
Placental Remote Control of Fetal Metabolism: Trophoblast mTOR Signaling Regulates Liver IGFBP-1 Phosphorylation and IGF-1 Bioavailability


 and
and 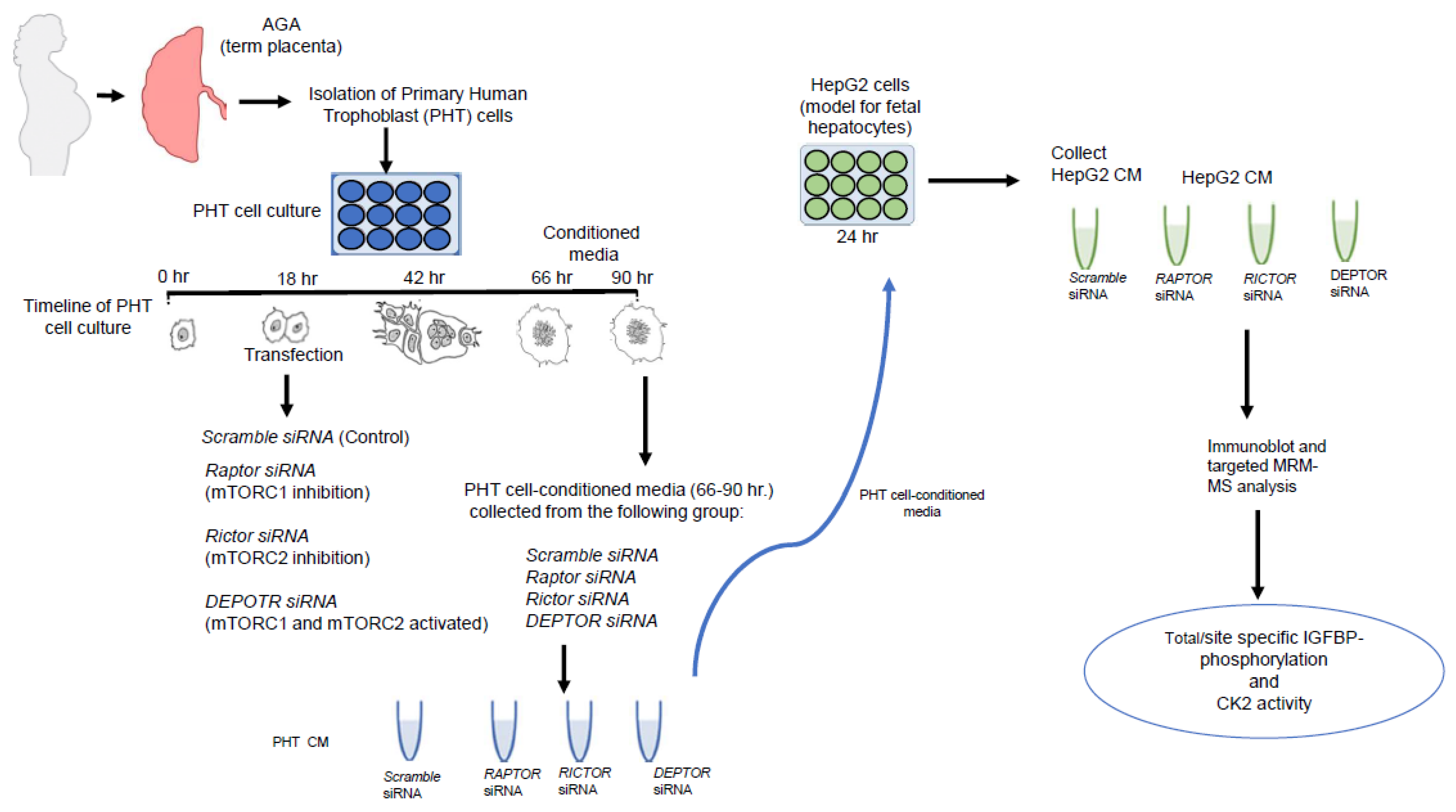{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Results
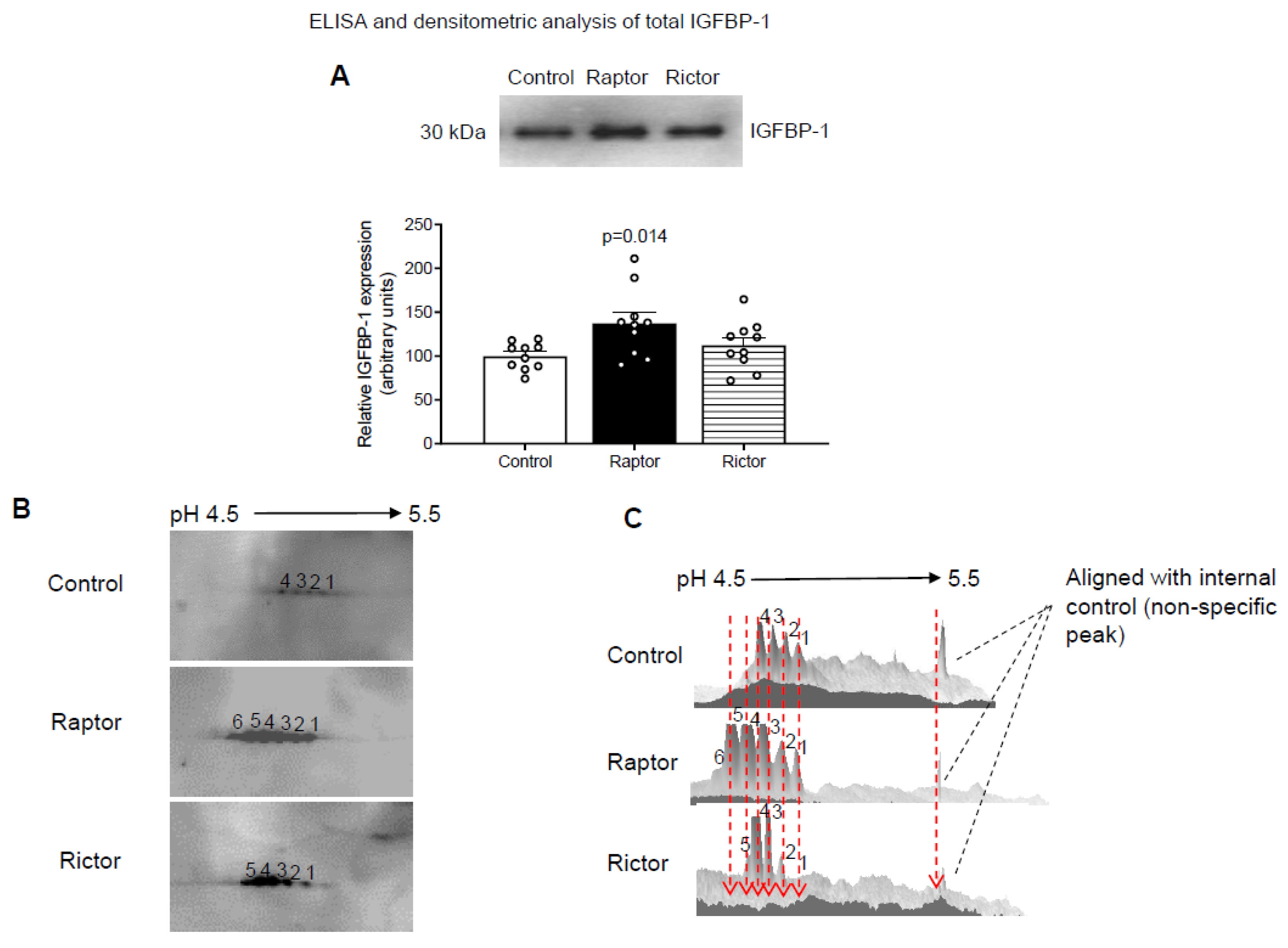
2.1. CM from PHT Cells with mTORC1 Inhibition Increased IGFBP-1 Secretion in HepG2 Cells
2.2. Increased IGFBP-1 Phosphorylation in HepG2 Cells Incubated in CM from PHT Cells with Either mTORC1 or mTORC2 as Indicated by 2-D Immunoblot Analysis
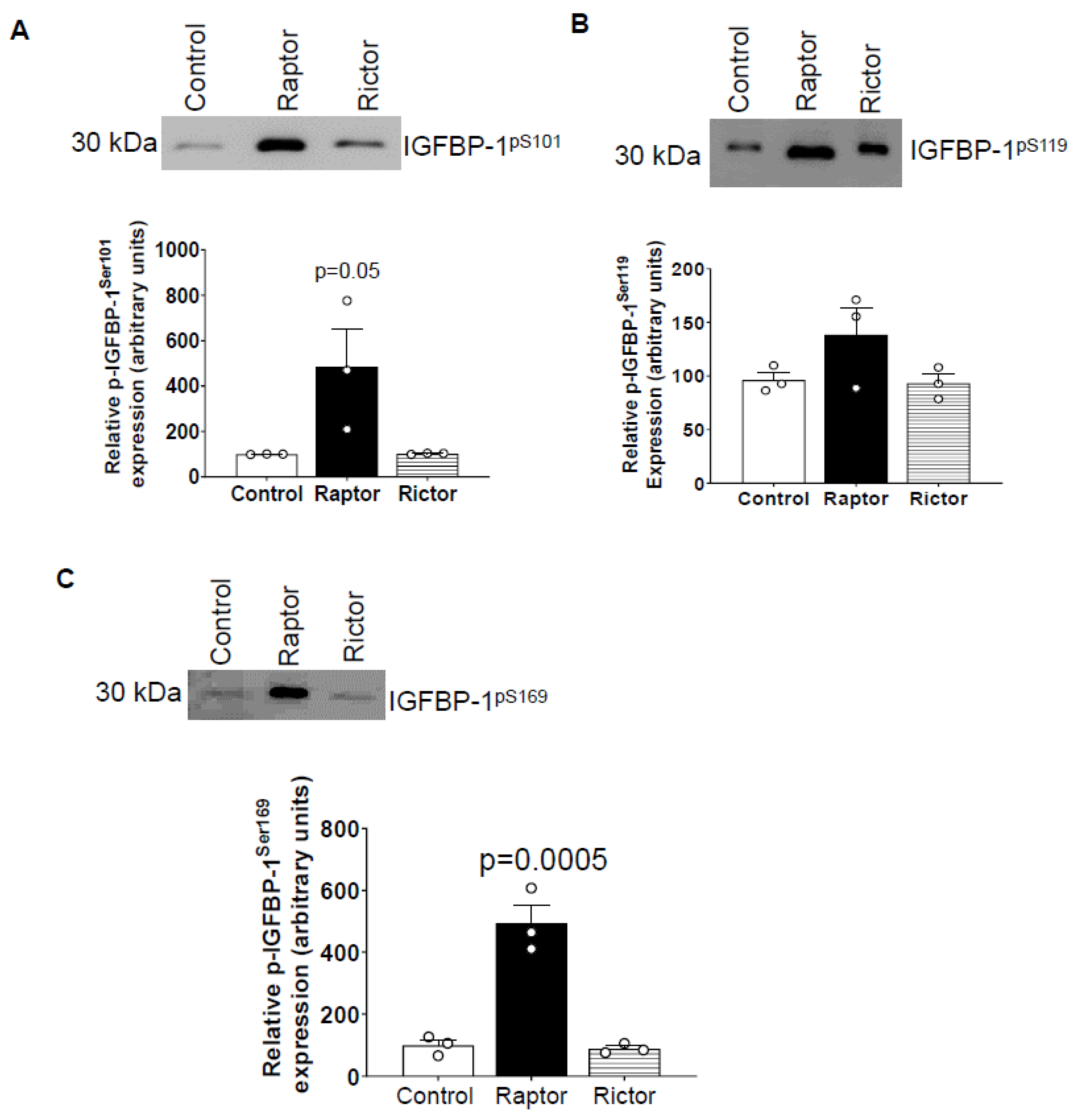
2.3. Incubation of HepG2 Cells in CM from PHT Cells with mTORC1 Increased Site-Specific IGFBP-1 Phosphorylation
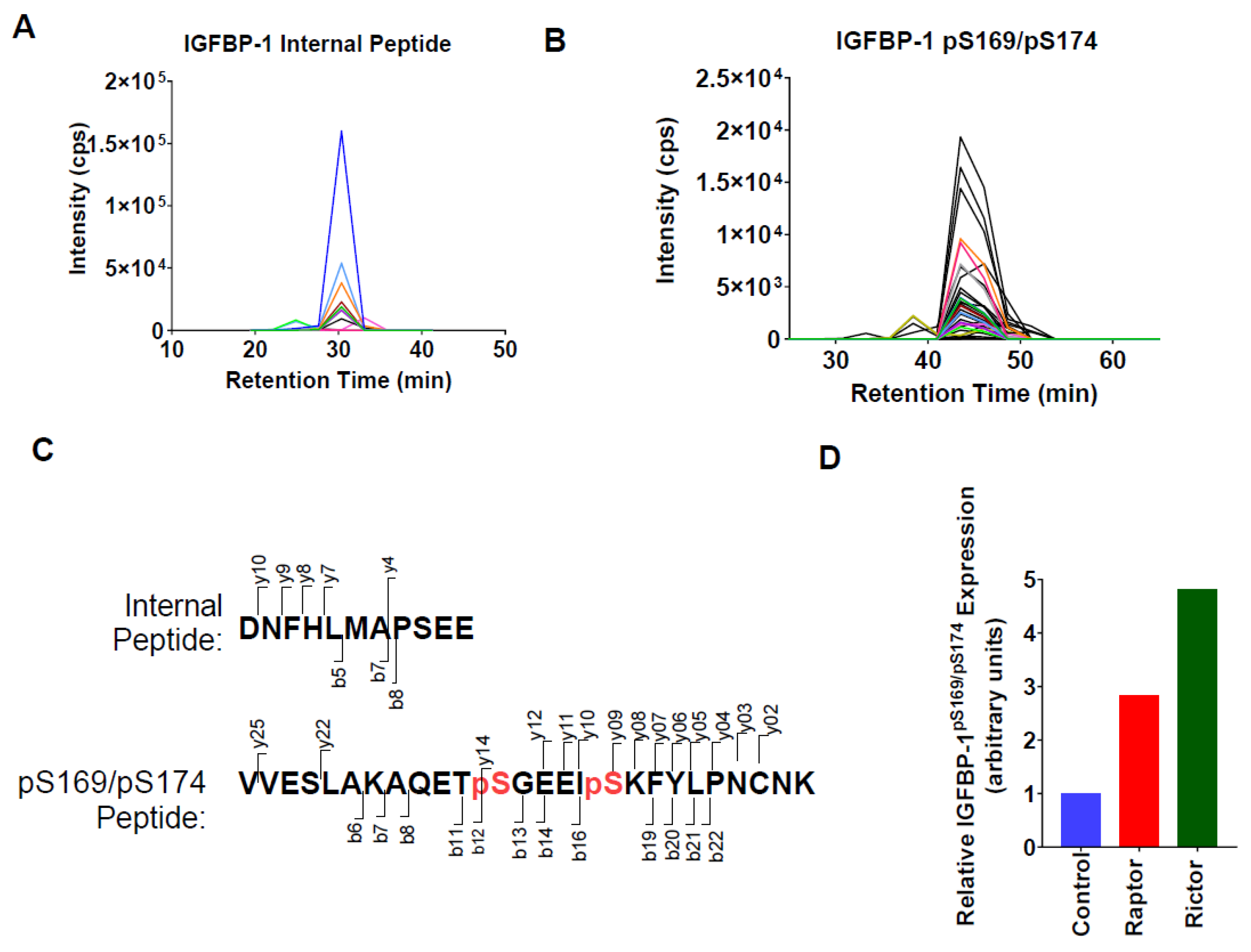
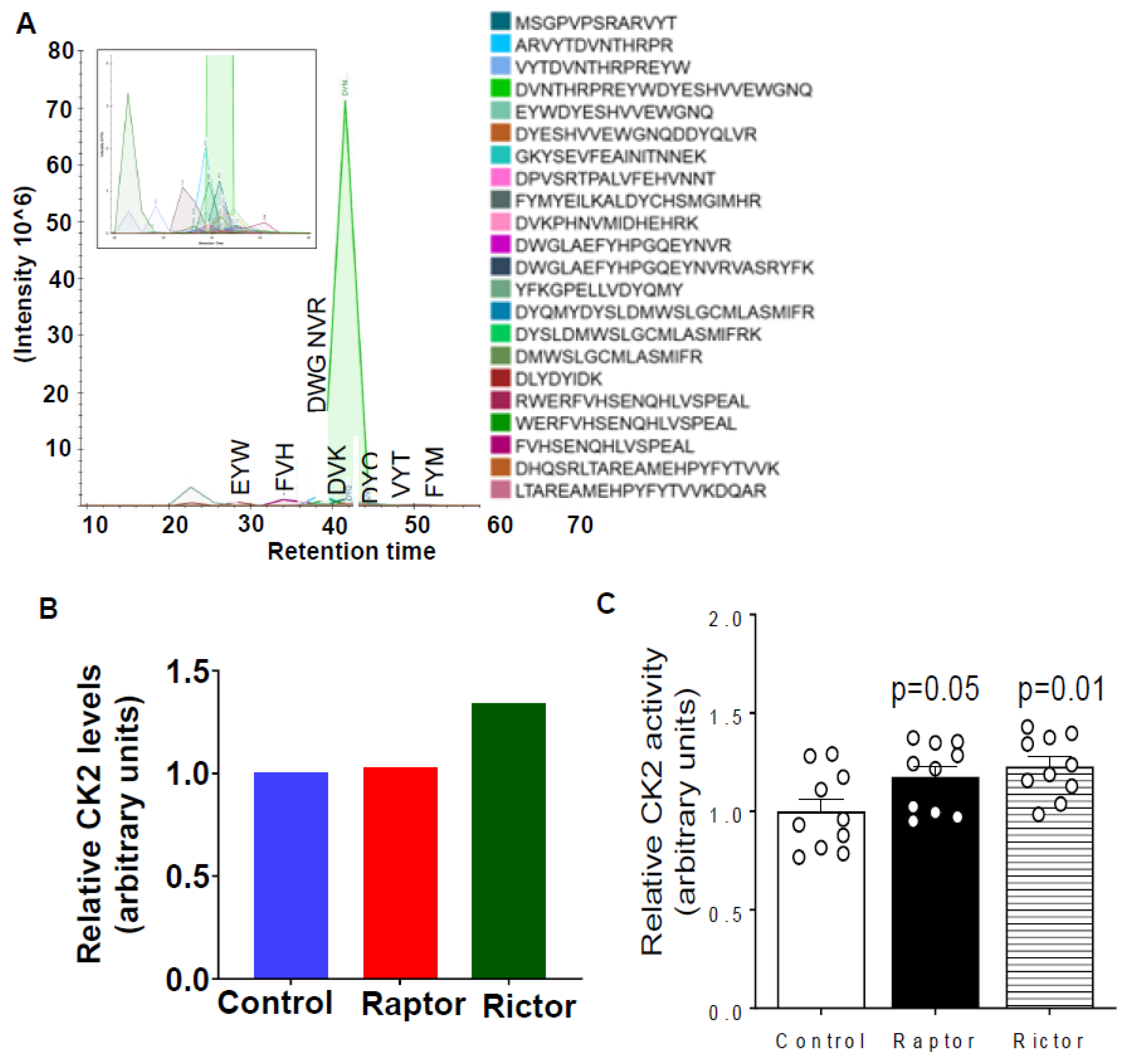
2.4. Parallel Reaction Monitoring Mass Spectrometry (PRM-MS) Analysis Identified Increased IGFBP-1 Phosphorylation at pSer169/pSer174 Following Incubation in CM from PHT Cells with mTOR Inhibition
2.5. HepG2 Cells Incubated in CM from PHT Cells with mTORC1 or mTORC2 Inhibition Activated CK2 Activity
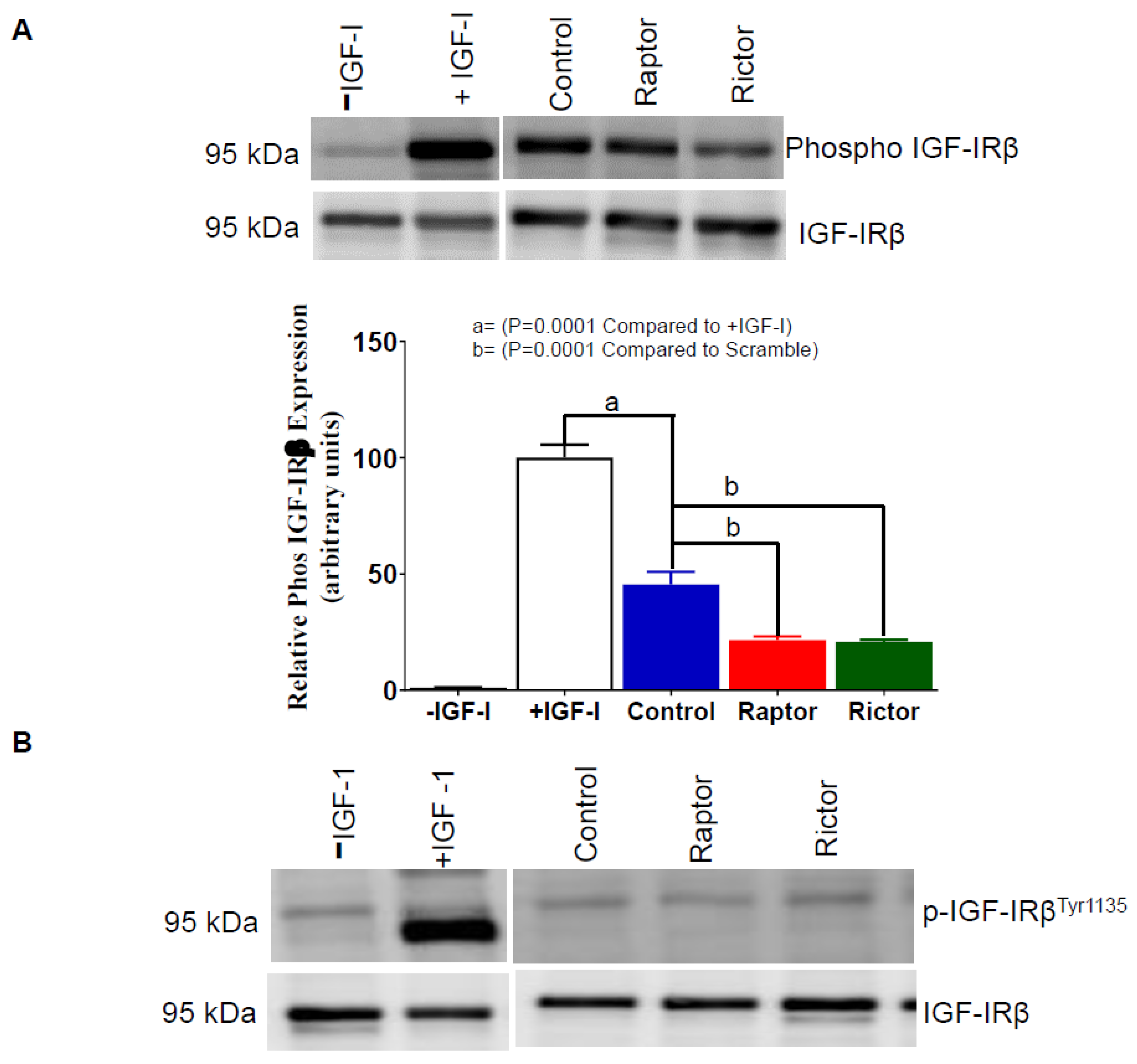
2.6. IGF-1R Autophosphorylation Decreased in HepG2 Cells Incubated in CM from PHT Cells with mTORC1 or mTORC2 Inhibition
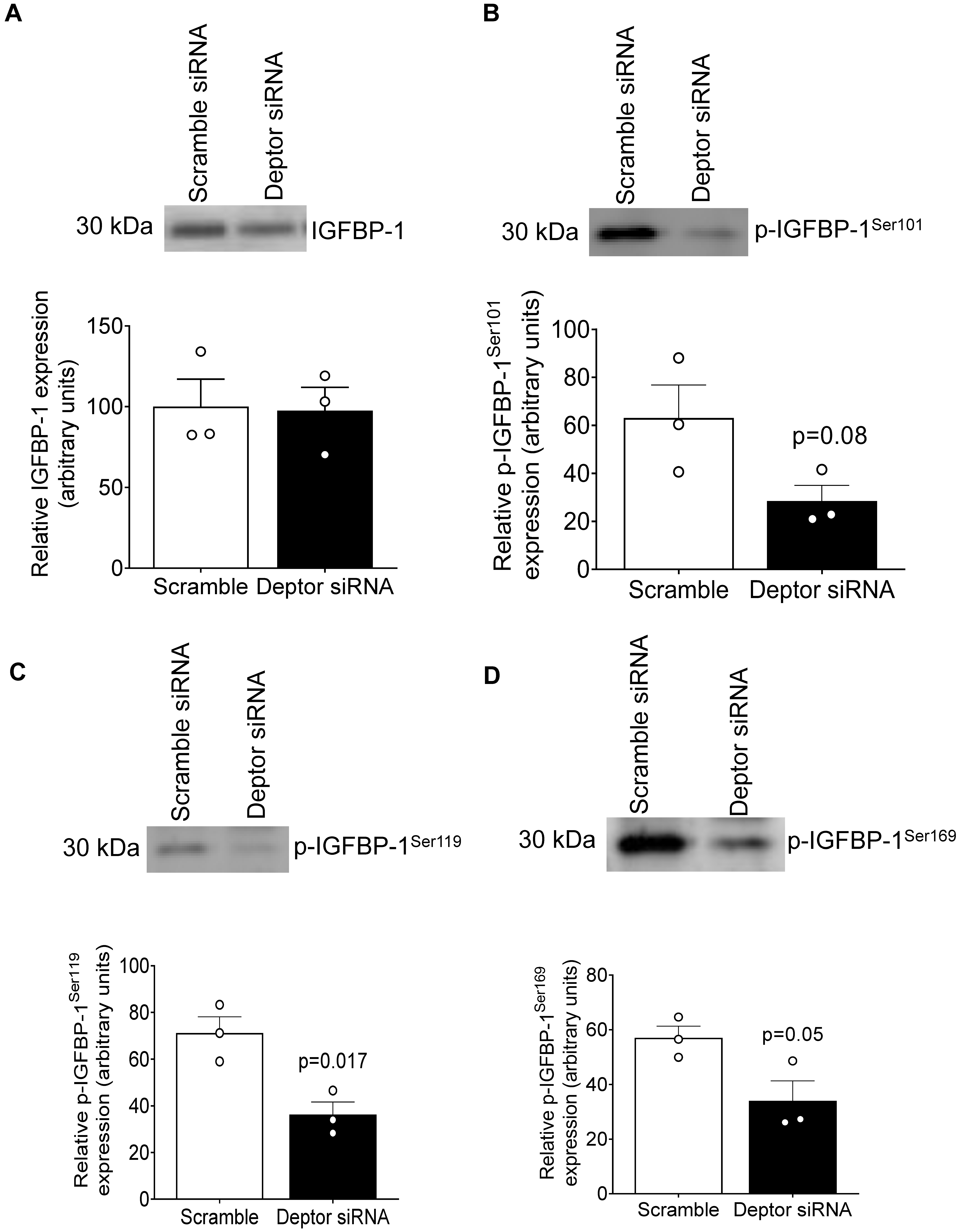
2.7. DEPTOR Silencing Markedly Inhibited IGFBP-1 Phosphorylation
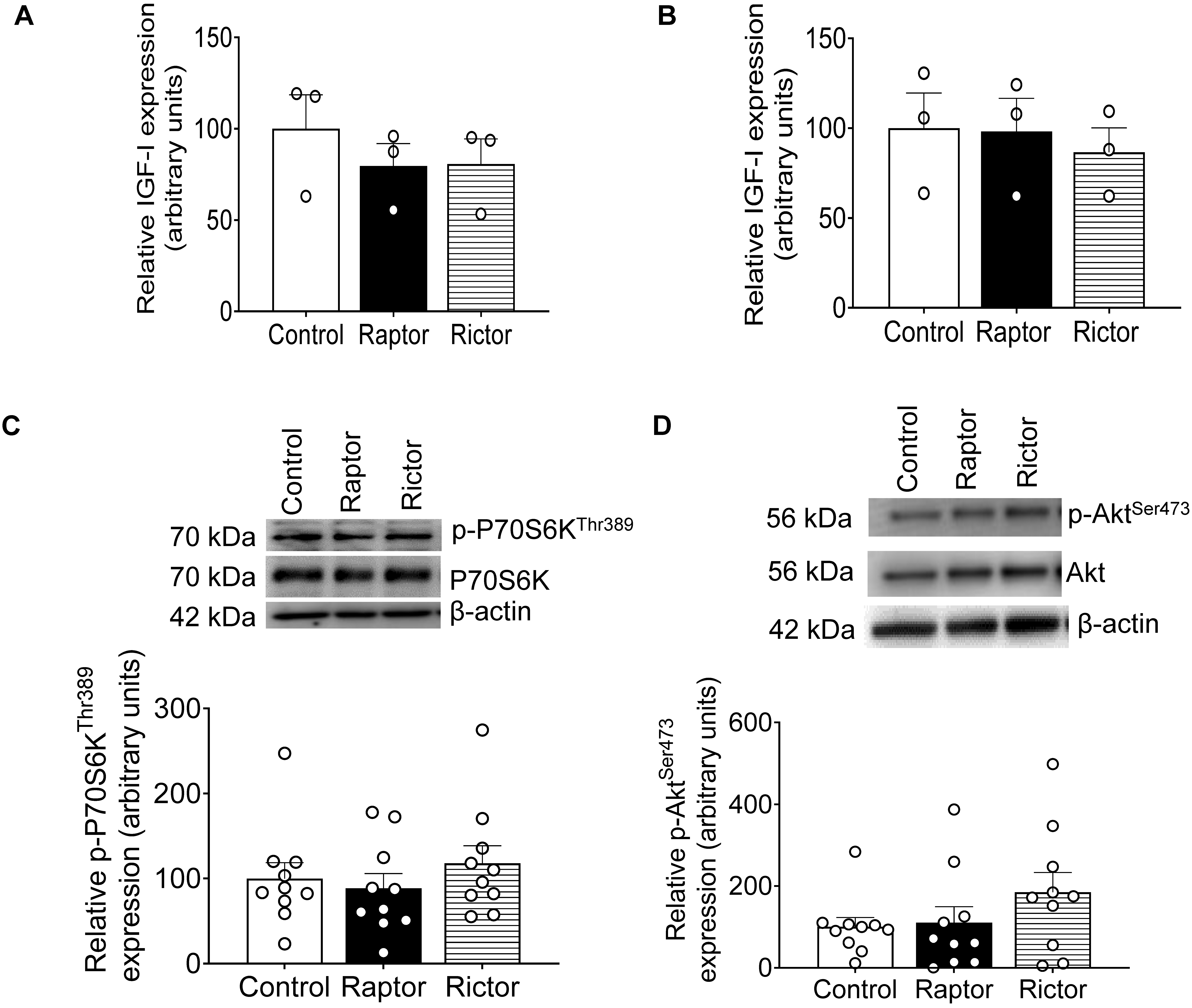
2.8. No Change in IGF-1 Secretion from PHT Cells with mTORC1 or mTORC2 Inhibition
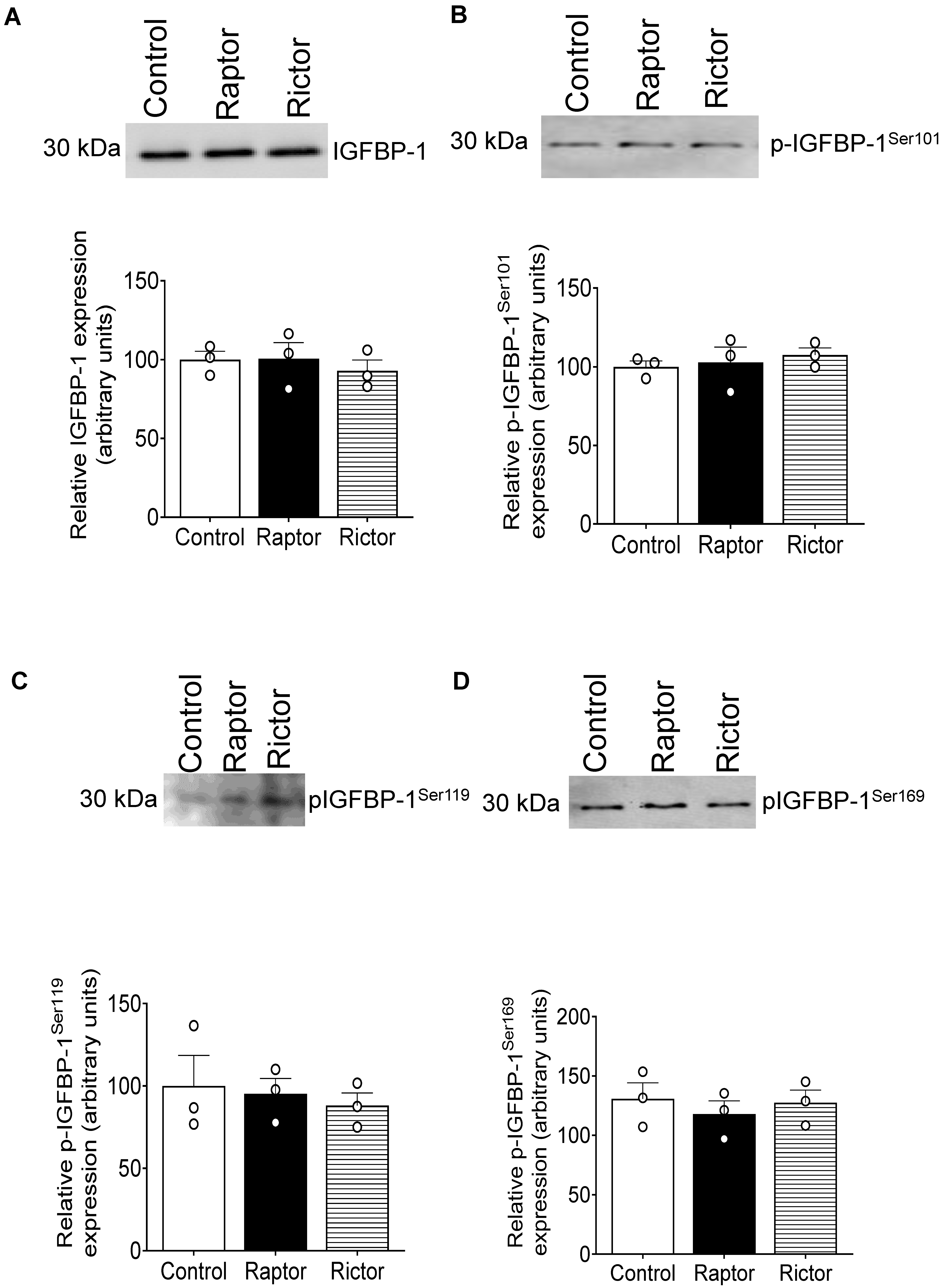
2.9. CM from RAPTOR- or RICTOR-Silenced PHT Cells Does Not Affect mTORC1 or mTORC2 Activity in HepG2 Cells
2.10. CM from Non-Trophoblast Cells with mTORC1 or mTORC2 Inhibition Did Not Affect HepG2 IGFBP-1 Phosphorylation
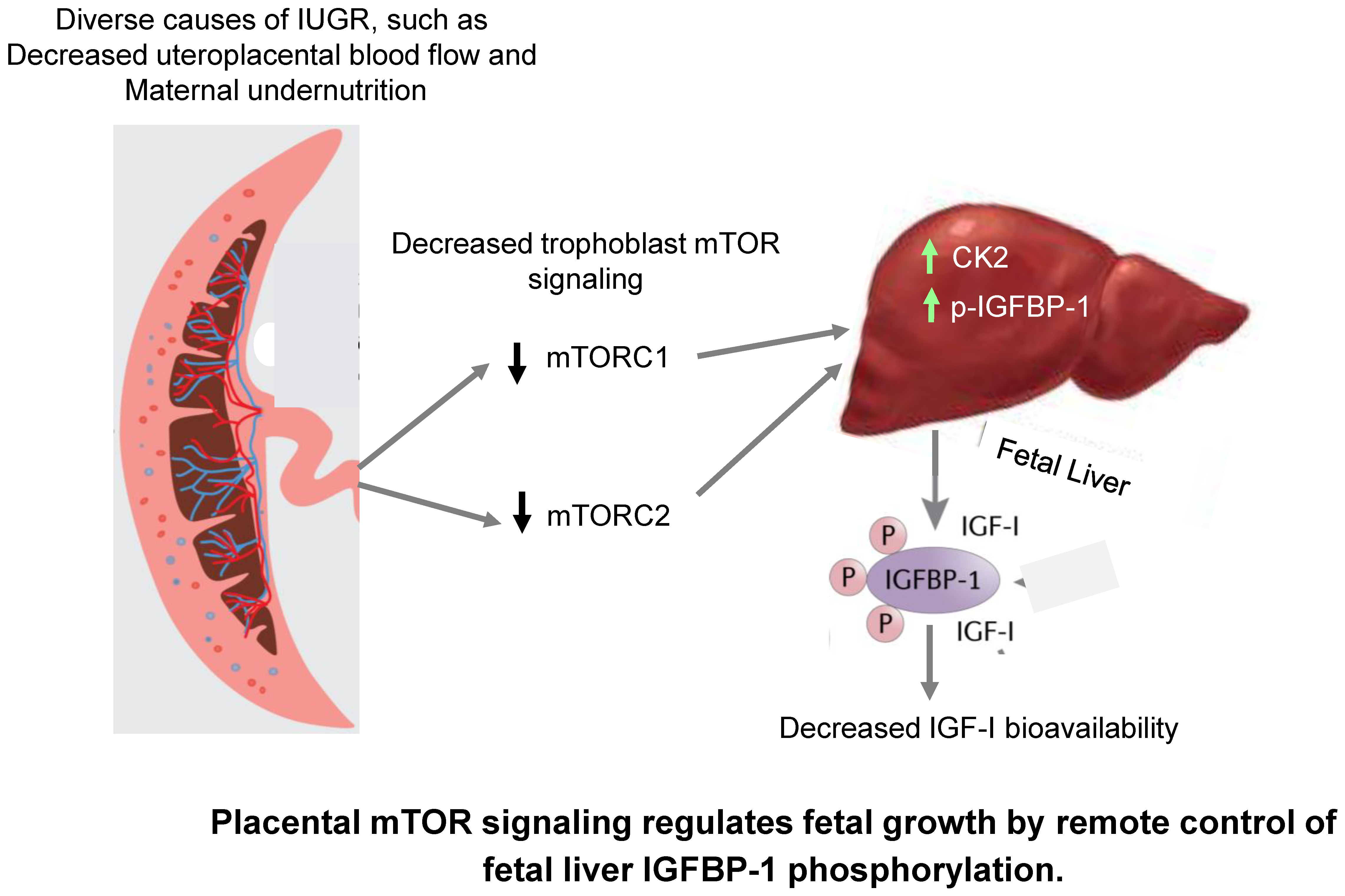
3. Discussion
4. Materials and Methods
4.1. Human Placenta Collection
4.2. Isolation and Culture of Primary Human Trophoblast PHT Cells
4.3. RNA Interference-Mediated Silencing (siRNA) in PHT Cells
4.4. HepG2 Cell Culture
4.5. Incubation of Cultured HepG2 Cells in CM from PHT Cells
4.6. SDS-PAGE and Western Blotting
4.7. Two-Dimensional (2-D) Immunoblot of IGFBP-1
4.8. Parallel Reaction Monitoring-Mass Spectrometry (PRM-MS)
4.8.1. Immunoprecipitation of IGFBP-1
4.8.2. Parallel Reaction Monitoring Mass Spectrometry (PRM-MS) Analyses of IGFBP-1 Peptides and CK2 co-Immunoprecipitation
4.9. IGF-1 Receptor Activation Assay
4.10. Casein Kinase CK2 Activity Assay
4.11. Enzyme-Linked Immunosorbent Assay (ELISA) for IGF-1 in CM Samples of HepG2 Cells
4.12. Non-Trophoblast Renal Carcinoma Cells
4.13. Data Presentation and Statistics
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Stremming, J.; Jansson, T.; Powell, T.L.; Rozance, P.J.; Brown, L.D. Reduced Na(+) K(+) -ATPase activity may reduce amino acid uptake in intrauterine growth restricted fetal sheep muscle despite unchanged ex vivo amino acid transporter activity. J. Physiol. 2020, 598, 1625–1639. [Google Scholar] [CrossRef] [PubMed]
- Lien, Y.-C.; E Pinney, S.; Lu, X.M.; A Simmons, R. Identification of Novel Regulatory Regions Induced by Intrauterine Growth Restriction in Rat Islets. Endocrinology 2021, 163, bqab251. [Google Scholar] [CrossRef] [PubMed]
- Shen, Z.; Zhu, W.; Du, L. Analysis of Gene Expression Profiles in the Liver of Rats With Intrauterine Growth Retardation. Front. Pediatr. 2022, 10, 801544. [Google Scholar] [CrossRef]
- Rosario, F.J.; Pardo, S.; Michelsen, T.M.; Erickson, K.; Moore, L.; Powell, T.L.; Weintraub, S.T.; Jansson, T. Characterization of the Primary Human Trophoblast Cell Secretome Using Stable Isotope Labeling With Amino Acids in Cell Culture. Front. Cell Dev. Biol. 2021, 9, 704781. [Google Scholar] [CrossRef] [PubMed]
- Goeden, N.; Velasquez, J.; Arnold, K.A.; Chan, Y.; Lund, B.T.; Anderson, G.M.; Bonnin, A. Maternal Inflammation Disrupts Fetal Neurodevelopment via Increased Placental Output of Serotonin to the Fetal Brain. J. Neurosci. 2016, 36, 6041–6049. [Google Scholar] [CrossRef] [PubMed]
- Hanswijk, S.; Spoelder, M.; Shan, L.; Verheij, M.; Muilwijk, O.; Li, W.; Liu, C.; Kolk, S.; Homberg, J. Gestational Factors throughout Fetal Neurodevelopment: The Serotonin Link. Int. J. Mol. Sci. 2020, 21, 5850. [Google Scholar] [CrossRef]
- Howerton, C.L.; Bale, T.L. Targeted placental deletion of OGT recapitulates the prenatal stress phenotype including hypothalamic mitochondrial dysfunction. Proc. Natl. Acad. Sci. USA 2014, 111, 9639–9644. [Google Scholar] [CrossRef]
- Howerton, C.L.; Morgan, C.P.; Fischer, D.B.; Bale, T.L. O-GlcNAc transferase (OGT) as a placental biomarker of maternal stress and reprogramming of CNS gene transcription in development. Proc. Natl. Acad. Sci. USA 2013, 110, 5169–5174. [Google Scholar] [CrossRef]
- Kelly, A.C.; Kramer, A.; Rosario, F.J.; Powell, T.L.; Jansson, T. Inhibition of mechanistic target of rapamycin signaling decreases levels of O-GlcNAc transferase and increases serotonin release in the human placenta. Clin. Sci. 2020, 134, 3123–3136. [Google Scholar] [CrossRef]
- Saxton, R.A.; Sabatini, D.M. mTOR Signaling in Growth, Metabolism, and Disease. Cell 2017, 169, 361–371. [Google Scholar] [CrossRef]
- Kim, J.; Guan, K.L. mTOR as a central hub of nutrient signalling and cell growth. Nat. Cell Biol. 2019, 21, 63–71. [Google Scholar] [CrossRef]
- Yung, H.-W.; Calabrese, S.; Hynx, D.; Hemmings, B.A.; Cetin, I.; Charnock-Jones, D.S.; Burton, G.J. Evidence of Placental Translation Inhibition and Endoplasmic Reticulum Stress in the Etiology of Human Intrauterine Growth Restriction. Am. J. Pathol. 2008, 173, 311–344. [Google Scholar] [CrossRef]
- Chen, Y.-Y.; Rosario, F.J.; Abu Shehab, M.; Powell, T.; Gupta, M.B.; Jansson, T. Increased ubiquitination and reduced plasma membrane trafficking of placental amino acid transporter SNAT-2 in human IUGR. Clin. Sci. 2015, 129, 1131–1141. [Google Scholar] [CrossRef]
- Hung, T.-H.; Hsieh, T.-T.; Wu, C.-P.; Li, M.-J.; Yeh, Y.-L.; Chen, S.-F. Mammalian target of rapamycin signaling is a mechanistic link between increased endoplasmic reticulum stress and autophagy in the placentas of pregnancies complicated by growth restriction. Placenta 2017, 60, 9–20. [Google Scholar] [CrossRef]
- Dimasuay, K.G.; Aitken, E.H.; Rosario, F.; Njie, M.; Glazier, J.; Rogerson, S.J.; Fowkes, F.J.I.; Beeson, J.G.; Powell, T.; Jansson, T.; et al. Inhibition of placental mTOR signaling provides a link between placental malaria and reduced birthweight. BMC Med. 2017, 15, 1–11. [Google Scholar] [CrossRef]
- Roos, S.; Jansson, N.; Palmberg, I.; Säljö, K.; Powell, T.; Jansson, T. Mammalian target of rapamycin in the human placenta regulates leucine transport and is down-regulated in restricted fetal growth. J. Physiol. 2007, 582, 449–459. [Google Scholar] [CrossRef]
- Rosario, F.J.; Jansson, N.; Kanai, Y.; Prasad, P.D.; Powell, T.; Jansson, T. Maternal Protein Restriction in the Rat Inhibits Placental Insulin, mTOR, and STAT3 Signaling and Down-Regulates Placental Amino Acid Transporters. Endocrinology 2011, 152, 1119–1129. [Google Scholar] [CrossRef]
- Kavitha, J.V.; Rosario, F.J.; Nijland, M.J.; McDonald, T.J.; Wu, G.; Kanai, Y.; Powell, T.L.; Nathanielsz, P.W.; Jansson, T. Down-regulation of placental mTOR, insulin/IGF-I signaling, and nutrient transporters in response to maternal nutrient restriction in the baboon. FASEB J. 2013, 28, 1294–1305. [Google Scholar] [CrossRef] [PubMed]
- Arroyo, J.A.; Brown, L.D.; Galan, H.L. Placental mammalian target of rapamycin and related signaling pathways in an ovine model of intrauterine growth restriction. Am. J. Obstet. Gynecol. 2009, 201, 616.e1–616.e7. [Google Scholar] [CrossRef] [PubMed]
- Rosario, F.J.; Schumacher, M.A.; Jiang, J.; Kanai, Y.; Powell, T.L.; Jansson, T. Chronic maternal infusion of full-length adiponectin in pregnant mice down-regulates placental amino acid transporter activity and expression and decreases fetal growth. J. Physiol. 2012, 590, 1495–1509. [Google Scholar] [CrossRef] [PubMed]
- Rosario, F.J.; Nathanielsz, P.W.; Powell, T.L.; Jansson, T. Maternal folate deficiency causes inhibition of mTOR signaling, down-regulation of placental amino acid transporters and fetal growth restriction in mice. Sci. Rep. 2017, 7, 3982. [Google Scholar] [CrossRef] [PubMed]
- Roos, S.; Lagerlöf, O.; Wennergren, M.; Powell, T.L.; Jansson, T. Regulation of amino acid transporters by glucose and growth factors in cultured primary human trophoblast cells is mediated by mTOR signaling. Am. J. Physiol. Cell 2009, 297, C723–C731. [Google Scholar] [CrossRef]
- Lager, S.; Gaccioli, F.; Ramirez, V.I.; Jones, H.N.; Jansson, T.; Powell, T.L. Oleic acid stimulates system A amino acid transport in primary human trophoblast cells mediated by toll-like receptor 4. J. Lipid Res. 2013, 54, 725–733. [Google Scholar] [CrossRef]
- Silva, E.; Rosario, F.J.; Powell, T.L.; Jansson, T. Mechanistic Target of Rapamycin Is a Novel Molecular Mechanism Linking Folate Availability and Cell Function. J. Nutr. 2017, 147, 1237–1242. [Google Scholar] [CrossRef]
- Rosario, F.J.; Powell, T.L.; Jansson, T. mTOR folate sensing links folate availability to trophoblast cell function. J. Physiol. 2017, 595, 4189–4206. [Google Scholar] [CrossRef] [PubMed]
- Vaughan, O.R.; Fisher, H.M.; Dionelis, K.N.; Jefferies, E.C.; Higgins, J.S.; Musial, B.; Sferruzzi-Perri, A.N.; Fowden, A.L. Corticosterone alters materno-fetal glucose partitioning and insulin signalling in pregnant mice. J. Physiol. 2015, 593, 1307–1321. [Google Scholar] [CrossRef] [PubMed]
- Aye, I.L.M.H.; Rosario, F.J.; Powell, T.L.; Jansson, T. Adiponectin supplementation in pregnant mice prevents the adverse effects of maternal obesity on placental function and fetal growth. Proc. Natl. Acad. Sci. USA 2015, 112, 12858–12863. [Google Scholar] [CrossRef]
- Jansson, T.; Castillo-Castrejon, M.; Gupta, M.B.; Powell, T.L.; Rosario, F.J. Down-regulation of placental Cdc42 and Rac1 links mTORC2 inhibition to decreased trophoblast amino acid transport in human intrauterine growth restriction. Clin. Sci. 2020, 134, 53–70. [Google Scholar] [CrossRef]
- Roos, S.; Kanai, Y.; Prasad, P.D.; Powell, T.L.; Jansson, T. Regulation of placental amino acid transporter activity by mammalian target of rapamycin. Am. J. Physiol. Physiol. 2009, 296, C142–C150. [Google Scholar] [CrossRef]
- Rosario, F.J.; Kanai, Y.; Powell, T.L.; Jansson, T. Mammalian target of rapamycin signalling modulates amino acid uptake by regulating transporter cell surface abundance in primary human trophoblast cells. J. Physiol. 2013, 591, 609–625. [Google Scholar] [CrossRef]
- Rosario, F.J.; Dimasuay, K.G.; Kanai, Y.; Powell, T.; Jansson, T. Regulation of amino acid transporter trafficking by mTORC1 in primary human trophoblast cells is mediated by the ubiquitin ligase Nedd4-2. Clin. Sci. 2016, 130, 499–512. [Google Scholar] [CrossRef] [PubMed]
- Rosario, F.J.; Powell, T.L.; Jansson, T. Mechanistic target of rapamycin (mTOR) regulates trophoblast folate uptake by modulating the cell surface expression of FR-α and the RFC. Sci. Rep. 2016, 6, 31705. [Google Scholar] [CrossRef]
- Rosario, F.J.; Gupta, M.B.; Myatt, L.; Powell, T.L.; Glenn, J.P.; Cox, L.; Jansson, T. Mechanistic Target of Rapamycin Complex 1 Promotes the Expression of Genes Encoding Electron Transport Chain Proteins and Stimulates Oxidative Phosphorylation in Primary Human Trophoblast Cells by Regulating Mitochondrial Biogenesis. Sci. Rep. 2019, 9, 246. [Google Scholar] [CrossRef] [PubMed]
- Baker, J.; Liu, J.P.; Robertson, E.J.; Efstratiadis, A. Role of insulin-like growth factors in embryonic and postnatal growth. Cell 1993, 75, 73–82. [Google Scholar] [CrossRef] [PubMed]
- Liu, J.-P.; Baker, J.; Perkins, A.S.; Robertson, E.J.; Efstratiadis, A. Mice carrying null mutations of the genes encoding insulin-like growth factor I (Igf-1) and type 1 IGF receptor (Igf1r). Cell 1993, 75, 59–72. [Google Scholar] [CrossRef]
- Lassarre, C.; Hardouin, S.; Daffos, F.; Forestier, F.; Frankenne, F.; Binoux, M. Serum Insulin-Like Growth Factors and Insulin-Like Growth Factor Binding Proteins in the Human Fetus. Relationships with Growth in Normal Subjects and in Subjects with Intrauterine Growth Retardation. Pediatr. Res. 1991, 29, 219–225. [Google Scholar] [CrossRef]
- Clemmons, D.R. Insulin-like growth factor binding proteins and their role in controlling IGF actions. Cytokine Growth Factor Rev. 1997, 8, 45–62. [Google Scholar] [CrossRef] [PubMed]
- Ben Lagha, N.; Seurin, D.; Le Bouc, Y.; Binoux, M.; Berdal, A.; Menuelle, P.; Babajko, S. Insulin-Like Growth Factor Binding Protein (IGFBP-1) Involvement in Intrauterine Growth Retardation: Study on IGFBP-1 Overexpressing Transgenic Mice. Endocrinology 2006, 147, 4730–4737. [Google Scholar] [CrossRef] [PubMed]
- Chard, T. Insulin-like growth factors and their binding proteins in normal and abnormal human fetal growth. Growth Regul. 1994, 4, 91–100. [Google Scholar]
- Murphy, V.E.; Smith, R.; Giles, W.B.; Clifton, V. Endocrine Regulation of Human Fetal Growth: The Role of the Mother, Placenta, and Fetus. Endocr. Rev. 2006, 27, 141–169. [Google Scholar] [CrossRef]
- Watson, C.S.; Bialek, P.; Anzo, M.; Khosravi, J.; Yee, S.-P.; Han, V.K.M. Elevated Circulating Insulin-Like Growth Factor Binding Protein-1 Is Sufficient to Cause Fetal Growth Restriction. Endocrinology 2006, 147, 1175–1186. [Google Scholar] [CrossRef] [PubMed]
- Rajkumar, K.; Barron, D.; LeWitt, M.S.; Murphy, L.J. Growth retardation and hyperglycemia in insulin-like growth factor binding protein-1 transgenic mice. Endocrinology 1995, 136, 4029–4034. [Google Scholar] [CrossRef] [PubMed]
- Jones, J.; Busby, W.; Wright, G.; Smith, C.; Kimack, N.; Clemmons, D. Identification of the sites of phosphorylation in insulin-like growth factor binding protein-1. Regulation of its affinity by phosphorylation of serine 101. J. Biol. Chem. 1993, 268, 1125–1131. [Google Scholar] [CrossRef] [PubMed]
- Gibson, M.; Aplin, J.; White, A.; Westwood, M. Regulation of IGF bioavailability in pregnancy. Mol. Hum. Reprod. 2001, 7, 79–87. [Google Scholar] [CrossRef] [PubMed]
- Giudice, L.C.; de Zegher, F.; Gargosky, S.E.; Dsupin, B.A.; De Las Fuentes, L.; Crystal, R.A.; Rosenfeld, R.G. Insulin-like growth factors and their binding proteins in the term and preterm human fetus and neonate with normal and extremes of intrauterine growth. J. Clin. Endocrinol. Metab. 1995, 80, 1548–1555. [Google Scholar] [PubMed]
- Abu Shehab, M.; Khosravi, J.; Han, V.K.M.; Shilton, B.H.; Gupta, M.B. Site-Specific IGFBP-1 Hyper-Phosphorylation in Fetal Growth Restriction: Clinical and Functional Relevance. J. Proteome Res. 2010, 9, 1873–1881. [Google Scholar] [CrossRef]
- Abu Shehab, M.; Inoue, S.; Han, V.K.M.; Gupta, M.B. Site Specific Phosphorylation of Insulin-Like Growth Factor Binding Protein-1 (IGFBP-1) for Evaluating Clinical Relevancy in Fetal Growth Restriction. J. Proteome Res. 2009, 8, 5325–5335. [Google Scholar] [CrossRef]
- Abu Shehab, M.; Damerill, I.; Shen, T.; Rosario, F.J.; Nijland, M.; Nathanielsz, P.W.; Kamat, A.; Jansson, T.; Gupta, M.B. Liver mTOR Controls IGF-I Bioavailability by Regulation of Protein Kinase CK2 and IGFBP-1 Phosphorylation in Fetal Growth Restriction. Endocrinology 2014, 155, 1327–1339. [Google Scholar] [CrossRef]
- Gupta, M.B.; Abu Shehab, M.; Nygard, K.; Biggar, K.; Singal, S.S.; Santoro, N.; Powell, T.L.; Jansson, T. IUGR Is Associated With Marked Hyperphosphorylation of Decidual and Maternal Plasma IGFBP-1. J. Clin. Endocrinol. Metab. 2018, 104, 408–422. [Google Scholar] [CrossRef]
- Kelly, J.H.; Darlington, G.J. Modulation of the liver specific phenotype in the human hepatoblastoma line Hep G2. Vitr. Cell. Dev. Biol. Plant 1989, 25, 217–222. [Google Scholar] [CrossRef]
- Wilkening, S.; Stahl, F.; Bader, A. Comparison of primary human hepatocytes and hepatoma cell line Hepg2 with regard to their biotransformation properties. Drug Metab. Dispos. 2003, 31, 1035–1042. [Google Scholar] [CrossRef]
- Abu Shehab, M.; Iosef, C.; Wildgruber, R.; Sardana, G.; Gupta, M.B. Phosphorylation of IGFBP-1 at Discrete Sites Elicits Variable Effects on IGF-I Receptor Autophosphorylation. Endocrinology 2013, 154, 1130–1143. [Google Scholar] [CrossRef]
- Frost, R.; Tseng, L. Insulin-like growth factor-binding protein-1 is phosphorylated by cultured human endometrial stromal cells and multiple protein kinases in vitro. J. Biol. Chem. 1991, 266, 18082–18088. [Google Scholar] [CrossRef] [PubMed]
- Ankrapp, D.; Jones, J.; Clemmons, D. Characterization of insulin-like growth factor binding protein-1 kinases from human hepatoma cells. J. Cell. Biochem. 1996, 60, 387–399. [Google Scholar] [CrossRef]
- Damerill, I.; Biggar, K.K.; Abu Shehab, M.; Li, S.S.-C.; Jansson, T.; Gupta, M.B. Hypoxia Increases IGFBP-1 Phosphorylation Mediated by mTOR Inhibition. Mol. Endocrinol. 2016, 30, 201–216. [Google Scholar] [CrossRef]
- Dolcini, L.; Sala, A.; Campagnoli, M.; Labò, S.; Valli, M.; Visai, L.; Minchiotti, L.; Monaco, H.L.; Galliano, M. Identification of the amniotic fluid insulin-like growth factor binding protein-1 phosphorylation sites and propensity to proteolysis of the isoforms. FEBS J. 2009, 276, 6033–6046. [Google Scholar] [CrossRef]
- Yu, J.; Iwashita, M.; Kudo, Y.; Takeda, Y. Phosphorylated insulin-like growth factor (IGF)-binding protein-1 (IGFBP-1) inhibits while non-hosphorylated IGFBP-1 stimulates IGF-I-induced amino acid uptake in cultured trophoblast cells. Growth Horm. IGF Res. 1998, 8, 65–70. [Google Scholar] [CrossRef] [PubMed]
- Siddals, K.; Westwood, M.; Gibson, M.; White, A. IGF-binding protein-1 inhibits IGF effects on adipocyte function: Implications for insulin-like actions at the adipocyte. J. Endocrinol. 2002, 174, 289–297. [Google Scholar] [CrossRef]
- Seferovic, M.D.; Ali, R.; Kamei, H.; Liu, S.; Khosravi, J.M.; Nazarian, S.; Han, V.K.M.; Duan, C.; Gupta, M.B. Hypoxia and Leucine Deprivation Induce Human Insulin-Like Growth Factor Binding Protein-1 Hyperphosphorylation and Increase Its Biological Activity. Endocrinology 2008, 150, 220–231. [Google Scholar] [CrossRef]
- Gupta, M.B. The role and regulation of IGFBP-1 phosphorylation in fetal growth restriction. J. Cell Commun. Signal. 2015, 9, 111–123. [Google Scholar] [CrossRef]
- Michelsen, T.M.; Henriksen, T.; Reinhold, D.; Powell, T.L.; Jansson, T. The human placental proteome secreted into the maternal and fetal circulations in normal pregnancy based on 4-vessel sampling. FASEB J. 2018, 33, 2944–2956. [Google Scholar] [CrossRef] [PubMed]
- Hessvik, N.P.; Llorente, A. Current knowledge on exosome biogenesis and release. Cell. Mol. Life Sci. 2018, 75, 193–208. [Google Scholar] [CrossRef]
- Guay, C.; Regazzi, R. Exosomes as new players in metabolic organ cross-talk. Diabetes Obes. Metab. 2017, 19 (Suppl. S1), 137–146. [Google Scholar] [CrossRef]
- Ying, W.; Riopel, M.; Bandyopadhyay, G.; Dong, Y.; Birmingham, A.; Seo, J.B.; Ofrecio, J.M.; Wollam, J.; Hernandez-Carretero, A.; Fu, W.; et al. Adipose Tissue Macrophage-Derived Exosomal miRNAs Can Modulate In Vivo and In Vitro Insulin Sensitivity. Cell 2017, 171, 372–384.e12. [Google Scholar] [CrossRef] [PubMed]
- Valadi, H.; Ekström, K.; Bossios, A.; Sjöstrand, M.; Lee, J.J.; Lötvall, J.O. Exosome-mediated transfer of mRNAs and microRNAs is a novel mechanism of genetic exchange between cells. Nat. Cell Biol. 2007, 9, 654–659. [Google Scholar] [CrossRef] [PubMed]
- Mitchell, M.D.; Peiris, H.N.; Kobayashi, M.; Koh, Y.Q.; Duncombe, G.; Illanes, S.E.; Rice, G.E.; Salomon, C. Placental exosomes in normal and complicated pregnancy. Am. J. Obstet. Gynecol. 2015, 213, S173–S181. [Google Scholar] [CrossRef]
- Adam, S.; Elfeky, O.; Kinhal, V.; Dutta, S.; Lai, A.; Jayabalan, N.; Nuzhat, Z.; Palma, C.; Rice, G.E.; Salomon, C. Review: Fetal-maternal communication via extracellular vesicles—Implications for complications of pregnancies. Placenta 2017, 54, 83–88. [Google Scholar] [CrossRef]
- Zou, W.; Lai, M.; Zhang, Y.; Zheng, L.; Xing, Z.; Li, T.; Zou, Z.; Song, Q.; Zhao, X.; Xia, L.; et al. Exosome Release Is Regulated by mTORC1. Adv. Sci. 2018, 6, 1801313. [Google Scholar] [CrossRef]
- Mahdipour, E.; Salmasi, Z.; Sabeti, N. Potential of stem cell-derived exosomes to regenerate β islets through Pdx-1 dependent mechanism in a rat model of type 1 diabetes. J. Cell. Physiol. 2019, 234, 20310–20321. [Google Scholar] [CrossRef]
- Jin, J.; Menon, R. Placental exosomes: A proxy to understand pregnancy complications. Am. J. Reprod. Immunol. 2017, 79, e12788. [Google Scholar] [CrossRef]
- Mincheva-Nilsson, L.; Baranov, V. The Role of Placental Exosomes in Reproduction. Am. J. Reprod. Immunol. 2010, 63, 520–533. [Google Scholar] [CrossRef]
- Salomon, C.; Rice, G.E. Role of Exosomes in Placental Homeostasis and Pregnancy Disorders. Prog. Mol. Biol. Transl. Sci. 2017, 145, 163–179. [Google Scholar]
- Sarker, S.; Scholz-Romero, K.; Perez, A.; Illanes, S.E.; Mitchell, M.D.; Rice, G.E.; Salomon, C. Placenta-derived exosomes continuously increase in maternal circulation over the first trimester of pregnancy. J. Transl. Med. 2014, 12, 204. [Google Scholar] [CrossRef] [PubMed]
- Miranda, J.; Paules, C.; Nair, S.; Lai, A.; Palma, C.; Scholz-Romero, K.; Rice, G.E.; Gratacos, E.; Crispi, F.; Salomon, C. Placental exosomes profile in maternal and fetal circulation in intrauterine growth restriction—Liquid biopsies to monitoring fetal growth. Placenta 2018, 64, 34–43. [Google Scholar] [CrossRef] [PubMed]
- Chang, G.; Mouillet, J.-F.; Mishima, T.; Chu, T.; Sadovsky, E.; Coyne, C.B.; Parks, W.T.; Surti, U.; Sadovsky, Y. Expression and trafficking of placental microRNAs at the feto-maternal interface. FASEB J. 2017, 31, 2760–2770. [Google Scholar] [CrossRef]
- Gupta, M.B.; Jansson, T. Novel roles of mechanistic target of rapamycin signaling in regulating fetal growth. Biol. Reprod. 2018, 100, 872–884. [Google Scholar] [CrossRef] [PubMed]
- Chassen, S.; Jansson, T. Complex, coordinated and highly regulated changes in placental signaling and nutrient transport capacity in IUGR. Biochim. et Biophys. Acta (BBA) Mol. Basis Dis. 2019, 1866, 165373. [Google Scholar] [CrossRef]
- Cao, B.; Parnell, L.A.; Diamond, M.S.; Mysorekar, I.U. Inhibition of autophagy limits vertical transmission of Zika virus in pregnant mice. J. Exp. Med. 2017, 214, 2303–2313. [Google Scholar] [CrossRef]
- Rosario, F.J.; Kelly, A.C.; Gupta, M.B.; Powell, T.L.; Cox, L.; Jansson, T. Mechanistic Target of Rapamycin Complex 2 Regulation of the Primary Human Trophoblast Cell Transcriptome. Front. Cell Dev. Biol. 2021, 9, 670980. [Google Scholar] [CrossRef]
- Nissum, M.; Abu Shehab, M.; Sukop, U.; Khosravi, J.M.; Wildgruber, R.; Eckerskorn, C.; Han, V.K.M.; Gupta, M.B. Functional and Complementary Phosphorylation State Attributes of Human Insulin-like Growth Factor-Binding Protein-1 (IGFBP-1) Isoforms Resolved by Free Flow Electrophoresis. Mol. Cell. Proteom. 2009, 8, 1424–1435. [Google Scholar] [CrossRef]
- Nandi, P.; Jang, C.E.; Biggar, K.; Halari, C.D.; Jansson, T.; Gupta, M.B. Mechanistic Target of Rapamycin Complex 1 Signaling Links Hypoxia to Increased IGFBP-1 Phosphorylation in Primary Human Decidualized Endometrial Stromal Cells. Biomolecules 2021, 11, 1382. [Google Scholar] [CrossRef] [PubMed]
- MacLean, B.; Tomazela, D.M.; Shulman, N.; Chambers, M.; Finney, G.L.; Frewen, B.; Kern, R.; Tabb, D.L.; Liebler, D.C.; MacCoss, M.J. Skyline: An open source document editor for creating and analyzing targeted proteomics experiments. Bioinformatics 2010, 26, 966–968. [Google Scholar] [CrossRef]
- Pietrzkowski, Z.; Sell, C.; Lammers, R.; Ullrich, A.; Baserga, R. Roles of insulinlike growth factor 1 (IGF-1) and the IGF-1 receptor in epidermal growth factor-stimulated growth of 3T3 cells. Mol. Cell. Biol. 1992, 12, 3883–3889. [Google Scholar]
- Vilk, G.; Saulnier, R.B.; Pierre, R.S.; Litchfield, D.W. Inducible Expression of Protein Kinase CK2 in Mammalian Cells. Evidence for functional specialization of CK2 isoforms. J. Biol. Chem. 1999, 274, 14406–14414. [Google Scholar] [CrossRef] [PubMed]
- Shanmugasundaram, K.; Block, K.; Nayak, B.K.; Livi, C.B.; Venkatachalam, M.A.; Sudarshan, S. PI3K regulation of the SKP-2/p27 axis through mTORC2. Oncogene 2013, 32, 2027–2036. [Google Scholar] [CrossRef] [PubMed]










Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Rosario, F.J.; Chopra, A.; Biggar, K.; Powell, T.L.; Gupta, M.B.; Jansson, T. Placental Remote Control of Fetal Metabolism: Trophoblast mTOR Signaling Regulates Liver IGFBP-1 Phosphorylation and IGF-1 Bioavailability. Int. J. Mol. Sci. 2023, 24, 7273. https://doi.org/10.3390/ijms24087273
Rosario FJ, Chopra A, Biggar K, Powell TL, Gupta MB, Jansson T. Placental Remote Control of Fetal Metabolism: Trophoblast mTOR Signaling Regulates Liver IGFBP-1 Phosphorylation and IGF-1 Bioavailability. International Journal of Molecular Sciences. 2023; 24(8):7273. https://doi.org/10.3390/ijms24087273
Chicago/Turabian StyleRosario, Fredrick J., Anand Chopra, Kyle Biggar, Theresa L. Powell, Madhulika B. Gupta, and Thomas Jansson. 2023. "Placental Remote Control of Fetal Metabolism: Trophoblast mTOR Signaling Regulates Liver IGFBP-1 Phosphorylation and IGF-1 Bioavailability" International Journal of Molecular Sciences 24, no. 8: 7273. https://doi.org/10.3390/ijms24087273
APA StyleRosario, F. J., Chopra, A., Biggar, K., Powell, T. L., Gupta, M. B., & Jansson, T. (2023). Placental Remote Control of Fetal Metabolism: Trophoblast mTOR Signaling Regulates Liver IGFBP-1 Phosphorylation and IGF-1 Bioavailability. International Journal of Molecular Sciences, 24(8), 7273. https://doi.org/10.3390/ijms24087273