
p53 and Myofibroblast Apoptosis in Organ Fibrosis

{kind=link}
{kind=link}
Abstract
1. Introduction
2. Physiological Wound Repair
3. Dysregulated Wound Repair
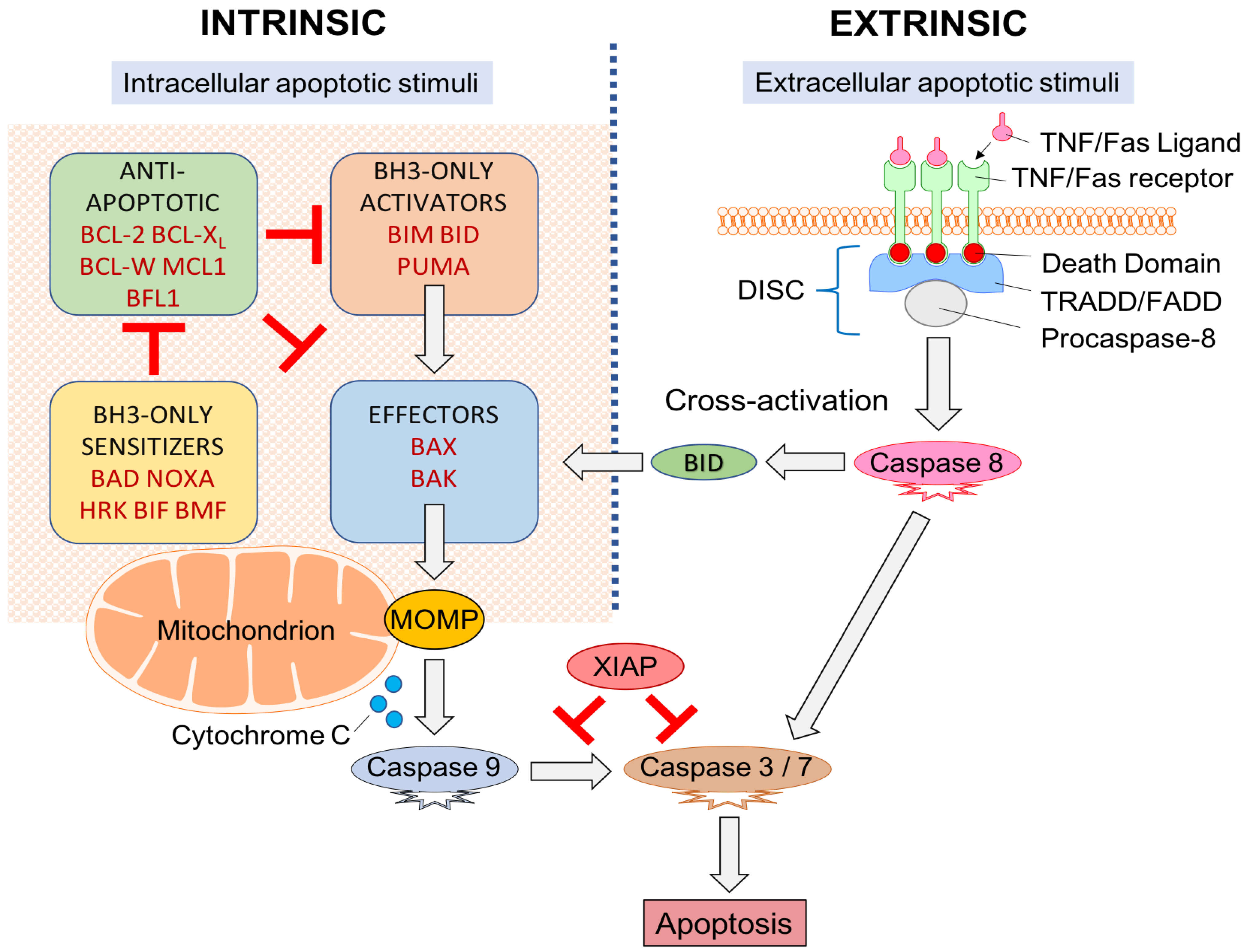
4. Apoptosis
5. p53
6. Lung Fibrosis
7. Liver Fibrosis
8. Renal Fibrosis
9. Cardiac Fibrosis
10. Glaucoma
11. Therapeutics
12. Conclusions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Wallace, D.M.; O’Brien, C.J. The role of lamina cribrosa cells in optic nerve head fibrosis in glaucoma. Exp. Eye Res. 2016, 142, 102–109. [Google Scholar] [CrossRef]
- Horowitz, J.C.; Thannickal, V.J. Mechanisms for the Resolution of Organ Fibrosis. Physiology 2018, 34, 43–55. [Google Scholar] [CrossRef]
- Wells, A.U.; Denton, C.P. Interstitial lung disease in connective tissue disease—Mechanisms and management. Nat. Rev. Rheumatol. 2014, 10, 728–739. [Google Scholar] [CrossRef] [PubMed]
- Chandler, C.; Liu, T.; Buckanovich, R.; Coffman, L.G. The double edge sword of fibrosis in cancer. Transl. Res. 2019, 209, 55–67. [Google Scholar] [CrossRef]
- Wynn, T.A. Fibrotic disease and the TH1/TH2 paradigm. Nat. Rev. Immunol. 2004, 4, 583–594. [Google Scholar] [CrossRef]
- Thannickal, V.J.; Zhou, Y.; Gaggar, A.; Duncan, S.R. Fibrosis: Ultimate and proximate causes. J. Clin. Invest. 2014, 124, 4673–4677. [Google Scholar] [CrossRef]
- Ehrlich, H.P. The physiology of wound healing. A summary of normal and abnormal wound healing processes. Adv. Wound Care J. Prev. 1998, 11, 326–328. [Google Scholar]
- Hardy, M.A. The biology of scar formation. Phys. Ther. 1989, 69, 1014–1024. [Google Scholar] [CrossRef] [PubMed]
- Eming, S.A.; Martin, P.; Tomic-Canic, M. Wound repair and regeneration: Mechanisms, signaling, and translation. Sci. Transl. Med. 2014, 6, 265sr6. [Google Scholar] [CrossRef]
- Gurtner, G.C.; Werner, S.; Barrandon, Y.; Longaker, M.T. Wound repair and regeneration. Nature 2008, 453, 314–321. [Google Scholar] [CrossRef]
- Hinz, B.; Lagares, D. Evasion of apoptosis by myofibroblasts: A hallmark of fibrotic diseases. Nat. Rev. Rheumatol. 2019, 16, 11–31. [Google Scholar] [CrossRef]
- Humphrey, J.D.; Dufresne, E.R.; Schwartz, M.A. Mechanotransduction and extracellular matrix homeostasis. Nat. Rev. Mol. Cell Biol. 2014, 15, 802–812. [Google Scholar] [CrossRef]
- Hinz, B.; Phan, S.H.; Thannickal, V.J.; Galli, A.; Bochaton-Piallat, M.L.; Gabbiani, G. The myofibroblast: One function, multiple origins. Am. J. Pathol. 2007, 170, 1807–1816. [Google Scholar] [CrossRef] [PubMed]
- Horowitz, J.C.; Thannickal, V.J. Epithelial–Mesenchymal Interactions in Pulmonary Fibrosis. Bone 2012, 23, 1–7. [Google Scholar] [CrossRef]
- Marangoni, R.; Korman, B.; Varga, J. Myofibroblasts in Murine Cutaneous Fibrosis Originate from Adiponectin-Positive Intradermal Progenitors. Arthritis Rheumatol. 2015, 67, 1062–1073. [Google Scholar] [CrossRef] [PubMed]
- Rajkumar, V.S.; Howell, K.; Csiszar, K.; Denton, C.P.; Black, C.M.; Abraham, D.J. Shared expression of phenotypic markers in systemic sclerosis indicates a convergence of pericytes and fibroblasts to a myofibroblast lineage in fibrosis. Arthritis Res. Ther. 2005, 7, R1113. [Google Scholar] [CrossRef] [PubMed]
- Philippeos, C.; Telerman, S.B.; Oulès, B.; Pisco, A.O.; Shaw, T.J.; Elgueta, R.; Lombardi, G.; Driskell, R.R.; Soldin, M.; Lynch, M.D.; et al. Spatial and Single-Cell Transcriptional Profiling Identifies Functionally Distinct Human Dermal Fibroblast Subpopulations. J. Invest. Dermatol. 2018, 138, 811–825. [Google Scholar] [CrossRef] [PubMed]
- Jiang, D.; Correa-Gallegos, D.; Christ, S.; Stefanska, A.; Liu, J.; Ramesh, P.; Rajendran, V.; De Santis, M.M.; Wagner, D.E.; Rinkevich, Y. Two succeeding fibroblastic lineages drive dermal development and the transition from regeneration to scarring. Nat. Cell Biol. 2018, 20, 422–431. [Google Scholar] [CrossRef] [PubMed]
- Desmoulière, A.; Chaponnier, C.; Gabbiani, G. Tissue repair, contraction, and the myofibroblast. Wound Repair Regen. 2005, 13, 7–12. [Google Scholar] [CrossRef]
- Hinz, B.; Gabbiani, G. Fibrosis: Recent advances in myofibroblast biology and new therapeutic perspectives. F1000 Biol. Rep. 2010, 2, 1–5. [Google Scholar] [CrossRef]
- Talele, N.P.; Fradette, J.; Davies, J.E.; Kapus, A.; Hinz, B. Expression of α-Smooth Muscle Actin Determines the Fate of Mesenchymal Stromal Cells. Stem. Cell Rep. 2015, 4, 1016–1030. [Google Scholar] [CrossRef] [PubMed]
- Ibrahim, M.; Chen, L.; Medina, M.; Levinson, H. Myofibroblasts Contribute to but are not Necessary for Wound Contraction. Lab. Investig. 2015, 95, 1429–1438. [Google Scholar] [CrossRef]
- Tomasek, J.J.; Gabbiani, G.; Hinz, B.; Chaponnier, C.; Brown, R.A. Myofibroblasts and mechano: Regulation of connective tissue remodeling. Nat. Rev. Mol. Cell Biol. 2002, 3, 349–363. [Google Scholar] [CrossRef] [PubMed]
- Gabbiani, G. Presence of Modified Fibroblasts in Granulation Tissue and their Possible Role in Wound Contraction. Specialia 1971, 27, 549–550. [Google Scholar] [CrossRef]
- Hinz, B. The role of myofibroblasts in wound healing. Curr. Res. Transl. Med. 2016, 64, 171–177. [Google Scholar] [CrossRef]
- Lambert, A.W.; Pattabiraman, D.R.; Weinberg, R.A. Emerging Biological Principles of Metastasis. Cell 2017, 168, 670–691. [Google Scholar] [CrossRef] [PubMed]
- Bonnans, C.; Chou, J.; Werb, Z. Remodelling the extracellular matrix in development and disease. Nat. Rev. Mol. Cell Biol. 2014, 15, 786–801. [Google Scholar] [CrossRef] [PubMed]
- Jun, J., II; Lau, L.F. Resolution of organ fibrosis. J. Clin. Invest. 2018, 128, 97–107. [Google Scholar] [CrossRef]
- Wynn, T.A.; Ramalingam, T.R. Mechanisms of fibrosis: Therapeutic translation for fibrotic disease. Nat. Med. 2013, 18, 1028–1040. [Google Scholar] [CrossRef]
- Horowitz, J.C.; Thannickal, V.J. Idiopathic pulmonary fibrosis: New concepts in pathogenesis and implications for drug therapy. Treat. Respir. Med. 2006, 5, 325–342. [Google Scholar] [CrossRef]
- Wells, A.U.; Brown, K.K.; Flaherty, K.R.; Kolb, M.; Thannickal, V.J. What’s in a name? That which we call IPF, by any other name would act the same. Eur. Respir. J. 2018, 51, 1–12. [Google Scholar] [CrossRef]
- Wick, G.; Grundtman, C.; Mayerl, C.; Wimpissinger, T.F.; Feichtinger, J.; Zelger, B.; Sgonc, R.; Wolfram, D. The Immunology of Fibrosis. Annu. Rev. Immunol. 2013, 31, 107–135. [Google Scholar] [CrossRef] [PubMed]
- Horowitz, J.C.; Rogers, D.S.; Simon, R.H.; Sisson, T.H.; Thannickal, V.J. Plasminogen activation-induced pericellular fibronectin proteolysis promotes fibroblast apoptosis. Am. J. Respir. Cell Mol. Biol. 2008, 38, 78–87. [Google Scholar] [CrossRef]
- Wheaton, A.K.; Agarwal, M.; Jia, S.; Kim, K.K. Lung epithelial cell focal adhesion kinase signaling inhibits lung injury and fibrosis. Am. J. Physiol. -Lung Cell Mol. Physiol. 2017, 312, L722–L730. [Google Scholar] [CrossRef]
- Jia, S.; Agarwal, M.; Yang, J.; Horowitz, J.C.; White, E.S.; Kim, K.K. Discoidin domain receptor 2 signaling regulates fibroblast apoptosis through PDK1/akt. Am. J. Respir. Cell Mol. Biol. 2018, 59, 295–305. [Google Scholar] [CrossRef] [PubMed]
- Sakai, N.; Tager, A.M. Fibrosis of two: Epithelial cell-fibroblast interactions in pulmonary fibrosis. Biochim. Biophys. Acta -Mol. Basis Dis. 2013, 1832, 911–921. [Google Scholar] [CrossRef] [PubMed]
- Selman, M.; Pardo, A. Idiopathic Pulmonary Fibrosis: Prevailing and evolving hypotheses about its pathogenesis and implications for therapy. Annu. Intern. Med. 2001, 134, 136–151. [Google Scholar] [CrossRef]
- Klahr, S.; Morrissey, J. Obstructive nephropathy and renal fibrosis. Am. J. Physiol. -Ren. Physiol. 2002, 283, 5. [Google Scholar] [CrossRef]
- Rebecca, G. Wells. Cellular Sources of Extracellular Matrix in Hepatic Fibrosis. Clin. Liver Dis. 2008, 12, 1–10. [Google Scholar]
- Piek, A.; de Boer, R.A.; Silljé, H.H.W. The fibrosis-cell death axis in heart failure. Heart Fail. Rev. 2016, 21, 199–211. [Google Scholar] [CrossRef]
- Abergel, P.; Uitto, J. Biochemical composition of the connective tissue in keloids and analysis of collagen metabolism in keloid fibroblast cultures. J. Invest. Dermatol. 1985, 84, 384–390. [Google Scholar] [CrossRef]
- Desmouliere, A.; Redard, M.; Darby, I.; Gabbiani, G. Apoptosis mediates the decrease in cellularity during the transition between granulation tissue and scar. Am. J. Pathol. 1995, 146, 56–66. [Google Scholar] [PubMed]
- Eyden, B. The myofibroblast: Phenotypic characterization as a prerequisite to understanding its functions in translational medicine: Translational Medicine. J. Cell Mol. Med. 2008, 12, 22–37. [Google Scholar] [CrossRef] [PubMed]
- Youle, R.J.; Strasser, A. The BCL-2 protein family: Opposing activities that mediate cell death. Nat. Rev. Mol. Cell Biol. 2008, 9, 47–59. [Google Scholar] [CrossRef]
- Czabotar, P.E.; Lessene, G.; Strasser, A.; Adams, J.M. Control of apoptosis by the BCL-2 protein family: Implications for physiology and therapy. Nat. Rev. Mol. Cell Biol. 2014, 15, 49–63. [Google Scholar] [CrossRef] [PubMed]
- Jorgensen, I.; Rayamajhi, M.; Miao, E.A. Programmed cell death as a defence against infection. Nat. Rev. Immunol. 2017, 17, 151–164. [Google Scholar] [CrossRef]
- Elliott, M.R.; Ravichandran, K.S. Clearance of apoptotic cells: Implications in health and disease. J. Cell Biol. 2010, 189, 1059–1070. [Google Scholar] [CrossRef]
- Nagata, S. Apoptosis and Clearance of Apoptotic Cells. Annu. Rev. Immunol. 2018, 36, 489–517. [Google Scholar] [CrossRef]
- Cellerino, A.; Bähr, M.; Isenmann, S. Apoptosis in the developing visual system. Cell Tissue Res. 2000, 301, 53–69. [Google Scholar] [CrossRef]
- Elmore, S. Apoptosis: A Review of Programmed Cell Death. Toxicol. Pathol. 2007, 35, 495–516. [Google Scholar] [CrossRef]
- Kerr, J.F.R.; Wyllie, A.H.; Currie, A.R. Apoptosis: A basic biological phenomenon with wide-ranging implications in tissue kinetics. Br. J. Cancer 1972, 26, 239–257. [Google Scholar] [CrossRef] [PubMed]
- Nicholson, D.W. Caspase structure, proteolytic substrates, and function during apoptotic cell death. Cell Death Differ. 1999, 6, 1028–1042. [Google Scholar] [CrossRef] [PubMed]
- Creagh, E.M.; Conroy, H.; Martin, S.J. Caspase-activation pathways in apoptosis and immunity. Immunol. Rev. 2003, 193, 10–21. [Google Scholar] [CrossRef]
- McIlwain, D.R.; Berger, T.; Mak, T.W. Caspase functions in cell death and disease. Cold Spring Harb. Perspect Biol. 2013, 5, 1–28. [Google Scholar] [CrossRef]
- Singh, R.; Letai, A.; Sarosiek, K. Regulation of apoptosis in health and disease: The balancing act of BCL-2 family proteins. Nat. Rev. Mol. Cell Biol. 2019, 20, 175–193. [Google Scholar] [CrossRef] [PubMed]
- Belkacemi, L. Exploiting the Extrinsic and the Intrinsic Apoptotic Pathways for Cancer Therapeutics. J. Cancer Cure 2018, 1, 1004. [Google Scholar]
- Lomonosova, E.; Chinnadurai, G. BH3-only proteins in apoptosis and beyond: An overview. Oncogene 2008, 27, S2–S19. [Google Scholar] [CrossRef] [PubMed]
- Vela, L.; Gonzalo, O.; Naval, J.; Marzo, I. Direct interaction of bax and bak proteins with Bcl-2 homology domain 3 (BH3)-only proteins in living cells revealed by fluorescence complementation. J. Biol. Chem. 2013, 288, 4935–4946. [Google Scholar] [CrossRef]
- Lagares, D. Targeted Apoptosis of Myofibroblasts with the BH3 Mimetic ABT-263 Reverses Established Fibrosis. Sci. Transl. Med. 2017, 9, eaal3765. [Google Scholar] [CrossRef]
- Thannickal, V.J.; Horowitz, J.C. Evolving concepts of apoptosis in idiopathic pulmonary fibrosis. Proc. Am. Thorac. Soc. 2006, 3, 350–356. [Google Scholar] [CrossRef]
- Liu, B.H.; Chen, L.; Li, S.R.; Wang, Z.X.; Cheng, W.G. Smac/DIABLO regulates the apoptosis of hypertrophic scar fibroblasts. Int. J. Mol. Med. 2013, 32, 615–622. [Google Scholar] [CrossRef]
- Savill, J.; Fadok, V. Corpse clearance defines the meaning of cell death [In Process Citation]. Nature 2000, 407, 784–788. [Google Scholar] [CrossRef] [PubMed]
- Kantari, C.; Walczak, H. Caspase-8 and Bid: Caught in the act between death receptors and mitochondria. Biochim. Biophys. Acta -Mol. Cell Res. 2011, 1813, 558–563. [Google Scholar] [CrossRef]
- Esposti, M.D. The roles of Bid. Apoptosis 2002, 7, 433–440. [Google Scholar] [CrossRef] [PubMed]
- Kuwana, T.; Mackey, M.R.; Perkins, G.; Ellisman, M.H.; Latterich, M.; Schneiter, R.; Green, D.R.; Newmeyer, D.D. Bid, Bax, and lipids cooperate to form supramolecular openings in the outer mitochondrial membrane. Cell 2002, 111, 331–342. [Google Scholar] [CrossRef]
- Fink, S.L.; Cookson, B.T. Apoptosis, Pyroptosis, and Necrosis: Mechanistic Description of Dead and Dying Eukaryotic Cells. Infect. Immun. 2005, 73, 1907–1916. [Google Scholar] [CrossRef]
- Ghobrial, I.M.; Witzig, T.E.; Adjei, A.A. Targeting Apoptosis Pathways in Cancer Therapy. CA Cancer J. Clin. 2005, 55, 178–194. [Google Scholar] [CrossRef] [PubMed]
- Iredale, J.P.; Benyon, C.; Pickering, J.; McCullen, M.; Northrop, M.; Pawley, S.; Hovell, C.; Arthur, M.J.P. Mechanisms of spontaneous resolution of rat liver fibrosis: Hepatic Stellate Cell Apoptosis and Reduced Hepatic Expression of Metalloproteinase Inhibitors. J. Clin. Invest. 1998, 102, 538–549. [Google Scholar] [CrossRef]
- Glasser, S.W.; Hagood, J.S.; Wong, S.; Taype, C.A.; Madala, S.K.; Hardie, W.D. Mechanisms of Lung Fibrosis Resolution. Am. J. Pathol. 2016, 186, 1066–1077. [Google Scholar] [CrossRef]
- Van Caam, A.; Vonk, M.; Van Den Hoogen, F.; Van Lent, P.; Van Der Kraan, P. Unraveling SSc pathophysiology; The myofibroblast. Front. Immunol. 2018, 9, 1–22. [Google Scholar] [CrossRef] [PubMed]
- Nagaraja, M.R.; Tiwari, N.; Shetty, S.K.; Marudamuthu, A.S.; Fan, L.; Ostrom, R.S.; Fu, J.; Gopu, V.; Radhakrishnan, V.; Idell, S.; et al. p53 Expression in Lung Fibroblasts Is Linked to Mitigation of Fibrotic Lung Remodeling. Am. J. Pathol. 2018, 188, 2207–2222. [Google Scholar] [CrossRef]
- Lane, D.P.; Crawford, L.V. T antigen is bound to a host protein in SY40-transformed cells [19]. Nature 1979, 278, 261–263. [Google Scholar] [CrossRef]
- Levine, A.J.; Oren, M. The first 30 years of p53: Growing ever more complex. Nat. Rev. Cancer 2009, 9, 749–758. [Google Scholar] [CrossRef]
- Nag, S.; Qin, J.; Srivenugopal, K.S.; Wang, M.; Zhang, R. The MDM2-p53 pathway revisited. J. Biomed. Res. 2013, 27, 254–271. [Google Scholar] [PubMed]
- Bieging, K.T.; Mello, S.S.; Attardi, L.D. Unravelling mechanisms of p53-mediated tumour suppression. Nat. Rev. Cancer 2014, 14, 359–370. [Google Scholar] [CrossRef] [PubMed]
- Nag, S.; Zhang, X.; Srivenugopal, K.S.; Wang, M.H.; Wang, W.; Zhang, R. Targeting MDM2-p53 Interaction for Cancer Therapy: Are We There Yet? Curr. Med. Chem. 2014, 21, 553–574. [Google Scholar] [CrossRef]
- Lane, D.P. P53, Guardian of the Genome. Nature 1992, 358, 15–16. [Google Scholar] [CrossRef]
- Taylor, W.R.; Stark, G.R. Regulation of the G2/M transition by p53. Oncogene 2001, 20, 1803–1815. [Google Scholar] [CrossRef] [PubMed]
- Sherr, C.J. G1 phase progression: Cycling on cue. Cell 1994, 79, 551–555. [Google Scholar] [CrossRef]
- Schafer, K.A. Cell cycle review. Vet. Pathology 1998, 35, 461–478. [Google Scholar] [CrossRef]
- Siliciano, J.D.; Canman, C.E.; Taya, Y.; Sakaguchi, K.; Appella, E.; Kastan, M.B. DNA damage induces phosphorylation of the amino terminus of p53. Genes Dev. 1997, 11, 3471–3481. [Google Scholar] [CrossRef] [PubMed]
- Sherr, C.J.; Roberts, J.M. Living with or without cyclins and cyclin-dependent kinases. Genes Dev. 2004, 18, 2699–2711. [Google Scholar] [CrossRef]
- Malumbres, M.; Barbacid, M. To cycle or not to cycle: A critical decision in cancer. Nat. Rev. Cancer 2001, 1, 222–231. [Google Scholar] [CrossRef] [PubMed]
- Chen, J. The Cell-Cycle Arrest and Apoptotic and Progression. Cold Spring Harb. Perspect. Biol. 2016, 6, a026104. [Google Scholar] [CrossRef] [PubMed]
- Nigg, E.A. Cyclin-dependent protein kinases: Key regulators of the eukaryotic cell cycle. BioEssays 1995, 17, 471–480. [Google Scholar] [CrossRef] [PubMed]
- Fridman, J.S.; Lowe, S.W. Control of apoptosis by p53. Oncogene 2003, 22, 9030–9040. [Google Scholar] [CrossRef]
- Riley, T.; Sontag, E.; Chen, P.; Levine, A. Transcriptional control of human p53-regulated genes. Nat. Rev. Mol. Cell Biol. 2008, 9, 402–412. [Google Scholar] [CrossRef]
- Aubrey, B.J.; Kelly, G.L.; Janic, A.; Herold, M.J.; Strasser, A. How does p53 induce apoptosis and how does this relate to p53-mediated tumour suppression? Cell Death Differ. 2018, 25, 104–113. [Google Scholar] [CrossRef]
- Tagscherer, K.E.; Fassl, A.; Sinkovic, T.; Combs, S.E.; Roth, W. p53-dependent regulation of Mcl-1 contributes to synergistic cell death by ionizing radiation and the Bcl-2/Bcl-XL inhibitor ABT-737. Apoptosis 2012, 17, 187–199. [Google Scholar] [CrossRef]
- Ho, T.; Tan, B.X.; Lane, D. How the other half lives: What p53 does when it is not being a transcription factor. Int. J. Mol. Sci. 2019, 21, 13. [Google Scholar] [CrossRef]
- Pietsch, E.C.; Sykes, S.M.; McMahon, S.B.; Murphy, M.E. The p53 family and programmed cell death. Oncogene 2008, 27, 6507–6521. [Google Scholar] [CrossRef] [PubMed]
- Vaseva, A.V.; Marchenko, N.D.; Ji, K.; Tsirka, S.E.; Holzmann, S.; Moll, U.M. P53 opens the mitochondrial permeability transition pore to trigger necrosis. Cell 2012, 149, 1536–1548. [Google Scholar] [CrossRef] [PubMed]
- Matlashewski, G.J.; Tuck, S.; Pim, D.; Lamb, P.; Schneider, J.; Crawford, L.V. Primary structure polymorphism at amino acid residue 72 of human p53. Mol. Cell Biol. 1987, 7, 961–963. [Google Scholar] [PubMed]
- Lodhi, N.; Singh, R.; Rajput, S.P.; Saquib, Q. SARS-CoV-2: Understanding the transcriptional regulation of ACE2 and TMPRSS2 and the role of single nucleotide polymorphism (SNP) at codon 72 of p53 in the innate immune response against virus infection. Int. J. Mol. Sci. 2021, 22, 8660. [Google Scholar] [CrossRef] [PubMed]
- Frank, A.K.; Leu, J.I.-J.; Zhou, Y.; Devarajan, K.; Nedelko, T.; Klein-Szanto, A.; Hollstein, M.; Murphy, M.E. The Codon 72 Polymorphism of p53 Regulates Interaction with NF-κB and Transactivation of Genes Involved in Immunity and Inflammation. Mol. Cell Biol. 2011, 31, 1201–1213. [Google Scholar] [CrossRef] [PubMed]
- Ko, L.J.; Prives, C. p53: Puzzle and paradigm. Genes Dev. 1996, 10, 1054–1072. [Google Scholar] [CrossRef] [PubMed]
- Jeong, B.-S.; Hu, W.; Belyi, V.; Rabadan, R.; Levine, A.J. Differential levels of transcription of p53-regulated genes by the arginine/proline polymorphism: p53 with arginine at codon 72 favors apoptosis. FASEB J. Off Publ. Fed. Am. Soc. Exp. Biol. 2010, 24, 1347–1353. [Google Scholar] [CrossRef] [PubMed]
- Li, X.; Dumont, P.; Della Pietra, A.; Shetler, G.; Murphy, M.E. The codon 47 polymorphism in p53 is functionally significant. J. Biol. Chem. 2005, 280, 24245–24251. [Google Scholar] [CrossRef] [PubMed]
- Walerych, D.; Napoli, M.; Collavin, L.; Del Sal, G. The rebel angel: Mutant p53 as the driving oncogene in breast cancer. Carcinogenesis 2012, 33, 2007–2017. [Google Scholar] [CrossRef] [PubMed]
- King, T.E.; Pardo, A.; Selman, M. Idiopathic pulmonary fibrosis. Lancet 2011, 378, 1949–1961. [Google Scholar] [CrossRef] [PubMed]
- Murray, L.A.; Rubinowitz, A.; Herzog, E.L. Interstitial lung disease: Is interstitial lung disease the same as scleroderma lung disease? Curr. Opin. Rheumatol. 2012, 24, 656–662. [Google Scholar] [CrossRef] [PubMed]
- Mora, A.L.; Rojas, M.; Pardo, A.; Selman, M. Emerging therapies for idiopathic pulmonary fibrosis, a progressive age-related disease. Nat. Rev. Drug Discov. 2017, 16, 755–772. [Google Scholar] [CrossRef] [PubMed]
- Liu, T.; De Los Santos, F.G.; Phan, S.H. The bleomycin model of pulmonary fibrosis. Methods Mol. Biol. 2017, 1627, 27–42. [Google Scholar] [PubMed]
- Moore, B.B.; Lawson, W.E.; Oury, T.D.; Sisson, T.H.; Raghavendran, K.; Hogaboam, C.M. Animal models of fibrotic lung disease. Am. J. Respir. Cell Mol. Biol. 2013, 49, 167–179. [Google Scholar] [CrossRef]
- Chen, F.; Gong, L.; Zhang, L.; Wang, H.; Qi, X.; Wu, X.; Xiao, Y.; Cai, Y.; Liu, L.; Li, X.; et al. Short courses of low dose dexamethasone delay bleomycin-induced lung fibrosis in rats. Eur. J. Pharmacol. 2006, 536, 287–295. [Google Scholar] [CrossRef]
- Kis, K.; Liu, X.; Hagood, J.S. Myofibroblast differentiation and survival in fibrotic disease. Expert Rev. Mol. Med. 2011, 13, 1–24. [Google Scholar] [CrossRef]
- Richeldi, L.; Collard, H.R.; Jones, M.G. Idiopathic pulmonary fibrosis. Lancet 2017, 389, 1941–1952. [Google Scholar] [CrossRef]
- Lee, C.M.; He, C.H.; Park, J.W.; Lee, J.H.; Kamle, S.; Ma, B.; Akosman, B.; Cotez, R.; Chen, E.; Zhou, Y.; et al. Chitinase 1 regulates pulmonary fibrosis by modulating TGF-β/SMAD7 pathway via TGFBRAP1 and FOXO3. Life Sci. Alliance 2019, 2, 1–14. [Google Scholar] [CrossRef] [PubMed]
- Hou, J.; Ma, T.; Cao, H.; Chen, Y.; Wang, C.; Chen, X.; Xiang, Z.; Han, X. TNF-α-induced NF-κB activation promotes myofibroblast differentiation of LR-MSCs and exacerbates bleomycin-induced pulmonary fibrosis. J. Cell Physiol. 2018, 233, 2409–2419. [Google Scholar] [CrossRef] [PubMed]
- Kishi, M.; Aono, Y.; Sato, S.; Koyama, K.; Azuma, M.; Abe, S.; Kawano, H.; Kishi, J.; Toyoda, Y.; Okazaki, H.; et al. Blockade of platelet-derived growth factor receptor-β, not receptor-α ameliorates bleomycin-induced pulmonary fibrosis in mice. PLoS ONE 2018, 13, 1–19. [Google Scholar] [CrossRef]
- Wu, Q.; Zhang, K.J.; Jiang, S.M.; Fu, L.; Shi, Y.; Tan, R.B.; Cui, J.; Zhou, Y. P53: A key protein that regulates pulmonary fibrosis. Oxidative Med. Cell. Longev. 2020, 2020, 6635794. [Google Scholar] [CrossRef]
- Selman, M.; Pardo, A. The leading role of epithelial cells in the pathogenesis of idiopathic pulmonary fibrosis. Cell. Signal. 2020, 66, 109482. [Google Scholar] [CrossRef]
- Selman, M.; Pardo, A. Role of epithelial cells in idiopathic pulmonary fibrosis: From innocent targets to serial killers. Proc. Am. Thorac. Soc. 2006, 3, 364–372. [Google Scholar] [CrossRef]
- Chapman, H.A. Epithelial-mesenchymal interactions in pulmonary fibrosis. Annu. Rev. Physiol. 2011, 73, 413–435. [Google Scholar] [CrossRef]
- Johnson, A.; Di Pietro, L.A. Apoptosis and angiogenesis: An evolving mechanism for fibrosis. FASEB J. 2013, 27, 3893–3901. [Google Scholar] [CrossRef] [PubMed]
- Hagimoto, N.; Kuwano, K.; Nomoto, Y.; Kunitake, R.; Hara, N. Apoptosis and expression of Fas/Fas ligand mRNA in bleomycin-induced pulmonary fibrosis in mice. Am. J. Respir. Cell Mol. Biol. 1997, 16, 91–101. [Google Scholar] [CrossRef] [PubMed]
- Kramann, R.; Schneider, R.K.; Dirocco, D.P.; Machado, F.; Fleig, S.; Bondzie, P.A.; Henderson, J.M.; Ebert, B.L.; Humphreys, B.D. Perivascular Gli1+ progenitors are key contributors to injury-induced organ fibrosis. Cell Stem Cell 2015, 16, 51–66. [Google Scholar] [CrossRef]
- Zolak, J.S.; Jagirdar, R.; Surolia, R.; Karki, S.; Oliva, O.; Hock, T.; Guroji, P.; Ding, Q.; Liu, R.M.; Bolisetty, S.; et al. Pleural mesothelial cell differentiation and invasion in fibrogenic lung injury. Am. J. Pathol. 2013, 182, 1239–1247. [Google Scholar] [CrossRef]
- Rock, J.R.; Barkauskas, C.E.; Cronce, M.J.; Xue, Y.; Harris, J.R.; Liang, J.; Noble, P.W.; Hogan, B.L.M. Multiple stromal populations contribute to pulmonary fibrosis without evidence for epithelial to mesenchymal transition. Proc. Natl. Acad. Sci. USA 2011, 108, E1475–E1483. [Google Scholar] [CrossRef]
- El Agha, E.; Moiseenko, A.; Kheirollahi, V.; De Langhe, S.; Crnkovic, S.; Kwapiszewska, G.; Kosanovic, D.; Schwind, F.; Schermuly, R.T.; Henneke, I.; et al. Two-Way Conversion between Lipogenic and Myogenic Fibroblastic Phenotypes Marks the Progression and Resolution of Lung Fibrosis. Cell Stem Cell 2017, 20, 261–273.e3. [Google Scholar] [CrossRef] [PubMed]
- Hung, C.; Linn, G.; Chow, Y.H.; Kobayashi, A.; Mittelsteadt, K.; Altemeier, W.A.; Gharib, S.A.; Schnapp, L.M.; Duffield, J.S. Role of lung Pericytes and resident fibroblasts in the pathogenesis of pulmonary fibrosis. Am. J. Respir. Crit. Care Med. 2013, 188, 820–830. [Google Scholar] [CrossRef] [PubMed]
- Tanaka, T.; Yoshimi, M.; Maeyama, T.; Hagimoto, N.; Kuwano, K.; Hara, N. Resistance to Fas-mediated apoptosis in human lung fibroblast. Eur. Respir. J. 2002, 20, 359–368. [Google Scholar] [CrossRef]
- Fattman, C.L. Apoptosis in pulmonary fibrosis: Too much or not enough? Antioxid. Redox Signal 2008, 10, 379–385. [Google Scholar] [CrossRef]
- Jinta, T.; Miyazaki, Y.; Kishi, M.; Akashi, T.; Takemura, T.; Inase, N.; Yoshizawa, Y. The Pathogenesis of Chronic Hypersensitivity Pneumonitis in Common With Idiopathic Pulmonary Fibrosis. Am. J. Clin. Pathol. 2010, 134, 613–620. [Google Scholar] [CrossRef]
- Maher, T.M.; Evans, I.C.; Bottoms, S.E.; Mercer, P.F.; Thorley, A.J.; Nicholson, A.G.; Laurent, G.J.; Tetley, T.D.; Chambers, R.C.; McAnulty, R.J. Diminished prostaglandin E2 contributes to the apoptosis paradox in idiopathic pulmonary fibrosis. Am. J. Respir. Crit. Care Med. 2010, 182, 73–82. [Google Scholar] [CrossRef]
- Larson-Casey, J.L.; Deshane, J.S.; Ryan, A.J.; Thannickal, V.J.; Carter, A.B. Macrophage Akt1 Kinase-Mediated Mitophagy Modulates Apoptosis Resistance and Pulmonary Fibrosis. Immunity 2016, 44, 582–596. [Google Scholar] [CrossRef]
- Kuhn, C.; McDonald, J.A. The roles of the myofibroblast in idiopathic pulmonary fibrosis: Ultrastructural and immunohistochemical features of sites of active extracellular matrix synthesis. Am. J. Pathol. 1991, 138, 1257–1265. [Google Scholar] [PubMed]
- Drakopanagiotakis, F.; Xifteri, A.; Polychronopoulos, V.; Bouros, D. Apoptosis in lung injury and fibrosis. Eur. Respir. J. 2008, 32, 1631–1638. [Google Scholar] [CrossRef]
- Uhal, B.D. The role of apoptosis in pulmonary fibrosis. Eur. Respir. Rev. 2008, 17, 138–144. [Google Scholar] [CrossRef]
- Wordinger, R.J.; Fleenor, D.L.; Hellberg, P.E.; Pang, I.H.; Tovar, T.O.; Zode, G.S.; Fuller, J.A.; Clark, A.F. Effects of TGF-β2, BMP-4, and gremlin in the trabecular meshwork: Implications for glaucoma. Investig. Ophthalmol. Vis. Sci. 2007, 48, 1191–1200. [Google Scholar] [CrossRef]
- Fuchshofer, R.; Birke, M.; Welge-Lussen, U.; Kook, D.; Lütjen-Drecoll, E. Transforming growth factor-β2 modulated extracellular matrix component expression in cultured human optic nerve head astrocytes. Investig. Ophthalmol. Vis. Sci. 2005, 46, 568–578. [Google Scholar] [CrossRef] [PubMed]
- Rockey, D.C.; Darwin Bell, P.; Hill, J.A. Fibrosis-a common pathway to organ injury and failure. N. Engl. J. Med. 2015, 372, 1138–1149. [Google Scholar] [CrossRef] [PubMed]
- Zhang, H.Y.; Phan, S.H. Inhibition of myofibroblast apoptosis by transforming growth factor β1. Am. J. Respir. Cell Mol. Biol. 1999, 21, 658–665. [Google Scholar] [CrossRef]
- Horowitz, J.C.; Ajayi, I.O.; Kulasekaran, P.; Rogers, D.S.; White, J.B.; Townsend, S.K.; White, E.S.; Nho, R.S.; Higgins, P.D.R. Survivin expression induced by endothelin-1 promotes myofibroblast resistance to apoptosis. Int. J. Biochem. Cell Biol. 2012, 44, 158–169. [Google Scholar] [CrossRef] [PubMed]
- Horowitz, J.C.; Rogers, D.S.; Sharma, V.; Vittal, R.; White, E.S.; Cui, Z.; Thannickal, V.J. Combinatorial activation of FAK and AKT by transforming growth factor-β1 confers an anoikis-resistant phenotype to myofibroblasts. Cell Signal 2007, 19, 761–771. [Google Scholar] [CrossRef]
- Horowitz, J.C.; Lee, D.Y.; Waghray, M.; Keshamouni, V.G.; Thomas, P.E.; Zhang, H.; Cui, Z.; Thannickal, V.J. Activation of the Pro-survival Phosphatidylinositol 3-Kinase/AKT Pathway by Transforming Growth Factor-β1 in Mesenchymal Cells Is Mediated by p38 MAPK-dependent Induction of an Autocrine Growth Factor. J. Biol. Chem. 2004, 279, 1359–1367. [Google Scholar] [CrossRef] [PubMed]
- Ajayi, I.O.; Sisson, T.H.; Higgins, P.D.R.; Booth, A.J.; Sagana, R.L.; Huang, S.K.; White, E.S.; King, J.E.; Moore, B.B.; Horowitz, J.C. X-linked inhibitor of apoptosis regulates lung fibroblast resistance to fas-mediated apoptosis. Am. J. Respir. Cell Mol. Biol. 2013, 49, 86–95. [Google Scholar] [CrossRef]
- Bai, L.; Bernard, K.; Tang, X.; Hu, M.; Horowitz, J.C.; Thannickal, V.J.; Sanders, Y.Y. Glutaminolysis epigenetically regulates antiapoptotic gene expression in idiopathic pulmonary fibrosis fibroblasts. Am. J. Respir. Cell Mol. Biol. 2019, 60, 49–57. [Google Scholar] [CrossRef]
- Dodi, A.E.; Ajayi, I.O.; Chang, C.; Beard, M.; Ashley, S.L.; Huang, S.K.; Thannickal, V.J.; Tschumperlin, D.J.; Sisson, T.H.; Horowitz, J.C. Regulation of fibroblast Fas expression by soluble and mechanical pro-fibrotic stimuli. Respir. Res. 2018, 19, 1–12. [Google Scholar] [CrossRef]
- Kulasekaran, P.; Scavone, C.A.; Rogers, D.S.; Arenberg, D.A.; Thannickal, V.J.; Horowitz, J.C. Endothelin-1 and transforming growth factor-β1 independently induce fibroblast resistance to apoptosis via AKT activation. Am. J. Respir. Cell Mol. Biol. 2009, 41, 484–493. [Google Scholar] [CrossRef]
- Riches, D.W.H.; Backos, D.S.; Redente, E.F. ROCK and rho: Promising therapeutic targets to ameliorate pulmonary fibrosis. Am. J. Pathol. 2015, 185, 909–912. [Google Scholar] [CrossRef] [PubMed]
- Kuehl, T.; Lagares, D. BH3 mimetics as anti-fibrotic therapy: Unleashing the mitochondrial pathway of apoptosis in myofibroblasts. Matrix Biol. 2018, 68–69, 94–105. [Google Scholar] [CrossRef] [PubMed]
- Del, V.; Moore, G.; Brown, J.R.; Certo, M.; Love, T.M.; Novina, C.D.; Letai, A. Chronic lymphocytic leukemia requires BCL2 to sequester prodeath BIM, explaining sensitivity to BCL2 antagonist ABT-737. J. Clin. Invest. 2007, 117, 112–121. [Google Scholar]
- Ryan, J.A.; Brunelle, J.K.; Letai, A. Heightened mitochondrial priming is the basis for apoptotic hypersensitivity of CD4+ CD8+ thymocytes. Proc. Natl. Acad. Sci. USA 2010, 107, 12895–12900. [Google Scholar] [CrossRef] [PubMed]
- Deng, J.; Carlson, N.; Takeyama, K.; Dal Cin, P.; Shipp, M.; Letai, A. BH3 Profiling Identifies Three Distinct Classes of Apoptotic Blocks to Predict Response to ABT-737 and Conventional Chemotherapeutic Agents. Cancer Cell 2007, 12, 171–185. [Google Scholar] [CrossRef]
- Ni Chongaile, T.; Sarosiek, K.A.; Vo, T.; Jeremy, A.; Tammareddi, A.; Del, V.; Moore, G.; Deng, J.; Anderson, K.C.; Richardson, P.; et al. Pretreatment Mitochondrial Priming Correlates with Clinical Response to Cytotoxic Chemotherapy. Science 2011, 334, 1129–1133. [Google Scholar] [CrossRef]
- Certo, M.; Moore, V.D.G.; Nishino, M.; Wei, G.; Korsmeyer, S.; Armstrong, S.A.; Letai, A. Mitochondria primed by death signals determine cellular addiction to antiapoptotic BCL-2 family members. Cancer Cell 2006, 9, 351–365. [Google Scholar] [CrossRef]
- Zhou, Y.; Huang, X.; Hecker, L.; Kurundkar, D.; Kurundkar, A.; Liu, H.; Jin, T.H.; Desai, L.; Bernard, K.; Thannickal, V.J. Inhibition of mechanosensitive signaling in myofibroblasts ameliorates experimental pulmonary fibrosis. J. Clin. Invest. 2013, 123, 1096–1108. [Google Scholar] [CrossRef]
- Hecker, L.; Logsdon, N.J.; Kurundkar, D.; Kurundkar, A.; Bernard, K.; Hock, T.; Meldrum, E.; Sanders, Y.Y.; Thannickal, V.J. Reversal of persistent fibrosis in aging by targeting Nox4-Nrf2 redox imbalance. Sci. Transl. Med. 2014, 6, 231ra47. [Google Scholar] [CrossRef]
- Ashley, S.L.; Sisson, T.H.; Wheaton, A.K.; Kim, K.K.; Wilke, C.A.; Ajayi, I.O.; Subbotina, N.; Wang, S.; Duckett, C.S.; Moore, B.B.; et al. Targeting inhibitor of apoptosis proteins protects from bleomycin-induced lung fibrosis. Am. J. Respir. Cell Mol. Biol. 2016, 54, 482–492. [Google Scholar] [CrossRef]
- Hohmann, M.S.; Habiel, D.M.; Coelho, A.L.; Verri, W.A.; Hogaboam, C.M. Quercetin enhances ligand-induced apoptosis in senescent idiopathic pulmonary fibrosis fibroblasts and reduces lung fibrosis in vivo. Am. J. Respir. Cell Mol. Biol. 2019, 60, 28–40. [Google Scholar] [CrossRef] [PubMed]
- Redente, E.F.; Keith, R.C.; Janssen, W.; Henson, P.M.; Ortiz, L.A.; Downey, G.P.; Bratton, D.L.; Riches, D.W.H. Tumor necrosis factor-α accelerates the resolution of established pulmonary fibrosis in mice by targeting profibrotic lung macrophages. Am. J. Respir. Cell Mol. Biol. 2014, 50, 825–837. [Google Scholar] [CrossRef]
- Pan, J.; Deguan, L.; Meng, A. Inhibition of Bcl-2/xl with ABT-263 Selectively Kills Senescent Type II Pneumocytes and Reverses Persistent Pulmonary Fibrosis Induced by Ionizing Radiation in Mice. Int. J. Radiat. Oncol. Biol. Phys. 2017, 99, 353–361. [Google Scholar] [CrossRef]
- Furihata, M.; Sonobe, H.; Ohtsuki, Y. The aberrant p53 protein. Int. J. Oncol. 1995, 6, 1209–1226. [Google Scholar] [PubMed]
- Hojo, S.; Fujita, J.; Yamadori, I.; Kamei, T.; Yoshinouchi, T.; Ohtsuki, Y.; Okada, H.; Bandoh, S.; Yamaji, Y.; Takahara, J.; et al. Heterogeneous point mutations of the p53 gene in pulmonary fibrosis. Eur. Respir. J. 1998, 12, 1404–1408. [Google Scholar] [CrossRef]
- Kuwano, K.; Kunitake, R.; Kawasaki, M.; Nomoto, Y.; Hagimoto, N.; Nakanishi, Y.; Hara, N. P21Waf1Cip1/Sdi1 and p53 Expression in Association with DNA Strand Breaks in Idiopathic Pulmonary Fibrosis. Pneumologie 1997, 51, 870. [Google Scholar]
- Lok, S.S.; Stewart, J.P.; Kelly, B.G.; Hasleton, P.S.; Egan, J.J. Epstein-Barr virus and wild p53 in idiopathic pulmonary fibrosis. Respir. Med. 2001, 95, 787–791. [Google Scholar] [CrossRef]
- Zaafan, M.A.; Haridy, A.R.; Abdelhamid, A.M. Amitriptyline attenuates bleomycin-induced pulmonary fibrosis: Modulation of the expression of NF-κβ, iNOS, and Nrf2. Naunyn-Schmiedeberg’s Arch. Pharmacol. 2019, 392, 279–286. [Google Scholar] [CrossRef]
- Chuang, C.Y.; Liu, H.C.; Wu, L.C.; Chen, C.Y.; Chang, J.T.; Hsu, S.L. Gallic acid induces apoptosis of lung fibroblasts via a reactive oxygen species-dependent ataxia telangiectasia mutated-p53 activation pathway. J. Agric. Food Chem. 2010, 58, 2943–2951. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.; Xu, P.; Wang, Y.; Wang, M.; Li, H.; Lin, S.; Mao, C.; Wang, B.; Song, X.; Lv, C. Astaxanthin prevents pulmonary fibrosis by promoting myofibroblast apoptosis dependent on Drp1-mediated mitochondrial fission. J. Cell Mol. Med. 2015, 19, 2215–2231. [Google Scholar] [CrossRef]
- Bhandary, Y.P.; Shetty, S.K.; Marudamuthu, A.S.; Ji, H.L. Neuenschwander PF, Boggaram V, Morris GF, Fu J, Idell S., Shetty S. Regulation of lung injury and fibrosis by p53-mediated changes in urokinase and plasminogen activator inhibitor-1. Am. J. Pathol. 2013, 183, 131–143. [Google Scholar] [CrossRef] [PubMed]
- Plataki, M.; Koutsopoulos, A.V.; Darivianaki, K.; Delides, G.; Siafakas, N.M.; Bouros, D. Expression of apoptotic and antiapoptotic markers in epithelial cells in idiopathic pulmonary fibrosis. Chest 2005, 127, 266–274. [Google Scholar] [CrossRef]
- Nakashima, N.; Kuwano, K.; Maeyama, T.; Hagimoto, N.; Yoshimi, M.; Hamada, N.; Yamada, M.; Nakanishi, Y. The p53-Mdm2 association in epithelial cells in idiopathic pulmonary fibrosis and non-specific interstitial pneumonia. J. Clin. Pathol. 2005, 58, 583–589. [Google Scholar] [CrossRef]
- Shetty, S.K.; Tiwari, N.; Marudamuthu, A.S.; Uthusseri, B.; Bhandary, Y.P.; Fu, J.; Levin, J.; Idell, S.; Shetty, S. p53 and miR-34a Feedback Promotes Lung Epithelial Injury and Pulmonary Fibrosis. Am. J. Pathol. 2017, 187, 1016–1034. [Google Scholar] [CrossRef] [PubMed]
- Pan, Y.; Fu, H.; Kong, Q.; Xiao, Y.; Shou, Q.; Chen, H.; Ke, Y.; Chen, M. Prevention of pulmonary fibrosis with salvianolic acid A by inducing fibroblast cell cycle arrest and promoting apoptosis. J. Ethnopharmacol. 2014, 155, 1589–1596. [Google Scholar] [CrossRef]
- Tiwari, N.; Nagaraja, M.; Shetty, S.; Marudamuthu, A.; Fan, L.; Ostrom, R.; Fu, J.; Gopu, V. p53 Expression in Lung Fibroblasts: Linkage to Fibrotic Lung Remodeling. In C73. FIBROBLAST BIOLOGY; American Thoracic Society: New York, NY, USA, 2018. [Google Scholar]
- Ellis, E.L.; Mann, D.A. Clinical evidence for the regression of liver fibrosis. J. Hepatol. 2012, 56, 1171–1180. [Google Scholar] [CrossRef]
- Higashi, T.; Friedman, S.L.; Hoshida, Y. Hepatic stellate cells as key target in liver fibrosis. Adv. Drug Deliv. Rev. 2017, 121, 27–42. [Google Scholar] [CrossRef]
- Lozano, R.; Naghavi, M.; Foreman, K.; Lim, S.; Shibuya, K.; Aboyans, V.; Abraham, J.; Adair, T.; Aggarwal, R.; Ahn, S.Y.; et al. Global and regional mortality from 235 causes of death for 20 age groups in 1990 and 2010: A systematic analysis for the Global Burden of Disease Study 2010. Lancet 2012, 380, 2095–2128. [Google Scholar] [CrossRef]
- Tsochatzis, E.A.; Bosch, J.; Burroughs, A.K. Liver cirrhosis. Lancet 2014, 383, 1749–1761. [Google Scholar] [CrossRef] [PubMed]
- Schuppan, D.; Afdhal, N. Liver cirrhosis. Lancet 2008, 371, 838–851. [Google Scholar] [CrossRef] [PubMed]
- Wells, R.G.; Schwabe, R. Origin and function of myofibroblasts in the liver. Semin. Liver Dis. 2015, 35, 97–106. [Google Scholar] [CrossRef]
- Moscoso, C.G.; Steer, C.J. “Let my liver rather heat with wine”—A review of hepatic fibrosis pathophysiology and emerging therapeutics. Hepatic Med. Evid. Res. 2019, 11, 109–129. [Google Scholar] [CrossRef]
- Coll, M.; Perea, L.; Boon, R.; Leite, S.B.; Vallverdú, J.; Mannaerts, I.; Smout, A.; El Taghdouini, A.; Blaya, D.; Rodrigo-Torres, D.; et al. Generation of Hepatic Stellate Cells from Human Pluripotent Stem Cells Enables In Vitro Modeling of Liver Fibrosis. Cell Stem Cell 2018, 23, 101–113.e7. [Google Scholar] [CrossRef]
- Leite, S.B.; Roosens, T.; El Taghdouini, A.; Mannaerts, I.; Smout, A.J.; Najimi, M.; Sokal, E.; Noor, F.; Chesne, C.; van Grunsven, L.A. Novel human hepatic organoid model enables testing of drug-induced liver fibrosis in vitro. Biomaterials 2016, 78, 1–10. [Google Scholar] [CrossRef]
- Luedde, T.; Kaplowitz, N.; Schwabe, R.F. Cell death and cell death responses in liver disease: Mechanisms and clinical relevance. Gastroenterology 2014, 147, 765–783.e4. [Google Scholar] [CrossRef]
- Wree, A.; Broderick, L.; Canbay, A.; Hoffman, H.M.; Feldstein, A.E. From NAFLD to NASH to cirrhosis-new insights into disease mechanisms. Nat. Rev. Gastroenterol. Hepatol. 2013, 10, 627–636. [Google Scholar] [CrossRef]
- Murphy, F.R.; Issa, R.; Zhou, X.; Ratnarajah, S.; Nagase, H.; Arthur, M.J.P.; Benyon, C.; Iredale, J.P. Inhibition of apoptosis of activated hepatic stellate cells by tissue inhibitor of metalloproteinase-1 is mediated via effects on matrix metalloproteinase inhibition. Implications for reversibility of liver fibrosis. J. Biol. Chem. 2002, 277, 11069–11076. [Google Scholar] [CrossRef] [PubMed]
- Iwaisako, K.; Jiang, C.; Zhang, M.; Cong, M.; Moore-Morris, T.J.; Park, T.J.; Liu, X.; Xu, J.; Wang, P.; Paik, Y.H.; et al. Origin of myofibroblasts in the fibrotic liver in mice. Proc. Natl. Acad. Sci. USA 2014, 111, E3297–E3305. [Google Scholar] [CrossRef]
- Mederacke, I.; Hsu, C.C.; Troeger, J.S.; Huebener, P.; Mu, X.; Dapito, D.H.; Pradere, J.P.; Schwabe, R.F. Fate-tracing reveals hepatic stellate cells as dominant contributors to liver fibrosis independent of its etiology. Nat. Commun. 2013, 4, 2823. [Google Scholar] [CrossRef] [PubMed]
- Michelotti, G.A.; Xie, G.; Swiderska, M.; Choi, S.S.; Karaca, G.; Krüger, L.; Premont, R.; Yang, L.; Syn, W.K.; Metzger, D.; et al. Smoothened is a master regulator of adult liver repair. J. Clin. Invest. 2013, 123, 2380–2394. [Google Scholar] [CrossRef] [PubMed]
- Canbay, A.; Taimr, P.; Torok, N.; Higuchi, H.; Friedman, S.; Gores, G.J. Apoptotic body engulfment by a human stellate cell line is profibrogenic. Lab. Investig. 2003, 83, 655–663. [Google Scholar] [CrossRef]
- Zhan, S.S.; Jiang, J.X.; Wu, J.; Halsted, C.; Friedman, S.L.; Zern, M.A.; Torok, N.J. Phagocytosis of apoptotic bodies by hepatic stellate cells induces NADPH oxidase and is associated with liver fibrosis in vivo. Hepatology 2006, 43, 435–443. [Google Scholar] [CrossRef] [PubMed]
- Povero, D.; Busletta, C.; Novo, E.; Valfrè, L.; Cannito, S.; Paternostro, C.; Parola, M. Liver fibrosis: A dynamic and potentially reversible process. Histol. Histopathol. 2010, 25, 1075–1091. [Google Scholar] [PubMed]
- Hernandez-Gea, V.; Friedman, S.L. Pathogenesis of liver fibrosis. Annu. Rev. Pathol. Mech. Dis. 2011, 6, 425–456. [Google Scholar] [CrossRef]
- Parsons, C.J.; Takashima, M.; Rippe, R.A. Molecular mechanisms of hepatic fibrogenesis. J. Gastroenterol. Hepatol. 2007, 22, 79–84. [Google Scholar] [CrossRef]
- Lemoinne, S.; Cadoret, A.; El Mourabit, H.; Thabut, D.; Housset, C. Origins and functions of liver myofibroblasts. Biochim. Biophys. Acta—Mol. Basis Dis. 2013, 1832, 948–954. [Google Scholar] [CrossRef] [PubMed]
- Kisseleva, T.; Brenner, D.A. Mechanisms of fibrogenesis. Exp. Biol. Med. 2008, 233, 109–122. [Google Scholar] [CrossRef]
- Kalluri, R.; Neilson, E.G.; Kalluri, R.; Neilson, E.G. Epithelial-mesenchymal transition and its implications for fibrosis Find the latest version: Epithelial-mesenchymal transition and its implications for fibrosis. J. Clin. Invest. 2003, 112, 1776–1784. [Google Scholar] [CrossRef]
- Li, Y.; Wang, J.; Asahina, K. Mesothelial cells give rise to hepatic stellate cells and myofibroblasts via mesothelial-mesenchymal transition in liver injury. Proc. Natl. Acad. Sci. USA 2013, 110, 2324–2329. [Google Scholar] [CrossRef]
- Mutsaers, S.E.; Birnie, K.; Lansley, S.; Herrick, S.E.; Lim, C.B.; Prêle, C.M. Mesothelial cells in tissue repair and fibrosis. Front. Pharmacol. 2015, 6, 1–12. [Google Scholar] [CrossRef]
- Lua, I.; Li, Y.; Pappoe, L.S. Myofibroblastic conversion and regeneration of mesothelial cells in peritoneal and liver fibrosis. Am. J. Pathol. 2015, 185, 3258–3273. [Google Scholar] [CrossRef]
- Kisseleva, T.; Cong, M.; Paik, Y.H.; Scholten, D.; Jiang, C.; Benner, C.; Iwaisako, K.; Moore-Morris, T.; Scott, B.; Tsukamoto, H.; et al. Myofibroblasts revert to an inactive phenotype during regression of liver fibrosis. Proc. Natl. Acad. Sci. USA 2012, 109, 9448–9453. [Google Scholar] [CrossRef]
- Hannivoort, R.A.; Hernandez-Gea, V.; Friedman, S.L. Genomics and proteomics in liver fibrosis and cirrhosis. Fibrogenes Tissue Repair. 2012, 5, 1. [Google Scholar] [CrossRef]
- Novo, E.; Marra, F.; Zamara, E.; Di Bonzo, L.V.; Monitillo, L.; Cannito, S.; Petrai, I.; Mazzocca, A.; Bonacchi, A.; De Franco, R.S.M.; et al. Overexpression of Bcl-2 by activated human hepatic stellate cells: Resistance to apoptosis as a mechanism of progressive hepatic fibrogenesis in humans. Gut 2006, 55, 1174–1182. [Google Scholar] [CrossRef] [PubMed]
- Kisseleva, T.; Brenner, D. Molecular and cellular mechanisms of liver fibrosis and its regression. Nat. Rev. Gastroenterol. Hepatol. 2021, 18, 151–166. [Google Scholar] [CrossRef] [PubMed]
- Glässner, A.; Eisenhardt, M.; Krämer, B.; Körner, C.; Coenen, M.; Sauerbruch, T.; Spengler, U.; Nattermann, J. NK cells from HCV-infected patients effectively induce apoptosis of activated primary human hepatic stellate cells in a TRAIL-, FasL-and NKG2D-dependent manner. Lab. Investig. 2012, 92, 967–977. [Google Scholar] [CrossRef]
- Pellicoro, A.; Ramachandran, P.; Iredale, J.P.; Fallowfield, J.A. Liver fibrosis and repair: Immune regulation of wound healing in a solid organ. Nat. Rev. Immunol. 2014, 14, 181–194. [Google Scholar] [CrossRef]
- Oh, Y.; Park, O.; Swierczewska, M.; Hamilton, J.P.; Park, J.S.; Kim, T.H.; Lim, S.M.; Eom, H.; Jo, D.G.; Lee, C.E.; et al. Systemic PEGylated TRAIL treatment ameliorates liver cirrhosis in rats by eliminating activated hepatic stellate cells. Hepatology 2016, 64, 209–223. [Google Scholar] [CrossRef] [PubMed]
- Kendall, T.J.; Hennedige, S.; Aucott, R.L.; Hartland, S.N.; Vernon, M.A.; Benyon, R.C.; Iredale, J.P. P75 neurotrophin receptor signaling regulates hepatic myofibroblast proliferation and apoptosis in recovery from rodent liver fibrosis. Hepatology 2009, 49, 901–910. [Google Scholar] [CrossRef]
- Watson, M.R.; Wallace, K.; Gieling, R.G.; Manas, D.M.; Jaffray, E.; Hay, R.T.; Mann, D.A.; Oakley, F. NF-κB is a critical regulator of the survival of rodent and human hepatic myofibroblasts. J. Hepatol. 2008, 48, 589–597. [Google Scholar] [CrossRef]
- Pradere, J.P.; Kluwe, J.; De Minicis, S.; Jiao, J.J.; Gwak, G.Y.; Dapito, D.H.; Jang, M.K.; Guenther, N.D.; Mederacke, I.; Friedman, R.; et al. Hepatic macrophages but not dendritic cells contribute to liver fibrosis by promoting the survival of activated hepatic stellate cells in mice. Hepatology 2013, 58, 1461–1473. [Google Scholar] [CrossRef]
- Lee, Y.A.; Wallace, M.C.; Friedman, S.L. Pathobiology of liver fibrosis: A translational success story. Gut 2015, 64, 830–841. [Google Scholar] [CrossRef] [PubMed]
- Karin, M.; Lin, A. NF-κB at the crossroads of life and death. Nat. Immunol. 2002, 3, 221–227. [Google Scholar] [CrossRef] [PubMed]
- Kucharczak, J.; Simmons, M.J.; Fan, Y.; Gélinas, C. To be, or not to be: NF-κB is the answer—Role of Rel/NF-κB in the regulation of apoptosis. Oncogene 2003, 22, 8961–8982. [Google Scholar] [CrossRef] [PubMed]
- Dhanasekaran, D.N.; Reddy, E.P. JNK signaling in apoptosis. Oncogene 2008, 27, 6245–6251. [Google Scholar] [CrossRef]
- Jiang, H.Q.; Zhang, X.L.; Liu, L.; Yang, C.C. Relationship between focal adhesion kinase and hepatic stellate cell proliferation during rat hepatic fibrogenesis. World J. Gastroenterol. 2004, 10, 3001–3005. [Google Scholar] [CrossRef]
- Parsons, C.J.; Bradford, B.U.; Pan, C.Q.; Cheung, E.; Schauer, M.; Knorr, A.; Krebs, B.; Kraft, S.; Zahn, S.; Brocks, B.; et al. Antifibrotic effects of a tissue inhibitor of metalloproteinase-1 antibody on established liver fibrosis in rats. Hepatology 2004, 40, 1106–1115. [Google Scholar] [CrossRef]
- Rizvi, S.; Mertens, J.C.; Bronk, S.F.; Hirsova, P.; Dai, H.; Roberts, L.R.; Kaufmann, S.H.; Gores, G.J. Platelet-derived growth factor primes cancer-associated fibroblasts for apoptosis. J. Biol. Chem. 2014, 289, 22835–22849. [Google Scholar] [CrossRef]
- Moncsek, A.; Al-Suraih, M.S.; Trussoni, C.E.; O’Hara, S.P.; Splinter, P.L.; Zuber, C.; Patsenker, E.; Valli, P.V.; Fingas, C.D.; Weber, A.; et al. Targeting senescent cholangiocytes and activated fibroblasts with B-cell lymphoma-extra large inhibitors ameliorates fibrosis in multidrug resistance 2 gene knockout (Mdr2−/−) mice. Hepatology 2018, 67, 247–259. [Google Scholar] [CrossRef]
- Feldstein, A.E.; Canbay, A.; Angulo, P.; Taniai, M.; Burgart, L.J.; Lindor, K.D.; Gores, G.J. Hepatocyte apoptosis and Fas expression are prominent features of human nonalcoholic steatohepatitis. Gastroenterology 2003, 125, 437–443. [Google Scholar] [CrossRef]
- Natori, S.; Rust, C.; Stadheim, L.M.; Srinivasan, A.; Burgart, L.J.; Gores, G.J. Hepatocyte apoptosis is a pathologic feature of human alcoholic hepatitis. J. Hepatol. 2001, 34, 248–253. [Google Scholar] [CrossRef] [PubMed]
- Weng, S.Y.; Yang, C.Y.; Li, C.C.; Sun, T.P.; Tung, S.Y.; Yen, J.J.Y.; Tsai, T.F.; Chen, C.M.; Chen, S.H.; Hsiao, M.; et al. Synergism between p53 and Mcl-1 in protecting from hepatic injury, fibrosis and cancer. J. Hepatol. 2011, 54, 685–694. [Google Scholar] [CrossRef] [PubMed]
- Kodama, T.; Takehara, T.; Hikita, H.; Shimizu, S.; Shigekawa, M.; Tsunematsu, H.; Li, W.; Miyagi, T.; Hosui, A.; Tatsumi, T.; et al. Increases in p53 expression induce CTGF synthesis by mouse and human hepatocytes and result in liver fibrosis in mice. J. Clin. Invest. 2011, 121, 3343–3356. [Google Scholar] [CrossRef]
- Yu, S.; Ji, G.; Zhang, L. The role of p53 in liver fibrosis. Front. Pharmacol. 2022, 13, 1–7. [Google Scholar] [CrossRef]
- Krizhanovsky, V.; Yon, M.; Dickins, R.A.; Hearn, S.; Simon, J.; Miething, C.; Yee, H.; Zender, L.; Lowe, S.W. Senescence of Activated Stellate Cells Limits Liver Fibrosis. Cell 2008, 134, 657–667. [Google Scholar] [CrossRef]
- Nishizawa, H.; Iguchi, G.; Fukuoka, H.; Takahashi, M.; Suda, K.; Bando, H.; Matsumoto, R.; Yoshida, K.; Odake, Y.; Ogawa, W.; et al. IGF-I induces senescence of hepatic stellate cells and limits fibrosis in a p53-dependent manner. Sci. Rep. 2016, 6, 1–11. [Google Scholar] [CrossRef] [PubMed]
- Schnabl, B.; Purbeck, C.A.; Choi, Y.H.; Hagedorn, C.H.; Brenner, D.A. Replicative senescence of activated human hepatic stellate cells is accompanied by a pronounced inflammatory but less fibrogenic phenotype. Hepatology 2003, 37, 653–664. [Google Scholar] [CrossRef] [PubMed]
- Lujambio, A.; Akkari, L.; Simon, J.; Grace, D.; Tschaharganeh, D.F.; Bolden, J.E.; Zhao, Z.; Thapar, V.; Joyce, J.A.; Krizhanovsky, V.; et al. Non-cell-autonomous tumor suppression by p53. Cell 2013, 153, 449–460. [Google Scholar] [CrossRef]
- Zeisberg, M.; Neilson, E.G. Mechanisms of tubulointerstitial fibrosis. J. Am. Soc. Nephrol. 2010, 21, 1819–1834. [Google Scholar] [CrossRef]
- Naghavi, M.; Wang, H.; Lozano, R.; Davis, A.; Liang, X.; Zhou, M.; Vollset, S.E.; Abbasoglu Ozgoren, A.; Abdalla, S.; Abd-Allah, F.; et al. Global, regional, and national age-sex specific all-cause and cause-specific mortality for 240 causes of death, 1990-2013: A systematic analysis for the Global Burden of Disease Study 2013. Lancet 2015, 385, 117–171. [Google Scholar]
- Bakris, G.; Vassalotti, J.; Ritz, E.; Wanner, C.; Stergiou, G.; Molitch, M.; Nesto, R.; Kaysen, G.A.; Sowers, J.R. National kidney foundation consensus conference on cardiovascular and kidney diseases and diabetes risk: An integrated therapeutic approach to reduce events. Kidney Int. 2010, 78, 726–736. [Google Scholar] [CrossRef] [PubMed]
- Wynn, T.A. Cellular and molecular mechanisms of fibrosis. J. Pathol. 2008, 214, 199–210. [Google Scholar] [CrossRef] [PubMed]
- Prakoura, N.; Hadchouel, J.; Chatziantoniou, C. Novel Targets for Therapy of Renal Fibrosis. J. Histochem. Cytochem. 2019, 67, 701–715. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y. Renal fibrosis: New insights into the pathogenesis and therapeutics. Kidney Int. 2006, 69, 213–217. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y. Cellular and molecular mechanisms of renal fibrosis. Nat. Rev. Nephrol. 2011, 7, 684–696. [Google Scholar] [CrossRef]
- Kaissling, B.; LeHir, M.; Kriz, W. Renal epithelial injury and fibrosis. Biochim. Biophys. Acta -Mol. Basis Dis. 2013, 1832, 931–939. [Google Scholar] [CrossRef]
- Nogueira, A.; Pires, M.J.; Oliveira, P.A. Pathophysiological mechanisms of renal fibrosis: A review of animal models and therapeutic strategies. In Vivo 2017, 31, 1–22. [Google Scholar] [CrossRef] [PubMed]
- Leung, K.C.W.; Tonelli, M.; James, M.T. Chronic kidney disease following acute kidney injury—Risk and outcomes. Nat. Rev. Nephrol. 2013, 9, 77–85. [Google Scholar] [CrossRef]
- Venkatachalam, M.A.; Griffin, K.A.; Lan, R.; Geng, H.; Saikumar, P.; Bidani, A.K. Acute kidney injury: A springboard for progression in chronic kidney disease. Am. J. Physiol. -Ren. Physiol. 2010, 298, 1078–1094. [Google Scholar] [CrossRef]
- Yang, L.; Besschetnova, T.Y.; Brooks, C.R.; Shah, J.V.; Bonventre, J.V. Epithelial cell cycle arrest in G2/M mediates kidney fibrosis after injury. Nat. Med. 2010, 16, 535–543. [Google Scholar] [CrossRef]
- Bonventre, J.V.; Yang, L.; Bonventre, J.V.; Yang, L. Cellular pathophysiology of ischemic acute kidney injury Find the latest version: Science in medicine Cellular pathophysiology of ischemic acute kidney injury. Sci. Med. 2011, 121, 4210–4221. [Google Scholar]
- Padanilam, B.J. Cell death induced by acute renal injury: A perspective on the contributions of apoptosis and necrosis. Am. J. Physiol. -Ren. Physiol. 2003, 284, 608–627. [Google Scholar] [CrossRef] [PubMed]
- Schelling, J.R.; Cleveland, R.P. Involvement of Fas-dependent apoptosis in renal tubular epithelial cell deletion in chronic renal failure. Kidney Int. 1999, 56, 1313–1316. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Galluzzi, L.; Vitale, I.; Abrams, J.M.; Alnemri, E.S.; Baehrecke, E.H.; Blagosklonny, M.V.; Dawson, T.M.; Dawson, V.L.; El-Deiry, W.S.; Fulda, S. Molecular definitions of cell death subroutines: Recommendations of the Nomenclature Committee on Cell Death 2012. Cell Death Differ. 2012, 19, 107–120. [Google Scholar] [CrossRef] [PubMed]
- Portilla, D. Apoptosis, fibrosis and senescence. Nephron. Clin. Pract. 2014, 127, 65–69. [Google Scholar] [CrossRef] [PubMed]
- Mao, H.; Li, Z.; Zhou, Y.; Li, Z.; Zhuang, S.; An, X.; Zhang, B.; Chen, W.; Nie, J.; Wang, Z.; et al. HSP72 attenuates renal tubular cell apoptosis and interstitial fibrosis in obstructive nephropathy. Am. J. Physiol. -Ren. Physiol. 2008, 295, 202–214. [Google Scholar] [CrossRef]
- Whelan, R.S.; Konstantinidis, K.; Wei, A.C.; Chen, Y.; Reyna, D.E.; Jha, S.; Yang, Y.; Calvert, J.W.; Lindsten, T.; Thompson, C.B. Bax regulates primary necrosis through mitochondrial dynamics. Proc. Natl. Acad. Sci. USA 2012, 109, 6566–6571. [Google Scholar] [CrossRef]
- Zhang, G.; Oldroyd, S.D.; Huang, L.H.; Yang, B.; Li, Y.; Ye, R.; El Nahas, A.M. Role of apoptosis and Bcl-2/Bax in the development of tubulointerstitial fibrosis during experimental obstructive nephropathy. Exp. Nephrol. 2001, 9, 71–80. [Google Scholar] [CrossRef]
- Wei, Q.; Dong, G.; Franklin, J.; Dong, Z. The pathological role of Bax in cisplatin nephrotoxicity. Kidney Int. 2007, 72, 53–62. [Google Scholar] [CrossRef]
- Wei, Q.; Dong, G.; Chen, J.K.; Ramesh, G.; Dong, Z. Bax and Bak have critical roles in ischemic acute kidney injury in global and proximal tubule-specific knockout mouse models. Kidney Int. 2013, 84, 138–148. [Google Scholar] [CrossRef]
- Jang, H.S.; Padanilam, B.J. Simultaneous deletion of bax and bak is required to prevent apoptosis and interstitial fibrosis in obstructive nephropathy. Am. J. Physiol. -Ren. Physiol. 2015, 309, F540–F550. [Google Scholar] [CrossRef]
- Mei, S.; Li, L.; Wei, Q.; Hao, J.; Su, Y.; Mei, C.; Dong, Z. Double knockout of Bax and Bak from kidney proximal tubules reduces unilateral urethral obstruction associated apoptosis and renal interstitial fibrosis. Sci. Rep. 2017, 7, 1–7. [Google Scholar] [CrossRef]
- Lovisa, S.; LeBleu, V.S.; Tampe, B.; Sugimoto, H.; Vadnagara, K.; Carstens, J.L.; Wu, C.C.; Hagos, Y.; Burckhardt, B.C.; Pentcheva-Hoang, T. Epithelial-to-mesenchymal transition induces cell cycle arrest and parenchymal damage in renal fibrosis. Nat. Med. 2015, 21, 998–1009. [Google Scholar] [CrossRef]
- Grande, M.T.; Sánchez-Laorden, B.; López-Blau, C.; De Frutos, C.A.; Boutet, A.; Arévalo, M.; Rowe, R.G.; Weiss, S.J.; López-Novoa, J.M.; Nieto, M.A. Snail1-induced partial epithelial-to-mesenchymal transition drives renal fibrosis in mice and can be targeted to reverse established disease. Nat. Med. 2015, 21, 989–997. [Google Scholar] [CrossRef] [PubMed]
- Wynn, T.A. Review series introduction Common and unique mechanisms regulate fibrosis in various fibroproliferative diseases. J. Cell Investig. 2007, 117, 524–529. [Google Scholar] [CrossRef]
- Brooks, C.; Wei, Q.; Cho, S.G.; Dong, Z. Regulation of mitochondrial dynamics in acute kidney injury in cell culture and rodent models. J. Clin. Invest. 2009, 119, 1275–1285. [Google Scholar] [CrossRef] [PubMed]
- Friedman, S.L.; Sheppard, D.; Duffield, J.S.; Violette, S. Therapy for fibrotic diseases: Nearing the starting line. Sci. Transl. Med. 2013, 5, 167sr1. [Google Scholar] [CrossRef] [PubMed]
- Ye, C.; Tang, H.; Zhao, Z.; Lei, C.T.; You, C.Q.; Zhang, J.; Gao, P.; He, F.F.; Chen, S.; Wang, Y.M.; et al. MDM2 mediates fibroblast activation and renal tubulointerstitial fibrosis via a p53-independent pathway. Am. J. Physiol. -Ren. Physiol. 2017, 312, F760–F768. [Google Scholar] [CrossRef]
- Laplante, P.; Sirois, I.; Raymond, M.A.; Kokta, V.; Beliveau, A.; Prat, A.; Pshezhetsky, A.V.; Hebert, M.J. Caspase-3-mediated secretion of connective tissue growth factor by apoptotic endothelial cells promotes fibrosis. Cell Death Differ. 2010, 17, 291–303. [Google Scholar] [CrossRef]
- Lebleu, V.S.; Taduri, G.; O’Connell, J.; Teng, Y.; Cooke, V.G.; Woda, C.; Sugimoto, H.; Kalluri, R. and function of myofibroblasts in kidney fibrosis. Nat. Med. 2013, 19, 1047–1053. [Google Scholar] [CrossRef]
- Meran, S.; Steadman, R. Fibroblasts and myofibroblasts in renal fibrosis. Int. J. Exp. Pathol. 2011, 92, 158–167. [Google Scholar] [CrossRef] [PubMed]
- Schrimpf, C.; Duffield, J.S. Mechanisms of fibrosis: The role of the pericyte. Curr. Opin. Nephrol. Hypertens. 2011, 20, 297–305. [Google Scholar] [CrossRef] [PubMed]
- Grande, M.T.; López-Novoa, J.M. Fibroblast activation and myofibroblast generation in obstructive nephropathy. Nat. Rev. Nephrol. 2009, 5, 319–328. [Google Scholar] [CrossRef]
- Barnes, J.L.; Gorin, Y. Myofibroblast differentiation during fibrosis: Role of NAD(P)H oxidases. Kidney Int. 2011, 79, 944–956. [Google Scholar] [CrossRef] [PubMed]
- Humphreys, B.D.; Lin, S.L.; Kobayashi, A.; Hudson, T.E.; Nowlin, B.T.; Bonventre, J.V.; Valerius, M.T.; McMahon, A.P.; Duffield, J.S. Fate tracing reveals the pericyte and not epithelial origin of myofibroblasts in kidney fibrosis. Am. J. Pathol. 2010, 176, 85–97. [Google Scholar] [CrossRef]
- Gewin, L.S. Renal fibrosis: Primacy of the proximal tubule. Matrix Biol. 2018, 68–69, 248–262. [Google Scholar] [CrossRef] [PubMed]
- Hayashida, T.; Wu, M.H.; Pierce, A.; Poncelet, A.C.; Varga, J.; Schnaper, H.W. MAP-kinase activity necessary for TGFβ1-stimulated mesangial cell type I collagen expression requires adhesion-dependent phosphorylation of FAK tyrosine 397. J. Cell Sci. 2007, 120, 4230–4240. [Google Scholar] [CrossRef]
- Kato, M.; Putta, S.; Wang, M.; Yuan, H.; Lanting, L.; Nair, I.; Gunn, A.; Nakagawa, Y.; Shimano, H.; Todorov, I. TGF-β activates Akt kinase through a microRNA-dependent amplifying circuit targeting PTEN. Nat. Cell Biol. 2009, 11, 881–889. [Google Scholar] [CrossRef]
- Docherty, N.G.; O’Sullivan, O.E.; Healy, D.A.; Fitzpatrick, J.M.; Watson, R.W.G. Evidence that inhibition of tubular cell apoptosis protects against renal damage and development of fibrosis following ureteric obstruction. Am. J. Physiol. -Ren. Physiol. 2006, 290, 4–13. [Google Scholar] [CrossRef] [PubMed]
- Liu, L.; Zhang, P.; Bai, M.; He, L.; Zhang, L.; Liu, T.; Yang, Z. p53 upregulated by HIF- 1 α promotes hypoxia- induced G 2/M arrest and renal fibrosis in vitro and in vivo. J. Mol. Sci. Biol. 2019, 11, 371–382. [Google Scholar]
- Kelly, K.J.; Plotkin, Z.; Vulgamott, S.L.; Dagher, P.C. P53 mediates the apoptotic response to GTP depletion after renal ischemia-reperfusion: Protective role of a p53 inhibitor. J. Am. Soc. Nephrol. 2003, 14, 128–138. [Google Scholar] [CrossRef] [PubMed]
- Dagher, P.C.; Mai, E.M.; Hato, T.; Lee, S.Y.; Anderson, M.D.; Karozos, S.C.; Mang, H.E.; Knipe, N.L.; Plotkin, Z.; Sutton, T.A. The p53 inhibitor pifithrin-α can stimulate fibrosis in a rat model of ischemic acute kidney injury. Am. J. Physiol. -Ren. Physiol. 2012, 302, 284–291. [Google Scholar] [CrossRef]
- Overstreet, J.M.; Gifford, C.C.; Tang, J.; Higgins, P.J.; Samarakoon, R. Emerging role of tumor suppressor p53 in acute and chronic kidney diseases. Cell Mol. Life Sci. 2022, 79, 1–12. [Google Scholar] [CrossRef]
- Higgins, S.P.; Tang, Y.; Higgins, C.E.; Mian, B.; Zhang, W.; Czekay, R.P.; Samarakoon, R.; Conti, D.J.; Higgins, P.J. TGF-β1/p53 signaling in renal fibrogenesis. Cell Signal 2018, 43, 1–10. [Google Scholar] [CrossRef]
- Higgins, C.E.; Tang, J.; Mian, B.M.; Higgins, S.P.; Gifford, C.C.; Conti, D.J.; Meldrum, K.K.; Samarakoon, R.; Higgins, P.J. TGF-β1–p53 cooperativity regulates a profibrotic genomic program in the kidney: Molecular mechanisms and clinical implications. FASEB J. 2019, 33, 10596–10606. [Google Scholar] [CrossRef] [PubMed]
- Huang, Y.; Border, W.A.; Noble, N.A. Perspectives on blockade of TGFβ overexpression. Kidney Int. 2006, 69, 1713–1714. [Google Scholar] [CrossRef]
- Travers, J.G.; Kamal, F.A.; Robbins, J.; Yutzey, K.E.; Blaxall, B.C. Cardiac fibrosis: The fibroblasts awakens. Circ. Res. 2016, 118, 1021–1040. [Google Scholar] [CrossRef] [PubMed]
- Ranjan, P.; Kumari, R.; Verma, S.K. Cardiac Fibroblasts and Cardiac Fibrosis: Precise Role of Exosomes. Front. Cell Dev. Biol. 2019, 7, 1–12. [Google Scholar] [CrossRef]
- Janicki, O.S.; Brower, G.L. The role of myocardial fibrillar collagen in ventricular remodeling and function. J. Card Fail. 2002, 8, 319–325. [Google Scholar] [CrossRef]
- Kong, P.; Christia, P.; Frangogiannis, N.G. The pathogenesis of cardiac fibrosis. Cell Mol. Life Sci. 2014, 71, 549–574. [Google Scholar] [CrossRef]
- Berk, B.C.; Fujiwara, K.; Lehoux, S. ECM remodeling in hypertensive heart disease Bradford C. Berk. J. Clin. Invest. 2007, 117, 568. [Google Scholar] [CrossRef] [PubMed]
- Liu, T.; Song, D.; Dong, J.; Zhu, P.; Liu, J.; Liu, W.; Ma, X.; Zhao, L.; Ling, S. Current understanding of the pathophysiology of myocardial fibrosis and its quantitative assessment in heart failure. Front. Physiol. 2017, 8, 1–13. [Google Scholar] [CrossRef] [PubMed]
- Duraes, A.R.; Bitar, Y.D.S.L.; Roever, L.; Neto, M.G. Endomyocardial fibrosis: Past, present, and future. Hear. Fail. Rev. 2019, 25, 725–730. [Google Scholar] [CrossRef]
- Frangogiannis, N.G. Cardiac fibrosis: Cell biological mechanisms, molecular pathways and therapeutic opportunities. Mol. Asp. Med. 2019, 65, 70–99. [Google Scholar] [CrossRef]
- Overbeek, M.J.; Mouchaers, K.T.B.; Niessen, H.M.; Hadi, A.M.; Kupreishvili, K.; Boonstra, A.; Voskuyl, A.E.; Belien, J.A.M.; Smit, E.F.; Dijkmans, B.C.; et al. Characteristics of Interstitial Fibrosis and Inflammatory Cell Infiltration in Right Ventricles of Systemic Sclerosis-Associated Pulmonary Arterial Hypertension. Available online: https://www.hindawi.com/journals/ijr/2010/604615/abs/ (accessed on 20 April 2019).
- Saraste, A.; Pulkki, K.; Kallajoki, M.; Henriksen, K.; Parvinen, M.; Voipio-Pulkki, L.-M. Apoptosis in Human Acute Myocardial Infarction. Circulation 1997, 95, 320–323. [Google Scholar] [CrossRef]
- Kajstura, J.; Cheng, W.; Reiss, K.; A Clark, W.; Sonnenblick, E.H.; Krajewski, S.; Reed, J.C.; Olivetti, G.; Anversa, P. Apoptotic and necrotic myocyte cell deaths are independent contributing variables of infarct size in rats. Lab. Investig. 1996, 74, 86–107. [Google Scholar]
- Bardales, R.H.; Hailey, L.S.; Xie, S.S.; Schaefer, R.F.; Hsu, S.M. In situ apoptosis assay for the detection of early acute myocardial infarction. Am. J. Pathol. 1996, 149, 821–829. [Google Scholar]
- Zhao, Y.; Tan, Y.; Xi, S.; Li, Y.; Li, C.; Cui, J.; Yan, X.; Li, X.; Wang, G.; Li, W.; et al. A novel mechanism by which sdf-1b protects cardiac cells from palmitate-induced endoplasmic reticulum stress and apoptosis via CXCR7 and AMPK/p38 MAPK-Mediated interleukin-6 generation. Diabetes 2013, 62, 2545–2558. [Google Scholar] [CrossRef]
- Shinde, A.V.; Frangogiannis, N.G. Mechanisms of Fibroblast Activation in the Remodeling Myocardium. Curr. Pathobiol. Rep. 2017, 5, 145–152. [Google Scholar] [CrossRef] [PubMed]
- Misao, J.; Hayakawa, Y.; Ohno, M.; Kato, S.; Fujiwara, T.; Fujiwara, H. Expression of bcl-2 Protein, an Inhibitor of Apoptosis, and Bax, an Accelerator of Apoptosis, in Ventricular Myocytes of Human Hearts With Myocardial Infarction. Circulation 1996, 94, 1506–1512. [Google Scholar] [CrossRef]
- Frangogiannis, N.G. Regulation of the Inflammatory Response in Cardiac Repair. Circ. Res. 2012, 110, 159–173. [Google Scholar] [CrossRef]
- Humeres, C.; Frangogiannis, N.G. Fibroblasts in the Infarcted, Remodeling, and Failing Heart. JACC Basic Transl. Sci. 2019, 4, 449–467. [Google Scholar] [CrossRef]
- Kawaguchi, M.; Takahashi, M.; Hata, T.; Kashima, Y.; Usui, F.; Morimoto, H.; Izawa, A.; Takahashi, Y.; Masumoto, J.; Koyama, J.; et al. Inflammasome activation of cardiac fibroblasts is essential for myocardial is-chemia/reperfusion injury. Circulation 2011, 123, 594–604. [Google Scholar] [CrossRef]
- Cleutjens, J.P.; Verluyten, M.J.; Smiths, J.F.; Daemen, M.J. Collagen remodeling after myocardial infarction in the rat heart. Am. J. Pathol. 1995, 147, 325–338. [Google Scholar]
- Cavalera, M.; Wang, J.; Frangogiannis, N.G. Obesity, metabolic dysfunction, and cardiac fibrosis: Pathophysiological pathways, molecular mechanisms, and therapeutic opportunities. Transl. Res. 2014, 164, 323–335. [Google Scholar] [CrossRef]
- Biernacka, A.; Frangogiannis, N.G. Aging and Cardiac Fibrosis. Aging Dis. 2011, 2, 158–173. [Google Scholar]
- Rathod, R.H.; Powell, A.J.; Geva, T. Myocardial Fibrosis in Congenital Heart Disease. Circ. J. 2016, 80, 1300–1307. [Google Scholar] [CrossRef] [PubMed]
- Kanisicak, O.; Khalil, H.; Ivey, M.J.; Karch, J.; Maliken, B.D.; Correll, R.N.; Brody, M.J.; Lin, S.-C.J.; Aronow, B.J.; Tallquist, M.D.; et al. Genetic lineage tracing defines myofibroblast origin and function in the injured heart. Nat. Commun. 2016, 7, 12260. [Google Scholar] [CrossRef]
- Pinto, A.R.; Ilinykh, A.; Ivey, M.J.; Kuwabara, J.T.; D’Antoni, M.L.; Debuque, R.; Chandran, A.; Wang, L.; Arora, K.; Rosenthal, N.; et al. Revisiting Cardiac Cellular Composition. Circ. Res. 2016, 118, 400–409. [Google Scholar] [CrossRef] [PubMed]
- Zhou, P.; Pu, W.T. Recounting Cardiac Cellular Composition. Circ. Res. 2017, 118, 368–370. [Google Scholar] [CrossRef] [PubMed]
- Moore-Morris, T.; Guimarães-Camboa, N.; Banerjee, I.; Zambon, A.C.; Kisseleva, T.; Velayoudon, A.; Stallcup, W.B.; Gu, Y.; Dalton, N.D.; Cedenilla, M.; et al. Resident fibroblast lineages mediate pressure overload–induced cardiac fibrosis. J. Clin. Investig. 2014, 124, 2921–2934. [Google Scholar] [CrossRef]
- Moore-Morris, T.; van Vliet, P.; Andelfinger, G.; Puceat, M. Role of Epigenetics in Cardiac Development and Congenital Diseases. Physiol. Rev. 2018, 98, 2453–2475. [Google Scholar] [CrossRef]
- Ali, S.R.; Ranjbarvaziri, S.; Talkhabi, M.; Zhao, P.; Subat, A.; Hojjat, A.; Kamran, P.; Müller, A.M.; Volz, K.S.; Tang, Z.; et al. Developmental Heterogeneity of Cardiac Fibroblasts Does Not Predict Pathological Proliferation and Activation. Circ. Res. 2014, 115, 625–635. [Google Scholar] [CrossRef]
- Zeisberg, E.M.; Tarnavski, O.; Zeisberg, M.; Dorfman, A.L.; McMullen, J.R.; Gustafsson, E.; Chandraker, A.; Yuan, X.; Pu, W.T.; Roberts, A.B.; et al. Endothelial-to-mesenchymal transition contributes to cardiac fibrosis. Nat. Med. 2007, 13, 952–961. [Google Scholar] [CrossRef]
- Aisagbonhi, O.; Rai, M.; Ryzhov, S.; Atria, N.; Feoktistov, I.; Hatzopoulos, A.K. Experimental myocardial infarction triggers canonical Wnt signaling and endothe-lial-to-mesenchymal transition. Dis. Model Mech. 2011, 4, 469–483. [Google Scholar] [CrossRef]
- Verma, S.K.; Garikipati, V.N.; Krishnamurthy, P.; Schumacher, S.M.; Grisanti, L.A.; Cimini, M.; Cheng, Z.; Khan, M.; Yue, Y.; Benedict, C.; et al. Interleukin-10 Inhibits Bone Marrow Fibroblast Progenitor Cell–Mediated Cardiac Fibrosis in Pressure-Overloaded Myocardium. Circulation 2017, 136, 940–953. [Google Scholar] [CrossRef] [PubMed]
- Dobaczewski, M.; Bujak, M.; Zymek, P.; Ren, G.; Entman, M.L.; Frangogiannis, N.G. Extracellular matrix remodeling in canine and mouse myocardial infarcts. Cell Tissue Res. 2006, 324, 475–488. [Google Scholar] [CrossRef] [PubMed]
- Takemura, G.; Ohno, M.; Hayakawa, Y.; Misao, J.; Kanoh, M.; Ohno, A.; Uno, Y.; Minatoguchi, S.; Fujiwara, T.; Fujiwara, H. Role of Apoptosis in the Disappearance of Infiltrated and Proliferated Interstitial Cells After Myocardial Infarction. Circ. Res. 1998, 82, 1130–1138. [Google Scholar] [CrossRef]
- Van Amerongen, M.; Bou-Gharios, G.; Popa, E.; Van Ark, J.; Petersen, A.; Van Dam, G.; Van Luyn, M.; Harmsen, M. Bone marrow-derived myofibroblasts contribute functionally to scar formation after myocardial infarction. J. Pathol. 2007, 214, 377–386. [Google Scholar] [CrossRef] [PubMed]
- Frangogiannis, N.G.; Michael, L.H.; Entman, M.L. Myofibroblasts in reperfused myocardial infarcts express the embryonic form of smooth muscle myosin heavy chain (SMemb). Cardiovasc. Res. 2000, 48, 89–100. [Google Scholar] [CrossRef]
- Dobaczewski, M.; Gonzalez-Quesada, C.; Frangogiannis, N.G. The extracellular matrix as a modulator of the inflammatory and reparative response following myocardial infarction. J. Mol. Cell. Cardiol. 2010, 48, 504–511. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Takemura, G.; Kosai, K.-I.; Takahashi, T.; Okada, H.; Miyata, S.; Yuge, K.; Nagano, S.; Esaki, M.; Khai, N.C.; et al. Critical Roles for the Fas/Fas Ligand System in Postinfarction Ventricular Remodeling and Heart Failure. Circ. Res. 2004, 95, 627–636. [Google Scholar] [CrossRef] [PubMed]
- Frangogiannis, N.G. Pathophysiology of Myocardial Infarction. Compr. Physiol. 2015, 5, 1841–1875. [Google Scholar] [CrossRef] [PubMed]
- Willems, I.E.M.G.; Havenith, M.G.; De Mey, J.G.R.; Daemen, M.J.A.P. The alpha-smooth muscle actin-positive cells in healing human my-ocardial scars. Am. J. Pathol. 1994, 145, 868–875. [Google Scholar] [PubMed]
- Turner, N.A.; Porter, K.E. Function and fate of myofibroblasts after myocardial infarction. Fibrogenesis Tissue Repair 2013, 6, 5. [Google Scholar] [CrossRef]
- Rosenkranz, S. TGF-β1 and angiotensin networking in cardiac remodeling. Cardiovasc. Res. 2004, 63, 423–432. [Google Scholar] [CrossRef]
- Brown, R.D.; Ambler, S.K.; Mitchell, M.D.; Long, C.S. THE CARDIAC FIBROBLAST: Therapeutic Target in Myocardial Remodeling and Failure. Annu. Rev. Pharmacol. Toxicol. 2005, 45, 657–687. [Google Scholar] [CrossRef]
- Chen, D.B.; Wang, L.; Wang, P.H. Insulin-like growth factor I retards apoptotic signaling induced by ethanol in cardiomyocytes. Life Sci. 2000, 67, 1683–1693. [Google Scholar] [CrossRef]
- Li, P.F.; Dietz, R.; Von Harsdorf, R. Superoxide induces apoptosis in cardiomyocytes, but proliferation and expression of transforming growth factor-β1 in cardiac fibroblasts. FEBS Lett. 1999, 448, 206–210. [Google Scholar] [CrossRef]
- Zhang, X.; Azhar, G.; Nagano, K.; Wei, J.Y. Differential vulnerability to oxidative stress in rat cardiac myocytes versus fibroblasts. J. Am. Coll. Cardiol. 2001, 38, 2055–2062. [Google Scholar] [CrossRef]
- Mayorga, M.; Bahi, N.; Ballester, M.; Comella, J.; Sanchis, D.; Junutula, J.R.; Schonteich, E.; Wilson, G.M.; Peden, A.A.; Scheller, R.H.; et al. Bcl-2 Is a Key Factor for Cardiac Fibroblast Resistance to Programmed Cell Death. J. Biol. Chem. 2004, 279, 34882–34889. [Google Scholar] [CrossRef]
- Murtha, L.A.; Morten, M.; Schuliga, M.J.; Mabotuwana, N.S.; Hardy, S.A.; Waters, D.W.; Burgess, J.K.; Ngo, D.T.; Sverdlov, A.L.; Knight, D.A.; et al. The Role of Pathological Aging in Cardiac and Pulmonary Fibrosis. Aging Dis. 2019, 10, 419–428. [Google Scholar] [CrossRef] [PubMed]
- Zhu, F.; Li, Y.; Zhang, J.; Piao, C.; Liu, T.; Li, H.-H.; Du, J. Senescent Cardiac Fibroblast Is Critical for Cardiac Fibrosis after Myocardial Infarction. PLoS ONE 2013, 8, e74535. [Google Scholar] [CrossRef] [PubMed]
- Meyer, K.; Hodwin, B.; Ramanujam, D.; Engelhardt, S.; Sarikas, A. Essential Role for Premature Senescence of Myofibroblasts in Myocardial Fibrosis. J. Am. Coll. Cardiol. 2016, 67, 2018–2028. [Google Scholar] [CrossRef] [PubMed]
- Nagpal, V.; Rai, R.; Place, A.T.; Murphy, S.B.; Verma, S.K.; Ghosh, A.K.; Vaughan, D.E. MiR-125b Is Critical for Fibroblast-to-Myofibroblast Transition and Cardiac Fibrosis. Circulation 2016, 133, 291–301. [Google Scholar] [CrossRef] [PubMed]
- Burke, R.M.; Lighthouse, J.K.; Quijada, P.; Dirkx, R.A.; Rosenberg, A.; Moravec, C.S.; Alexis, J.D.; Small, E.M. Small proline-rich protein 2B drives stress-dependent p53 degradation and fibroblast proliferation in heart failure. Proc. Natl. Acad. Sci. USA 2018, 115, E3436–E3445. [Google Scholar] [CrossRef]
- Pang, S.; Chen, Y.; Dai, C.; Liu, T.; Zhang, W.; Wang, J.; Cui, X.; Guo, X.; Jiang, F. Anti-fibrotic effects of p53 activation induced by RNA polymerase I inhibitor in primary cardiac fibroblasts. Eur. J. Pharmacol. 2021, 907, 174303. [Google Scholar] [CrossRef]
- Quigley, H.A. Glaucoma. Lancet 2011, 377, 1367–1377. [Google Scholar] [CrossRef]
- Quigley, H.; Broman, A.T. The number of people with glaucoma worldwide in 2010 and 2020. Br. J. Ophthalmol. 2006, 90, 262–267. [Google Scholar] [CrossRef]
- Hernandez, M.R. Extracellular Matrix Macromolecules of the Lamina Cribrosa: A Pressure-sensitive Connective Tissue. Eur. J. Gastroenterol. Hepatol. 1993, 2, 50–57. [Google Scholar] [CrossRef]
- Hernandez, M.R.; Pena, J.D.O. The Optic Nerve Head in Glaucomatous Optic Neuropathy. Arch. Ophthalmol. 1997, 115, 389–395. [Google Scholar] [CrossRef]
- Anderson, D.R. Ultrastructure of Human and Monkey Lamina Cribrosa and Optic Nerve Head. Arch. Ophthalmol. 1969, 82, 800–814. [Google Scholar] [CrossRef] [PubMed]
- Ogden, T.E.; Duggan, J.; Danley, K.; Wilcox, M.; Minckler, D.S. Morphometry of nerve fiber bundle pores in the optic nerve head of the human. Exp. Eye Res. 1988, 46, 559–568. [Google Scholar] [CrossRef] [PubMed]
- Jonas, J.B.; Mardin, C.Y.; Schlötzer-Schrehardt, U.; Naumann, G.O. Morphometry of the human lamina cribrosa surface. Investig. Opthalmology Vis. Sci. 1991, 32, 401–405. [Google Scholar]
- Burgoyne, C.F.; Downs, J.C.; Bellezza, A.J.; Suh, J.-K.F.; Hart, R.T. The optic nerve head as a biomechanical structure: A new paradigm for understanding the role of IOP-related stress and strain in the pathophysiology of glaucomatous optic nerve head damage. Prog. Retin. Eye Res. 2005, 24, 39–73. [Google Scholar] [CrossRef]
- Weinreb, R.N.; Aung, T.; Medeiros, F.A. The Pathophysiology and Treatment of Glaucoma: A review. JAMA 2014, 311, 1901–1911. [Google Scholar] [CrossRef] [PubMed]
- Quigley, H.A. Optic Nerve Damage in Human Glaucoma. Arch. Ophthalmol. 1981, 99, 635–649. [Google Scholar] [CrossRef]
- Yang, H.; Williams, G.; Downs, J.C.; Sigal, I.A.; Roberts, M.D.; Thompson, H.; Burgoyne, C.F. Posterior (Outward) Migration of the Lamina Cribrosa and Early Cupping in Monkey Experimental Glaucoma. Investig. Opthalmology Vis. Sci. 2011, 52, 7109–7121. [Google Scholar] [CrossRef]
- Yang, H.; Thompson, H.; Roberts, M.D.; Sigal, I.A.; Downs, J.C.; Burgoyne, C.F. Deformation of the Early Glaucomatous Monkey Optic Nerve Head Connective Tissue after Acute IOP Elevation in 3-D Histomorphometric Reconstructions. Investig. Opthalmology Vis. Sci. 2011, 52, 345–363. [Google Scholar] [CrossRef]
- Downs, J.C.; Roberts, M.D.; Sigal, I.A. Glaucomatous cupping of the lamina cribrosa: A review of the evidence for active progressive remodeling as a mechanism. Exp. Eye Res. 2011, 93, 133–140. [Google Scholar] [CrossRef]
- Quigley, H.A.; Hohman, R.M.; Addicks, E.M.; Massof, R.W.; Green, W.R. Morphologic Changes in the Lamina Cribrosa Correlated with Neural Loss in Open-Angle Glaucoma. Am. J. Ophthalmol. 1983, 95, 673–691. [Google Scholar] [CrossRef]
- Minckler, D.S. Optic nerve damage in glaucoma: I. Obstruction to axoplasmic flow. Surv. Ophthalmol. 1981, 26, 128–136. [Google Scholar] [CrossRef] [PubMed]
- Downs, J.C.; Girkin, C.A. Lamina cribrosa in glaucoma. Curr. Opin. Ophthalmol. 2017, 28, 113–119. [Google Scholar] [CrossRef] [PubMed]
- Hernandez, M.R.; Andrzejewska, W.M.; Neufeld, A.H. Changes in the Extracellular Matrix of the Human Optic Nerve Head in Primary Open-Angle Glaucoma. Am. J. Ophthalmol. 1990, 109, 180–188. [Google Scholar] [CrossRef] [PubMed]
- Hernandez, M.R. Ultrastructural immunocytochemical analysis of elastin in the human lamina cribrosa. Changes in elastic fibers in primary open-angle glaucoma. Investig. Opthalmology Vis. Sci. 1992, 33, 2891–2903. [Google Scholar]
- Pena, J.D.O.; Taylor, A.W.; Ricard, C.S.; Vidal, I.; Hernandez, M.R. Transforming growth factor β isoforms in human optic nerve heads. Br. J. Ophthalmol. 1999, 83, 209–218. [Google Scholar] [CrossRef]
- Wallace, D.M.; Clark, A.F.; Lipson, K.E.; Andrews, D.; Crean, J.K.; O’Brien, C.J. Anti-Connective Tissue Growth Factor Antibody Treatment Reduces Extracellular Matrix Production in Trabecular Meshwork and Lamina Cribrosa Cells. Investig. Opthalmology Vis. Sci. 2013, 54, 7836–7848. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Fukuchi, T.; Sawaguchi, S.; Hara, H.; Shirakashi, M.; Iwata, K. Extracellular matrix changes of the optic nerve lamina cribrosa in monkey eyes with experimentally chronic glaucoma. Graefe’s Arch. Clin. Exp. Ophthalmol. 1992, 230, 421–427. [Google Scholar] [CrossRef]
- Jonas, J.B.; Berenshtein, E.; Holbach, L. Anatomic Relationship between Lamina Cribrosa, Intraocular Space, and Cerebrospinal Fluid Space. Investig. Opthalmology Vis. Sci. 2003, 44, 5189–5195. [Google Scholar] [CrossRef]
- Anderson, D.R.; Hendrickson, A. Effect of intraocular pressure on rapid axoplasmic transport in monkey optic nerve. Investig. Ophthalmol. 1974, 13, 771–783. [Google Scholar]
- Liu, B.; McNally, S.; Kilpatrick, J.I.; Jarvis, S.P.; O’Brien, C.J. Aging and ocular tissue stiffness in glaucoma. Surv. Ophthalmol. 2018, 63, 56–74. [Google Scholar] [CrossRef] [PubMed]
- Halpern, D.L.; Grosskreutz, C.L. Glaucomatous optic neuropathy: Mechanisms of disease. Ophthalmol. Clin. North Am. 2002, 15, 61–68. [Google Scholar] [CrossRef] [PubMed]
- Quigley, H.A.; Green, W.R. The Histology of Human Glaucoma Cupping and Optic Nerve Damage: Clinicopathologic Corre-lation in 21 Eyes. Ophthalmology 1979, 86, 1803–1827. [Google Scholar] [CrossRef]
- Kerrigan, L.A.; Zack, D.J.; Quigley, H.A.; Smith, S.D.; Pease, M. TUNEL-Positive Ganglion Cells in Human Primary Open-angle Glaucoma. Arch. Ophthalmol. 1997, 115, 1031–1035. [Google Scholar] [CrossRef] [PubMed]
- Berkelaar, M.; Clarke, D.; Wang, Y.; Bray, G.; Aguayo, A. Axotomy results in delayed death and apoptosis of retinal ganglion cells in adult rats. J. Neurosci. 1994, 14, 4368–4374. [Google Scholar] [CrossRef]
- Quigley, H.A.; Nickells, R.W.; Kerrigan, L.A.; Pease, M.E.; Thibault, D.J.; Zack, D.J. Retinal ganglion cell death in experimental glaucoma and after axotomy occurs by apoptosis. Investig. Ophthalmol. Vis. Sci. 1995, 36, 774–786. [Google Scholar]
- Garcia-Valenzuela, E.; Shareef, S.; Walsh, J.; Sharma, S.C. Programmed cell death of retinal ganglion cells during experimental glau-coma. Exp. Eye Res. 1995, 61, 33–44. [Google Scholar] [CrossRef]
- Okisaka, S.; Murakami, A.; Mizukawa, A.; Junji, I. Apoptosis in retinal ganglion cell decrease in human glaucomatous eyes. Jpn. J. Ophthalmol. 1997, 41, 84–88. [Google Scholar] [CrossRef]
- Morrison, J.C.; Dorman-Pease, M.E.; Dunkelberger, G.R.; Quigley, H.A. Optic Nerve Head Extracellular Matrix in Primary Optic Atrophy and Experimental Glaucoma. Arch. Ophthalmol. 1990, 108, 1020–1024. [Google Scholar] [CrossRef] [PubMed]
- Kirwan, R.P.; Fenerty, C.H.; Crean, J.; Wordinger, R.J.; Clark, A.F.; O’Brien, C.J. Influence of cyclical mechanical strain on extracellular matrix gene expression in human lamina cribrosa cells in vitro. Mol. Vis. 2005, 11, 798–810. [Google Scholar]
- Hinz, B. Myofibroblasts. Exp. Eye Res. 2015, 142, 56–70. [Google Scholar] [CrossRef]
- Lambert, W.S.; Clark, A.F.; Wordinger, R.J. Neurotrophin and Trk expression by cells of the human lamina cribrosa following oxygen-glucose deprivation. BMC Neurosci. 2004, 5, 51. [Google Scholar] [CrossRef] [PubMed]
- Wordinger, R.J.; Agarwal, R.; Talati, M.; Fuller, J.; Lambert, W.; Clark, A.F. Expression of bone morphogenetic proteins (BMP), BMP receptors, and BMP associated proteins in human trabecular meshwork and optic nerve head cells and tissues. Mol. Vis. 2002, 8, 1162–1170. [Google Scholar]
- Kirwan, R.P.; Leonard, M.O.; Murphy, M.; Clark, A.F.; O’Brien, C.J. Transforming Growth factor-β-regulated gene transcription and protein ex-pression in human GFAP-negative lamina cribrosa cells. Glia 2005, 52, 309–324. [Google Scholar] [CrossRef] [PubMed]
- Rogers, R.; Dharsee, M.; Ackloo, S.; Flanagan, J.G. Proteomics analyses of activated human optic nerve head lamina cribrosa cells fol-lowing biomechanical strain. Investig. Ophthalmol. Vis. Sci. 2012, 53, 3806–3816. [Google Scholar] [CrossRef] [PubMed]
- Kirwan, R.P.; Crean, J.K.; Fenerty, C.H.; Clark, A.F.; O’Brien, C.J. Effect of cyclical mechanical stretch and exogenous transforming growth fac-tor-β1 on matrix metalloproteinase-2 activity in lamina cribrosa cells from the human optic nerve head. J. Glaucoma 2004, 13, 327–334. [Google Scholar] [CrossRef]
- Quill, B.; Docherty, N.G.; Clark, A.F.; O’Brien, C.J. The Effect of Graded Cyclic Stretching on Extracellular Matrix–Related Gene Expression Profiles in Cultured Primary Human Lamina Cribrosa Cells. Investig. Opthalmology Vis. Sci. 2011, 52, 1908–1915. [Google Scholar] [CrossRef]
- Irnaten, M.; Zhdanov, A.; Brennan, D.; Crotty, T.; Clark, A.; Papkovsky, D.; O’Brien, C. Activation of the NFAT–Calcium Signaling Pathway in Human Lamina Cribrosa Cells in Glaucoma. Investig. Opthalmology Vis. Sci. 2018, 59, 831–842. [Google Scholar] [CrossRef]
- McElnea, E.; Quill, B.; Docherty, N.; Irnaten, M.; Siah, W.; Clark, A.; O’Brien, C.; Wallace, D. Oxidative stress, mitochondrial dysfunction and calcium overload in human lamina cribrosa cells from glaucoma donors. Mol. Vis. 2011, 17, 1182–1191. [Google Scholar]
- Irnaten, M.; Barry, R.C.; Quill, B.; Clark, A.F.; Harvey, B.J.P.; O’Brien, C.J. Activation of Stretch-Activated Channels and Maxi-K+Channels by Membrane Stress of Human Lamina Cribrosa Cells. Investig. Opthalmology Vis. Sci. 2009, 50, 194–202. [Google Scholar] [CrossRef]
- Kirwan, R.P.; Felice, L.; Clark, A.F.; Leonard, M.O.; O’Brien, C.J. Hypoxia Regulated Gene Transcription in Human Optic Nerve Lamina Cribrosa Cells in Culture. Investig. Opthalmology Vis. Sci. 2012, 53, 2243–2255. [Google Scholar] [CrossRef] [PubMed]
- Duffy, M.J.; Synnott, N.C.; Crown, J. p53 in cancer: Ready for therapeutic targeting? Transl. Cancer Res. 2016, 5, 627–631. [Google Scholar] [CrossRef]
- Karni-Schmidt, O.; Lokshin, M.; Prives, C. The Roles of MDM2 and MDMX in Cancer. Annu. Rev. Pathol. Mech. Dis. 2016, 11, 617–644. [Google Scholar] [CrossRef] [PubMed]
- Fang, S.; Jensen, J.P.; Ludwig, R.L.; Vousden, K.H.; Weissman, A.M. Mdm2 Is a RING Finger-dependent Ubiquitin Protein Ligase for Itself and p53. J. Biol. Chem. 2000, 275, 8945–8951. [Google Scholar] [CrossRef]
- Momand, J.; Jung, D.; Wilczynski, S.; Niland, J. The MDM2 gene amplification database. Nucleic Acids Res. 1998, 26, 3453–3459. [Google Scholar] [CrossRef]
- Haupt, Y.; Maya, R.; Kazaz, A.; Oren, M. Mdm2 promotes the rapid degradation of p53. Nature 1997, 387, 296–299. [Google Scholar] [CrossRef]
- Cross, B.; Chen, L.; Cheng, Q.; Li, B.; Yuan, Z.-M.; Chen, J. Inhibition of p53 DNA Binding Function by the MDM2 Protein Acidic Domain. J. Biol. Chem. 2011, 286, 16018–16029. [Google Scholar] [CrossRef]
- Oliner, J.D.; Pietenpol, J.A.; Thiagalingam, S.; Gyuris, J.; Kinzler, K.W.; Vogelstein, B. Oncoprotein MDM2 conceals the activation domain of tumour suppressor p53. Nature 1993, 362, 857–860. [Google Scholar] [CrossRef]
- Shangary, S.; Wang, S. Small-Molecule Inhibitors of the MDM2-p53 Protein-Protein Interaction to Reactivate p53 Function: A Novel Approach for Cancer Therapy. Annu. Rev. Pharmacol. Toxicol. 2009, 49, 223–241. [Google Scholar] [CrossRef]
- Kussie, P.H.; Gorina, S.; Marechal, V.; Elenbaas, B.; Moreau, J.; Levine, A.J.; Pavletich, N.P. Structure of the MDM2 oncoprotein bound to the p53 tumor suppressor transac-tivation domain. Science 1996, 274, 948–953. [Google Scholar] [CrossRef]
- Klein, C.; Vassilev, L.T. Targeting the p53–MDM2 interaction to treat cancer. Br. J. Cancer 2004, 91, 1415–1419. [Google Scholar] [CrossRef] [PubMed]
- Burgess, A.; Chia, K.M.; Haupt, S.; Thomas, D.; Haupt, Y.; Lim, E. Clinical overview of MDM2/X-targeted therapies. Front. Oncol. 2016, 6, 1–7. [Google Scholar] [CrossRef]
- Andreeff, M.; Kelly, K.R.; Yee, K.; Assouline, S.; Strair, R.; Popplewell, L.; Bowen, D.; Martinelli, G.; Drummond, M.W.; Vyas, P.; et al. Results of the Phase I Trial of RG7112, a Small-Molecule MDM2 Antagonist in Leukemia. Clin. Cancer Res. 2016, 22, 868–876. [Google Scholar] [CrossRef]
- Vu, B.; Wovkulich, P.; Pizzolato, G.; Lovey, A.; Ding, Q.; Jiang, N.; Liu, J.-J.; Zhao, C.; Glenn, K.; Wen, Y.; et al. Discovery of RG7112: A Small-Molecule MDM2 Inhibitor in Clinical Development. ACS Med. Chem. Lett. 2013, 4, 466–469. [Google Scholar] [CrossRef]
- Her, N.-G.; Oh, J.-W.; Oh, Y.J.; Han, S.; Cho, H.J.; Lee, Y.; Ryu, G.H.; Nam, D.-H. Potent effect of the MDM2 inhibitor AMG232 on suppression of glioblastoma stem cells. Cell Death Dis. 2018, 9, 1–12. [Google Scholar] [CrossRef] [PubMed]
- Khurana, A.; Shafer, D.A. MDM2 antagonists as a novel treatment option for acute myeloid leukemia: Perspectives on the therapeutic potential of idasanutlin (RG7388). OncoTargets Ther. 2019, ume 12, 2903–2910. [Google Scholar] [CrossRef]
- Konopleva, M.Y.; Röllig, C.; Cavenagh, J.; Deeren, D.; Girshova, L.; Krauter, J.; Martinelli, G.; Montesinos, P.; Schäfer, J.A.; Ottmann, O.G.; et al. Idasanutlin Plus Cytarabine in Relapsed or Refractory Acute Myeloid Leukemia: Results of the MIRROS Trial. Blood Adv. 2022, 6, 4147–4156. [Google Scholar] [CrossRef]
- Koo, N.; Sharma, A.K.; Narayan, S. Therapeutics Targeting p53-MDM2 Interaction to Induce Cancer Cell Death. Int. J. Mol. Sci. 2022, 23, 5005. [Google Scholar] [CrossRef] [PubMed]
- Tisato, V.; Voltan, R.; Gonelli, A.; Secchiero, P.; Zauli, G. MDM2/X inhibitors under clinical evaluation: Perspectives for the management of hematological malignancies and pediatric cancer. J. Hematol. Oncol. 2017, 10, 1–17. [Google Scholar] [CrossRef]
- Wang, X.; Jiang, X. Mdm2 and MdmX partner to regulate p53. FEBS Lett. 2012, 586, 1390–1396. [Google Scholar] [CrossRef]
- Huang, L.; Yan, Z.; Liao, X.; Li, Y.; Yang, J.; Wang, Z.G.; Zuo, Y.; Kawai, H.; Shadfan, M.; Ganapathy, S.; et al. The p53 inhibitors MDM2/MDMX complex is required for control of p53 activity in vivo. Proc. Natl. Acad. Sci. USA 2011, 108, 12001–12006. [Google Scholar] [CrossRef]
- Shadfan, M.; Lopez-Pajares, V.; Yuan, Z.-M. MDM2 and MDMX: Alone and together in regulation of p53. Transl. Cancer Res. 2012, 1, 88–89. [Google Scholar]
- Prodosmo, A.; Giglio, S.; Moretti, S.; Mancini, F.; Barbi, F.; Avenia, N.; Di Conza, G.; Schünemann, H.J.; Pistola, L.; Ludovini, V.; et al. Analysis of human MDM4 variants in papillary thyroid carcinomas reveals new potential markers of cancer properties. J. Mol. Med. 2008, 86, 585–596. [Google Scholar] [CrossRef] [PubMed]
- Saleh, M.N.; Patel, M.R.; Bauer, T.M.; Goel, S.; Falchook, G.S.; Shapiro, G.I.; Chung, K.Y.; Infante, J.R.; Conry, R.M.; Rabinowits, G.; et al. Correction: Phase I Trial of ALRN-6924, a Dual Inhibitor of MDMX and MDM2, in Patients with Solid Tumors and Lymphomas Bearing Wild-type TP53. Clin. Cancer Res. 2022, 28, 429. [Google Scholar] [CrossRef] [PubMed]
- Chang, Y.S.; Graves, B.; Guerlavais, V.; Tovar, C.; Packman, K.; To, K.-H.; Olson, K.A.; Kesavan, K.; Gangurde, P.; Mukherjee, A.; et al. Stapled α−helical peptide drug development: A potent dual inhibitor of MDM2 and MDMX for p53-dependent cancer therapy. Proc. Natl. Acad. Sci. USA 2013, 110, E3445–E3454. [Google Scholar] [CrossRef]
- Carvajal, L.A.; Ben Neriah, D.; Senecal, A.; Benard, L.; Thiruthuvanathan, V.; Yatsenko, T.; Narayanagari, S.-R.; Wheat, J.C.; Todorova, T.I.; Mitchell, K.; et al. Dual inhibition of MDMX and MDM2 as a therapeutic strategy in leukemia. Sci. Transl. Med. 2018, 10, eaao3003. [Google Scholar] [CrossRef] [PubMed]
- Zhu, H.; Gao, H.; Ji, Y.; Zhou, Q.; Du, Z.; Tian, L.; Jiang, Y.; Yao, K.; Zhou, Z. Targeting p53–MDM2 interaction by small-molecule inhibitors: Learning from MDM2 inhibitors in clinical trials. J. Hematol. Oncol. 2022, 15, 1–23. [Google Scholar] [CrossRef] [PubMed]
- Hauck, L.; Stanley-Hasnain, S.; Fung, A.; Grothe, D.; Rao, V.; Mak, T.W.; Billia, F. Cardiac-specific ablation of the E3 ubiquitin ligase Mdm2 leads to oxidative stress, broad mitochondrial deficiency and early death. PLoS ONE 2017, 12, e0189861. [Google Scholar] [CrossRef]
- Saito, R.; Rocanin-Arjo, A.; You, Y.-H.; Darshi, M.; Van Espen, B.; Miyamoto, S.; Pham, J.; Pu, M.; Romoli, S.; Natarajan, L.; et al. Systems biology analysis reveals role of MDM2 in diabetic nephropathy. J. Clin. Investig. 2016, 1, e87877. [Google Scholar] [CrossRef]
- Dezor, M.; Dorszewska, J.; Florczak, J.; Kempisty, B.; Jaroszewska-Kolecka, J.; Rozycka, A.; Polrolniczak, A.; Bugaj, R.; Jagodzinski, P.P.; Kozubski, W. Expression of 8-oxoguanine DNA glycosylase 1 (OGG1) and the level of p53 and TNF-αlpha proteins in peripheral lymphocytes of patients with Alzheimer’s disease. Folia Neuropathol. 2011, 49, 123–131. [Google Scholar]
- Mulay, S.R.; Thomasova, D.; Ryu, M.; Anders, H.-J. MDM2 (murine double minute-2) links inflammation and tubular cell healing during acute kidney injury in mice. Kidney Int. 2012, 81, 1199–1211. [Google Scholar] [CrossRef] [PubMed]
- Kon, N.; Wang, D.; Li, T.; Jiang, L.; Qiang, L.; Gu, W. Inhibition of Mdmx (Mdm4) in vivo induces anti-obesity effects. Oncotarget 2018, 9, 7282–7297. [Google Scholar] [CrossRef] [PubMed]
- Assmann, G.; Voswinkel, J.; Mueller, M.; Bittenbring, J.; Koenig, J.; Menzel, A.; Pfreundschuh, M.; Roemer, K.; Melchers, I. Association of rheumatoid arthritis with Mdm2 SNP309 and genetic evidence for an allele-specific interaction between MDM2 and p53 P72R variants: A case control study. Ann. Rheum. Dis. 2009, 27, 615–619. [Google Scholar]
- Zhang, C.-X.; Chen, J.; Cai, L.; Wu, J.; Wang, J.-Y.; Cao, L.-F.; Zhou, W.; Chen, T.-X. DNA induction of MDM2 promotes proliferation of human renal mesangial cells and alters peripheral B cells subsets in pediatric systemic lupus erythematosus. Mol. Immunol. 2018, 94, 166–175. [Google Scholar] [CrossRef]
- Mulay, S.R.; Romoli, S.; Desai, J.; Honarpisheh, M.M.; Kumar, S.V.; Anders, H.J.; Thomasova, D. Murine Double Minute-2 Inhibition Ameliorates Established Crescentic Glomerulone-phritis. Am. J. Pathol. 2016, 186, 1442–1453. [Google Scholar] [CrossRef]
- Zhao, H.; Shen, R.; Dong, X.; Shen, Y. Murine Double Minute-2 Inhibition Attenuates Cardiac Dysfunction and Fibrosis by Mod-ulating NF-κB Pathway After Experimental Myocardial Infarction. Inflammation 2017, 40, 232–239. [Google Scholar] [CrossRef]
- Schreckenberg, R.; Bencsik, P.; Weber, M.; Abdallah, Y.; Csonka, C.; Gömöri, K.; Kiss, K.; Pálóczi, J.; Pipis, J.; Sárközy, M.; et al. Adverse Effects on β-Adrenergic Receptor Coupling: Ischemic Postconditioning Failed to Preserve Long-Term Cardiac Function. J. Am. Hear. Assoc. 2017, 6, e006809. [Google Scholar] [CrossRef]
- He, F.-F.; You, R.-Y.; Ye, C.; Lei, C.-T.; Tang, H.; Su, H.; Zhang, C. Inhibition of SIRT2 Alleviates Fibroblast Activation and Renal Tubulointerstitial Fibrosis via MDM2. Cell. Physiol. Biochem. 2018, 46, 451–460. [Google Scholar] [CrossRef]


Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
McElhinney, K.; Irnaten, M.; O’Brien, C. p53 and Myofibroblast Apoptosis in Organ Fibrosis. Int. J. Mol. Sci. 2023, 24, 6737. https://doi.org/10.3390/ijms24076737
McElhinney K, Irnaten M, O’Brien C. p53 and Myofibroblast Apoptosis in Organ Fibrosis. International Journal of Molecular Sciences. 2023; 24(7):6737. https://doi.org/10.3390/ijms24076737
Chicago/Turabian StyleMcElhinney, Kealan, Mustapha Irnaten, and Colm O’Brien. 2023. "p53 and Myofibroblast Apoptosis in Organ Fibrosis" International Journal of Molecular Sciences 24, no. 7: 6737. https://doi.org/10.3390/ijms24076737
APA StyleMcElhinney, K., Irnaten, M., & O’Brien, C. (2023). p53 and Myofibroblast Apoptosis in Organ Fibrosis. International Journal of Molecular Sciences, 24(7), 6737. https://doi.org/10.3390/ijms24076737