
Engineering the Active Site Lid Dynamics to Improve the Catalytic Efficiency of Yeast Cytosine Deaminase

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Results
2.1. Construct yCD with a Helix Tail
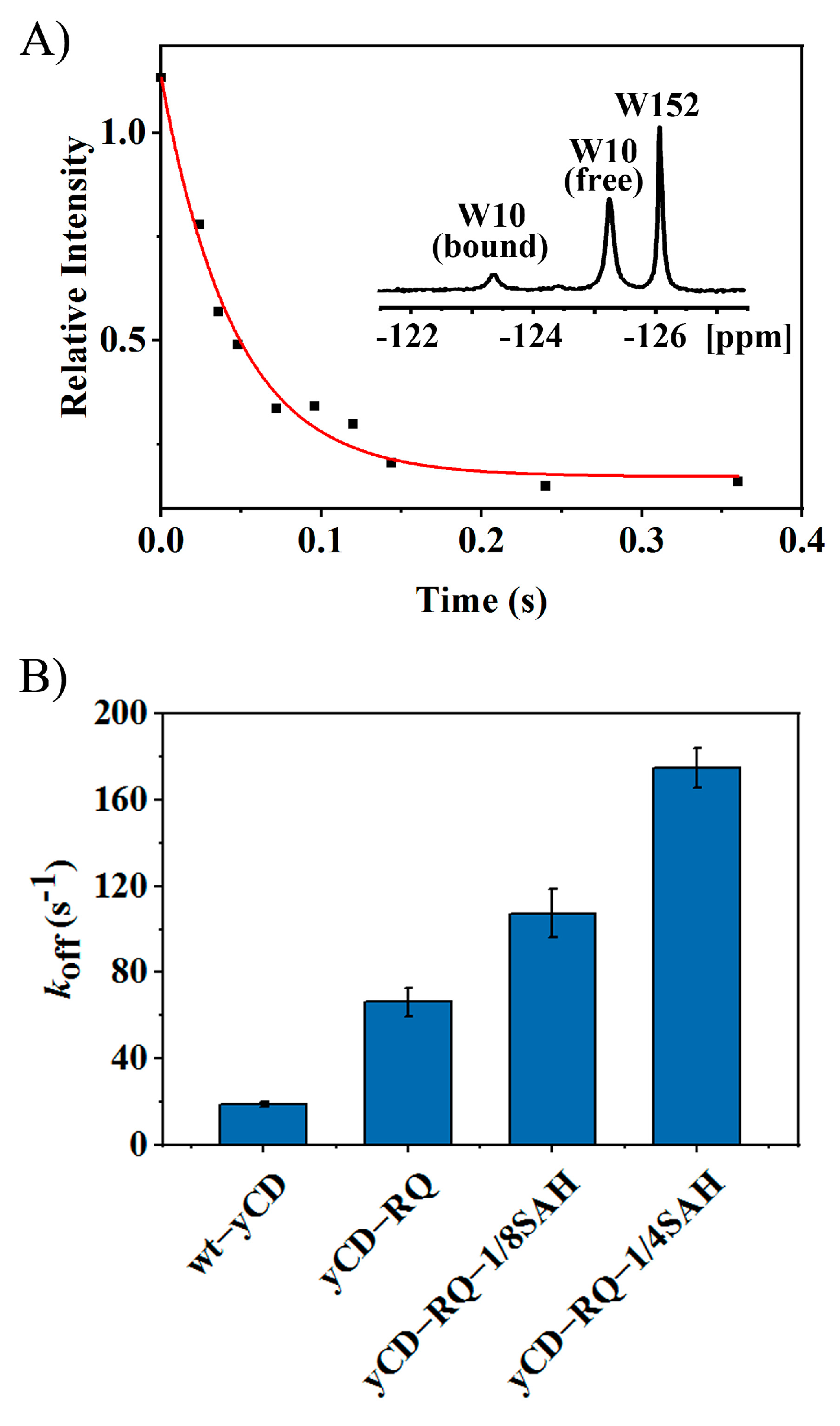
2.2. Product Release Rate of yCD Variants
2.3. Activation Enthalpy and Entropy of yCD Variants
2.4. Structure and Active Site Interactions of yCD-RQ and yCD-RQ-1/8SAH
2.5. Conformational Dynamics from H/D Exchanges
3. Discussion
4. Materials and Methods
4.1. Construction of Mutagenesis Library
4.2. Screening Procedure
4.3. Expression and Purification of yCD Fusion Proteins
4.4. Kinetic Measurements and Data Fitting
4.5. 19F NMR Spectroscopy and the Product Release Rate
4.6. H/D Exchange Rate of yCD and Its Variants
4.7. Crystallization, Data Collection, and Structure Refinement
5. Patents
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Tzeng, S.R.; Kalodimos, C.G. Protein activity regulation by conformational entropy. Nature 2012, 488, 236–240. [Google Scholar] [CrossRef] [PubMed]
- Carroll, M.J.; Mauldin, R.V.; Gromova, A.V.; Singleton, S.F.; Collins, E.J.; Lee, A.L. Evidence for dynamics in proteins as a mechanism for ligand dissociation. Nat. Chem. Biol. 2012, 8, 246–252. [Google Scholar] [CrossRef] [PubMed]
- Bhabha, G.; Ekiert, D.C.; Jennewein, M.; Zmasek, C.M.; Tuttle, L.M.; Kroon, G.; Dyson, H.J.; Godzik, A.; Wilson, I.A.; Wright, P.E. Divergent evolution of protein conformational dynamics in dihydrofolate reductase. Nat. Struct. Mol. Biol. 2013, 20, 1243–1249. [Google Scholar] [CrossRef] [PubMed]
- Oyen, D.; Fenwick, R.B.; Stanfield, R.L.; Dyson, H.J.; Wright, P.E. Cofactor-Mediated Conformational Dynamics Promote Product Release From Escherichia coli Dihydrofolate Reductase via an Allosteric Pathway. J. Am. Chem. Soc. 2015, 137, 9459–9468. [Google Scholar] [CrossRef]
- Palmer, A.G., 3rd. Enzyme dynamics from NMR spectroscopy. Acc. Chem. Res. 2015, 48, 457–465. [Google Scholar] [CrossRef]
- Papaleo, E.; Saladino, G.; Lambrughi, M.; Lindorff-Larsen, K.; Gervasio, F.L.; Nussinov, R. The Role of Protein Loops and Linkers in Conformational Dynamics and Allostery. Chem. Rev. 2016, 116, 6391–6423. [Google Scholar] [CrossRef]
- Wei, G.; Xi, W.; Nussinov, R.; Ma, B. Protein Ensembles: How Does Nature Harness Thermodynamic Fluctuations for Life? The Diverse Functional Roles of Conformational Ensembles in the Cell. Chem. Rev. 2016, 116, 6516–6551. [Google Scholar] [CrossRef]
- Lisi, G.P.; Loria, J.P. Solution NMR Spectroscopy for the Study of Enzyme Allostery. Chem. Rev. 2016, 116, 6323–6369. [Google Scholar] [CrossRef]
- Kovermann, M.; Rogne, P.; Wolf-Watz, M. Protein dynamics and function from solution state NMR spectroscopy. Q. Rev. Biophys. 2016, 49, e6. [Google Scholar] [CrossRef]
- Otten, R.; Liu, L.; Kenner, L.R.; Clarkson, M.W.; Mavor, D.; Tawfik, D.S.; Kern, D.; Fraser, J.S. Rescue of conformational dynamics in enzyme catalysis by directed evolution. Nat. Commun. 2018, 9, 1314. [Google Scholar] [CrossRef]
- Wang, Y.; V, S.M.; Kim, J.; Li, G.; Ahuja, L.G.; Aoto, P.; Taylor, S.S.; Veglia, G. Globally correlated conformational entropy underlies positive and negative cooperativity in a kinase’s enzymatic cycle. Nat. Commun. 2019, 10, 799. [Google Scholar] [PubMed]
- Gao, S.; Thompson, E.J.; Barrow, S.L.; Zhang, W.; Iavarone, A.T.; Klinman, J.P. Hydrogen-Deuterium Exchange within Adenosine Deaminase, a TIM Barrel Hydrolase, Identifies Networks for Thermal Activation of Catalysis. J. Am. Chem. Soc. 2020, 142, 19936–19949. [Google Scholar] [PubMed]
- Gao, S.; Klinman, J.P. Functional roles of enzyme dynamics in accelerating active site chemistry: Emerging techniques and changing concepts. Curr. Opin. Struct. Biol. 2022, 75, 102434. [Google Scholar] [CrossRef] [PubMed]
- Agarwal, P.K. A Biophysical Perspective on Enzyme Catalysis. Biochemistry-Us 2019, 58, 438–449. [Google Scholar]
- Yang, L.Q.; Sang, P.; Tao, Y.; Fu, Y.X.; Zhang, K.Q.; Xie, Y.H.; Liu, S.Q. Protein dynamics and motions in relation to their functions: Several case studies and the underlying mechanisms. J. Biomol. Struct. Dyn. 2014, 32, 372–393. [Google Scholar] [CrossRef]
- Maria-Solano, M.A.; Serrano-Hervas, E.; Romero-Rivera, A.; Iglesias-Fernandez, J.; Osuna, S. Role of conformational dynamics in the evolution of novel enzyme function. Chem. Commun. 2018, 54, 6622–6634. [Google Scholar]
- Campbell, E.C.; Correy, G.J.; Mabbitt, P.D.; Buckle, A.M.; Tokuriki, N.; Jackson, C.J. Laboratory evolution of protein conformational dynamics. Curr. Opin. Struct. Biol. 2018, 50, 49–57. [Google Scholar] [CrossRef]
- Crean, R.M.; Gardner, J.M.; Kamerlin, S.C.L. Harnessing Conformational Plasticity to Generate Designer Enzymes. J. Am. Chem. Soc. 2020, 142, 11324–11342. [Google Scholar] [CrossRef]
- Agarwal, P.K.; Bernard, D.N.; Bafna, K.; Doucet, N. Enzyme dynamics: Looking beyond a single structure. ChemCatChem 2020, 12, 4704–4720. [Google Scholar]
- Schenkmayerova, A.; Pinto, G.P.; Toul, M.; Marek, M.; Hernychova, L.; Planas-Iglesias, J.; Liskova, V.D.; Pluskal, D.; Vasina, M.; Emond, S.; et al. Engineering the protein dynamics of an ancestral luciferase. Nat. Commun. 2021, 12, 3616. [Google Scholar]
- Bata, Z.; Molnar, Z.; Madaras, E.; Molnar, B.; Santa-Bell, E.; Varga, A.; Leveles, I.; Qian, R.Z.; Hammerschmidt, F.; Paizs, C.; et al. Substrate Tunnel Engineering Aided by X-ray Crystallography and Functional Dynamics Swaps the Function of MIO-Enzymes. Acs. Catal. 2021, 11, 4538–4549. [Google Scholar] [CrossRef]
- Jackson, C.J.; Foo, J.L.; Tokuriki, N.; Afriat, L.; Carr, P.D.; Kim, H.K.; Schenk, G.; Tawfik, D.S.; Ollis, D.L. Conformational sampling, catalysis, and evolution of the bacterial phosphotriesterase. Proc. Natl. Acad. Sci. USA 2009, 106, 21631–21636. [Google Scholar] [CrossRef] [PubMed]
- Campbell, E.; Kaltenbach, M.; Correy, G.J.; Carr, P.D.; Porebski, B.T.; Livingstone, E.K.; Afriat-Jurnou, L.; Buckle, A.M.; Weik, M.; Hollfelder, F.; et al. The role of protein dynamics in the evolution of new enzyme function. Nat. Chem. Biol. 2016, 12, 944–950. [Google Scholar] [CrossRef] [PubMed]
- Caro, J.A.; Harpole, K.W.; Kasinath, V.; Lim, J.; Granja, J.; Valentine, K.G.; Sharp, K.A.; Wand, A.J. Entropy in molecular recognition by proteins. Proc. Natl. Acad. Sci. USA 2017, 114, 6563–6568. [Google Scholar] [CrossRef] [PubMed]
- Li, C.; Winnard, P.T., Jr.; Takagi, T.; Artemov, D.; Bhujwalla, Z.M. Multimodal image-guided enzyme/prodrug cancer therapy. J. Am. Chem. Soc. 2006, 128, 15072–15073. [Google Scholar] [CrossRef]
- Yata, V.K.; Gopinath, P.; Ghosh, S.S. Emerging implications of nonmammalian cytosine deaminases on cancer therapeutics. Appl. Biochem. Biotechnol. 2012, 167, 2103–2116. [Google Scholar] [CrossRef]
- Karjoo, Z.; Chen, X.; Hatefi, A. Progress and problems with the use of suicide genes for targeted cancer therapy. Adv. Drug. Deliv. Rev. 2016, 99, 113–128. [Google Scholar] [CrossRef]
- Warren, T.D.; Patel, K.; Eshleman, J.R.; Ostermeier, M. Protein-Programmed Accumulation of Yeast Cytosine Deaminase in Cancer Cells in Response to Mock-Hypoxia. ACS. Synth. Biol. 2019, 8, 948–954. [Google Scholar] [CrossRef]
- Stolworthy, T.S.; Korkegian, A.M.; Willmon, C.L.; Ardiani, A.; Cundiff, J.; Stoddard, B.L.; Black, M.E. Yeast cytosine deaminase mutants with increased thermostability impart sensitivity to 5-fluorocytosine. J. Mol. Biol. 2008, 377, 854–869. [Google Scholar] [CrossRef]
- Hiraoka, K.; Inagaki, A.; Kato, Y.; Huang, T.T.; Mitchell, L.A.; Kamijima, S.; Takahashi, M.; Matsumoto, H.; Hacke, K.; Kruse, C.A.; et al. Retroviral replicating vector-mediated gene therapy achieves long-term control of tumor recurrence and leads to durable anticancer immunity. Neuro-Oncology 2017, 19, 918–929. [Google Scholar]
- Mitchell, L.A.; Espinoza, F.L.; Mendoza, D.; Kato, Y.; Inagaki, A.; Hiraoka, K.; Kasahara, N.; Gruber, H.E.; Jolly, D.J.; Robbins, J.M. Toca 511 gene transfer and treatment with the prodrug, 5-fluorocytosine, promotes durable antitumor immunity in a mouse glioma model. Neuro-Oncology 2017, 19, 930–939. [Google Scholar] [CrossRef] [PubMed]
- Sklenak, S.; Yao, L.S.; Cukier, R.I.; Yan, H.G. Catalytic mechanism of yeast cytosine deaminase: An ONIOM computational study. J. Am. Chem. Soc. 2004, 126, 14879–14889. [Google Scholar] [CrossRef] [PubMed]
- Yao, L.S.; Li, Y.; Wu, Y.; Liu, A.Z.; Yan, H.G. Product release is rate-limiting in the activation of the prodrug 5-fluorocytosine by yeast cytosine deaminase. Biochemistry 2005, 44, 5940–5947. [Google Scholar] [CrossRef] [PubMed]
- Yao, L.S.; Sklenak, S.; Yan, H.G.; Cukier, R.I. A molecular dynamics exploration of the catalytic mechanism of yeast cytosine deaminase. J. Phys. Chem. B 2005, 109, 7500–7510. [Google Scholar] [CrossRef]
- Wang, J.; Sklenak, S.; Liu, A.; Felczak, K.; Wu, Y.; Li, Y.; Yan, H. Role of glutamate 64 in the activation of the prodrug 5-fluorocytosine by yeast cytosine deaminase. Biochemistry 2012, 51, 475–486. [Google Scholar] [CrossRef][Green Version]
- Ko, T.P.; Lin, J.J.; Hu, C.Y.; Hsu, Y.H.; Wang, A.H.; Liaw, S.H. Crystal structure of yeast cytosine deaminase. Insights into enzyme mechanism and evolution. J. Biol. Chem. 2003, 278, 19111–19117. [Google Scholar] [CrossRef]
- Ireton, G.C.; Black, M.E.; Stoddard, B.L. The 1.14 A crystal structure of yeast cytosine deaminase: Evolution of nucleotide salvage enzymes and implications for genetic chemotherapy. Structure 2003, 11, 961–972. [Google Scholar] [CrossRef]
- Spink, B.J.; Sivaramakrishnan, S.; Lipfert, J.; Doniach, S.; Spudich, J.A. Long single alpha-helical tail domains bridge the gap between structure and function of myosin VI. Nat. Struct. Mol. Biol. 2008, 15, 591–597. [Google Scholar] [CrossRef]
- Sivaramakrishnan, S.; Sung, J.; Ali, M.; Doniach, S.; Flyvbjerg, H.; Spudich, J.A. Combining single-molecule optical trapping and small-angle x-ray scattering measurements to compute the persistence length of a protein ER/K alpha-helix. Biophys. J. 2009, 97, 2993–2999. [Google Scholar] [CrossRef]
- Barnes, C.A.; Shen, Y.; Ying, J.; Takagi, Y.; Torchia, D.A.; Sellers, J.R.; Bax, A. Remarkable Rigidity of the Single alpha-Helical Domain of Myosin-VI As Revealed by NMR Spectroscopy. J. Am. Chem. Soc. 2019, 141, 9004–9017. [Google Scholar] [CrossRef]
- Lewandowski, J.R.; Halse, M.E.; Blackledge, M.; Emsley, L. Protein dynamics. Direct observation of hierarchical protein dynamics. Science 2015, 348, 578–581. [Google Scholar] [CrossRef] [PubMed]
- Yao, L.S.; Yan, H.G.; Cukier, R.I. A molecular dynamics study of the ligand release path inyeast cytosine deaminase. Biophys. J. 2007, 92, 2301–2310. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Dempsey, C.E. Hydrogen exchange in peptides and proteins using NMR-spectroscopy. Prog. Nucl. Magn. Reson. Spec. 2001, 39, 135–170. [Google Scholar] [CrossRef]
- Persson, F.; Halle, B. How amide hydrogens exchange in native proteins. Proc. Natl. Acad. Sci. USA 2015, 112, 10383–10388. [Google Scholar] [CrossRef] [PubMed]
- Hernandez, G.; Jenney, F.E.; Adams, M.W.W.; LeMaster, D.M. Millisecond time scale conformational flexibility in a hyperthermophile protein at ambient temperature. Proc. Natl. Acad. Sci. USA 2000, 97, 3166–3170. [Google Scholar] [CrossRef] [PubMed]
- Li, J.J.; Chang, S.F.; Liau, I.-I.; Chan, P.C.; Liu, R.S.; Yen, S.H.; Wang, H.E.; Chang, C.A. Targeted antitumor prodrug therapy using CNGRC-yCD fusion protein in combination with 5-fluorocytosine. J. Biomed. Sci. 2016, 23, 15. [Google Scholar] [CrossRef]
- Delaglio, F.; Grzesiek, S.; Vuister, G.W.; Zhu, G.; Pfeifer, J.; Bax, A. NMRPIPE—A MULTIDIMENSIONAL SPECTRAL PROCESSING SYSTEM BASED ON UNIX PIPES. J. Biomol. NMR 1995, 6, 277–293. [Google Scholar] [CrossRef]
- Yu, F.; Wang, Q.S.; Li, M.J.; Zhou, H.; Liu, K.; Zhang, K.H.; Wang, Z.J.; Xu, Q.; Xu, C.Y.; Pan, Q.Y.; et al. Aquarium: An automatic data-processing and experiment information management system for biological macromolecular crystallography beamlines. J. Appl. Crystallogr. 2019, 52, 472–477. [Google Scholar] [CrossRef]
- Adams, P.D.; Afonine, P.V.; Bunkoczi, G.; Chen, V.B.; Davis, I.W.; Echols, N.; Headd, J.J.; Hung, L.W.; Kapral, G.J.; Grosse-Kunstleve, R.W.; et al. PHENIX: A comprehensive Python-based system for macromolecular structure solution. Acta Crystallogr. D 2010, 66, 213–221. [Google Scholar] [CrossRef]
- Emsley, P.; Lohkamp, B.; Scott, W.G.; Cowtan, K. Features and development of Coot. Acta Crystallogr. D 2010, 66, 486–501. [Google Scholar] [CrossRef]
- Echols, N.; Moriarty, N.W.; Klei, H.E.; Afonine, P.V.; Bunkoczi, G.; Headd, J.J.; McCoy, A.J.; Oeffner, R.D.; Read, R.J.; Terwilliger, T.C.; et al. Automating crystallographic structure solution and refinement of protein-ligand complexes. Acta Crystallogr. D 2014, 70, 144–154. [Google Scholar] [CrossRef] [PubMed]
- Chen, V.B.; Arendall, W.B.; Headd, J.J.; Keedy, D.A.; Immormino, R.M.; Kapral, G.J.; Murray, L.W.; Richardson, J.S.; Richardson, D.C. MolProbity: All-atom structure validation for macromolecular crystallography. Acta Crystallogr. D 2010, 66, 12–21. [Google Scholar] [CrossRef] [PubMed]







Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Deng, H.; Qin, M.; Liu, Z.; Yang, Y.; Wang, Y.; Yao, L. Engineering the Active Site Lid Dynamics to Improve the Catalytic Efficiency of Yeast Cytosine Deaminase. Int. J. Mol. Sci. 2023, 24, 6592. https://doi.org/10.3390/ijms24076592
Deng H, Qin M, Liu Z, Yang Y, Wang Y, Yao L. Engineering the Active Site Lid Dynamics to Improve the Catalytic Efficiency of Yeast Cytosine Deaminase. International Journal of Molecular Sciences. 2023; 24(7):6592. https://doi.org/10.3390/ijms24076592
Chicago/Turabian StyleDeng, Hanzhong, Mingming Qin, Zhijun Liu, Ying Yang, Yefei Wang, and Lishan Yao. 2023. "Engineering the Active Site Lid Dynamics to Improve the Catalytic Efficiency of Yeast Cytosine Deaminase" International Journal of Molecular Sciences 24, no. 7: 6592. https://doi.org/10.3390/ijms24076592
APA StyleDeng, H., Qin, M., Liu, Z., Yang, Y., Wang, Y., & Yao, L. (2023). Engineering the Active Site Lid Dynamics to Improve the Catalytic Efficiency of Yeast Cytosine Deaminase. International Journal of Molecular Sciences, 24(7), 6592. https://doi.org/10.3390/ijms24076592