
Structure-Based Design and Pharmacophore-Based Virtual Screening of Combinatorial Library of Triclosan Analogues Active against Enoyl-Acyl Carrier Protein Reductase of Plasmodium falciparum with Favourable ADME Profiles

 ,
,
Abstract
1. Introduction
- -
- We built and validated a Hansch-type QSAR model to correlate the relative Gibbs free energy (rGFE) of TCL binding to PfENR with the observed inhibitory potencies (identification of the bound conformation of TCL inhibitors).
- -
- The robustness of the built model is confirmed by a 3D-QSAR pharmacophore (PH4) model based on the bound conformations of the training set of inhibitors (generation of the PH4 model).
- -
- In addition, we built a virtual library of 33,480 analogues of TCLs and screened them with the help of PH4 to identify hits for inhibition of PfENR (virtual screening).
- -
- Finally, the ADME profile of the best designed analogues was predicted and compared with those of current antimalarial drugs and compounds undergoing clinical trials (postprocessing step 1).
- -
- Each of the hits was cross-checked for predicted inhibitory potency by computed rGFE of the formation of the PfENR-TCLx complex, which led to the identification of a handful of prospective novel TCLs that exceeded the potency of known TCLs against PfENR (postprocessing step 2).
- -
- The top 5 TCL hits and TCL11 underwent molecular dynamics simulations to explore the stability of the PfENR-TCLx complexes (postprocessing step 3).
2. Results
2.1. Training and Validation Sets of TCL Inhibitors
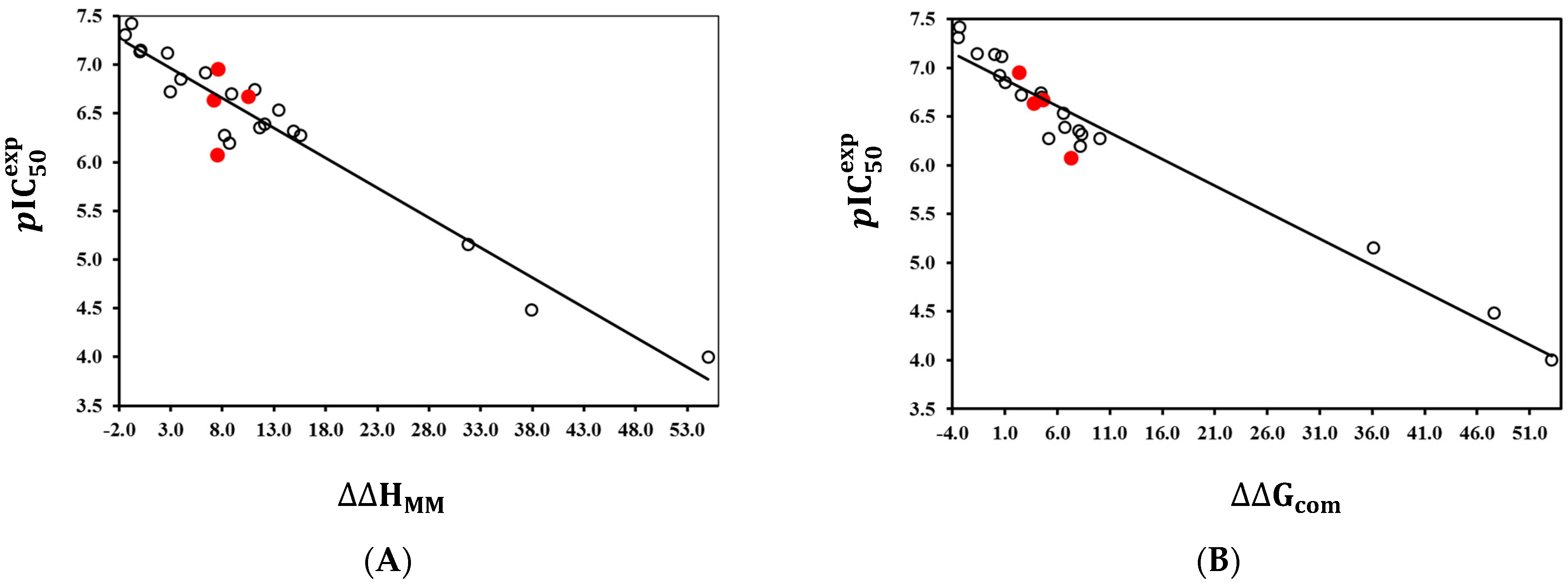
2.2. QSAR Model of PfENR Inhibition
2.2.1. Single-Descriptor QSAR Model of PfENR Inhibition by TCLs
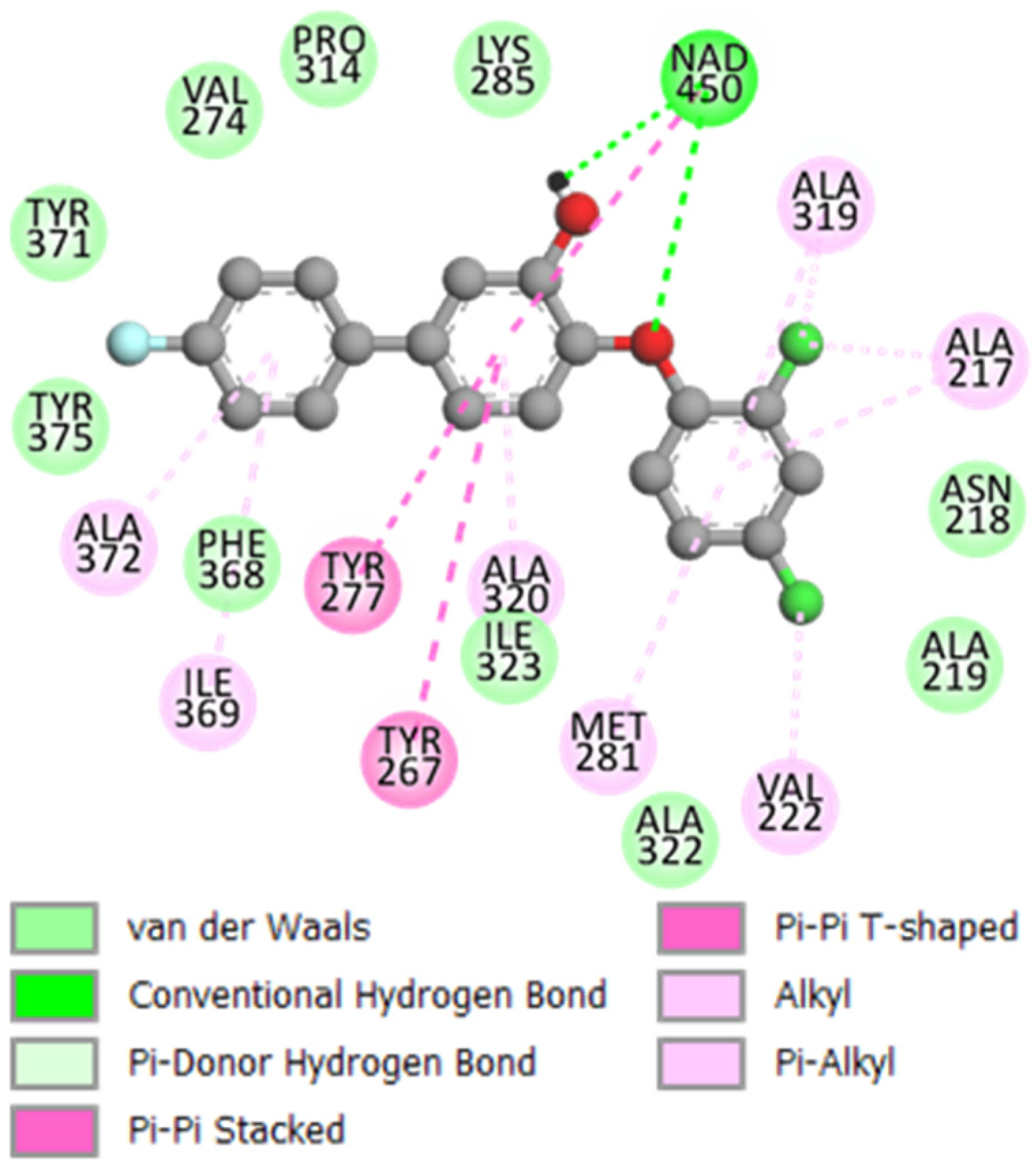
2.2.2. Binding Mode of TCLs to PfENR
2.3. 3D-QSAR Pharmacophore Model of PfENR Inhibition
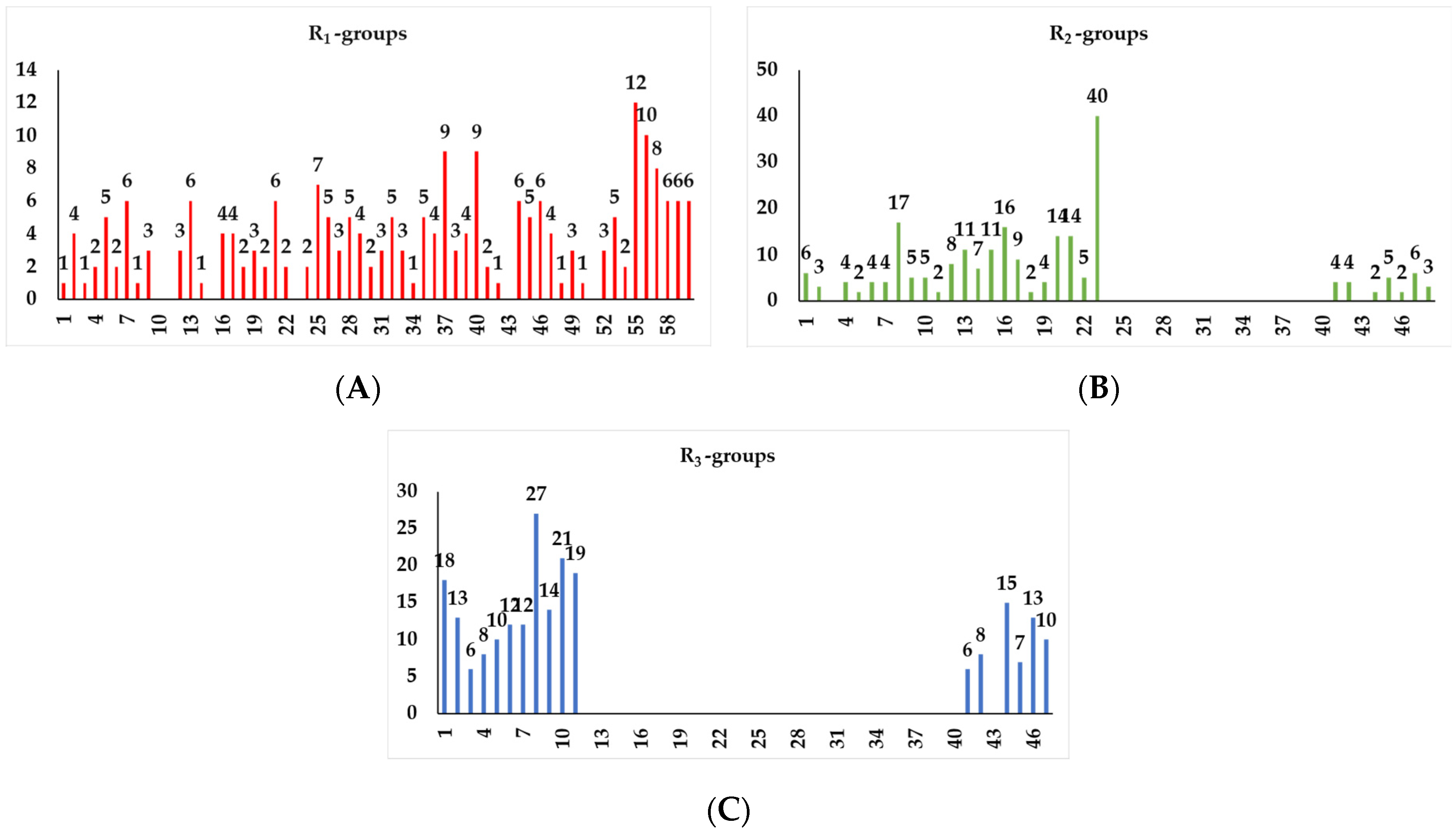
2.4. Virtual Library Generation and In Silico Screening of PfENR Inhibitors
2.4.1. Virtual Library
2.4.2. In Silico Screening
2.5. Novel TCLs against PfENR
2.6. Pharmacokinetic Profile of Novel TCL Analogues
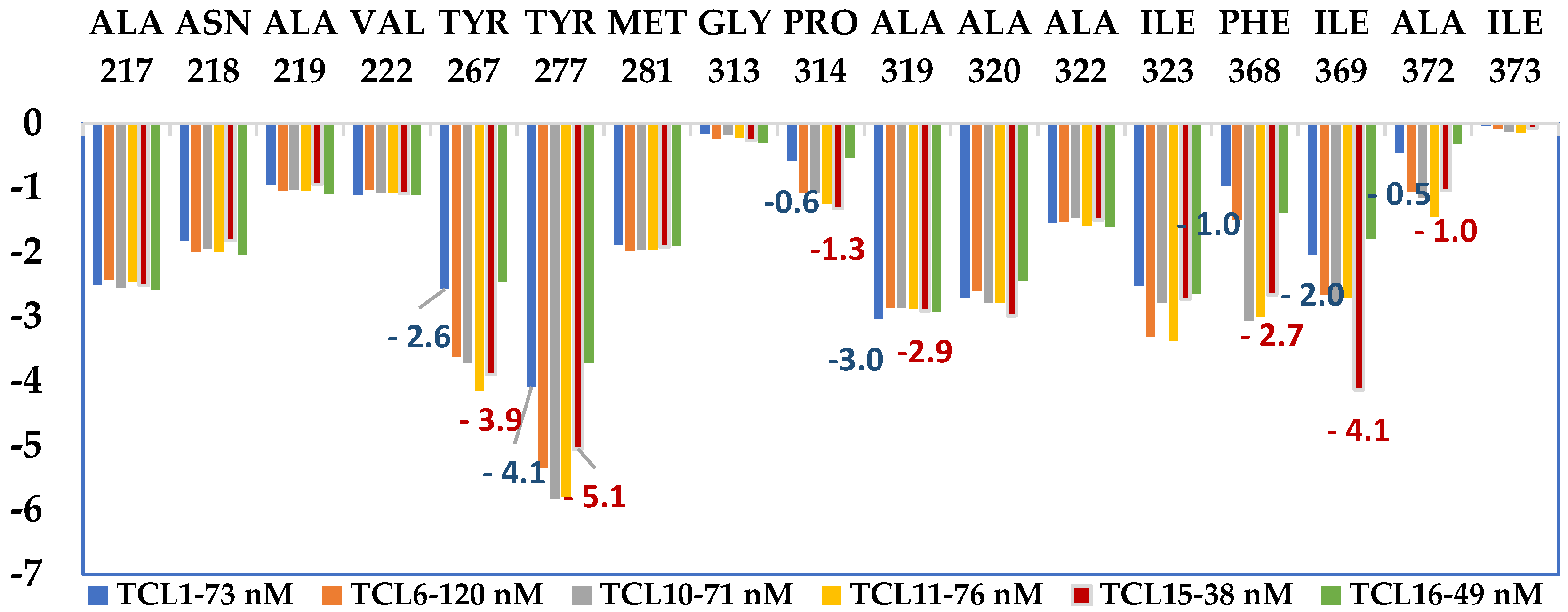
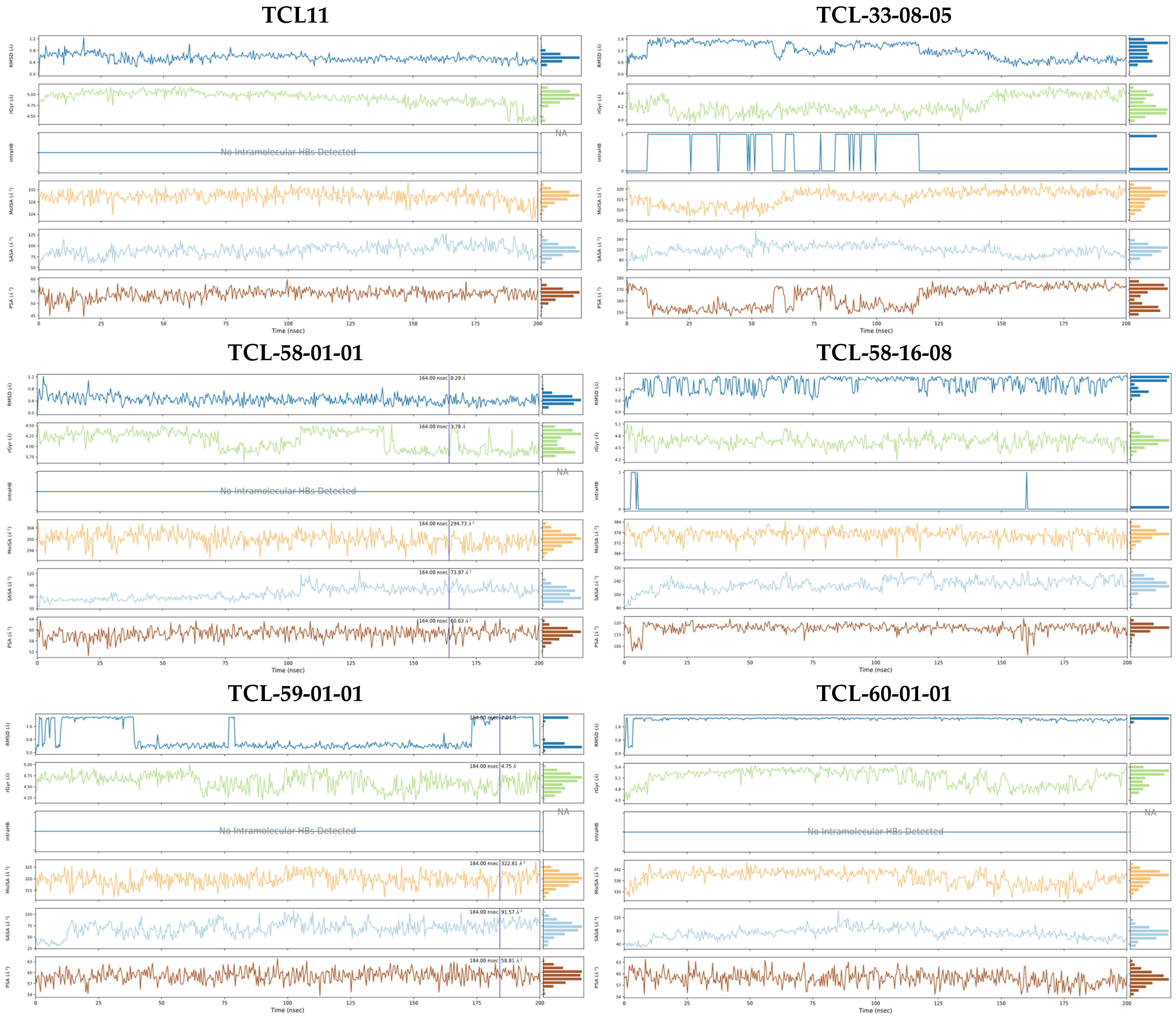
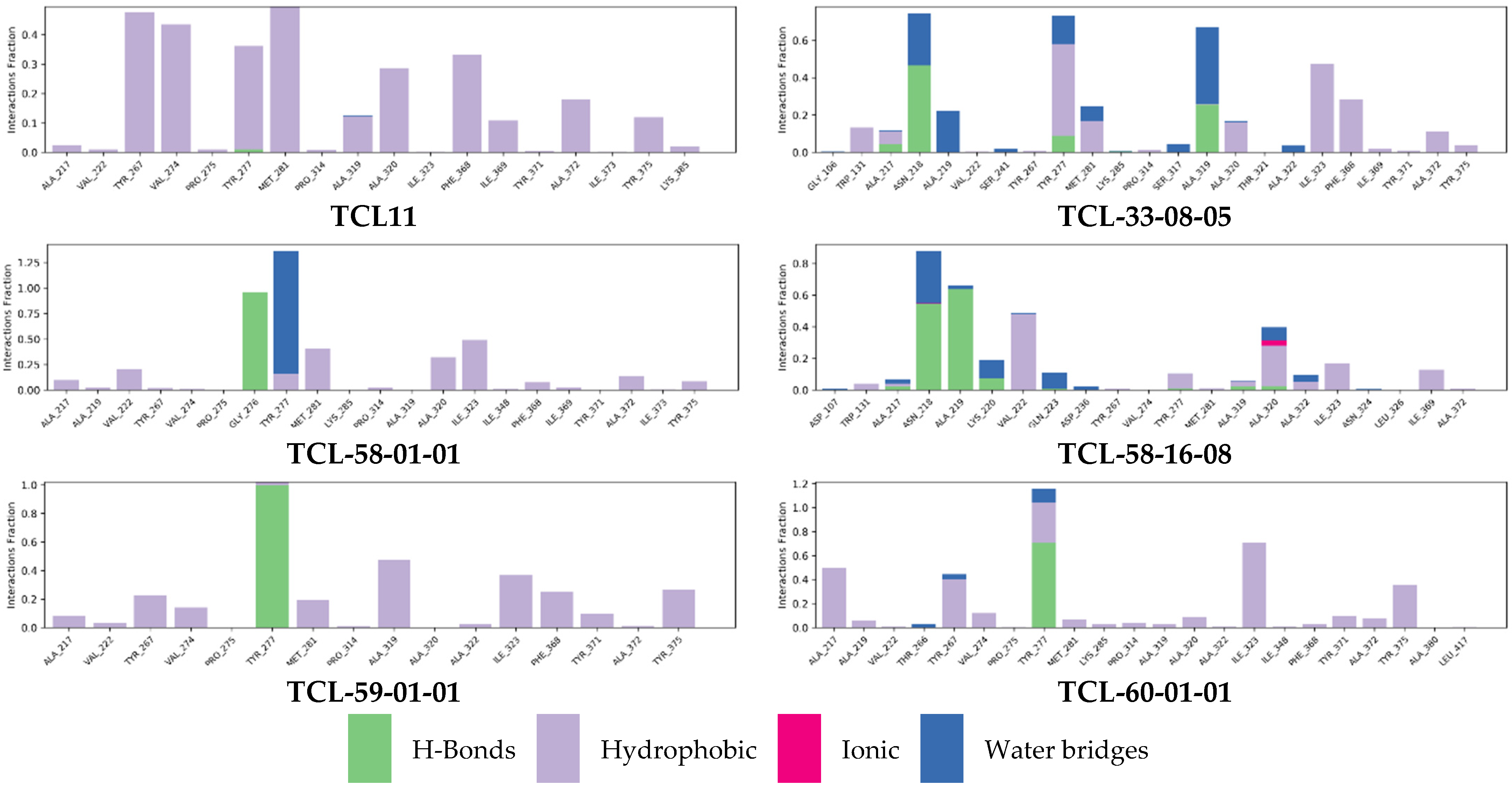
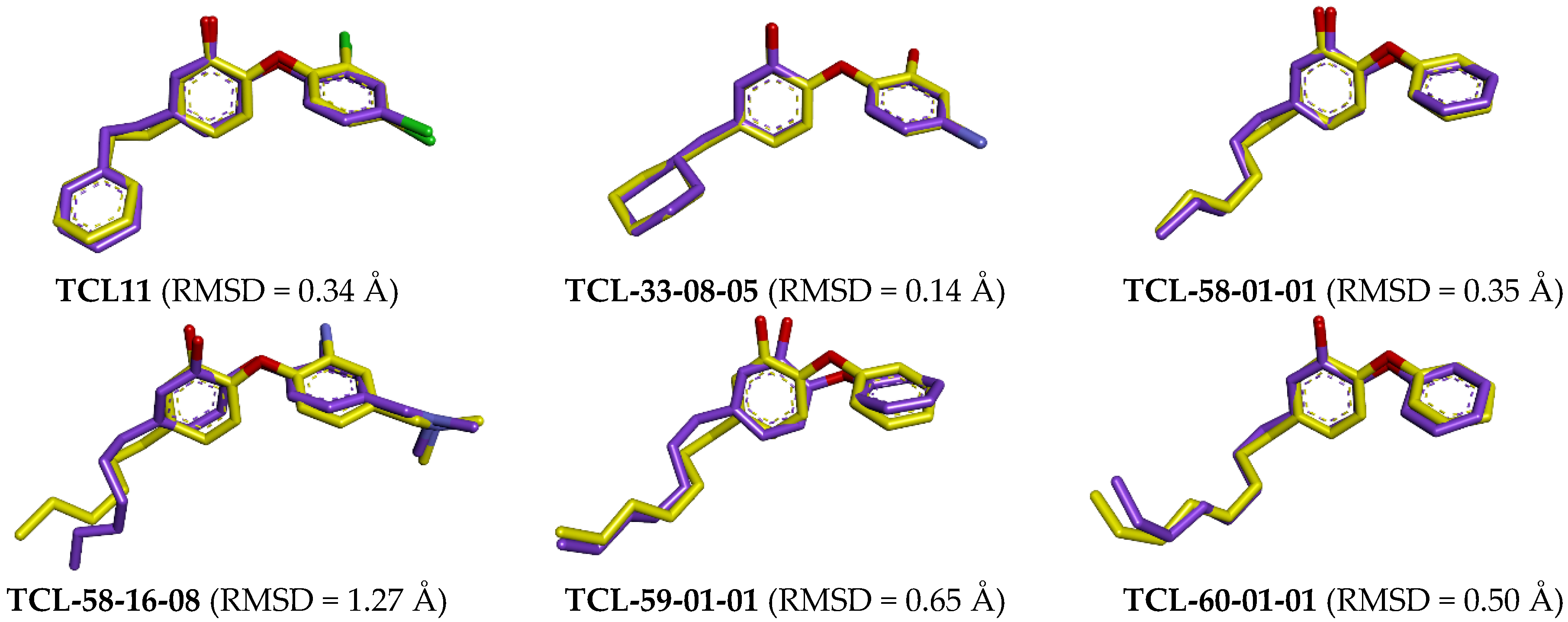
2.7. Molecular Dynamics Simulations
3. Discussion
3.1. Binding Mode of TCL
3.2. Molecular Dynamics Simulations
4. Materials and Methods
4.1. Training and Validation Sets of TCL Inhibitors
4.2. Model Building
4.3. Molecular Mechanics
4.4. Conformational Search
4.5. Solvation Gibbs Free Energies
4.6. Calculation of the Entropic Contribution
4.7. Calculation of Binding Affinity and QSAR Model
4.8. Interaction Energy
4.9. Pharmacophore (PH4) Generation
- -
- Cost(error) increases as the Root Mean Square difference between the estimated and experimental activities for the training set compounds;
- -
- increases in a Gaussian form as the feature weight deviates from an ideal value (2.0);
- -
- The configuration cost is a fixed cost depending on the complexity of the hypothesis space being optimised. It is equal to the entropy of the hypothesis space (log2P, P: the number of hypotheses initially created in a constructive phase that emerged through the subtractive one). In the standard HypoGen mode, its value should not exceed 17.
4.10. ADME Properties
4.11. Virtual Library Generation
4.12. ADME-Based Library Focussing
4.13. Pharmacophore-Based Library Searching
4.14. Inhibitory Potency Prediction
4.15. Molecular Dynamics Simulations
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- World Malaria Report 2021. World Health Organization. Geneva. 2021. Available online: https://apps.who.int/iris/handle/10665/350147 (accessed on 11 March 2023).
- Greenwood, B.; Mutabingwa, T. Malaria in 2002. Nature 2002, 415, 670–672. [Google Scholar] [CrossRef] [PubMed]
- Uwimana, A.; Legrand, E.; Stokes, B.H.; Ndikumana, J.M.; Umulisa, N.; Ngamije, D.; Munyaneza, T.; Mazarati, J.; Munguti, K.; Menard, A.M.D. Emergence and clonal expansion of in vitro artemisinin-resistant Plasmodium falciparum kelch13 R561H mutant parasites in Rwanda. Nat. Med. 2020, 6, 1602–1608. [Google Scholar] [CrossRef] [PubMed]
- Zhu, L.; Van der Pluijm, R.W.; Kucharski, M.; Nayak, S.; Tripathi, J.; White, N.J.; Day, N.P.J.; Faiz, A.; Phyo, A.P.; Amaratunga, C.; et al. Artemisinin resistance in the malaria parasite, Plasmodium falciparum, originates from its initial transcriptional response. Commun. Biol. 2022, 5, 274. [Google Scholar] [CrossRef] [PubMed]
- Paludisme: La Trithérapie Envisagée pour Freiner la Résistance aux Médicaments. Available online: https://www.scidev.net/afrique-sub-saharienne/news/la-tritherapie-envisagee-pour-freiner-la-resistance-aux-antipaludiques/ (accessed on 11 March 2023).
- Patrick, G.L. Antimalarial Agents Design and Mechanism of Action; Elsevier: Amsterdam, The Netherlands, 2020. [Google Scholar]
- Taft, C.A. Current Methods in Medicinal Chemistry And Biological Physics; Research Signpost: Thiruvananthapuram, India, 2008; Volume 2, 247p. [Google Scholar]
- Yu, M.; Kumar, T.R.S.; Nkrumah, L.J.; Copp, A.; Retzlaff, S.; Li, C.D.; Kelly, B.J.; Moura, P.A.; Lakshmanan, V.; Freundlich, J.S.; et al. The fatty acid biosynthesis enzyme FabI plays a key role in the development of liver-stage malarial parasites. Cell Host Microbe 2008, 4, 567–578. [Google Scholar] [CrossRef] [PubMed]
- ASingh, P.; Surolia, N.; Surolia, A. Triclosan inhibit the growth of the late liver-stage of Plasmodium. IUBMB Life 2009, 61, 923–928. [Google Scholar] [CrossRef]
- Ralph, S.A.; Van Dooren, G.G.; Waller, R.F.; Crawford, M.J.; Fraunholz, M.J.; Foth, B.J.; Tonkin, C.J.; Roos, D.S.; McFadden, G.I. Metabolic maps and functions of the Plasmodium falciparum apicoplast. Nature Rev. Microbiol. 2004, 2, 203–216. [Google Scholar] [CrossRef]
- Lindert, S.; Tallorin, L.C.; Nguyen, Q.G.; Burkart, M.D.; McCammon, J.A. In silico screening for Plasmodium falciparum Enoyl-ACP Reductase inhibitors. J. Comput. Aided Mol. Des. 2015, 29, 79–87. [Google Scholar] [CrossRef]
- Kapoor, M.; Dar, M.J.; Surolia, A.; Surolia, N. Kinetic Determinants of the Interaction of Enoyl-ACP Reductase from Plasmodium falciparum with Its Substrates and Inhibitors. Biochem. Biophys. Res. Commun. 2001, 289, 832–837. [Google Scholar] [CrossRef]
- Singh, P.; Srivastava, V. In-silico Identification of Triclosan Analogs as Novel Inhibitors of Enoyl-ACP Reductase from Plasmodium falciparum. Biosci. Biotech. Res. Comm. 2021, 14, 836–841. [Google Scholar] [CrossRef]
- Girling, D.J. The hepatic toxicity of antituberculosis regimens containing isoniazid, rifampicin and pyrazinamide. Tubercle 1977, 59, 13–32. [Google Scholar] [CrossRef]
- Asif, M. A review on potent antitubercular agent isoniazid and its analogues. Int. J. Pharm. Chem. 2013, 2, 110–120. [Google Scholar] [CrossRef]
- Surolia, N.; Surolia, A. Triclosan offers protection against blood stages of malaria by inhibiting enoyl-ACP reductase of Plasmodium falciparum. Nat. Med. 2001, 7, 167–173. [Google Scholar] [CrossRef]
- McLeod, R.; Muench, S.P.; Rafferty, J.B.; Kyle, D.E.; Mui, E.J.; Kirisits, M.J.; Mack, D.G.; Roberts, C.W.; Samuel, B.U.; Lyons, R.E.; et al. Triclosan inhibits the growth of Plasmodium falciparum and Toxoplasma gondii by inhibition of Apicomplexan Fab I. Int. J. Parasitol. 2001, 31, 109–113. [Google Scholar] [CrossRef]
- Perozzo, R.; Kuo, M.; Sidhu, A.B.S.; Valiyaveettil, J.T.; Bittman, R.; Jacobs, W.R.; Fidock, D.A.; Sacchettini, J.C. Structural Elucidation of the Specificity of the Antibacterial Agent Triclosan for Malarial Enoyl Acyl Carrier Protein Reductase. J. Biol. Chem. 2002, 277, 13106–13114. [Google Scholar] [CrossRef]
- Bhargava, H.N.; Leonard, P.A. Triclosan: Applications and safety. Am. J. Infect. Cont. 1996, 24, 209–218. [Google Scholar] [CrossRef]
- Weatherly, L.M.; Gosse, J.A. Triclosan Exposure, Transformation, and Human Health Effects. J. Toxicol. Environ. Health B Crit. Rev. 2017, 20, 447–469. [Google Scholar] [CrossRef]
- Freundlich, J.S.; Wang, F.; Tsai, H.-C.; Kuo, M.; Shieh, H.-M.; Anderson, J.W.; Nkrumah, L.J.; Valderramos, J.C.; Yu, M.; Kumar, T.R.S.; et al. X-ray Structural Analysis of Plasmodium falciparum Enoyl Acyl Carrier Protein Reductase as a Pathway toward the Optimization of Triclosan Antimalarial Efficacy. J. Biol. Chem. 2007, 282, 25436–25444. [Google Scholar] [CrossRef]
- Chhibber, M.; Kumar, G.; Parasuraman, P.; Ramya, T.N.C.; Surolia, N.; Surolia, A. Novel diphenyl ethers: Design, docking studies, synthesis and inhibition of enoyl ACP reductase of Plasmodium falciparum and Escherichia coli. Bioorg. Med. Chem. 2006, 14, 8086–8098. [Google Scholar] [CrossRef]
- Freundlich, J.S.; Yu, M.; Lucumi, E.; Kuo, M.; Tsai, H.-C.; Valderramos, J.-C.; Karagyozov, L.; Jacobs, W.R.; Schiehser, G.A.; Fidock, D.A.; et al. Sacchettini. Synthesis and biological activity of diaryl ether inhibitors of malarial enoyl acyl carrier protein reductase. Part 2: 2′-Substituted triclosan derivatives. Bioorg. Med. Chem. Lett. 2006, 16, 2163–2169. [Google Scholar] [CrossRef]
- Kuo, M.; Morbidoni, H.R.; Alland, D.; Sneddon, S.F.; Gourlie, B.B.; Staveski, M.M.; Leonard, M.; Gregory, J.S.; Janjigian, A.D.; Yee, C.; et al. Targeting Tuberculosis and Malaria through Inhibition of Enoyl Reductase. J. Biol. Chem. 2003, 278, 20851–20859. [Google Scholar] [CrossRef]
- Freundlich, J.S.; Wang, F.; Vilchèze, V.; Gulten, G.; Langley, R.; Schiehser, G.A.; Jacobus, D.P.; Jacobs, W.R., Jr.; Sacchettini, J.C. Triclosan Derivatives: Towards Potent Inhibitors of Drug-Sensitive and Drug-Resistant Mycobacterium tuberculosis. Chem. Med. Chem. 2009, 4, 241–248. [Google Scholar] [CrossRef] [PubMed]
- TDR Targets: Tropical Disease Research Is a Special Programme within World Health Organization Hosting a Database Facilitating Prioritization of Drug Targets. Available online: https://tdrtargets.org/targets (accessed on 11 March 2023).
- Freundlich, J.S.; Anderson, J.W.; Sarantakis, D.; Shieh, H.-M.; Yu, M.; Valderramos, J.-C.; Lucumi, E.; Kuo, M.; Jacobs, W.R.; Fidock, D.A.; et al. Sacchettiniet. Synthesis, biological activity, and X-ray crystal structural analysis of diaryl ether inhibitors of malarial enoyl acyl carrier protein reductase. Part 1: 4′-Substituted triclosan derivatives. Bioorg. Med. Chem. Lett. 2005, 15, 5247–5252. [Google Scholar] [CrossRef] [PubMed]
- Sullivan, T.J.; Truglio, J.J.; Boyne, M.E.; Novichenok, P.; Zhang, X.; Stratton, C.F.; Li, H.-J.; Kaur, T.; Amin, A.; Johnson, F.; et al. High Affinity InhA Inhibitors with Activity against Drug-Resistant Strains of Mycobacterium tuberculosis. ACS Chem. Biol. 2006, 1, 43–53. [Google Scholar] [CrossRef] [PubMed]
- Mishra, S.; Karmodiya, K.; Parasuraman, P.; Surolia, A. Design, synthesis, and application of novel triclosan prodrugs as potential antimalarial and antibacterial agents. Bioorg. Med. Chem. 2008, 16, 5536–5546. [Google Scholar] [CrossRef] [PubMed]
- Shah, P.; Siddiqi, M.I. 3D-QSAR studies on triclosan derivatives as Plasmodium falciparum enoyl acyl carrier reductase inhibitors. SAR QSAR Environ. Res. 2010, 21, 527–545. [Google Scholar] [CrossRef]
- Kumar, S.P.; George, L.B.; Jasrai, Y.T.; Pandya, H.A. Prioritization of active antimalarials using structural interaction profile of Plasmodium falciparum enoyl-acyl carrier protein reductase (PfENR)-triclosan derivatives. SAR QSAR Environ. Res. 2015, 26, 61–77. [Google Scholar] [CrossRef]
- Frecer, V.; Megnassan, E.; Miertus, S. Design and in silico screening of combinatorial library of antimalarial analogs of triclosan inhibiting Plasmodium falciparum enoyl-acyl carrier protein reductase. Eur. J. Med. Chem. 2009, 44, 3009–3019. [Google Scholar] [CrossRef]
- Salifu, E.Y.; Abugri, J.; Rashid, I.A.; Osei, F.; Ayariga, J.A. In silico identification of potential inhibitors of acyl carrier protein reductase and acetyl CoA carboxylase of Plasmodium falciparum in antimalarial therapy. Front. Drug Discov. 2023, 3, 1. [Google Scholar] [CrossRef]
- Accelrys Inc. Discovery Studio Molecular Modeling and Simulation Program, Release 2.5; Accelrys Inc.: San Diego, CA, USA, 2009.
- Kouassi, A.F.; Kone, M.; Keita, M.; Esmel, A.; Megnassan, E.; N’Guessan, Y.T.; Frecer, V.; Miertus, S. Computer-Aided Design of Orally Bioavailable Pyrrolidine Carboxamide Inhibitors of Enoyl-Acyl Carrier Protein Reductase of Mycobacterium tuberculosis with Favorable Pharmacokinetic Profiles. Int. J. Mol. Sci. 2015, 16, 29744–29771. [Google Scholar] [CrossRef]
- John, S.; Thangapandian, S.; Sakkiah, S.; Lee, K.W. Potent BACE-1 inhibitor design using pharmacophore modeling, in silico screening and molecular docking studies. BMC Bioinform. 2011, 12, S28. [Google Scholar] [CrossRef]
- OECD. Guidance Document on the Validation of (Quantitative) Structure-Activity Relationship [(Q)SAR] Models. 2014. Available online: https://www.oecd.org/chemicalsafety/risk-assessment/validationofqsarmodels.htm (accessed on 11 March 2023).
- BIOVIA. Available Chemicals Directory (ACD). 2022. Available online: https://www.psds.ac.uk/acd (accessed on 11 March 2023).
- Schrödinger. QikProp, 6.5 (Release 139); Schrödinger LLC: New York, NY, USA, 2019.
- Jorgensen, W.L.; Duffy, E.M. Prediction of drug solubility from Monte Carlo simulations. Bioorg. Med. Chem. Lett. 2000, 10, 1155–1158. [Google Scholar] [CrossRef]
- Jorgensen, W.L.; Duffy, E.M. Prediction of drug solubility from structure. Adv. Drug Deliv. Rev. 2002, 54, 355–366. [Google Scholar] [CrossRef]
- Maestro-Desmond Interoperability Tools. In Desmond Molecular Dynamics System; Version, 3.6; Maestro-Desmond Interoperability Tools, Schrödinger; D. E. Shaw Research: New York, NY, USA, 2021.
- Berman, H.M.; Westbrook, J.; Feng, Z.; Gilliland, G.; Bhat, T.N.; Weissig, H.; Shindyalov, I.N.; Bourne, P.E. The Protein Data Bank. Nucleic Acids Res. 2000, 28, 235–242. [Google Scholar] [CrossRef]
- Accelrys Inc. Insight-II and Discover Molecular Modeling and Simulation Package; Release 2005; Accelrys Inc.: San Diego, CA, USA, 2005. [Google Scholar]
- Owono, L.C.; Keita, M.; Megnassan, E.; Frecer, V.; Miertus, S. Design of Thymidine Analogues Targeting Thymidilate Kinase of Mycobacterium tuberculosis. Tuberc. Res. Treat. 2013, 2013, 670836. [Google Scholar] [CrossRef]
- Frecer, V.; Miertus, S.; Tossi, A.; Romeo, D. Rational design of inhibitors for drug resistant HIV-1 aspartic protease mutants. Drug Des. Disc. 1998, 15, 211–231. [Google Scholar]
- Frecer, V.; Seneci, P.; Miertus, S. Computer-assisted combinatorial design of bicyclic thymidine analogs as inhibitors of Mycobacterium tuberculosis thymidine monophosphate kinase. J. Comput. Aidded Mol. Des. 2011, 25, 31–49. [Google Scholar] [CrossRef]
- Frecer, V.; Berti, F.; Benedetti, F.; Miertus, S. Design of peptidomimetic inhibitors of aspartic protease of HIV-1 containing –PheΨPro– core and displaying favourable ADME-related properties. J. Mol. Graph. Model. 2008, 27, 376–387. [Google Scholar] [CrossRef]
- Dali, B.; Keita, M.; Megnassan, E.; Frecer, V.; Miertus, S. Insight into Selectivity of Peptidomimetic Inhibitors with Modified Statine Core for Plasmepsin II of Plasmodium falciparum over Human Cathepsin D. Chem. Biol. Drug Des. 2012, 79, 411–430. [Google Scholar] [CrossRef]
- Frecer, V.; Majekova, M.; Miertus, S. Approximate methods for solvent effect calculations on biomolecules. J. Mol. Struct. THEOCHEM 1989, 52, 403–419. [Google Scholar] [CrossRef]
- Gilson, M.K.; Honig, B. The inclusion of electrostatic hydration energies in molecular mechanics calculations. J. Comput. Aid. Mol. Des. 1991, 5, 5–20. [Google Scholar] [CrossRef]
- Rocchia, W.; Sridharan, S.; Nicholls, A.; Alexov, E.; Chiabrera, A.; Honig, B. Rapid grid-based construction of the mo-lecular surface and the use of induced surface charge to calculate reaction field energies: Applications to the molecular systems and geometric objects. J. Comput. Chem. 2002, 23, 128–137. [Google Scholar] [CrossRef] [PubMed]
- Böttcher, C.J.F. Theory of Electric Polarization; Elsevier: Amsterdam, The Netherlands, 1973. [Google Scholar]
- Miertus, S.; Scrocco, E.; Tomasi, J. Electrostatic interaction of a solute with a continuum. A direct utilizaion of ab initio molecular potentials for the prevision of solvent effects. Chem. Phys. 1981, 55, 117–129. [Google Scholar] [CrossRef]
- Frecer, V.; Miertus, S. Polarizable continuum model of solvation for biopolymers. Int. J. Quant. Chem. 1992, 42, 1449–1468. [Google Scholar] [CrossRef]
- Fischer, S.; Smith, J.C.; Verma, C.S. Dissecting the vibrational entropy change on protein/ligand binding: Burial of a water molecule in bovine pancreatic trypsin inhibitor. J. Phys. Chem. B 2001, 105, 8050–8055. [Google Scholar] [CrossRef]
- Wermuth, C.G.; Ganellin, C.R.; Lindberg, P.; Mitscher, L.A. Glossary of terms used in medicinal chemistry (IUPAC recommendations 1998). Pure Appl. Chem. 1998, 70, 1129–1143. [Google Scholar] [CrossRef]
- Güner, O.F.; Bowen, J.P. Setting the record straight: The origin of the pharmacophore concept. J. Chem. Inf. Model. 2014, 54, 1269–1283. [Google Scholar] [CrossRef]
- Schueler, F.W. Chemobiodynamics and Drug Design; McGraw-Hill: New York, NY, USA, 1960. [Google Scholar]
- Güner, O.F. History and Evolution of the Pharmacophore Concept in Computer-Aided Drug Design. Curr. Med. Chem. 2002, 2, 1321–1332. [Google Scholar] [CrossRef]
- Li, H.; Sutter, J.; Hoffmann, R. Pharmacophore Perception, Development and Use in Drug Design; Güner, O.F., Ed.; International University Line: La Jolla, CA, USA, 2000; pp. 171–189. [Google Scholar]
- Kurogi, Y.; Güner, O.F. Pharmacophore Modeling and Three-dimensional Database Searching for Drug Design Using Catalyst. Curr. Med. Chem. 2001, 8, 1035–1055. [Google Scholar] [CrossRef]
- Binyamin, H.; Senderowitz, H. Photovoltaphores: Pharmacophore models for identifying metal-free dyes for dye-sensitized solar cells. NPJ Comput. Mater. 2022, 8, 142. [Google Scholar] [CrossRef]
- Molecular Operating Environment (MOE), Version 2014; Chemical Computing Group Inc.: Montreal, QC, Canada, 2014.
- Frecer, V.; Miertus, S. Design, structure-based focusing and in silico screening of combinatorial library of peptidomimetic inhibitors of dengue virus NS2B-NS3 protease. J. Comput. Aided Mol. Des. 2010, 24, 195–212. [Google Scholar] [CrossRef]
- Willett, P. Molecular Similarity in Drug Design; Dean, P.M., Ed.; Chapman and Hall: Glasgow, Scotland, 1994; pp. 110–137. [Google Scholar]
- Frecer, V.; Miertus, S. Antiviral agents against COVID-19: Structure-based design of specific peptidomimetic inhibitors of SARS-CoV-2 main protease. RSC Adv. 2020, 10, 40244–40263. [Google Scholar] [CrossRef]
- Lu, C.; Wu, C.; Ghoreishi, D.; Chen, W.; Wang, L.; Damm, W.; Ross, G.A.; Dahlgren, M.K.; Russell, E.; Von Bargen, C.D.; et al. OPLS4: Improving Force Field Accuracy on Challenging Regimes of Chemical Space. J. Chem. Theor. Comput. 2021, 17, 4291–4300. [Google Scholar] [CrossRef]
- Martyna, G.J.; Klein, M.L.; Tuckerman, M. Nosé–Hoover chains: The canonical ensemble via continuous dynamics. J. Chem. Phys. 1992, 97, 2635–2643. [Google Scholar] [CrossRef]
- Martyna, G.J.; Tobias, D.J.; Klein, M.L. Constant pressure molecular dynamics algorithms. J. Chem. Phys. 1994, 101, 4177–4189. [Google Scholar] [CrossRef]















{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
 | ||||
|---|---|---|---|---|
| Training Set (TS) | [nM] | |||
| TCL1 (Triclosan) | 73 | |||
| TCL2 | 200 | |||
| TCL3 | 530 | |||
| TCL4 | 480 | |||
| TCL5 | 180 | |||
| TCL6 | 120 | |||
| TCL7 | 290 | |||
| TCL8 | 640 | |||
| TCL9 | 530 | |||
| TCL10 | 71 | |||
| TCL11 | 76 | |||
| TCL12 | 140 | |||
| TCL13 | 440 | |||
| TCL14 | 410 | |||
| TCL15 | 38 | |||
| TCL16 | 49 | |||
| TCL17 | 190 | |||
| TCL18 | … | …NH2 | 7000 | |
| TCL19 | …2H-tetrazol-5-yl | … Cl | … Cl | 100,000 |
| TCL20 | … 3-pyridyl | …Cl | …Cl | 33,000 |
| Validation Set (VS) | [nM] | |||
| TCL21 | 210 | |||
| TCL22 | 110 | |||
| TCL23 | 840 | |||
| TCL24 | 230 | |||
| Training Set a | b | c | d | e | f, g |
|---|---|---|---|---|---|
| TCL1 | 0 | 0 | 0 | 0 | 73 |
| TCL2 | 8.88 | −2.30 | 2.06 | 4.52 | 200 |
| TCL3 | 15.58 | −0.16 | 5.38 | 10.04 | 530 |
| TCL4 | 14.87 | −2.65 | 3.90 | 8.31 | 480 |
| TCL5 | 11.16 | −2.60 | 4.16 | 4.40 | 180 |
| TCL6 | 6.35 | −0.94 | 4.90 | 0.51 | 120 |
| TCL7 | 13.45 | −2.34 | 4.53 | 6.58 | 290 |
| TCL8 | 8.68 | 0.12 | 0.62 | 8.17 | 640 |
| TCL9 | 8.21 | −0.20 | 2.86 | 5.15 | 530 |
| TCL10 | 0.11 | 0.62 | 2.38 | −1.65 | 71 |
| TCL11 | 2.71 | 0.19 | 2.25 | 0.66 | 76 |
| TCL12 | 4.01 | 0.03 | 2.99 | 1.04 | 140 |
| TCL13 | 11.64 | −1.93 | 1.69 | 8.02 | 440 |
| TCL14 | 12.08 | −1.21 | 4.18 | 6.69 | 410 |
| TCL15 | −0.78 | −0.73 | 1.81 | −3.33 | 38 |
| TCL16 | −1.38 | −1.61 | 0.46 | −3.44 | 49 |
| TCL17 | 2.97 | −0.45 | −0.04 | 2.56 | 190 |
| TCL18 | 31.80 | 6.56 | 2.24 | 36.13 | 7 000 |
| TCL19 | 55.03 | −0.34 | 1.58 | 53.11 | 100,000 |
| TCL20 | 37.93 | 12.97 | 3.3 | 47.61 | 33,000 |
| Validation Set a | b | c | d | e | f |
| TCL21 | 10.47 | −1.95 | 3.94 | 4.75 | 1.00 |
| TCL22 | 7.53 | −1.41 | 3.80 | 2.32 | 1.02 |
| TCL23 | 7.51 | 0.89 | 1.14 | 7.26 | 0.93 |
| TCL24 | 7.17 | −1.19 | 2.29 | 3.70 | 0.99 |
| Statistical Data of Regression | ||
|---|---|---|
| Number of compounds in TS | 20 | 20 |
| Squared correlation coefficient of regression | 0.94 | 0.95 |
| Cross-validated squared correlation coefficient | 0.92 | 0.93 |
| Standard error of regression σ | 0.21 | 0.18 |
| Statistical significance of regression, Fisher F-test | 327.3 | 426.16 |
| Level of statistical significance α | >95% | >95% |
| Range of experimental activities [nM] | 38–100,000 | |
| Hypothesis | RMSD a | b | Total Cost c | Costs Difference d | Closest Random e | Features f |
|---|---|---|---|---|---|---|
| Hypo1 | 6.565 | 0.96 | 409.3 | 5013.9 | 1021.7 | HBA, HYD-AL, HYD-AL, HYD-Ar, HYD |
| Hypo2 | 9.310 | 0.93 | 803.5 | 4619.7 | 1035.5 | HBA, HYD-AL, HYD-AL, HYD, HYD |
| Hypo3 | 9.428 | 0.92 | 822.7 | 4600.5 | 1058.9 | HBA, HYD-AL, HYD-Ar, HYD, HYD |
| Hypo4 | 9.587 | 0.92 | 850.3 | 4572.9 | 1068.1 | HBA, HYD-AL, HYD, HYD, HYD |
| Hypo5 | 10.776 | 0.90 | 1069.8 | 4353.4 | 1076.6 | HYD-AL, HYD-AL, HYD-Ar, HYD-Ar, HYD |
| Hypo6 | 10.780 | 0.90 | 1071.3 | 4352.0 | 1079.7 | HYD-AL, HYD-AL, HYD-Ar, HY, HY |
| Hypo7 | 18.776 | 0.64 | 3195.9 | 2227.3 | 1088.2 | HYD-AL, HYD-Ar, HYD-Ar, HY, HY |
| Hypo8 | 11.420 | 0.89 | 1195.3 | 4227.9 | 1090.9 | HBA, HYD-AL, HY, HYD, HYD |
| Hypo9 | 18.835 | 0.64 | 3216.9 | 2206.4 | 1099.1 | HYD-AL, HYD-Ar, HYD, HYD, HYD |
| Hypo10 | 11.583 | 0.88 | 1231.8 | 4191.4 | 1102.3 | HBA, HYD-AL, HYD-Ar, HYD, HYD |
 | |||||
|---|---|---|---|---|---|
| R-Groups | |||||
| 1 |  | 2 |  | 3 |  |
| 4 |  | 5 |  | 6 |  |
| 7 |  | 8 |  | 9 |  |
| 10 |  | 11 |  | 12 |  |
| 13 |  | 14 |  | 15 |  |
| 16 |  | 17 |  | 18 |  |
| 19 |  | 20 |  | 21 |  |
| 22 |  | 23 |  | 24 |  |
| 25 |  | 26 |  | 27 |  |
| 28 |  | 29 |  | 30 |  |
| 31 |  | 32 |  | 33 |  |
| 34 |  | 35 |  | 36 |  |
| 37 |  | 38 |  | 39 |  |
| 40 |  | 41 |  | 42 |  |
| 43 |  | 44 |  | 45 |  |
| 46 |  | 47 |  | 48 |  |
| 49 |  | 50 |  | 51 |  |
| 52 |  | 53 |  | 54 |  |
| 55 |  | 56 |  | 57 |  |
| 58 |  | 59 |  | 60 |  |
| No. | TCL Analogue | a | b | c | d | e | f [nM] |
|---|---|---|---|---|---|---|---|
| Ref. | TCL1 TCL-03-03-03 | 0 | 0 | 0 | 0 | 7.14 * | 73 * |
| 1 | TCL-58-01-01 | −17.8 | 8.1 | 7.6 | −17.3 | 7.87 | 13.4 |
| 2 | TCL-59-01-01 | −20.6 | 8.1 | 6.5 | −19.0 | 7.97 | 10.8 |
| 3 | TCL-60-01-01 | −21.8 | 8.9 | 7.5 | −20.4 | 8.04 | 9.1 |
| 8 | TCL-26-16-01 | −20.5 | 9.4 | 6.5 | −17.6 | 7.89 | 12.8 |
| 47 | TCL-33-08-05 | −18.9 | 0.4 | 8.3 | −26.7 | 8.39 | 4.1 |
| 51 | TCL-59-08-05 | −13.8 | 3.0 | 6.5 | −17.3 | 7.87 | 13.4 |
| 52 | TCL-60-08-05 | −15.2 | 2.6 | 6.2 | −18.8 | 7.96 | 11.1 |
| 85 | TCL-60-08-08 | −49.9 | 25.8 | 9.0 | −33.0 | 8.73 | 1.9 |
| 91 | TCL-33-16-08 | −15.45 | 2.28 | 12.14 | −25.3 | 8.31 | 4.9 |
| 92 | TCL-58-16-08 | −11.3 | 3.2 | 12.2 | −20.3 | 8.04 | 9.2 |
| 94 | TCL-60-16-08 | −14.8 | 7.3 | 12.1 | −19.6 | 8.00 | 10.1 |
| 112 | TCL-55-19-09 | 0.8 | −2.7 | 14.2 | −16.1 | 7.81 | 15.5 |
| 118 | TCL-58-23-09 | −29.2 | 11.2 | 11.2 | −29.2 | 8.52 | 3.1 |
| 119 | TCL-59-23-09 | −11.6 | 6.1 | 12.0 | −17.4 | 7.88 | 13.2 |
| 135 | TCL-36-23-10 | −17.3 | 5.1 | 6.2 | −18.4 | 7.93 | 11.6 |
| 154 | TCL-56-17-11 | −12.5 | 4.5 | 11.7 | −19.6 | 8.00 | 10.1 |
| 155 | TCL-35-20-11 | −4.3 | −1.7 | 11.6 | −17.7 | 7.90 | 12.7 |
| 191 | TCL-32-08-45 | −17.1 | 3.5 | 4.3 | −18.0 | 7.91 | 12.2 |
| 200 | TCL-56-16-46 | −14.0 | 4.2 | 7.6 | −17.4 | 7.88 | 13.2 |
| 205 | TCL-56-23-46 | −21.7 | 6.1 | 7.1 | −22.7 | 8.17 | 6.8 |
| Analogues a | # Star b | Smol d | Smol, hfo e | Vm f | RotB g | HBD h | HBA i | logPo/w j | logSwat k | logKHSA l | logB/B m | BIPcaco n | # Meta o | PfENR [nM] | HOA q | %HOA r | |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| 58-01-01 | 1 | 270 | 592.4 | 259.8 | 1009.0 | 8 | 1.0 | 1.3 | 5.0 | −5.2 | 0.8 | −0.5 | 2933.7 | 2 | 13.3 | 3 | 100 |
| 59-01-01 | 1 | 284 | 622.3 | 291.2 | 1070.5 | 9 | 1.0 | 1.3 | 5.4 | −5.7 | 0.9 | −0.5 | 2936.4 | 2 | 10.8 | 3 | 100 |
| 60-01-01 | 1 | 298 | 654.7 | 322.1 | 1126.8 | 10 | 1.0 | 1.3 | 5.8 | −6.1 | 1.0 | −0.6 | 2933.7 | 2 | 9.1 | 3 | 100 |
| 26-16-01 | 0 | 337 | 652.3 | 181.2 | 1130.1 | 6 | 1.0 | 3.3 | 4.7 | −5.0 | 0.8 | 0.2 | 759.3 | 3 | 12.9 | 3 | 100 |
| 29-16-01 | 0 | 320 | 635.4 | 181.3 | 1099.0 | 6 | 1.0 | 4.8 | 3.5 | −3.9 | 0.4 | −0.2 | 416.7 | 3 | 22.0 | 3 | 94 |
| 02-21-01 | 1 | 274 | 557.6 | 218.1 | 953.5 | 6 | 1.0 | 1.3 | 4.8 | −5.2 | 0.7 | −0.2 | 3200.1 | 2 | 20.2 | 3 | 100 |
| 05-21-01 | 0 | 272 | 562.4 | 218.0 | 961.2 | 7 | 2.0 | 2.0 | 4.7 | −4.3 | 0.4 | −0.9 | 965.7 | 3 | 18.5 | 3 | 100 |
| 55-23-01 | 1 | 411 | 787.1 | 107.3 | 1375.0 | 10 | 3.0 | 3.5 | 5.3 | −6.0 | 1.0 | −1.1 | 168.6 | 5 | 38.2 | 2 | 85 |
| 56-04-02 | 0 | 353 | 674.0 | 194.7 | 1166.8 | 8 | 2.0 | 5.8 | 3.2 | −4.1 | 0.3 | −0.7 | 181.1 | 6 | 73.3 | 3 | 86 |
| 40-23-02 | 3 | 448 | 809.8 | 106.3 | 1416.7 | 9 | 2.0 | 2.8 | 6.9 | −7.6 | 1.5 | −0.1 | 562.2 | 4 | 32.1 | 1 | 100 |
| 55-45-02 | 0 | 383 | 704.6 | 199.7 | 1228.9 | 10 | 3.0 | 4.3 | 4.0 | −4.5 | 0.5 | −0.8 | 183.5 | 6 | 35.5 | 3 | 91 |
| 34-14-03 | 0 | 341 | 629.0 | 88.1 | 1078.7 | 7 | 3.0 | 3.8 | 3.2 | −3.9 | 0.3 | −0.7 | 115.8 | 6 | 67.2 | 3 | 83 |
| 38-10-04 | 0 | 390 | 735.5 | 353.2 | 1292.5 | 8 | 3.0 | 4.5 | 4.3 | −5.1 | 0.7 | −0.5 | 333.5 | 8 | 67.2 | 3 | 100 |
| 57-20-04 | 0 | 392 | 776.1 | 391.3 | 1373.8 | 10 | 2.0 | 5.3 | 4.6 | −5.5 | 0.8 | −0.8 | 274.6 | 8 | 30.5 | 3 | 100 |
| 33-08-05 | 0 | 313 | 607.7 | 243.6 | 1060.9 | 7 | 3.5 | 3.0 | 3.2 | −4.5 | 0.4 | −1.5 | 265.6 | 4 | 4.1 | 3 | 89 |
| 58-08-05 | 0 | 301 | 624.3 | 259.8 | 1066.5 | 10 | 3.5 | 3.0 | 3.2 | −4.3 | 0.2 | −1.8 | 236.0 | 4 | 19.4 | 3 | 88 |
| 59-08-05 | 0 | 315 | 656.9 | 291.5 | 1126.4 | 11 | 3.5 | 3.0 | 3.6 | −4.8 | 0.3 | −1.9 | 244.3 | 4 | 13.3 | 3 | 90 |
| 60-08-05 | 0 | 329 | 686.6 | 322.1 | 1184.3 | 12 | 3.5 | 3.0 | 3.9 | −5.1 | 0.5 | −2.0 | 236.0 | 4 | 11.1 | 3 | 92 |
| 37-20-06 | 0 | 394 | 758.8 | 256.5 | 1334.3 | 10 | 2.8 | 3.3 | 5.6 | −6.0 | 1.0 | −0.2 | 560.6 | 6 | 51.6 | 3 | 96 |
| 49-16-07 | 0 | 423 | 708.2 | 180.3 | 1231.8 | 7 | 1.0 | 4.8 | 4.4 | −6.2 | 0.7 | −0.6 | 184.3 | 3 | 26.2 | 3 | 93 |
| 32-19-07 | 1 | 357 | 711.7 | 227.3 | 1248.1 | 8 | 1.0 | 2.8 | 5.6 | −8.0 | 1.2 | −1.2 | 729.9 | 3 | 41.7 | 1 | 100 |
| 58-08-08 | 0 | 302 | 628.7 | 259.8 | 1074.4 | 10 | 4 | 3.5 | 3.0 | −4.2 | 0.2 | −1.9 | 202.2 | 5 | 89.6 | 3 | 85 |
| 59-08-08 | 0 | 316 | 692.1 | 310.1 | 1164.7 | 11 | 4 | 3.5 | 3.6 | −5.2 | 0.3 | −2.0 | 223.4 | 5 | 40.2 | 3 | 89 |
| 60-08-08 | 0 | 330 | 690.1 | 322.1 | 1190.5 | 12 | 4 | 3.5 | 3.7 | −5.0 | 0.4 | −2.1 | 206.6 | 5 | 1.9 | 3 | 90 |
| 58-16-08 | 0 | 343 | 702.5 | 426.6 | 1230.8 | 11 | 2.5 | 4.3 | 3.7 | −4.2 | 0.5 | −1.0 | 169.6 | 6 | 9.2 | 3 | 88 |
| 59-16-08 | 0 | 357 | 756.6 | 479.9 | 1321.3 | 12 | 2.5 | 4.3 | 4.4 | −5.0 | 0.7 | −1.0 | 219.6 | 6 | 22.6 | 3 | 94 |
| 60-16-08 | 0 | 371 | 763.8 | 488.9 | 1347.0 | 13 | 2.5 | 4.3 | 4.5 | −5.0 | 0.8 | −1.2 | 173.2 | 6 | 10.0 | 3 | 93 |
| 40-13-09 | 0 | 428 | 777.9 | 268.0 | 1377.1 | 11 | 2.8 | 3.3 | 6.1 | −6.5 | 1.1 | −0.1 | 616.6 | 7 | 38.2 | 1 | 100 |
| 54-20-09 | 3 | 422 | 818.2 | 402.0 | 1465.9 | 11 | 2.0 | 2.8 | 6.7 | −7.2 | 1.5 | −0.3 | 606.3 | 6 | 16.9 | 1 | 100 |
| 32-17-10 | 0 | 339 | 669.4 | 453.1 | 1201.7 | 6 | 2.0 | 2.3 | 5.3 | −6.4 | 1.1 | −0.6 | 1787.7 | 6 | 57.1 | 1 | 100 |
| 07-22-10 | 0 | 310 | 650.9 | 307.0 | 1111.8 | 8 | 2.0 | 3.8 | 3.7 | −6.3 | 0.5 | −1.5 | 410.8 | 5 | 22.3 | 1 | 95 |
| 58-23-09 | 4 | 374 | 739.2 | 355.2 | 1326.4 | 10 | 1 | 1.3 | 7.2 | −7.7 | 1.6 | −0.5 | 3666.3 | 3 | 3.0 | 1 | 100 |
| 59-23-09 | 4 | 388 | 754.3 | 380.4 | 1382.8 | 11 | 1 | 1.3 | 7.6 | −7.8 | 1.7 | −0.6 | 3622.7 | 3 | 13.2 | 1 | 100 |
| 60-23-09 | 4 | 402 | 825.5 | 442.5 | 1470.6 | 12 | 1 | 1.3 | 8.1 | −9.0 | 1.9 | −0.8 | 3267.4 | 3 | 21.0 | 1 | 100 |
| 46-23-10 | 0 | 333 | 647.9 | 150.2 | 1118.0 | 7 | 2.0 | 4.3 | 3.7 | −5.3 | 0.5 | −1.4 | 407.7 | 4 | 50.3 | 3 | 96 |
| 58-23-10 | 1 | 375 | 754.9 | 338.5 | 1332.6 | 10 | 2 | 2.3 | 6.3 | −7.3 | 1.3 | −0.9 | 1715.4 | 4 | 45.0 | 1 | 100 |
| 59-23-10 | 2 | 389 | 781.0 | 368.7 | 1386.8 | 11 | 2 | 2.3 | 6.8 | −7.7 | 1.4 | −0.8 | 2514.6 | 4 | 32.9 | 1 | 100 |
| 60-23-10 | 3 | 403 | 824.9 | 407.6 | 1459.3 | 12 | 2 | 2.3 | 7.2 | −8.3 | 1.5 | −1.1 | 1825.5 | 4 | 22.9 | 1 | 100 |
| 40-09-11 | 0 | 391 | 782.8 | 365.6 | 1382.3 | 10 | 2.0 | 3.8 | 5.6 | −6.1 | 1.1 | −0.5 | 520.7 | 7 | 45.5 | 3 | 95 |
| 37-15-41 | 0 | 427 | 735.0 | 193.1 | 1275.9 | 9 | 3.0 | 4.3 | 4.1 | −4.1 | 0.7 | 0.0 | 86.9 | 6 | 52.3 | 3 | 86 |
| 27-16-41 | 0 | 433 | 703.5 | 183.0 | 1218.1 | 6 | 1.0 | 3.3 | 5.6 | −6.4 | 1.1 | 0.4 | 740.0 | 3 | 66.3 | 1 | 100 |
| 32-08-45 | 0 | 321 | 605.6 | 114.4 | 1055.1 | 7 | 2.5 | 3.0 | 4.0 | −4.8 | 0.5 | −0.9 | 879.5 | 4 | 12.2 | 3 | 100 |
| 25-07-46 | 1 | 343 | 668.1 | 143.9 | 1151.1 | 7 | 1.0 | 4.8 | 3.5 | −6.6 | 0.5 | −1.9 | 136.3 | 3 | 16.5 | 1 | 86 |
| 28-13-47 | 0 | 373 | 656.3 | 93.6 | 1135.4 | 7 | 2.8 | 2.5 | 5.1 | −6.3 | 0.7 | −0.6 | 994.2 | 5 | 13.0 | 1 | 100 |
| Chloroquine | 1 | 294 | 594.1 | 188.9 | 982.9 | 6 | 0.0 | 3.0 | 4.6 | −5.3 | 0.4 | −0.1 | 3718.1 | 0 | - | 3 | 100 |
| Amodiaquine | 1 | 334 | 603.2 | 131.7 | 1018.7 | 6 | 0.0 | 5.0 | 3.6 | −4.4 | 0.0 | −0.4 | 1689.1 | 0 | - | 3 | 100 |
| Dapsone | 1 | 236 | 431.6 | 0.0 | 687.9 | 2 | 0.0 | 7.0 | −0.4 | −0.5 | −1.3 | −0.9 | 289.1 | 0 | - | 3 | 69 |
| Trimethoprim | 0 | 272 | 500.2 | 223.9 | 835.9 | 5 | 0.0 | 6.5 | 0.6 | −1.5 | −0.9 | −1.2 | 282.8 | 3 | - | 3 | 74 |
| Mefloquine | 2 | 362 | 533.1 | 0.0 | 925.1 | 2 | 0.0 | 4.0 | 4.1 | −4.9 | 0.2 | 0.5 | 2903.1 | 0 | - | 3 | 100 |
| Pamaquine | 0 | 315 | 654.8 | 443.4 | 1148.1 | 9 | 1.0 | 4.8 | 4.0 | −3.8 | 0.4 | 0.2 | 1475.2 | 5 | - | 3 | 100 |
| Sulfametopyrazine | 1 | 268 | 473.4 | 77.8 | 773.3 | 4 | 0.0 | 9.0 | −1.0 | 0.2 | −1.7 | −1.3 | 195.8 | 1 | - | 2 | 62 |
| Quinacrine | 0 | 370 | 680.5 | 268.8 | 1163.6 | 7 | 0.0 | 3.5 | 5.6 | −6.5 | 0.8 | −0.1 | 4435.7 | 1 | - | 1 | 100 |
| Tetracycline | 5 | 422 | 604.5 | 173.1 | 1111.8 | 2 | 0.0 | 7.0 | −3.4 | 1.1 | −2.5 | −2.6 | 6.8 | 5 | - | 1 | 22 |
| Lumefantrine | 5 | 497 | 819.1 | 160.7 | 1437.5 | 7 | 0.0 | 3.0 | 8.3 | −10.0 | 1.7 | 0.2 | 4337.2 | 0 | - | 1 | 100 |
| Bulaquine | 0 | 369 | 560.2 | 360.2 | 1097.8 | 9 | 1.0 | 5.8 | 3.6 | −3.0 | 0.1 | −0.4 | 3099.7 | 7 | - | 3 | 100 |
| Hydroxychloroquine | 1 | 310 | 609.5 | 119.5 | 1006.5 | 6 | 0.0 | 5.0 | 3.4 | −4.5 | −0.1 | −0.7 | 1023.7 | 0 | - | 3 | 100 |
| Sulfadoxine | 1 | 296 | 510.6 | 152.3 | 849.5 | 5 | 0.0 | 9.5 | −0.8 | −0.1 | −1.7 | −1.4 | 213.4 | 2 | - | 2 | 64 |
| Halofantrine | 5 | 470 | 785.4 | 160.2 | 1351.8 | 5 | 0.0 | 3.0 | 7.6 | −9.9 | 1.5 | 0.2 | 2844.1 | 0 | - | 1 | 100 |
| Proguanil | 1 | 238 | 478.2 | 125.3 | 768.6 | 6 | 0.0 | 6.0 | 1.1 | −1.5 | −1.1 | −0.7 | 834.6 | 0 | - | 3 | 86 |
| Doxycycline | 4 | 422 | 602.2 | 174.1 | 1104.2 | 2 | 0.0 | 17.2 | −4.0 | 1.7 | −2.9 | −2.5 | 9.2 | 4 | - | 1 | 21 |
| Artemether | 1 | 312 | 531.1 | 506.1 | 970.2 | 2 | 0.0 | 5.7 | 2.7 | −2.99 | −0.20 | 0.20 | 5731.8 | 0 | - | 3 | 100 |
| Dihydroartemisinine | 1 | 284 | 477.4 | 395.7 | 864.6 | 1 | 1.0 | 5.7 | 1.8 | −2.92 | −0.10 | −0.10 | 1664.9 | 0 | - | 3 | 95 |
| Artemisinin | 0 | 282 | 456.6 | 380.6 | 848.4 | 0 | 0.0 | 5.3 | 1.7 | −2.10 | −0.30 | 0.00 | 1886.0 | 1 | - | 3 | 96 |
| Inhibitor | Structure | a | b | c [nM] |
|---|---|---|---|---|
| TCL11 * |  | −113,962.1 | −142,462.7 | 76 |
| TCL-33-08-05 |  | −114,523.3 | −143,133.8 | 4.1 |
| TCL-58-01-01 |  | −116,055.6 | −145,058.2 | 13.3 |
| TCL-58-16-08 |  | −114,528.9 | −143,152.9 | 9.2 |
| TCL-59-01-01 |  | −114,445.5 | −143,063.9 | 10.8 |
| TCL-60-01-01 |  | −114,415.0 | −143,027.9 | 9.1 |
| Inhibitors of PfENR | Number of Water Molecules | Number of Counterions | Number of Atoms of the System |
|---|---|---|---|
| TCL11 | 13,747 | 5 Cl− | 45,561 |
| TCL-33-08-05 | 13,806 | 6 Cl− | 46,746 |
| TCL-58-01-01 | 14,028 | 5 Cl− | 47,406 |
| TCL-58-16-08 | 13,810 | 6 Cl− | 46,767 |
| TCL-59-01-01 | 13,812 | 5 Cl− | 46,761 |
| TCL-60-01-01 | 13,807 | 5 Cl− | 46,749 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Bieri, C.; Esmel, A.; Keita, M.; Owono, L.C.O.; Dali, B.; Megnassan, E.; Miertus, S.; Frecer, V. Structure-Based Design and Pharmacophore-Based Virtual Screening of Combinatorial Library of Triclosan Analogues Active against Enoyl-Acyl Carrier Protein Reductase of Plasmodium falciparum with Favourable ADME Profiles. Int. J. Mol. Sci. 2023, 24, 6916. https://doi.org/10.3390/ijms24086916
Bieri C, Esmel A, Keita M, Owono LCO, Dali B, Megnassan E, Miertus S, Frecer V. Structure-Based Design and Pharmacophore-Based Virtual Screening of Combinatorial Library of Triclosan Analogues Active against Enoyl-Acyl Carrier Protein Reductase of Plasmodium falciparum with Favourable ADME Profiles. International Journal of Molecular Sciences. 2023; 24(8):6916. https://doi.org/10.3390/ijms24086916
Chicago/Turabian StyleBieri, Cecile, Akori Esmel, Melalie Keita, Luc Calvin Owono Owono, Brice Dali, Eugene Megnassan, Stanislav Miertus, and Vladimir Frecer. 2023. "Structure-Based Design and Pharmacophore-Based Virtual Screening of Combinatorial Library of Triclosan Analogues Active against Enoyl-Acyl Carrier Protein Reductase of Plasmodium falciparum with Favourable ADME Profiles" International Journal of Molecular Sciences 24, no. 8: 6916. https://doi.org/10.3390/ijms24086916
APA StyleBieri, C., Esmel, A., Keita, M., Owono, L. C. O., Dali, B., Megnassan, E., Miertus, S., & Frecer, V. (2023). Structure-Based Design and Pharmacophore-Based Virtual Screening of Combinatorial Library of Triclosan Analogues Active against Enoyl-Acyl Carrier Protein Reductase of Plasmodium falciparum with Favourable ADME Profiles. International Journal of Molecular Sciences, 24(8), 6916. https://doi.org/10.3390/ijms24086916