
Chemical and Biological Evaluation of Novel 1H-Chromeno[3,2-c]pyridine Derivatives as MAO Inhibitors Endowed with Potential Anticancer Activity

 ,
,  , , ,
, , ,  , ,
, ,  , , and
, , and 
Abstract

1. Introduction
2. Results and Discussion
2.1. Chemistry
2.2. Biological Evaluation
2.2.1. Inhibition of Monoamine Oxidases and Cholinesterases
2.2.2. Antiproliferative Activity on Tumor Cell Lines
2.2.3. Structure–Activity Relationships
3. Materials and Methods
3.1. Chemistry
3.1.1. General Methods
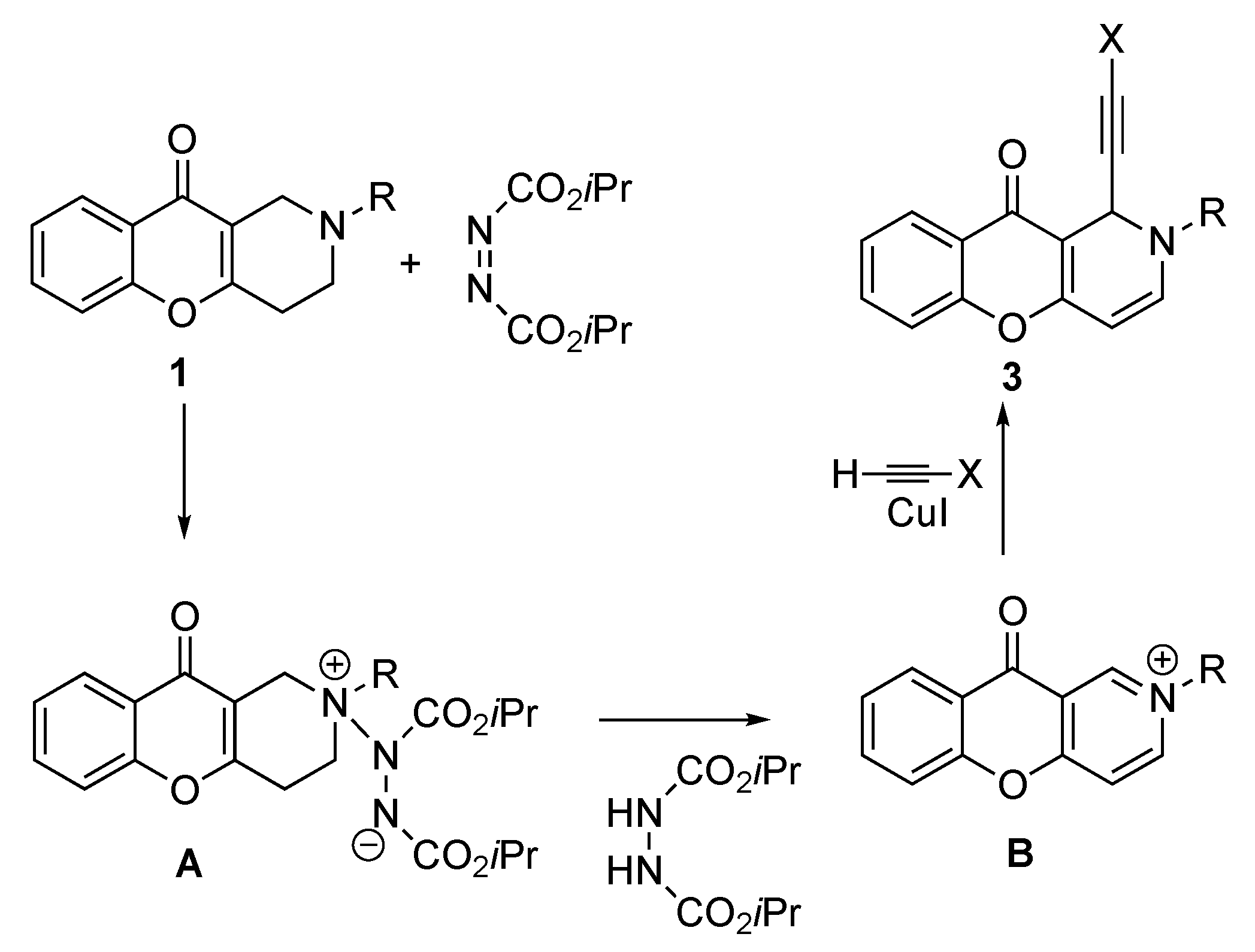
3.1.2. Synthesis of Tetrahydro- and Dihydrochromeno[3,2-c]pyridines 1 and 2
3.1.3. Synthesis of 2-Alkyl-1-(ethinyl)-1H-chromeno[3,2-c]pyridine-10(2H)-ones (3a–e)
3.1.4. Synthesis of 8-Bromo-2-methyl-10-(1-methyl-1H-pyrrole-2-yl)-2,3,4,10-tetrahydro-1H-chromeno[3,2-c]pyridine (4)
3.1.5. Synthesis of 8-Bromo-2-methyl-10-(nitromethyl)-2,3,4,10-tetrahydro-1H-chromeno[3,2-c]pyridine (5)
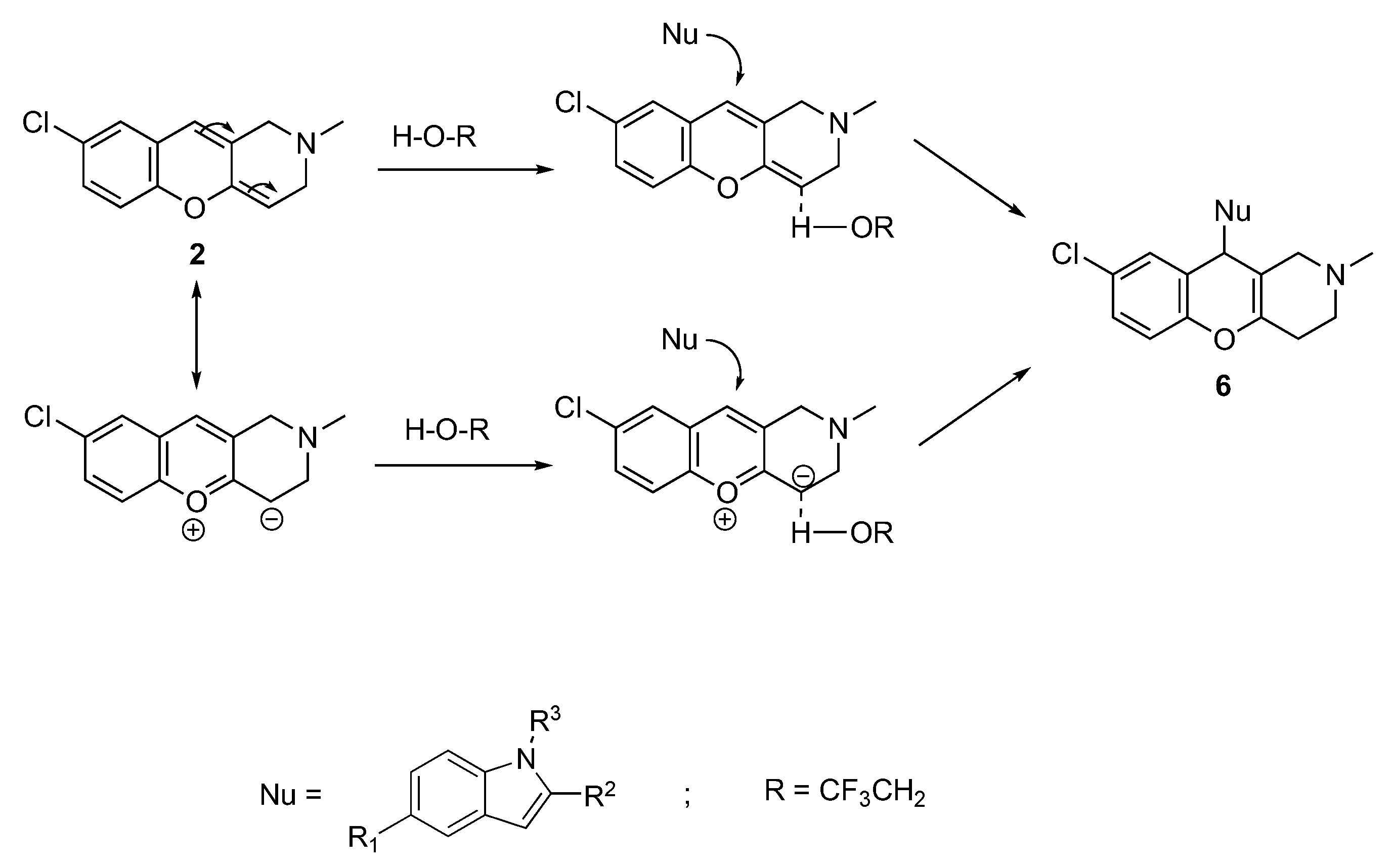
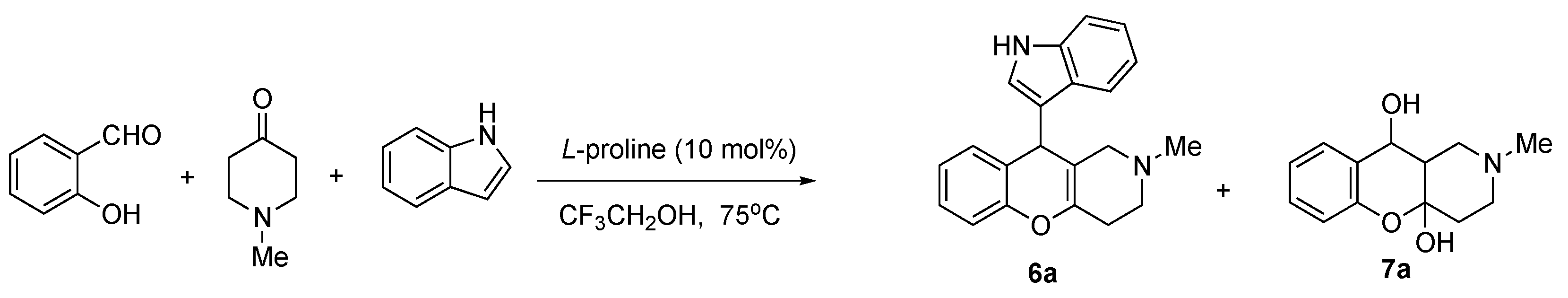
3.1.6. Synthesis of 8-Chloro-10-(1H-indol-3-yl)-2-methyl-2,3,4,10-tetrahydro-1H-chromeno[3,2-c]pyridines 6a–c
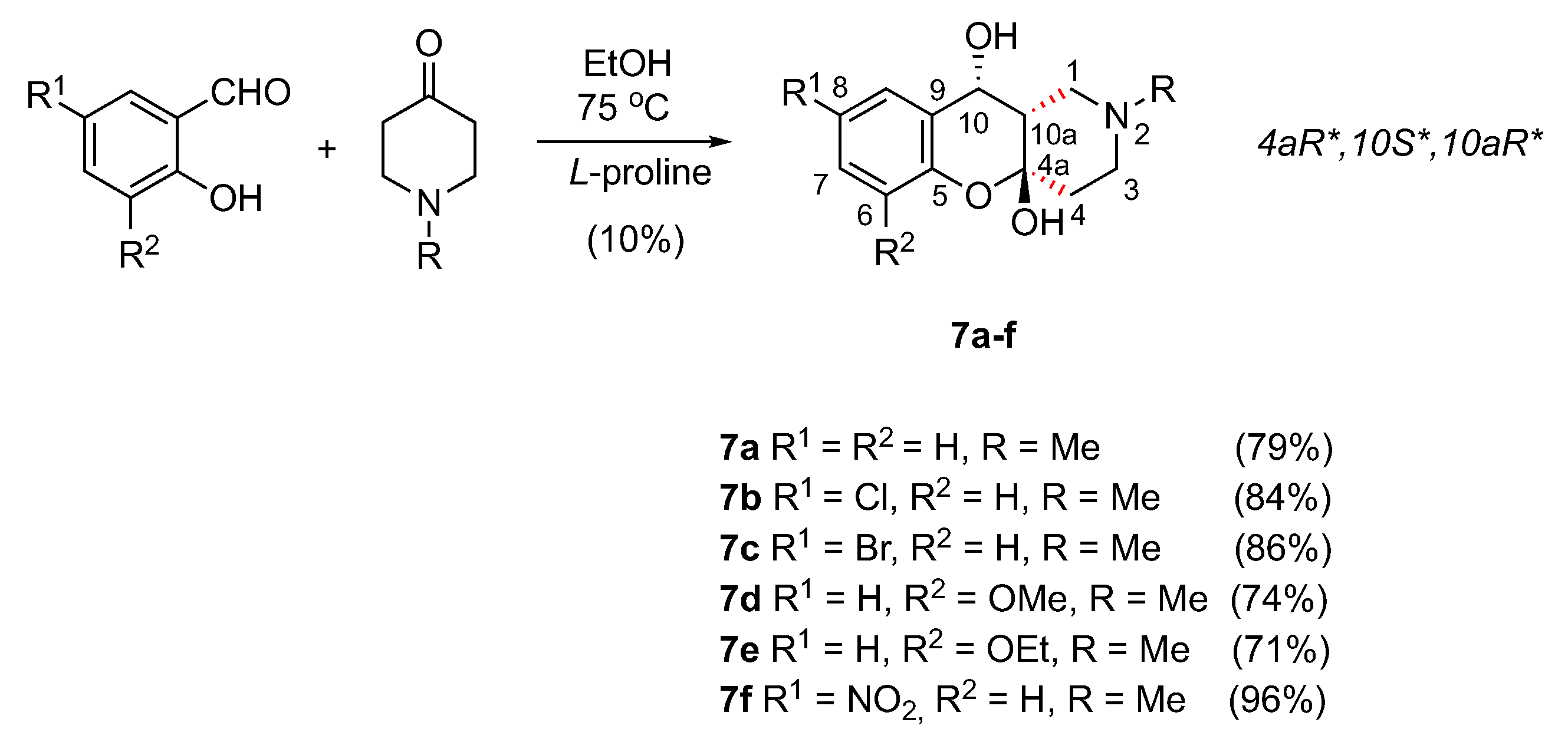
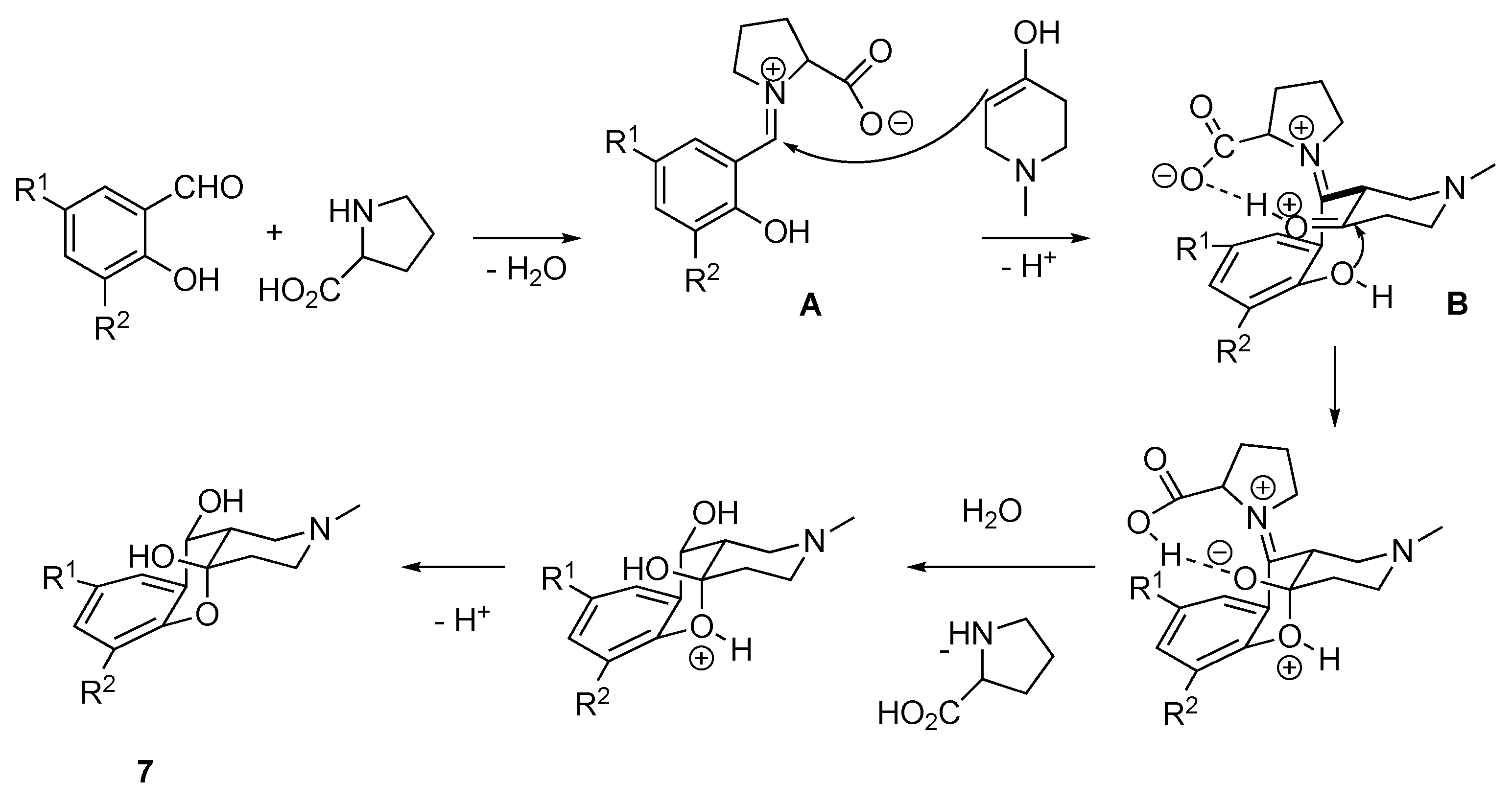
3.1.7. Synthesis of (4aR*,10S*,10aR*)-2-Alkyl-1,2,3,4,10,10a-hexahydro-4aH-chromeno[3,2-c]pyridine-4a,10-diol 7a–f
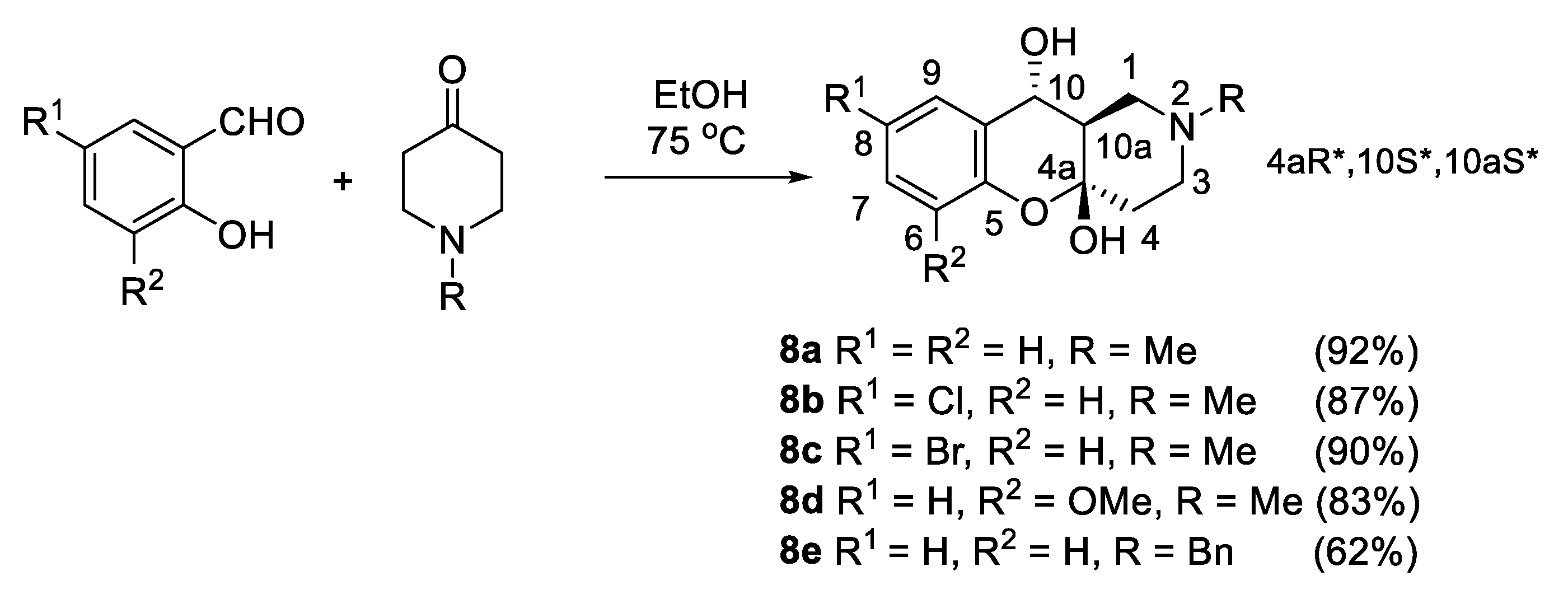
3.1.8. Synthesis of (4aR*,10S*,10aS*)-2-Alkyl-1,2,3,4,10,10a-hexahydro-4aH-chromeno[3,2-c]pyridine-4a,10-diols 8a–e
3.2. Biological Assays
3.2.1. Inhibition of Monoamine Oxidases and Cholinesterases
3.2.2. Cell Viability Assays
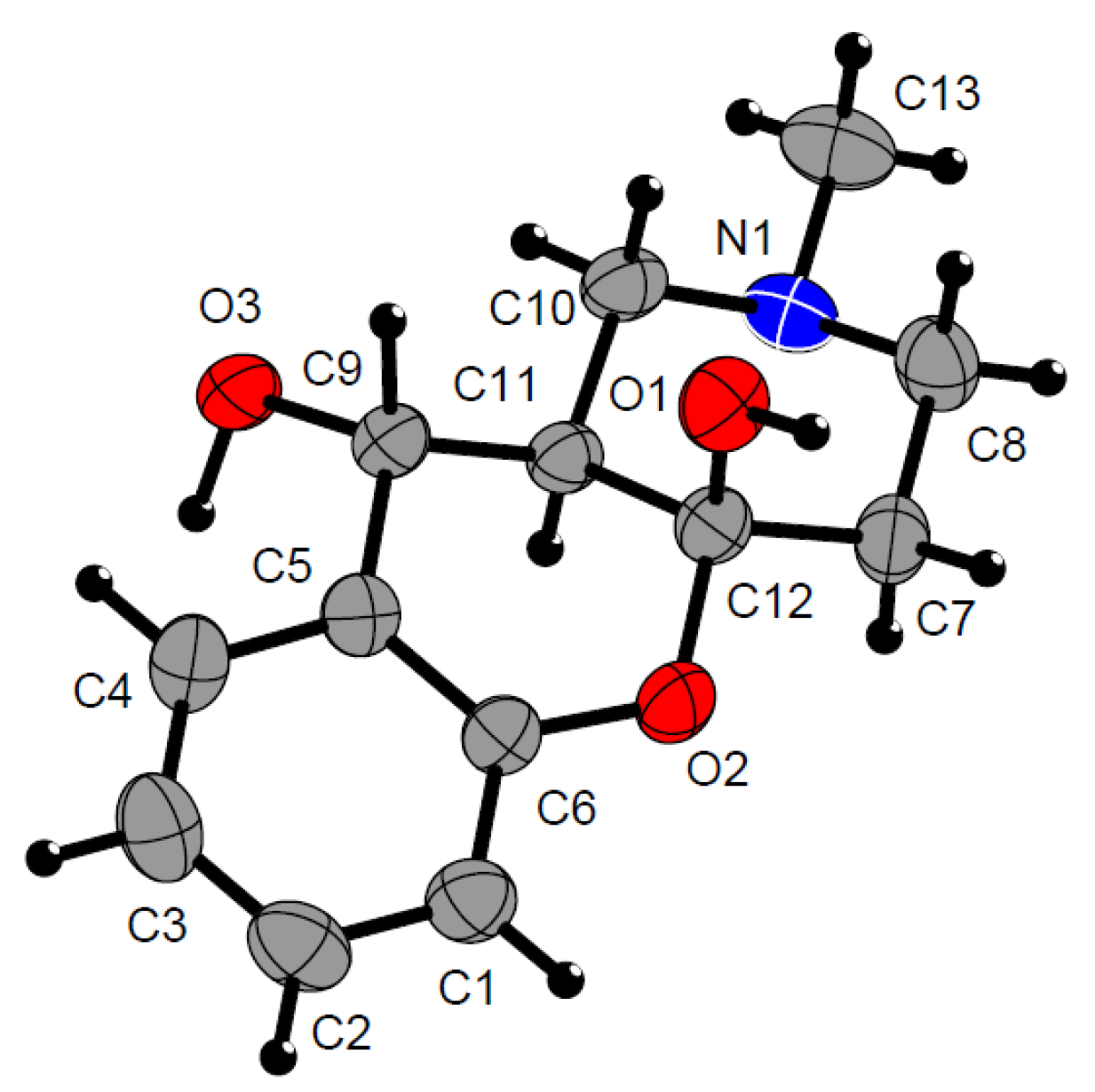
3.3. Accession Codes
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Benny, A.T.; Arikkatt, S.D.; Vazhappilly, C.G.; Kannadasan, S.; Thomas, R.; Leelabaiamma, M.S.N.; Radhakrishnan, E.K.; Shanmugam, P. Chromone, a privileged scaffold in drug discovery: Developments in the synthesis and bioactivity. Mini-Rev. Med. Chem. 2022, 22, 1030–1063. [Google Scholar] [CrossRef] [PubMed]
- Reis, J.; Cagide, F.; Valencia, M.E.; Teixeira, J.; Bagetta, D.; Pérez, C.; Uriarte, E.; Oliveira, P.J.; Ortuso, F.; Alcaro, S.; et al. Multi-target-directed ligands for Alzheimer’s disease: Discovery of chromone-based monoamine oxidase/cholinesterase inhibitors. Eur. J. Med. Chem. 2018, 158, 781–800. [Google Scholar] [CrossRef] [PubMed]
- Madhav, H.; Jameel, E.; Rehan, M.; Hoda, N. Recent advancements in chromone as a privileged scaffold towards the development of small molecules for neurodegenerative therapeutics. RSC Med. Chem. 2022, 13, 258–279. [Google Scholar] [CrossRef] [PubMed]
- Patil, V.M.; Masand, N.; Verma, S.; Masand, V. Chromones: Privileged scaffold in anticancer drug discovery. Chem. Biol. Drug Des. 2021, 98, 943–953. [Google Scholar] [CrossRef] [PubMed]
- Purgatorio, R.; Kulikova, L.N.; Pisani, L.; Catto, M.; Candia, M.; Carrieri, A.; Cellamare, S.; De Palma, A.; Beloglazkin, A.A.; Reza Raesi, G.; et al. Scouting around 1,2,3,4-tetrahydrochromeno[3,2-c]pyridin-10-ones for single- and multitarget ligands directed towards relevant Alzheimer’s targets. ChemMedChem 2020, 15, 1947–1955. [Google Scholar] [CrossRef]
- Kulikova, L.N.; Borisov, R.S.; Voskressensky, L.G. Ring opening in 1,2,3,4-tetrahydrochromeno[3,2-c]pyridines under the action of electron-deficient alkynes. Mendeleev Commun. 2017, 27, 640–641. [Google Scholar] [CrossRef]
- Makhaeva, G.F.; Boltneva, N.P.; Lushchekina, S.V.; Rudakova, E.V.; Serebryakova, O.G.; Kulikova, L.N.; Beloglazkin, A.A.; Borisov, R.S.; Richardson, R.J. Synthesis, molecular docking, and biological activity of 2-vinyl chromones: Toward selective butyrylcholinesterase inhibitors for potential Alzheimer’s disease therapeutics. Bioorg. Med. Chem. 2018, 26, 4716–4725. [Google Scholar] [CrossRef]
- Singh, K.; Singh, P.; Kaur, A.; Singh, P. C-1 alkynylation of N-methyltetrahydroisoquinolines through CDC: A direct access to phenethylisoquinoline alkaloids. Synlett 2012, 23, 760–764. [Google Scholar] [CrossRef]
- Yue, X.; Festa, A.A.; Storozhenko, O.A.; Varlamov, A.V.; Subramani, K.; Boccarelli, A.; Purgatorio, R.; Altomare, C.D.; Voskressensky, L.G. Reductive domino reaction to access chromeno[2,3-c]isoquinoline-5-amines with antiproliferative activities against human tumor cells. Bioorg. Chem. 2020, 104, 104169. [Google Scholar] [CrossRef]
- Kasralikar, H.; Jadhavar, S.; Bhusare, S. Synthesis and molecular docking study of novel chromeno-chromenones as anti-HIV-1 NNRT inhibitors. Synlett 2015, 26, 1969–1972. [Google Scholar] [CrossRef]
- Nourisefat, M.; Panahi, F.; Nabipour, M.; Heidari, S.; Khalafi-Nezhad, A. L-Cysteine-functionalized magnetic nanoparticles (LCMNP): As a magnetic reusable organocatalyst for one-pot synthesis of 9-(1H-indol-3-yl)xanthen-4-(9H)-ones. J. Iran Chem. Soc. 2016, 13, 1853–1865. [Google Scholar] [CrossRef]
- Sangsuwan, R.; Sangher, S.; Aree, T.; Mahidol, C.; Ruchirawat, S.; Kittakoop, P. An organocatalyst from renewable materials for the synthesis of coumarins and chromenes: Three-component reaction and multigram scale synthesis. RSC Adv. 2014, 4, 13708–13718. [Google Scholar] [CrossRef]
- Kulikova, L.N.; Raesi, G.R.; Levickaya, D.D.; Purgatorio, R.; La Spada, G.; Catto, M.; Altomare, C.D.; Voskressensky, L.G. Synthesis of novel benzo[b][1,6]naphthyridine derivatives and investigation of their potential as scaffolds of MAO inhibitors. Molecules 2023, 28, 1662. [Google Scholar] [CrossRef] [PubMed]
- Wang, K.; Luo, J.; Yeh, S.; You, B.; Meng, J.; Chang, P.; Niu, Y.; Li, G.; Lu, C.; Zhu, Y.; et al. The MAO inhibitors phenelzine and clorgyline revert enzalutamide resistance in castration resistant prostate cancer. Nat. Commun. 2020, 11, 2689. [Google Scholar] [CrossRef] [PubMed]
- Spearman, C. The Proof and measurement of association between two things. Am. J. Psychol. 1987, 100, 441–471. [Google Scholar] [CrossRef]
- Gidaro, M.C.; Astorino, C.; Petzer, A.; Carradori, S.; Alcaro, F.; Costa, G.; Artese, A.; Rafele, G.; Russo, F.M.; Petzer, J.P.; et al. Kaempferol as selective human MAO—A inhibitor: Analytical Detection in calabrian red wines, biological and molecular modeling studies. J. Agric. Food Chem. 2016, 64, 6–1394. [Google Scholar] [CrossRef]
- Celano, M.; Schenone, S.; Cosco, D.; Navarra, M.; Puxeddu, E.; Racanicchi, L.; Brullo, C.; Varano, E.; Alcaro, S.; Ferretti, E.; et al. Cytotoxic effects of a novel pyrazolopyrimidine derivative entrapped in liposomes in anaplastic thyroid cancer cells in vitro and in xenograft tumors in vivo. Endocr. Relat. Cancer 2008, 15, 499–510. [Google Scholar] [CrossRef] [PubMed]
- Titov, A.A.; Purgatorio, R.; Obydennik, A.Y.; Listratova, A.V.; Borisova, T.N.; de Candia, M.; Catto, M.; Altomare, C.D.; Varlamov, A.V.; Voskressensky, L.G. Synthesis of isomeric 3-Benzazecines decorated with endocyclic allene moiety and exocyclic conjugated double bond and evaluation of their anticholinesterase activity. Molecules 2022, 27, 6276. [Google Scholar] [CrossRef]
- Purgatorio, R.; de Candia, M.; Catto, M.; Rullo, M.; Pisani, L.; Denora, N.; Carrieri, A.; Nevskaya, A.A.; Voskressensky, L.G.; Altomare, C.D. Evaluation of water-soluble mannich base prodrugs of 2,3,4,5-tetrahydroazepino[4,3-b]indol-1(6H)-one as multitarget-directed agents for Alzheimer’s disease. ChemMedChem 2021, 16, 589–5984. [Google Scholar] [CrossRef]
- Skehan, P.; Storeng, R.; Scudiero, D.; Monks, A.; McMahon, J.; Vistica, D.; Warren, J.T.; Bokesch, H.; Kenney, S.; Boyd, M.R. New colorimetric cytotoxicity assay for anticancer-drug screening. J. Natl. Cancer. Inst. 1990, 82, 1107–1112. [Google Scholar] [CrossRef]
- Puzzo, D.; Fiorito, J.; Purgatorio, R.; Gulisano, W.; Palmeri, A.; Arancio, O.; Nicholls, R. Molecular mechanisms of learning and memory. In Genes, Environment and Alzheimer’s Disease; Lazarov, O., Tesco, G., Eds.; Academic Press: Cambridge, MA, USA, 2016; pp. 1–27. ISBN 9780128028513. [Google Scholar] [CrossRef]
- Teich, A.F.; Nicholls, R.E.; Puzzo, D.; Fiorito, J.; Purgatorio, R.; Fa’, M.; Arancio, O. Synaptic therapy in Alzheimer’s disease: A CREB-centric approach. Neurotherapeutics 2015, 12, 29–41. [Google Scholar] [CrossRef] [PubMed]
- Pisani, L.; Catto, M.; Muncipinto, G.; Nicolotti, O.; Carrieri, A.; Rullo, M.; Stefanachi, A.; Leonetti, F.; Altomare, C.D. A twenty-year journey exploring coumarin-based derivatives as bioactive molecules. Front. Chem. 2022, 10, 1002547. [Google Scholar] [CrossRef]
- Pisani, L.; de Candia, M.; Rullo, M.; Altomare, C.D. Hansch-type QSAR models for the rational design of MAO inhibitors: Basic principles and methodology. In Monoamine Oxidase: Methods in Molecular Biology; Binda, C., Ed.; Humana: New York, NY, USA, 2023; Volume 2558, pp. 207–220. [Google Scholar] [CrossRef]
- Catto, M.; Arnesano, F.; Palazzo, G.; De Stradis, A.; Calò, V.; Losacco, M.; Purgatorio, R.; Campagna, F. Investigation on the influence of (Z)-3-(2-(3-chlorophenyl)hydrazono)-5,6-dihydroxyindolin-2-one (PT2) on β-amyloid(1–40) aggregation and toxicity. Arch. Biochem. Biophys. 2014, 560, 73–82. [Google Scholar] [CrossRef]
- Purgatorio, R.; Gambacorta, N.; Catto, M.; de Candia, M.; Pisani, L.; Espargaró, A.; Sabaté, R.; Cellamare, S.; Nicolotti, O.; Altomare, C.D. Pharmacophore modeling and 3D-QSAR study of indole and Isatin derivatives as antiamyloidogenic agents targeting Alzheimer’s disease. Molecules 2020, 25, 5773. [Google Scholar] [CrossRef] [PubMed]
- Binde, C.D.; Tvete, I.F.; Gasemyr, J.; Natvig, B.; Klemp, M. A multiple treatment comparison meta-analysis of monoamine oxidase type B inhibitors for Parkinson’s disease. Br. J. Clin. Pharmacol. 2018, 84, 1917–1927. [Google Scholar] [CrossRef]
- Youdim, M.B.H. Monoamine oxidase inhibitors, and iron chelators in depressive illness and neurodegenerative diseases. J. Neural. Transm. 2018, 125, 1719–1733. [Google Scholar] [CrossRef]
- Aljanabi, R.; Alsous, L.; Sabbah, D.A.; Gul, H.I.; Gul, M.; Bardaweel, S.K. Monoamine oxidase (MAO) as a potential target for anticancer drug design and development. Molecules 2021, 26, 6019. [Google Scholar] [CrossRef] [PubMed]
















{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Conditions (Solvent, Temperature) | Yield of 7 | Yield of 6 |
|---|---|---|
| EtOH, 75 °C, l-proline (10 mol%) | 69% | - |
| H2O, 100 °C, l-proline (10 mol%) | 67% | - |
| H2O, oleic acid (15 mol%) | 58% | trace amounts |
| CF3COOH–EtOH (1:10), 20 °C | 10% | trace amounts |
| CF3CH2OH, 75 °C, l-proline (10 mol%) | 49% | 12% |
| Py-EtOH (1:3), 100 °C | 9% | - |
| Compounds | Tm, °C 7 | Tm, °C 8 |
|---|---|---|
 | 192–194 | 187–188 |
 | 172–173 | 167–168 |
 | 160–161 | 173–174 |
 | 134–135 | 148–149 |
| No. | R | R1 | R2 | X | hMAO-A | hMAO-B | hAChE | hBChE |
|---|---|---|---|---|---|---|---|---|
| 1a | Me | H | - | - | (15 ± 4%) | (29 ± 3%) | (34 ± 5%) | n.i. |
| 2a | Me | 8-Cl | - | - | 1.18 ± 0.07 | (45 ± 5%) | (25 ± 4%) | n.i. |
| 2b | Me | 8-Br | - | - | 0.703 ± 0.012 | 7.88 ± 0.02 | (31± 5%) | n.i. |
| 3a | Me | H | - | Ph | (35 ± 1%) | 0.510 ± 0.021 | 6.79 ± 0.42 | 8.42 ± 0.25 |
| 3b | Me | 6-OMe | - | Ph | (32 ± 3%) | 0.626 ± 0.059 | (39 ± 1%) | (19 ± 2%) |
| 3c | Et | H | - | CF3 | (20± 2%) | (26 ± 9%) | n.i. | (15 ± 2%) |
| 3d | iPr | 6-OEt | - | Ph | (36 ± 5%) | (22 ± 3%) | n.i. | (10 ± 3%) |
| 6a | Me | 8-Cl | H | - | (21 ± 4%) | 7.30 ± 0.65 | (53 ± 3%) | (38 ± 3%) |
| 6b | Me | 8-Cl | OMe | - | (21 ± 5%) | 4.72 ± 0.10 | (53 ± 1%) | (15 ± 2%) |
| 6c | Me | 8-Cl | Br | - | (29 ± 5%) | 3.51 ± 0.20 | (37 ± 4%) | (23 ± 4%) |
| Pargyline | 10.9 ± 0.6 | 2.69 ± 0.48 | ||||||
| Galantamine | 0.721 ± 0.152 | 8.78 ± 0.36 | ||||||
| No. | R | R1 | R2 | hMAO-A | hMAO-B | hAChE | hBChE |
|---|---|---|---|---|---|---|---|
| 7a | Me | H | H | (14 ± 5%) | (22 ± 5%) | (24 ± 4%) | n.i. |
| 7c | Me | Br | H | (21 ± 13%) | (35 ± 6%) | n.i. | (12 ± 2%) |
| 7e | Me | H | OEt | (19 ± 2%) | (25 ± 9%) | n.i. | n.i. |
| 8a | Me | H | H | (8 ± 3%) | (42 ± 4%) | (22 ± 4%) | n.i. |
| 8c | Me | Br | H | (14 ± 2%) | (15 ± 4%) | (45 ± 2%) | n.i. |
| 8e | Bn | H | H | (12 ± 4%) | (41 ± 3%) | (25 ± 4%) | (19 ± 4%) |
| No. | MCF-7 | HCT116 | SK-OV-3 |
|---|---|---|---|
| 2a | 48.1 ± 4.21 | 54.2 ± 18.1 | >50 |
| 2b | 42.9 ± 4.82 | >50 | >50 |
| 3b | 39.7 ± 11.3 | 36.1 ± 2.10 | >50 |
| 3c | 27.8 ± 9.60 | 47.2 ± 5.42 | >50 |
| 6a | 4.80 ± 0.81 | 8.62 ± 2.21 | 14.7 ± 2.81 |
| 6b | 6.62 ± 2.70 | 18.6 ± 2.22 | 22.3 ± 1.42 |
| 6c | 4.83 ± 0.79 | 9.40 ± 0.22 | 11.3 ± 0.32 |
| Cisplatin | 4.80 ± 2.20 | 5.02 ± 2.12 | 4.43 ± 0.32 |
| Doxorubicin | 0.18 ± 0.02 | 0.38 ± 0.03 | 2.20 ± 0.02 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kulikova, L.N.; Purgatorio, R.; Beloglazkin, A.A.; Tafeenko, V.A.; Reza, R.G.; Levickaya, D.D.; Sblano, S.; Boccarelli, A.; de Candia, M.; Catto, M.; et al. Chemical and Biological Evaluation of Novel 1H-Chromeno[3,2-c]pyridine Derivatives as MAO Inhibitors Endowed with Potential Anticancer Activity. Int. J. Mol. Sci. 2023, 24, 7724. https://doi.org/10.3390/ijms24097724
Kulikova LN, Purgatorio R, Beloglazkin AA, Tafeenko VA, Reza RG, Levickaya DD, Sblano S, Boccarelli A, de Candia M, Catto M, et al. Chemical and Biological Evaluation of Novel 1H-Chromeno[3,2-c]pyridine Derivatives as MAO Inhibitors Endowed with Potential Anticancer Activity. International Journal of Molecular Sciences. 2023; 24(9):7724. https://doi.org/10.3390/ijms24097724
Chicago/Turabian StyleKulikova, Larisa N., Rosa Purgatorio, Andrey A. Beloglazkin, Viktor A. Tafeenko, Raesi Gh. Reza, Daria D. Levickaya, Sabina Sblano, Angelina Boccarelli, Modesto de Candia, Marco Catto, and et al. 2023. "Chemical and Biological Evaluation of Novel 1H-Chromeno[3,2-c]pyridine Derivatives as MAO Inhibitors Endowed with Potential Anticancer Activity" International Journal of Molecular Sciences 24, no. 9: 7724. https://doi.org/10.3390/ijms24097724
APA StyleKulikova, L. N., Purgatorio, R., Beloglazkin, A. A., Tafeenko, V. A., Reza, R. G., Levickaya, D. D., Sblano, S., Boccarelli, A., de Candia, M., Catto, M., Voskressensky, L. G., & Altomare, C. D. (2023). Chemical and Biological Evaluation of Novel 1H-Chromeno[3,2-c]pyridine Derivatives as MAO Inhibitors Endowed with Potential Anticancer Activity. International Journal of Molecular Sciences, 24(9), 7724. https://doi.org/10.3390/ijms24097724