
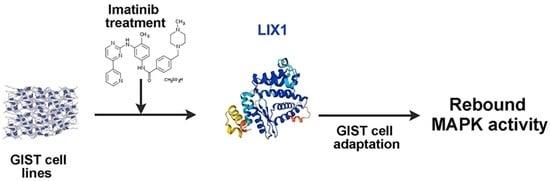
LIX1 Controls MAPK Signaling Reactivation and Contributes to GIST-T1 Cell Resistance to Imatinib
 ,
,  ,
,  , and
, and
Abstract
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Results
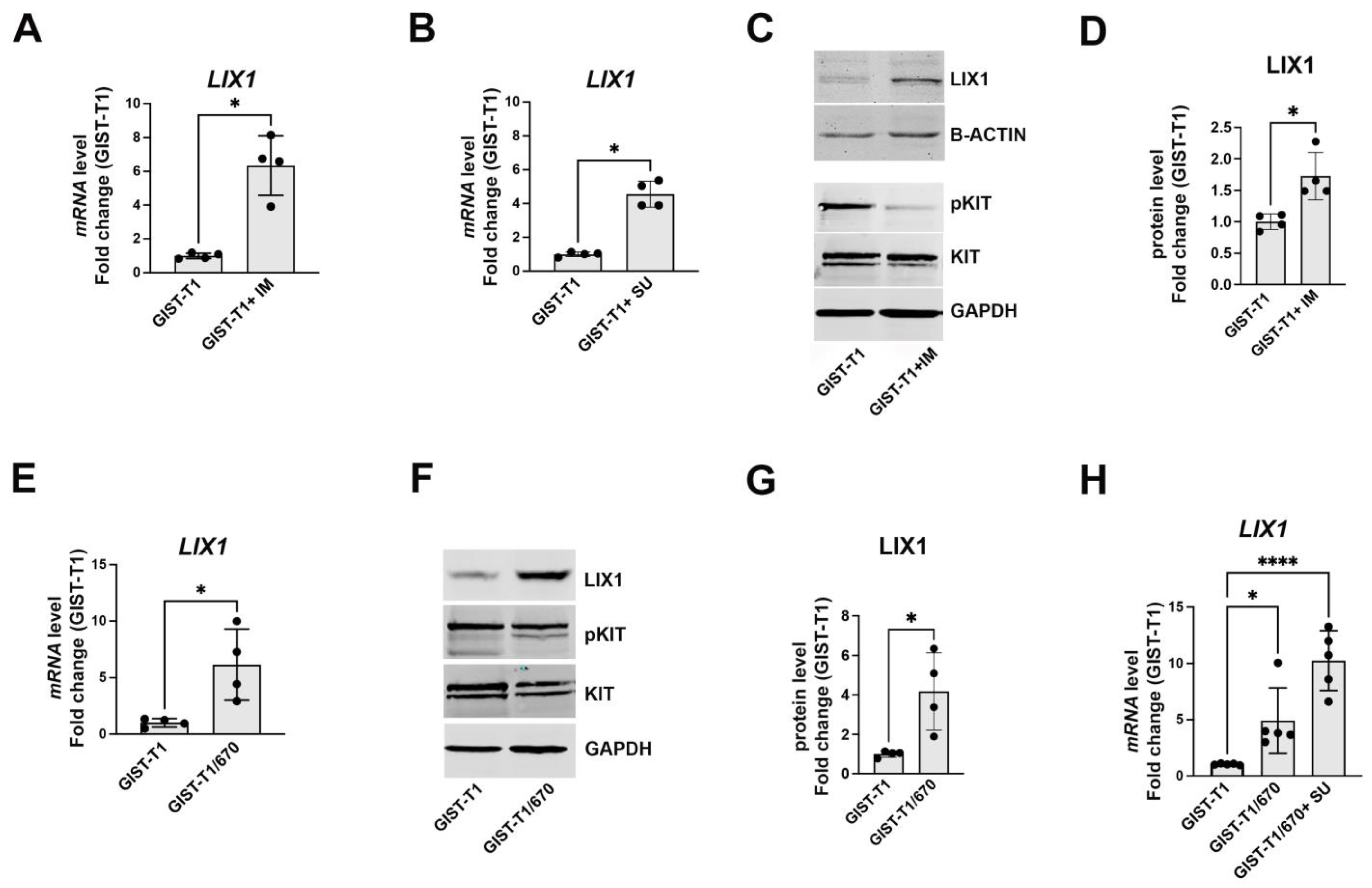
2.1. KIT-Signaling Abrogation Results in a Significant Increase in LIX1 Expression in GIST-T1 Cells
2.2. LIX1 Expression and MAPK Signaling Changes Following Incubation with Imatinib
2.3. LIX1 Promotes MAPK-Signaling Reactivation Following KIT Inhibition
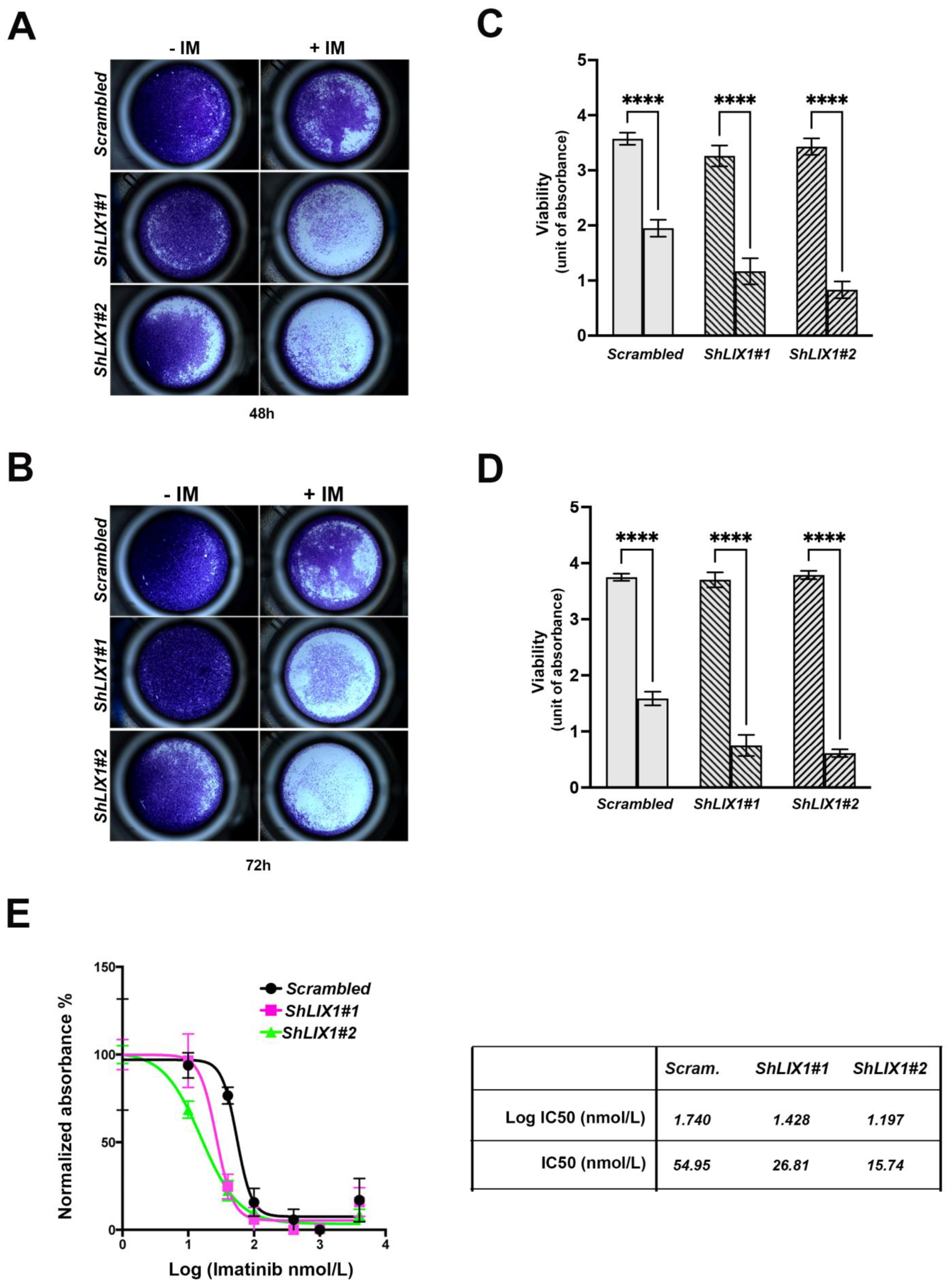
2.4. LIX1 Blockade Re-Sensitizes GIST-T1 Cells to Imatinib
3. Discussion
4. Materials and Methods
4.1. Cell Culture and Reagents
4.2. Immunoblot Analysis
4.3. Reverse Transcription and Quantitative Polymerase Chain Reaction (RT-qPCR)
4.4. Cell Viability Measurement
4.5. Statistical Analysis
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Acknowledgments
Conflicts of Interest
References
- Demetri, G.D.; von Mehren, M.; Antonescu, C.R.; DeMatteo, R.P.; Ganjoo, K.N.; Maki, R.G.; Pisters, P.W.T.; Raut, C.P.; Riedel, R.F.; Schuetze, S.; et al. NCCN Task Force Report: Update on the Management of Patients with Gastrointestinal Stromal Tumors. J. Natl. Compr. Canc Netw. 2010, 8 (Suppl. S2), S1–S41, quiz S42–S44. [Google Scholar] [CrossRef] [PubMed]
- Serrano, C.; George, S. Gastrointestinal Stromal Tumor: Challenges and Opportunities for a New Decade. Clin. Cancer Res. 2020, 26, 5078–5085. [Google Scholar] [CrossRef] [PubMed]
- Kondo, J.; Huh, W.J.; Franklin, J.L.; Heinrich, M.C.; Rubin, B.P.; Coffey, R.J. A Smooth Muscle-Derived, Braf-Driven Mouse Model of Gastrointestinal Stromal Tumor (GIST): Evidence for an Alternative GIST Cell-of-Origin. J. Pathol. 2020, 252, 441–450. [Google Scholar] [CrossRef] [PubMed]
- Hapkova, I.; Skarda, J.; Rouleau, C.; Thys, A.; Notarnicola, C.; Janikova, M.; Bernex, F.; Rypka, M.; Vanderwinden, J.-M.; Faure, S.; et al. High Expression of the RNA-Binding Protein RBPMS2 in Gastrointestinal Stromal Tumors. Exp. Mol. Pathol. 2013, 94, 314–321. [Google Scholar] [CrossRef] [PubMed]
- Szucs, Z.; Jones, R.L. Perspectives on the Evolving State of the Art Management of Gastrointestinal Stromal Tumours. Transl. Gastroenterol. Hepatol. 2018, 3, 21. [Google Scholar] [CrossRef]
- Miettinen, M.; Makhlouf, H.; Sobin, L.H.; Lasota, J. Gastrointestinal Stromal Tumors of the Jejunum and Ileum: A Clinicopathologic, Immunohistochemical, and Molecular Genetic Study of 906 Cases before Imatinib with Long-Term Follow-Up. Am. J. Surg. Pathol. 2006, 30, 477–489. [Google Scholar] [CrossRef]
- Klug, L.R.; Bannon, A.E.; Javidi-Sharifi, N.; Town, A.; Fleming, W.H.; VanSlyke, J.K.; Musil, L.S.; Fletcher, J.A.; Tyner, J.W.; Heinrich, M.C. LMTK3 Is Essential for Oncogenic KIT Expression in KIT-Mutant GIST and Melanoma. Oncogene 2019, 38, 1200–1210. [Google Scholar] [CrossRef]
- Chi, P.; Chen, Y.; Zhang, L.; Guo, X.; Wongvipat, J.; Shamu, T.; Fletcher, J.A.; Dewell, S.; Maki, R.G.; Zheng, D.; et al. ETV1 Is a Lineage Survival Factor That Cooperates with KIT in Gastrointestinal Stromal Tumours. Nature 2010, 467, 849–853. [Google Scholar] [CrossRef]
- Bauer, S.; Duensing, A.; Demetri, G.D.; Fletcher, J.A. KIT Oncogenic Signaling Mechanisms in Imatinib-Resistant Gastrointestinal Stromal Tumor: PI3-Kinase/AKT Is a Crucial Survival Pathway. Oncogene 2007, 26, 7560–7568. [Google Scholar] [CrossRef]
- Bosbach, B.; Rossi, F.; Yozgat, Y.; Loo, J.; Zhang, J.Q.; Berrozpe, G.; Warpinski, K.; Ehlers, I.; Veach, D.; Kwok, A.; et al. Direct Engagement of the PI3K Pathway by Mutant KIT Dominates Oncogenic Signaling in Gastrointestinal Stromal Tumor. Proc. Natl. Acad. Sci. USA 2017, 114, E8448–E8457. [Google Scholar] [CrossRef]
- García-Valverde, A.; Rosell, J.; Serna, G.; Valverde, C.; Carles, J.; Nuciforo, P.; Fletcher, J.A.; Arribas, J.; Politz, O.; Serrano, C. Preclinical Activity of PI3K Inhibitor Copanlisib in Gastrointestinal Stromal Tumor. Mol. Cancer Ther. 2020, 19, 1289–1297. [Google Scholar] [CrossRef] [PubMed]
- Tuveson, D.A.; Willis, N.A.; Jacks, T.; Griffin, J.D.; Singer, S.; Fletcher, C.D.; Fletcher, J.A.; Demetri, G.D. STI571 Inactivation of the Gastrointestinal Stromal Tumor C-KIT Oncoprotein: Biological and Clinical Implications. Oncogene 2001, 20, 5054–5058. [Google Scholar] [CrossRef] [PubMed]
- Joensuu, H. Predicting Recurrence-Free Survival after Surgery for GIST. Lancet Oncol. 2009, 10, 1025. [Google Scholar] [CrossRef] [PubMed]
- Blanke, C.D.; Demetri, G.D.; von Mehren, M.; Heinrich, M.C.; Eisenberg, B.; Fletcher, J.A.; Corless, C.L.; Fletcher, C.D.M.; Roberts, P.J.; Heinz, D.; et al. Long-Term Results from a Randomized Phase II Trial of Standard-versus Higher-Dose Imatinib Mesylate for Patients with Unresectable or Metastatic Gastrointestinal Stromal Tumors Expressing KIT. J. Clin. Oncol. 2008, 26, 620–625. [Google Scholar] [CrossRef]
- Liegl, B.; Kepten, I.; Le, C.; Zhu, M.; Demetri, G.D.; Heinrich, M.C.; Fletcher, C.D.M.; Corless, C.L.; Fletcher, J.A. Heterogeneity of Kinase Inhibitor Resistance Mechanisms in GIST. J. Pathol. 2008, 216, 64–74. [Google Scholar] [CrossRef] [PubMed]
- Wardelmann, E.; Thomas, N.; Merkelbach-Bruse, S.; Pauls, K.; Speidel, N.; Büttner, R.; Bihl, H.; Leutner, C.C.; Heinicke, T.; Hohenberger, P. Acquired Resistance to Imatinib in Gastrointestinal Stromal Tumours Caused by Multiple KIT Mutations. Lancet Oncol. 2005, 6, 249–251. [Google Scholar] [CrossRef]
- Schaefer, I.-M.; DeMatteo, R.P.; Serrano, C. The GIST of Advances in Treatment of Advanced Gastrointestinal Stromal Tumor. Am. Soc. Clin. Oncol. Educ. Book. 2022, 42, 885–899. [Google Scholar] [CrossRef]
- Demetri, G.D.; van Oosterom, A.T.; Garrett, C.R.; Blackstein, M.E.; Shah, M.H.; Verweij, J.; McArthur, G.; Judson, I.R.; Heinrich, M.C.; Morgan, J.A.; et al. Efficacy and Safety of Sunitinib in Patients with Advanced Gastrointestinal Stromal Tumour after Failure of Imatinib: A Randomised Controlled Trial. Lancet 2006, 368, 1329–1338. [Google Scholar] [CrossRef]
- Demetri, G.D.; Reichardt, P.; Kang, Y.-K.; Blay, J.-Y.; Rutkowski, P.; Gelderblom, H.; Hohenberger, P.; Leahy, M.; von Mehren, M.; Joensuu, H.; et al. Efficacy and Safety of Regorafenib for Advanced Gastrointestinal Stromal Tumours after Failure of Imatinib and Sunitinib (GRID): An International, Multicentre, Randomised, Placebo-Controlled, Phase 3 Trial. Lancet 2013, 381, 295–302. [Google Scholar] [CrossRef]
- Serrano, C.; George, S.; Valverde, C.; Olivares, D.; García-Valverde, A.; Suárez, C.; Morales-Barrera, R.; Carles, J. Novel Insights into the Treatment of Imatinib-Resistant Gastrointestinal Stromal Tumors. Target. Oncol. 2017, 12, 277–288. [Google Scholar] [CrossRef]
- Ivanyi, P.; Eggers, H.; Hornig, M.; Kasper, B.; Heissner, K.; Kopp, H.-G.; Kirstein, M.; Ganser, A.; Grünwald, V. Hepatic Toxicity during Regorafenib Treatment in Patients with Metastatic Gastrointestinal Stromal Tumors. Mol. Clin. Oncol. 2020, 13, 72. [Google Scholar] [CrossRef] [PubMed]
- Westerdijk, K.; Krens, S.D.; van der Graaf, W.T.A.; Mulder, S.F.; van Herpen, C.M.L.; Smilde, T.; van Erp, N.P.; Desar, I.M.E. The Relationship between Sunitinib Exposure and Both Efficacy and Toxicity in Real-World Patients with Renal Cell Carcinoma and Gastrointestinal Stromal Tumour. Br. J. Clin. Pharmacol. 2021, 87, 326–335. [Google Scholar] [CrossRef] [PubMed]
- Li, F.; Huynh, H.; Li, X.; Ruddy, D.A.; Wang, Y.; Ong, R.; Chow, P.; Qiu, S.; Tam, A.; Rakiec, D.P.; et al. FGFR-Mediated Reactivation of MAPK Signaling Attenuates Anti-tumor Effects of Imatinib in Gastrointestinal Stromal Tumors. Cancer Discov. 2015, 5, 438–451. [Google Scholar] [CrossRef] [PubMed]
- Javidi-Sharifi, N.; Traer, E.; Martinez, J.; Gupta, A.; Taguchi, T.; Dunlap, J.; Heinrich, M.C.; Corless, C.L.; Rubin, B.P.; Druker, B.J.; et al. Crosstalk between KIT and FGFR3 Promotes Gastrointestinal Stromal Tumor Cell Growth and Drug Resistance. Cancer Res. 2015, 75, 880–891. [Google Scholar] [CrossRef] [PubMed]
- Boichuk, S.; Galembikova, A.; Dunaev, P.; Valeeva, E.; Shagimardanova, E.; Gusev, O.; Khaiboullina, S. A Novel Receptor Tyrosine Kinase Switch Promotes Gastrointestinal Stromal Tumor Drug Resistance. Molecules 2017, 22, 2152. [Google Scholar] [CrossRef]
- Chi, P.; Qin, L.-X.; Nguyen, B.; Kelly, C.M.; D’Angelo, S.P.; Dickson, M.A.; Gounder, M.M.; Keohan, M.L.; Movva, S.; Nacev, B.A.; et al. Phase II Trial of Imatinib Plus Binimetinib in Patients with Treatment-Naive Advanced Gastrointestinal Stromal Tumor. J. Clin. Oncol. 2022, 40, 997–1008. [Google Scholar] [CrossRef]
- Gupta, A.; Singh, J.; García-Valverde, A.; Serrano, C.; Flynn, D.L.; Smith, B.D. Ripretinib and MEK Inhibitors Synergize to Induce Apoptosis in Preclinical Models of GIST and Systemic Mastocytosis. Mol. Cancer Ther. 2021, 20, 1234–1245. [Google Scholar] [CrossRef]
- McKey, J.; Martire, D.; de Santa Barbara, P.; Faure, S. LIX1 Regulates YAP1 Activity and Controls the Proliferation and Differentiation of Stomach Mesenchymal Progenitors. BMC Biol. 2016, 14, 34. [Google Scholar] [CrossRef]
- Guérin, A.; Martire, D.; Trenquier, E.; Lesluyes, T.; Sagnol, S.; Pratlong, M.; Lefebvre, E.; Chibon, F.; de Santa Barbara, P.; Faure, S. LIX1 Regulates YAP Activity and Controls Gastrointestinal Cancer Cell Plasticity. J. Cell. Mol. Med. 2020, 24, 9244–9254. [Google Scholar] [CrossRef]
- Guérin, A.; Angebault, C.; Kinet, S.; Cazevieille, C.; Rojo, M.; Fauconnier, J.; Lacampagne, A.; Mourier, A.; Taylor, N.; de Santa Barbara, P.; et al. LIX1-Mediated Changes in Mitochondrial Metabolism Control the Fate of Digestive Mesenchyme-Derived Cells. Redox Biol. 2022, 56, 102431. [Google Scholar] [CrossRef]
- Heinrich, M.C.; Corless, C.L.; Blanke, C.D.; Demetri, G.D.; Joensuu, H.; Roberts, P.J.; Eisenberg, B.L.; von Mehren, M.; Fletcher, C.D.M.; Sandau, K.; et al. Molecular Correlates of Imatinib Resistance in Gastrointestinal Stromal Tumors. J. Clin. Oncol. 2006, 24, 4764–4774. [Google Scholar] [CrossRef]
- Rubin, B.P.; Duensing, A. Mechanisms of Resistance to Small Molecule Kinase Inhibition in the Treatment of Solid Tumors. Lab. Investig. 2006, 86, 981–986. [Google Scholar] [CrossRef] [PubMed]
- Gupta, P.B.; Pastushenko, I.; Skibinski, A.; Blanpain, C.; Kuperwasser, C. Phenotypic Plasticity: Driver of Cancer Initiation, Progression, and Therapy Resistance. Cell. Stem Cell. 2019, 24, 65–78. [Google Scholar] [CrossRef] [PubMed]
- Serrano, C.; Mariño-Enríquez, A.; Tao, D.L.; Ketzer, J.; Eilers, G.; Zhu, M.; Yu, C.; Mannan, A.M.; Rubin, B.P.; Demetri, G.D.; et al. Complementary Activity of Tyrosine Kinase Inhibitors against Secondary Kit Mutations in Imatinib-Resistant Gastrointestinal Stromal Tumours. Br. J. Cancer 2019, 120, 612–620. [Google Scholar] [CrossRef]
- Kelly, C.M.; Shoushtari, A.N.; Qin, L.-X.; D’Angelo, S.P.; Dickson, M.A.; Gounder, M.M.; Keohan, M.L.; Mcfadyen, C.; Sjoberg, A.; Singer, S.; et al. A Phase Ib Study of BGJ398, a Pan-FGFR Kinase Inhibitor in Combination with Imatinib in Patients with Advanced Gastrointestinal Stromal Tumor. Investig. New. Drugs 2019, 37, 282–290. [Google Scholar] [CrossRef] [PubMed]
- Torihashi, S.; Nishi, K.; Tokutomi, Y.; Nishi, T.; Ward, S.; Sanders, K.M. Blockade of Kit Signaling Induces Transdifferentiation of Interstitial Cells of Cajal to a Smooth Muscle Phenotype. Gastroenterology 1999, 117, 140–148. [Google Scholar] [CrossRef]
- Chandel, N.S. Mitochondria as Signaling Organelles. BMC Biol. 2014, 12, 34. [Google Scholar] [CrossRef] [PubMed]
- Vyas, S.; Zaganjor, E.; Haigis, M.C. Mitochondria and Cancer. Cell 2016, 166, 555–566. [Google Scholar] [CrossRef]
- Huang, W.-K.; Gao, J.; Chen, Z.; Shi, H.; Yuan, J.; Cui, H.L.; Yeh, C.-N.; Bränström, R.; Larsson, C.; Li, S.; et al. Heterogeneity of Metabolic Vulnerability in Imatinib-Resistant Gastrointestinal Stromal Tumor. Cells 2020, 9, 1333. [Google Scholar] [CrossRef]
- Vitiello, G.A.; Medina, B.D.; Zeng, S.; Bowler, T.G.; Zhang, J.Q.; Loo, J.K.; Param, N.J.; Liu, M.; Moral, A.J.; Zhao, J.N.; et al. Mitochondrial Inhibition Augments the Efficacy of Imatinib by Resetting the Metabolic Phenotype of Gastrointestinal Stromal Tumor. Clin. Cancer Res. 2018, 24, 972–984. [Google Scholar] [CrossRef]
- Taguchi, T.; Sonobe, H.; Toyonaga, S.; Yamasaki, I.; Shuin, T.; Takano, A.; Araki, K.; Akimaru, K.; Yuri, K. Conventional and Molecular Cytogenetic Characterization of a New Human Cell Line, GIST-T1, Established from Gastrointestinal Stromal Tumor. Lab. Investig. 2002, 82, 663–665. [Google Scholar] [CrossRef] [PubMed]
- Pasquet, L.; Bellard, E.; Rols, M.P.; Golzio, M.; Teissie, J. Post-Pulse Addition of Trans-Cyclohexane-1,2-Diol Improves Electrotransfer Mediated Gene Expression in Mammalian Cells. Biochem. Biophys. Rep. 2016, 7, 287–294. [Google Scholar] [CrossRef] [PubMed]




Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ruiz-Demoulin, S.; Trenquier, E.; Dekkar, S.; Deshayes, S.; Boisguérin, P.; Serrano, C.; de Santa Barbara, P.; Faure, S. LIX1 Controls MAPK Signaling Reactivation and Contributes to GIST-T1 Cell Resistance to Imatinib. Int. J. Mol. Sci. 2023, 24, 7138. https://doi.org/10.3390/ijms24087138
Ruiz-Demoulin S, Trenquier E, Dekkar S, Deshayes S, Boisguérin P, Serrano C, de Santa Barbara P, Faure S. LIX1 Controls MAPK Signaling Reactivation and Contributes to GIST-T1 Cell Resistance to Imatinib. International Journal of Molecular Sciences. 2023; 24(8):7138. https://doi.org/10.3390/ijms24087138
Chicago/Turabian StyleRuiz-Demoulin, Salomé, Eva Trenquier, Sanaa Dekkar, Sébastien Deshayes, Prisca Boisguérin, César Serrano, Pascal de Santa Barbara, and Sandrine Faure. 2023. "LIX1 Controls MAPK Signaling Reactivation and Contributes to GIST-T1 Cell Resistance to Imatinib" International Journal of Molecular Sciences 24, no. 8: 7138. https://doi.org/10.3390/ijms24087138
APA StyleRuiz-Demoulin, S., Trenquier, E., Dekkar, S., Deshayes, S., Boisguérin, P., Serrano, C., de Santa Barbara, P., & Faure, S. (2023). LIX1 Controls MAPK Signaling Reactivation and Contributes to GIST-T1 Cell Resistance to Imatinib. International Journal of Molecular Sciences, 24(8), 7138. https://doi.org/10.3390/ijms24087138