focal adhesion kinase. J. Biol. Chem. 2003, 278, 12384–12389. [Google Scholar] [CrossRef] [PubMed]
- Wnuk, D.; Lasota, S.; Paw, M.; Madeja, Z.; Michalik, M. Asthma-derived fibroblast to myofibroblast transition is enhanced in comparison to fibroblasts derived from non-asthmatic patients in 3D in vitro culture due to Smad2/3 signalling. Acta Biochim. Pol. 2020, 67, 441–448. [Google Scholar] [CrossRef] [PubMed]
- Wojcik-Pszczola, K.; Jakiela, B.; Plutecka, H.; Koczurkiewicz, P.; Madeja, Z.; Michalik, M.; Sanak, M. Connective tissue growth factor regulates transition of primary bronchial fibroblasts to myofibroblasts in asthmatic subjects. Cytokine 2018, 102, 187–190. [Google Scholar] [CrossRef] [PubMed]
- Furumatsu, T.; Tsuda, M.; Yoshida, K.; Taniguchi, N.; Ito, T.; Hashimoto, M.; Ito, T.; Asahara, H. Sox9 and p300 cooperatively regulate chromatin-mediated transcription. J. Biol. Chem. 2005, 280, 35203–35208. [Google Scholar] [CrossRef]
- Furumatsu, T.; Tsuda, M.; Taniguchi, N.; Tajima, Y.; Asahara, H. Smad3 induces chondrogenesis through the activation of SOX9 via CREB-binding protein/p300 recruitment. J. Biol. Chem. 2005, 280, 8343–8350. [Google Scholar] [CrossRef]
- Ito, J.T.; Lourenço, J.D.; Righetti, R.F.; Tibério, I.F.L.C.; Prado, C.M.; Lopes, F.D.T.Q.S. Extracellular Matrix Component Remodeling in Respiratory Diseases: What Has Been Found in Clinical and Experimental Studies? Cells 2019, 8, 342. [Google Scholar] [CrossRef]
- Juel, C.T.-B.; Ali, Z.; Nilas, L.; Ulrik, C.S. Asthma and obesity: Does weight loss improve asthma control? a systematic review. J. Asthma Allergy 2012, 5, 21–26. [Google Scholar] [CrossRef]
- Li, L.; Emmett, N.; Mann, D.; Zhao, X. Fenofibrate attenuates tubulointerstitial fibrosis and inflammation through suppression of nuclear factor-κB and transforming growth factor-β1/Smad3 in diabetic nephropathy. Exp. Biol. Med. 2010, 235, 383–391. [Google Scholar] [CrossRef]
- Chen, Q.; Jiang, N.; Zhang, Y.; Ye, S.; Liang, X.; Wang, X.; Lin, X.; Zong, R.; Chen, H.; Liu, Z. Fenofibrate Inhibits Subretinal Fibrosis Through Suppressing TGF-β—Smad2/3 signaling and Wnt signaling in Neovascular Age-Related Macular Degeneration. Front. Pharmacol. 2020, 11, 580884. [Google Scholar] [CrossRef]
- Hashim, Y.; Abdel Baky, N.A.; Kamal, M.M.; Gad, A. Aliskiren and Fenofibrate Constrict Liver Fibrosis by means of Focusing on TGF-β1/Smad Signaling Pathway and Actuating HGF Expression. Azhar Int. J. Pharm. Med. Sci. 2021, 1, 73–86. [Google Scholar] [CrossRef]
- Trifilieff, A.; Bench, A.; Hanley, M.; Bayley, D.; Campbell, E.; Whittaker, P. PPAR-α and -γ but not -δ agonists inhibit airway inflammation in a murine model of asthma: In vitro evidence for an NF-κB-independent effect. Br. J. Pharmacol. 2003, 139, 163–171. [Google Scholar] [CrossRef]
- Gu, Y.; Li, X.; He, T.; Jiang, Z.; Hao, P.; Tang, X. The Antifibrosis Effects of Peroxisome Proliferator-Activated Receptor δ on Rat Corneal Wound Healing after Excimer Laser Keratectomy. PPAR Res. 2014, 2014, 464935. [Google Scholar] [CrossRef]
- Ham, S.A.; Hwang, J.S.; Yoo, T.; Lee, W.J.; Paek, K.S.; Oh, J.-W.; Park, C.-K.; Kim, J.-H.; Do, J.T.; Kim, J.-H.; et al. Ligand-activated PPARδ upregulates α-smooth muscle actin expression in human dermal fibroblasts: A potential role for PPARδ in wound healing. J. Dermatol. Sci. 2015, 80, 186–195. [Google Scholar] [CrossRef]
- Kostadinova, R.; Montagner, A.; Gouranton, E.; Fleury, S.; Guillou, H.; Dombrowicz, D.; Desreumaux, P.; Wahli, W. GW501516-activated PPARβ/δ promotes liver fibrosis via p38-JNK MAPK-induced hepatic stellate cell proliferation. Cell Biosci. 2012, 2, 34. [Google Scholar] [CrossRef]
- Iwaisako, K.; Haimerl, M.; Paik, Y.-H.; Taura, K.; Kodama, Y.; Sirlin, C.; Yu, E.; Yu, R.T.; Downes, M.; Evans, R.M.; et al. Protection from liver fibrosis by a peroxisome proliferator-activated receptor δ agonist. Proc. Natl. Acad. Sci. USA 2012, 109, E1369–E1376. [Google Scholar] [CrossRef]
- Fedorova, L.V.; Sodhi, K.; Gatto-Weis, C.; Puri, N.; Hinds, T.D.J.; Shapiro, J.I.; Malhotra, D. Peroxisome proliferator-activated receptor δ agonist, HPP593, prevents renal necrosis under chronic ischemia. PLoS ONE 2013, 8, e64436. [Google Scholar] [CrossRef]
- Sarna, M.; Wojcik, K.A.; Hermanowicz, P.; Wnuk, D.; Burda, K.; Sanak, M.; Czyz, J.; Michalik, M. Undifferentiated bronchial fibroblasts derived from asthmatic patients display higher elastic modulus than their non-asthmatic counterparts. PLoS ONE 2015, 10, e0116840. [Google Scholar] [CrossRef]
- Stephen, J.; Delvecchio, C.; Spitale, N.; Giesler, A.; Radford, K.; Bilan, P.; Cox, P.G.; Capone, J.P.; Nair, P. PPAR ligands decrease human airway smooth muscle cell migration and extracellular matrix synthesis. Eur. Respir. J. 2013, 41, 425–432. [Google Scholar] [CrossRef]
- Liu, G.; Li, X.; Li, Y.; Tang, X.; Xu, J.; Li, R.; Hao, P.; Sun, Y. PPARδ agonist GW501516 inhibits PDGF-stimulated pulmonary arterial smooth muscle cell function related to pathological vascular remodeling. Biomed. Res. Int. 2013, 2013, 903947. [Google Scholar] [CrossRef] [PubMed]
- Yan, Z.; Ni, Y.; Wang, P.; Chen, J.; He, H.; Sun, J.; Cao, T.; Chen, J.; Zhao, Z.; Luo, Z.; et al. Peroxisome proliferator-activated receptor delta protects against obesity-related glomerulopathy through the P38 MAPK pathway. Obesity 2013, 21, 538–545. [Google Scholar] [CrossRef] [PubMed]
- Poleni, P.E.; Bianchi, A.; Etienne, S.; Koufany, M.; Sebillaud, S.; Netter, P.; Terlain, B.; Jouzeau, J.Y. Agonists of peroxisome proliferators-activated receptors (PPAR) alpha, beta/delta or gamma reduce transforming growth factor (TGF)-beta-induced proteoglycans’ production in chondrocytes. Osteoarthr. Cartil. 2007, 15, 493–505. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Tu, K.; Liu, D.; Guo, L.; Chen, Y.; Li, Q.; Maiers, J.L.; Liu, Z.; Shah, V.H.; Dou, C.; et al. p300 Acetyltransferase Is a Cytoplasm-to-Nucleus Shuttle for SMAD2/3 and TAZ Nuclear Transport in Transforming Growth Factor β-Stimulated Hepatic Stellate Cells. Hepatology 2019, 70, 1409–1423. [Google Scholar] [CrossRef]
- Zhang, M.; Zhang, Z.; Pan, H.-Y.; Wang, D.-X.; Deng, Z.-T.; Ye, X.-L. TGF-beta1 induces human bronchial epithelial cell-to-mesenchymal transition in vitro. Lung 2009, 187, 187–194. [Google Scholar] [CrossRef]
- Janknecht, R.; Wells, N.J.; Hunter, T. TGF-beta-stimulated cooperation of smad proteins with the coactivators CBP/p300. Genes Dev. 1998, 12, 2114–2119. [Google Scholar] [CrossRef]
- Shen, X.; Hu, P.P.; Liberati, N.T.; Datto, M.B.; Frederick, J.P.; Wang, X.F. TGF-beta-induced phosphorylation of Smad3 regulates its interaction with coactivator p300/CREB-binding protein. Mol. Biol. Cell 1998, 9, 3309–3319. [Google Scholar] [CrossRef]
- Coricor, G.; Serra, R. TGF-β regulates phosphorylation and stabilization of Sox9 protein in chondrocytes through p38 and Smad dependent mechanisms. Sci. Rep. 2016, 6, 38616. [Google Scholar] [CrossRef]
- Bhattacharyya, S.; Ghosh, A.K.; Pannu, J.; Mori, Y.; Takagawa, S.; Chen, G.; Trojanowska, M.; Gilliam, A.C.; Varga, J. Fibroblast expression of the coactivator p300 governs the intensity of profibrotic response to transforming growth factor beta. Arthritis Rheum. 2005, 52, 1248–1258. [Google Scholar] [CrossRef]
- Scharf, G.M.; Kilian, K.; Cordero, J.; Wang, Y.; Grund, A.; Hofmann, M.; Froese, N.; Wang, X.; Kispert, A.; Kist, R.; et al. Inactivation of Sox9 in fibroblasts reduces cardiac fibrosis and inflammation. JCI Insight 2019, 4, e126721. [Google Scholar] [CrossRef]
- Raza, S.; Jokl, E.; Pritchett, J.; Martin, K.; Su, K.; Simpson, K.; Birchall, L.; Mullan, A.F.; Athwal, V.S.; Doherty, D.T.; et al. SOX9 is required for kidney fibrosis and activates NAV3 to drive renal myofibroblast function. Sci. Signal. 2021, 14, eabb4282. [Google Scholar] [CrossRef]
- Li, H.; Cai, H.; Deng, J.; Tu, X.; Sun, Y.; Huang, Z.; Ding, Z.; Dong, L.; Chen, J.; Zang, Y.; et al. TGF-β-mediated upregulation of Sox9 in fibroblast promotes renal fibrosis. Biochim. Biophys. Acta-Mol. Basis Dis. 2018, 1864, 520–532. [Google Scholar] [CrossRef]
- Athwal, V.S.; Pritchett, J.; Martin, K.; Llewellyn, J.; Scott, J.; Harvey, E.; Zaitoun, A.M.; Mullan, A.F.; Zeef, L.A.H.; Friedman, S.L.; et al. SOX9 regulated matrix proteins are increased in patients serum and correlate with severity of liver fibrosis. Sci. Rep. 2018, 8, 17905. [Google Scholar] [CrossRef]
- Gajjala, P.R.; Kasam, R.K.; Soundararajan, D.; Sinner, D.; Huang, S.K.; Jegga, A.G.; Madala, S.K. Dysregulated overexpression of Sox9 induces fibroblast activation in pulmonary fibrosis. JCI Insight 2021, 6, e152503. [Google Scholar] [CrossRef]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Gene | Sequence F′ | Sequence R′ |
| ACTA2 | CTGTTCCAGCCATCCTTCAT | CCGTGATCTCCTTCTGCATT |
| COL1A1 | CTTTGCATTCATCTCTCAAACTTAGTTTT | CCCCGCATGGGTCTTCA |
| COL1A2 | TGCTGCTGGTCAACCTGGTGC | ACTTCCAGCAGGACCGGGGG |
| GAPDH | GAAGGTGAAGGTCGGAGT | GAAGATGGTGATGGGATTTC |
| TLN | CCCTGATGTGCGGCTTCG | TGTCCTGTCAACTGCTGCTTC |
| TNC | GGTCCACACCTGGGCATTT | TTGCTGAATCAAACAACAAAACAGA |
| GJA1 | AGGAGTTCAATCACTTGGCG | GAGTTTGCCTAAGGCGCTC |
| PPARD | GGGCATGTCACACAACGCTAT | GCATTGTAGATGTGCTTGGAGAA |
| P300 | ACTTCTAATGGCCCTCTACCTGA | GTGCTGAAGAGGAGGGGTTT |
| SOX9 | CAAGAAGGACCACCCGGATT | AAGATGGCGTTGGGGGAGAT |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Paw, M.; Wnuk, D.; Madeja, Z.; Michalik, M. PPARδ Agonist GW501516 Suppresses the TGF-β-Induced Profibrotic Response of Human Bronchial Fibroblasts from Asthmatic Patients. Int. J. Mol. Sci. 2023, 24, 7721. https://doi.org/10.3390/ijms24097721
Paw M, Wnuk D, Madeja Z, Michalik M. PPARδ Agonist GW501516 Suppresses the TGF-β-Induced Profibrotic Response of Human Bronchial Fibroblasts from Asthmatic Patients. International Journal of Molecular Sciences. 2023; 24(9):7721. https://doi.org/10.3390/ijms24097721
Chicago/Turabian StylePaw, Milena, Dawid Wnuk, Zbigniew Madeja, and Marta Michalik. 2023. "PPARδ Agonist GW501516 Suppresses the TGF-β-Induced Profibrotic Response of Human Bronchial Fibroblasts from Asthmatic Patients" International Journal of Molecular Sciences 24, no. 9: 7721. https://doi.org/10.3390/ijms24097721
APA StylePaw, M., Wnuk, D., Madeja, Z., & Michalik, M. (2023). PPARδ Agonist GW501516 Suppresses the TGF-β-Induced Profibrotic Response of Human Bronchial Fibroblasts from Asthmatic Patients. International Journal of Molecular Sciences, 24(9), 7721. https://doi.org/10.3390/ijms24097721