

Identification of Key Ferroptosis-Related Genes in the Peripheral Blood of Patients with Relapsing-Remitting Multiple Sclerosis and Its Diagnostic Value

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Results
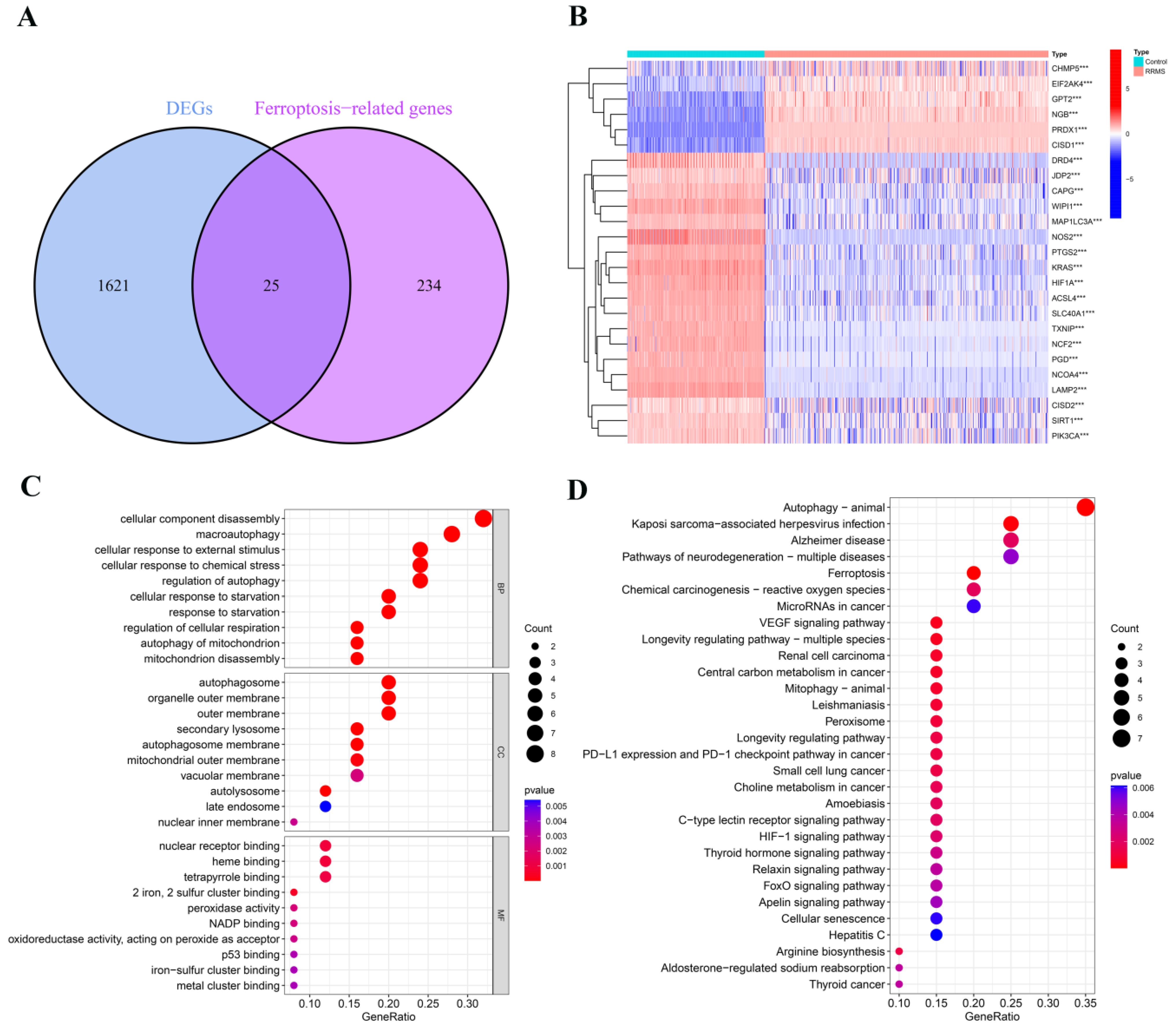
2.1. Detection of Differentially Expressed Ferroptosis-Related Genes in RRMS
2.2. Analysis of Dysregulated Genes Associated with Ferroptosis in RRMS
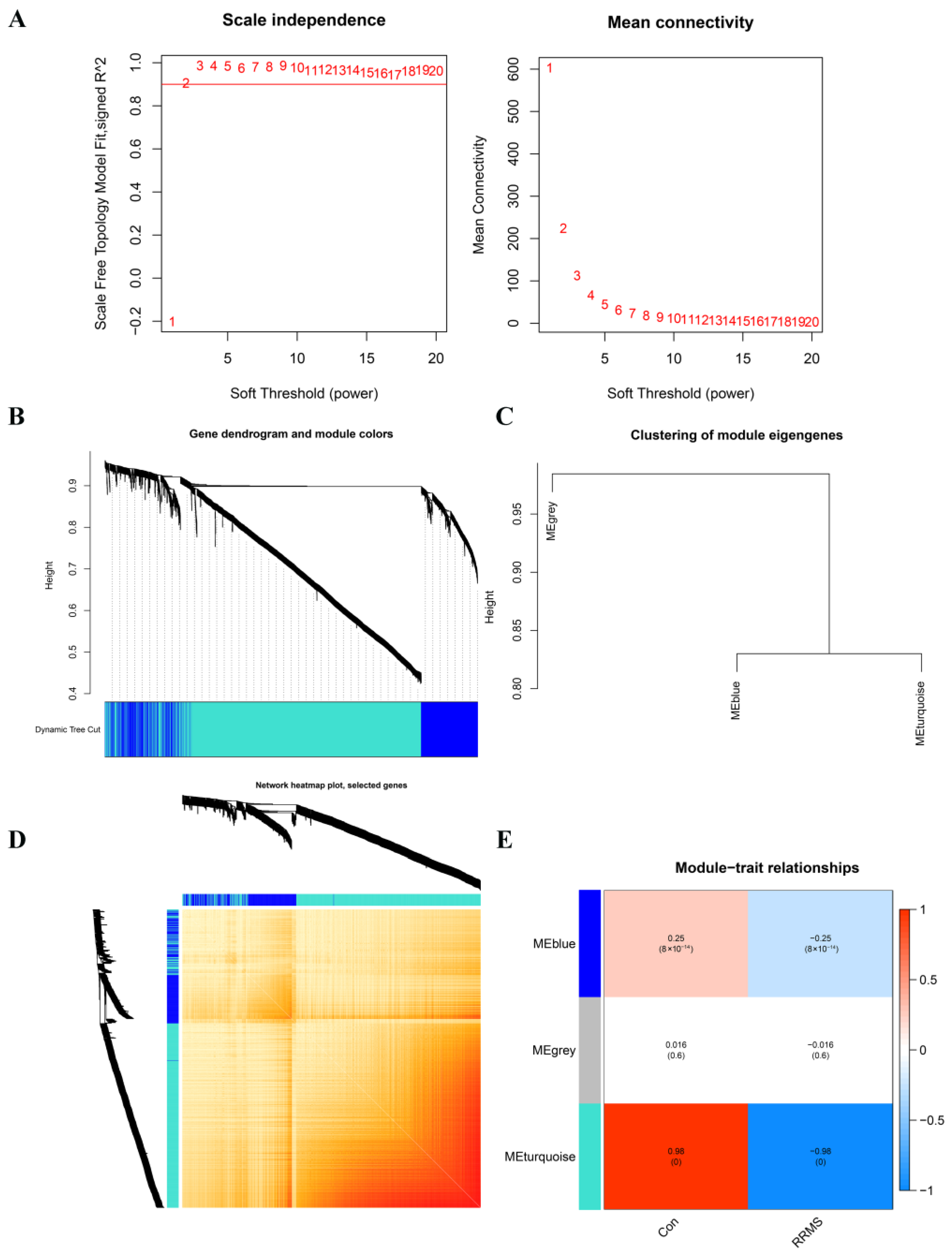
2.3. Construction of Co-Expression Network and Module Trait Screening
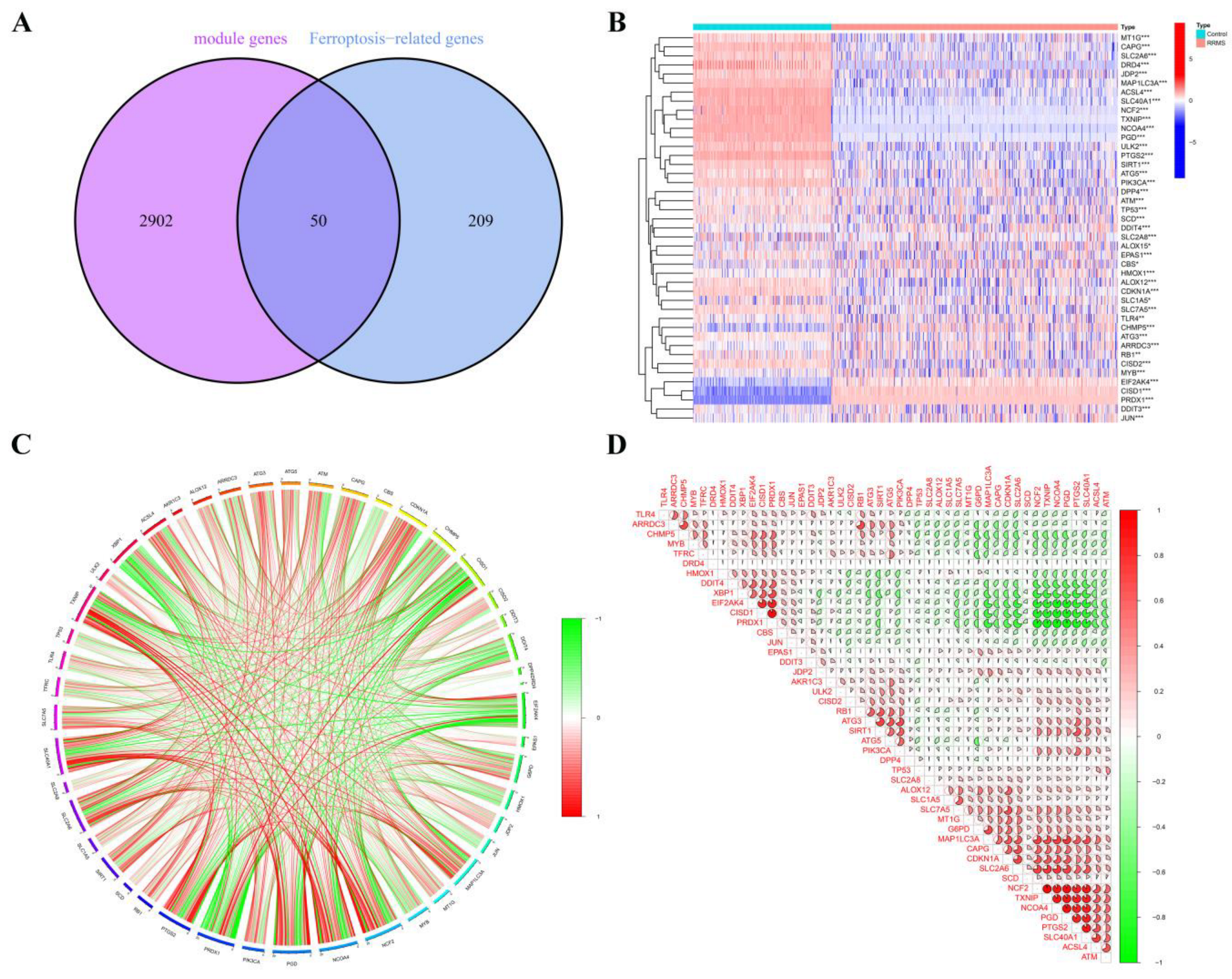
2.4. RRMS-Related Module Genes Overlapped with Ferroptosis-Related Genes
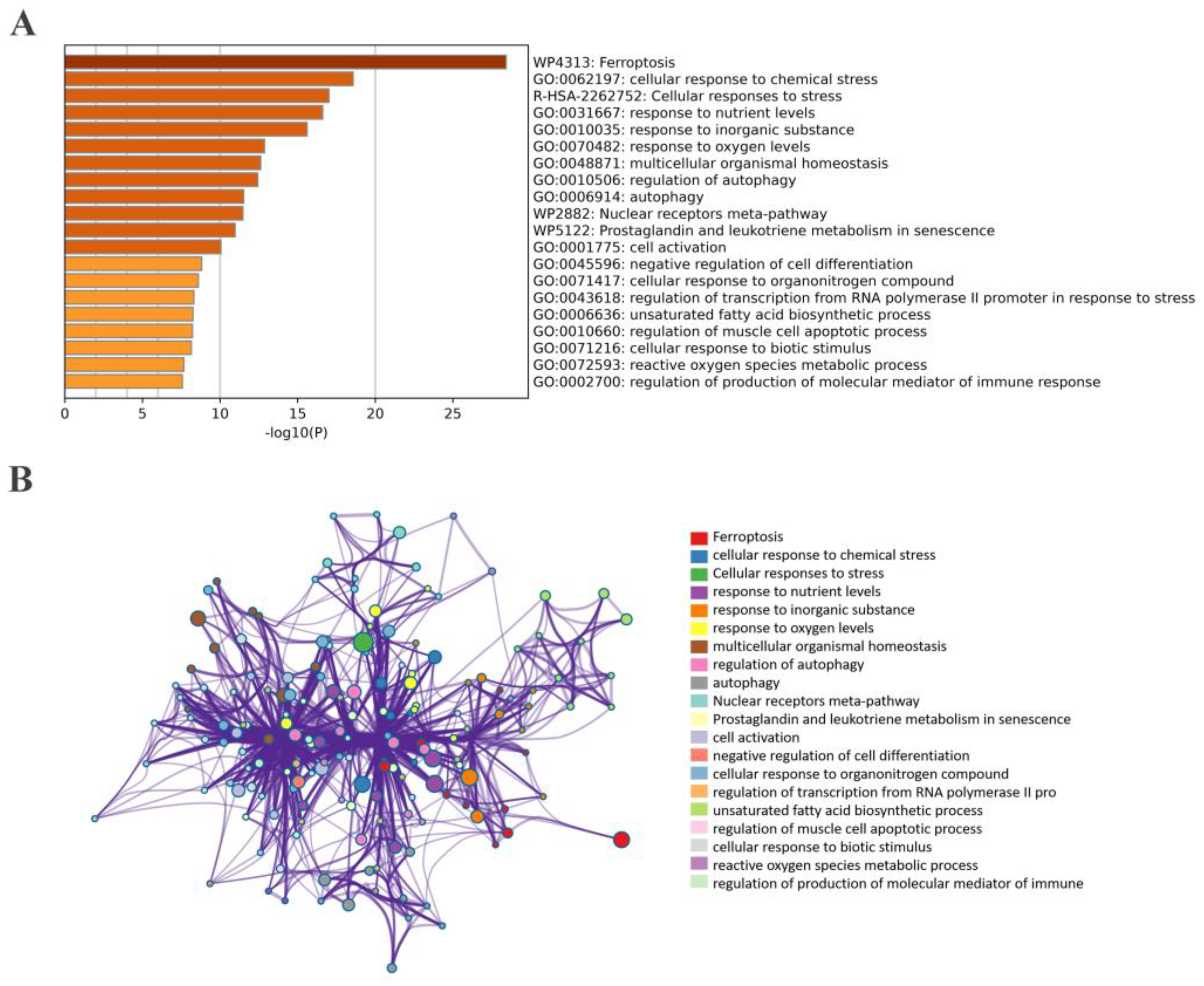
2.5. Correlation and Functional Enrichment Analyses of Overlapped Genes
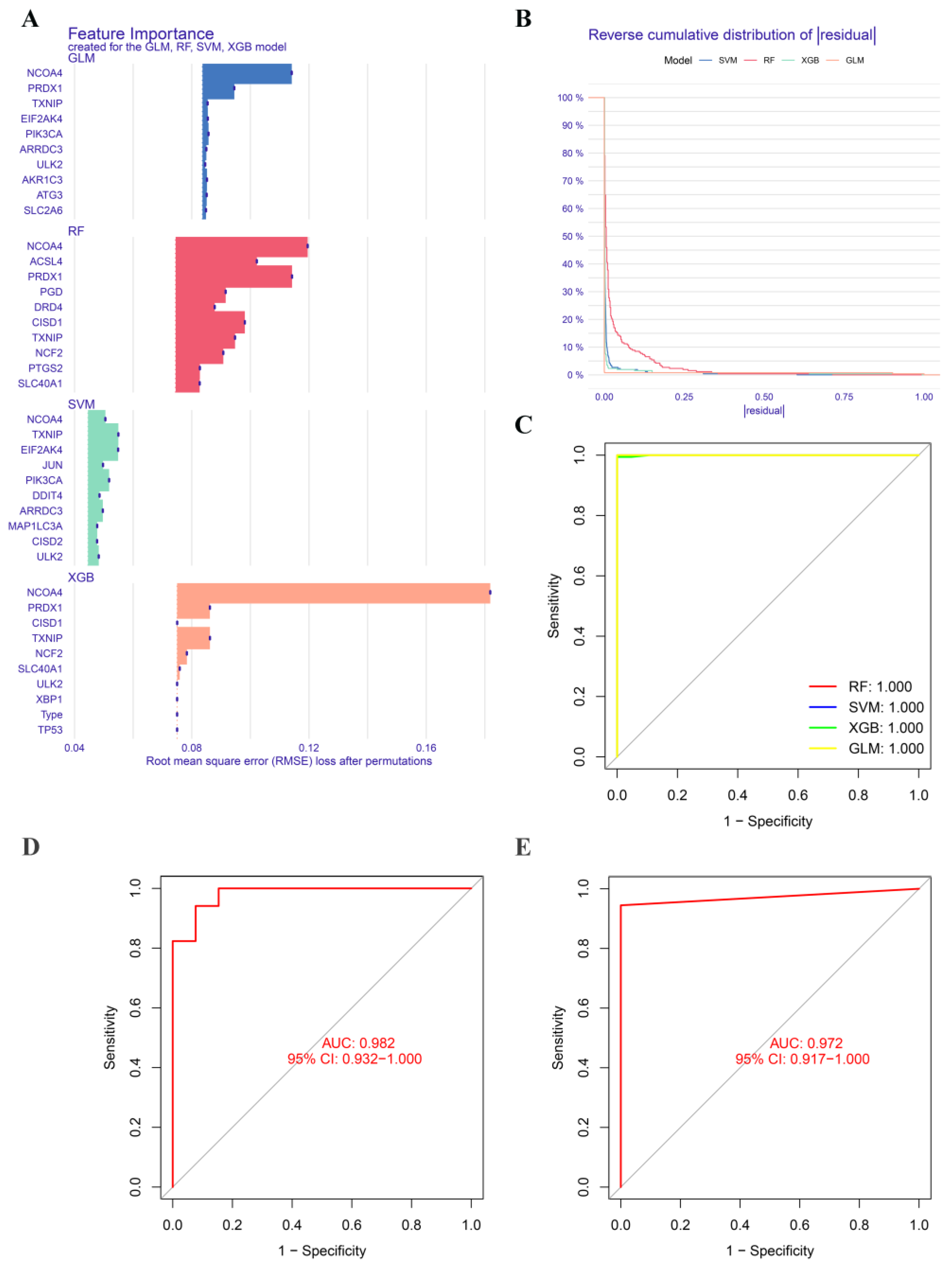
2.6. Construction and Validation of Significative Diagnostic Model Using Machine Learning Methods
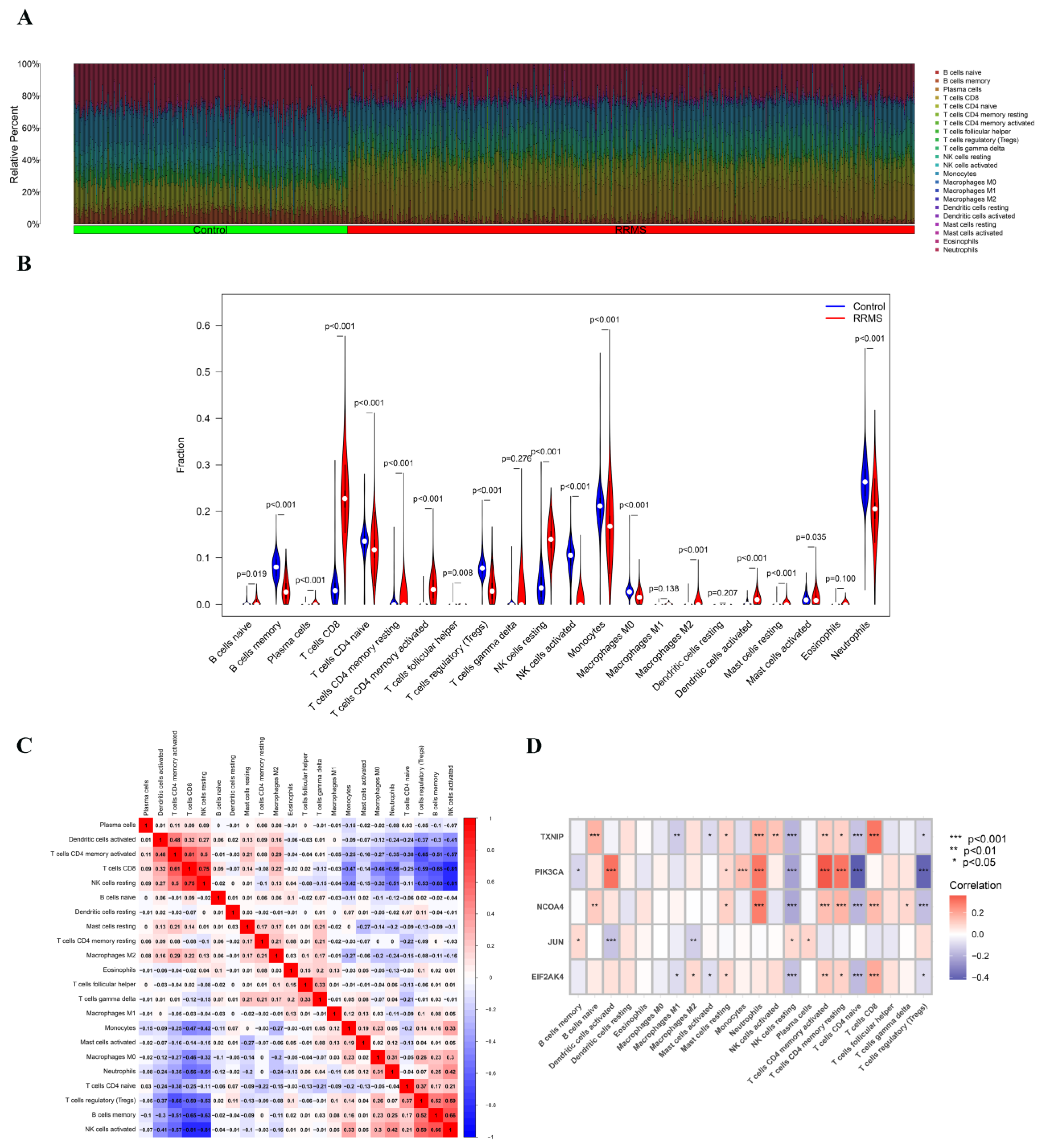
2.7. Immune Cell Infiltration Analysis of Overlapped Genes
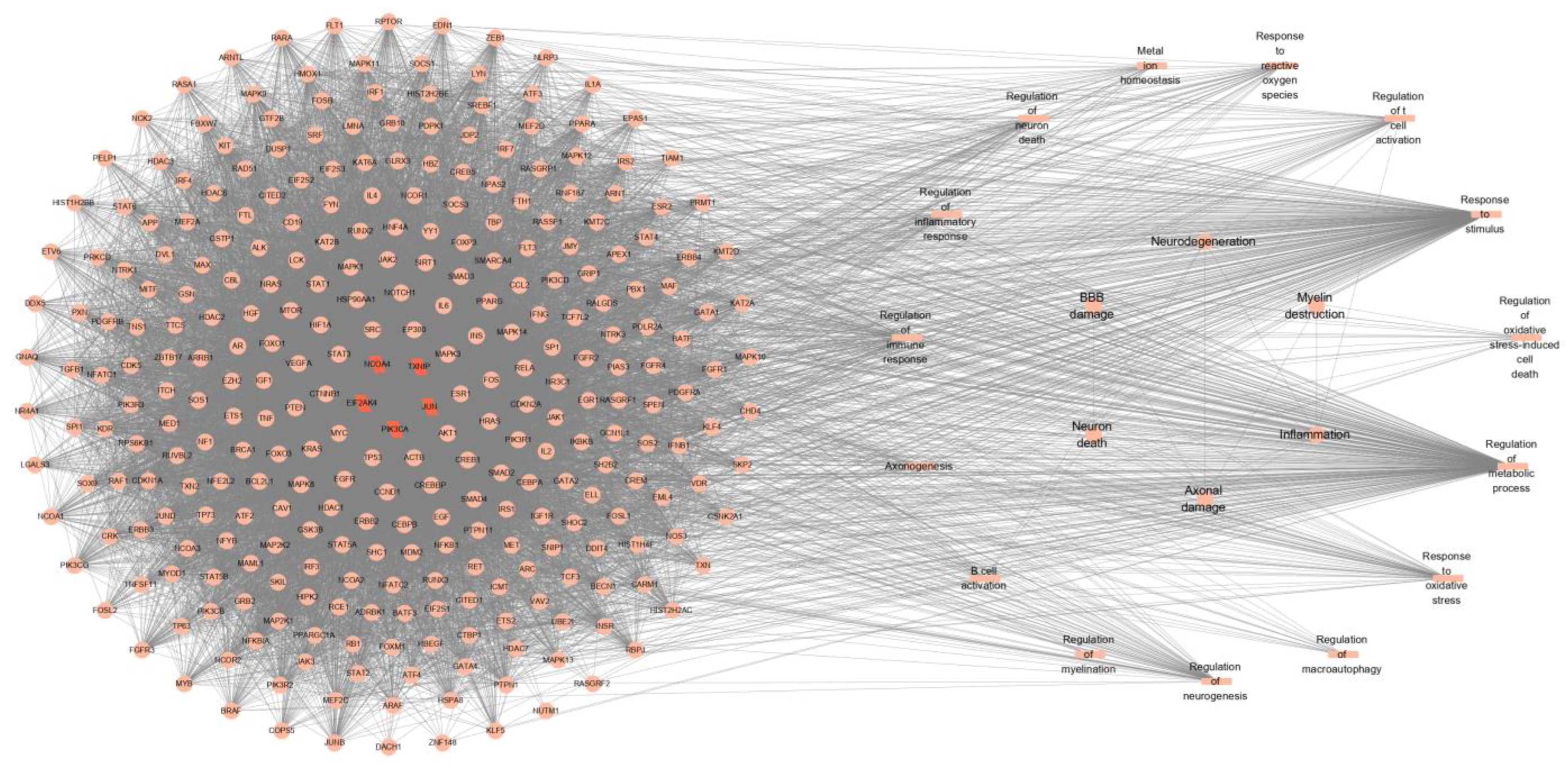
2.8. PPI Network Construction
3. Discussion
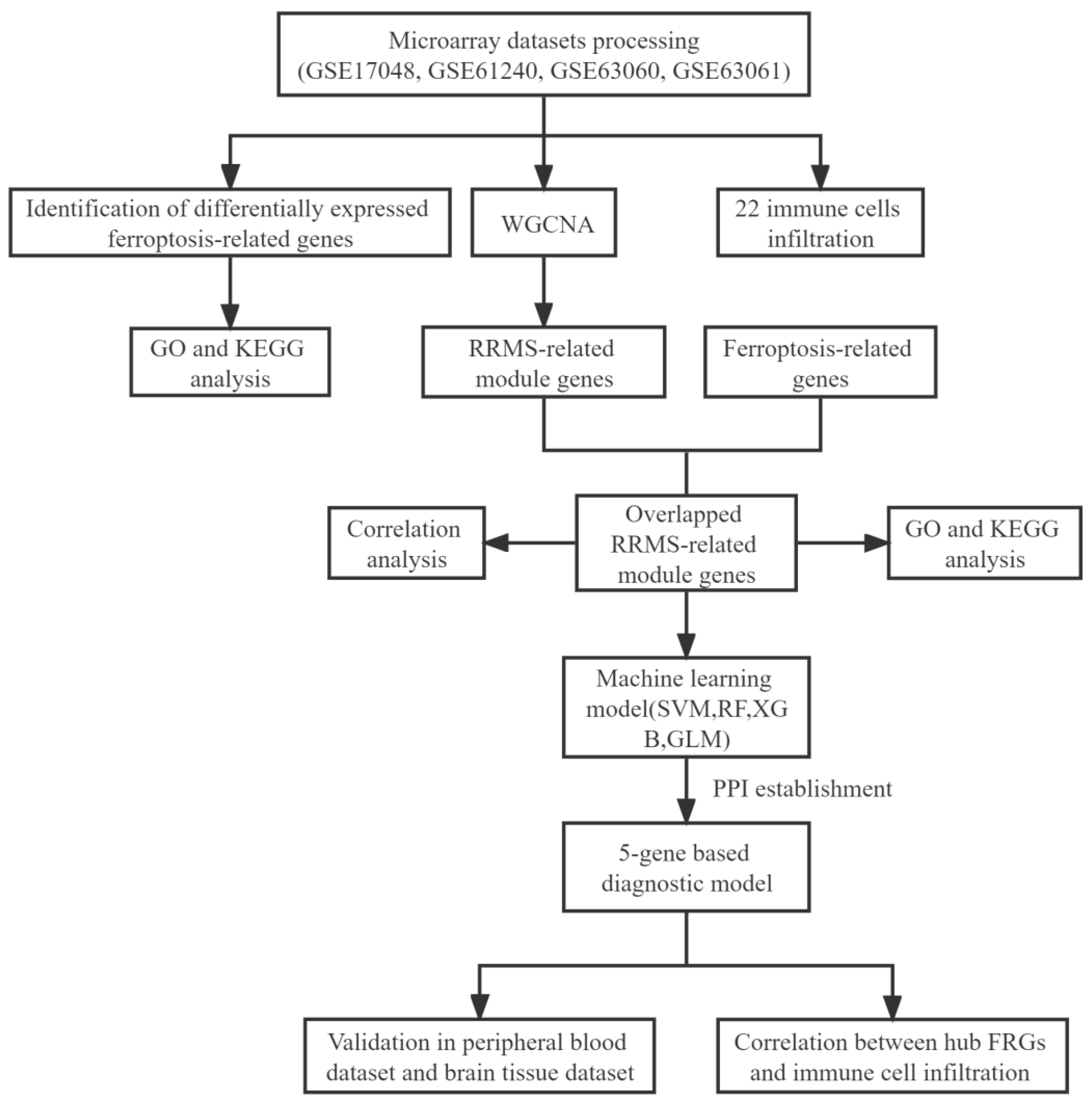
4. Materials and Methods
4.1. Dataset Extraction and Pre-Processing
4.2. Differential Expression Analysis
4.3. Functional Enrichment Analysis
4.4. Analysis of Weighted Gene Co-Expression Network
4.5. RRMS-Related Module Genes Overlapped with Ferroptosis-Related Genes
4.6. Construction and Validation of Diagnostic Model Based on Four Machine Learning Methods
4.7. Immune Cell Infilteration and Correlation Analysis
4.8. Construction and Analysis of PPI Network
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Baranzini, S.E. Revealing the genetic basis of multiple sclerosis: Are we there yet? Curr. Opin. Genet. Dev. 2011, 21, 317–324. [Google Scholar] [CrossRef] [PubMed]
- Cui, Y.; Zhang, Z.; Zhou, X.; Zhao, Z.; Zhao, R.; Xu, X.; Kong, X.; Ren, J.; Yao, X.; Wen, Q. Microglia and macrophage exhibit attenuated inflammatory response and ferroptosis resistance after RSL3 stimulation via increasing Nrf2 expression. J. Neuroinflamm. 2021, 18, 249. [Google Scholar] [CrossRef] [PubMed]
- Dixon, S.J.; Stockwell, B.R. The role of iron and reactive oxygen species in cell death. Nat. Chem. Biol. 2014, 10, 9–17. [Google Scholar] [CrossRef]
- Ruiz, F.; Vigne, S.; Pot, C.A.-O. Resolution of inflammation during multiple sclerosis. Semin. Immunopathol. 2019, 41, 711–726. [Google Scholar] [CrossRef]
- Acquaviva, M.; Menon, R.; Di Dario, M.; Dalla Costa, G.; Romeo, M.; Sangalli, F.; Colombo, B.; Moiola, L.; Martinelli, V.; Comi, G.; et al. Inferring Multiple Sclerosis Stages from the Blood Transcriptome via Machine Learning. Cell Rep. Med. 2020, 1, 100053. [Google Scholar] [CrossRef]
- Dusek, P.; Hofer, T.; Alexander, J.; Roos, P.; Aaseth, J. Cerebral Iron Deposition in Neurodegeneration. Biomolecules 2022, 12, 714. [Google Scholar] [CrossRef]
- Ferreira, H.; Neves, B.; Guerra, I.; Moreira, A.; Melo, T.; Paiva, A.; Domingues, M. An overview of lipidomic analysis in different human matrices of multiple sclerosis. Mult. Scler. Relat. Disord. 2020, 44, 102189. [Google Scholar] [CrossRef]
- Cree, B.A.C. Chapter 9—Multiple sclerosis genetics. In Handbook of Clinical Neurology; Goodin, D.S., Ed.; Elsevier: Amsterdam, The Netherlands, 2014; Volume 122, pp. 193–209. [Google Scholar]
- Hametner, S.; Wimmer, I.; Haider, L.; Pfeifenbring, S.; Brück, W.; Lassmann, H. Iron and neurodegeneration in the multiple sclerosis brain. Ann. Neurol. 2013, 74, 848–861. [Google Scholar] [CrossRef] [PubMed]
- Carsten, S.; David, P.; Yi, W. Iron in Multiple Sclerosis and Its Noninvasive Imaging with Quantitative Susceptibility Mapping. Int. J. Mol. Sci. 2016, 17, 100. [Google Scholar]
- Zhang, D.; Li, Y.; Du, C.; Sang, L.; Liu, L.; Li, Y.; Wang, F.; Fan, W.; Tang, P.; Zhang, S. Evidence of pyroptosis and ferroptosis extensively involved in autoimmune diseases at the single-cell transcriptome level. J. Transl. Med. 2022, 20, 363. [Google Scholar] [CrossRef]
- Hu, C.L.; Nydes, M.; Shanley, K.L.; Morales Pantoja, I.E.; Howard, T.A.; Bizzozero, O.A. Reduced expression of the ferroptosis inhibitor glutathione peroxidase-4 in multiple sclerosis and experimental autoimmune encephalomyelitis. J. Neurochem. Off. J. Int. Soc. Neurochem. 2019, 148, 426–439. [Google Scholar] [CrossRef]
- Constantinescu, C.S.; Farooqi, N.; O’Brien, K.; Gran, B. Experimental autoimmune encephalomyelitis (EAE) as a model for multiple sclerosis (MS). Br. J. Pharmacol. 2011, 164, 1079–1106. [Google Scholar] [CrossRef] [PubMed]
- Jhelum, P.; Santos-Nogueira, E.; Teo, W.; Haumont, A.; David, S. Ferroptosis Mediates Cuprizone-Induced Loss of Oligodendrocytes and Demyelination. J. Neurosci. 2020, 40, 9327–9341. [Google Scholar] [CrossRef] [PubMed]
- Galluzzi, L.; Vitale, I.; Aaronson, S.A.; Abrams, J.M.; Adam, D.; Agostinis, P. Molecular mechanisms of cell death: Recommendations of the Nomenclature Committee on Cell Death 2018. Cell Death Differ. 2018, 25, 486–541. [Google Scholar] [CrossRef]
- Gianfranca, C.; Lara, C.; Emanuele, P.; Alessandra, C.; Enrico, T.; Lidia, B.; Alessandro, C.; Silvia, B.; Angelo, A.M.; Pietro, A.; et al. Polarization dictates iron handling by inflammatory and alternatively activated macrophages. Haematologica 2010, 95, 1814–1822. [Google Scholar] [CrossRef]
- Achiron, A.; Gurevich, M. Peripheral blood gene expression signature mirrors central nervous system disease: The model of multiple sclerosis. Autoimmun. Rev. 2006, 5, 517–522. [Google Scholar] [CrossRef] [PubMed]
- Ransohoff, R.M. How neuroinflammation contributes to neurodegeneration. Science 2016, 353, 777–783. [Google Scholar] [CrossRef]
- Prat, A.; Biernacki, K.; Lavoie, J.F.; Poirier, J.; Duquette, P.; Antel, J.P. Migration of multiple sclerosis lymphocytes through brain endothelium. Arch. Neurol. 2002, 59, 391–397. [Google Scholar] [CrossRef]
- Prat, A.; Antel, J. Pathogenesis of multiple sclerosis. Curr. Opin. Neurol. 2005, 18, 225–230. [Google Scholar] [CrossRef]
- D’Ambrosio, A.; Pontecorvo, S.; Colasanti, T.; Zamboni, S.; Francia, A.; Margutti, P. Peripheral blood biomarkers in multiple sclerosis. Autoimmun. Rev. 2015, 14, 1097–1110. [Google Scholar] [CrossRef]
- Razia, R.; Majeed, F.; Amin, R.; Mukhtar, S.; Mehmood, K.; Baig, D.N. The analysis of dynamic gene expression patterns in peripheral blood of multiple sclerosis patients indicates possible diagnostic and prognostic biomarkers. Mol. Immunol. 2022, 147, 147–156. [Google Scholar] [CrossRef] [PubMed]
- Golabi, M.; Fathi, F.; Samadi, M.; Hesamian, M.S.; Eskandari, N.A.-O. Identification of Potential Biomarkers in the Peripheral Blood Mononuclear Cells of Relapsing-Remitting Multiple Sclerosis Patients. Inflammation 2022, 45, 1815–1828. [Google Scholar] [CrossRef] [PubMed]
- Chen, X.; Pang, X.; Yeo, A.J.; Xie, S.; Xiang, M.; Shi, B.; Yu, G.; Li, C. The Molecular Mechanisms of Ferroptosis and Its Role in Blood-Brain Barrier Dysfunction. Front. Cell. Neurosci. 2022, 16, 889765. [Google Scholar] [CrossRef] [PubMed]
- Sun, Y.; Xia, X.; Basnet, D.; Zheng, J.C.; Huang, J.; Liu, J. Mechanisms of Ferroptosis and Emerging Links to the Pathology of Neurodegenerative Diseases. Front. Aging Neurosci. 2022, 14, 904152. [Google Scholar] [CrossRef]
- Ropele, S.; Enzinger, C.; Fazekas, F. Iron Mapping in Multiple Sclerosis. Neuroimaging Clin. N. Am. 2017, 27, 335–342. [Google Scholar] [CrossRef]
- Ferreira, K.; Oliveira, S.; Kallaur, A.; Kaimen-Maciel, D.; Lozovoy, M.; de Almeida, E.; Morimoto, H.; Mezzaroba, L.; Dichi, I.; Reiche, E.; et al. Disease progression and oxidative stress are associated with higher serum ferritin levels in patients with multiple sclerosis. J. Neurol. Sci. 2017, 373, 236–241. [Google Scholar] [CrossRef]
- Iranmanesh, M.; Iranmanesh, F.; Sadeghi, H. Serum level of iron, zinc and copper in patients with multiple sclerosis. Pars Jahrom Univ. Med Sci. 2012, 10, 1–5. [Google Scholar] [CrossRef]
- Stankiewicz, J.M.; Neema, M.; Ceccarelli, A. Iron and multiple sclerosis. Neurobiol. Aging 2014, 35, S51–S58. [Google Scholar] [CrossRef]
- Ortiz, G.G.; Pacheco-Moisés, F.P.; Macías-Islas, M.; Flores-Alvarado, L.J.; Mireles-Ramírez, M.A.; González-Renovato, E.D.; Hernández-Navarro, V.E.; Sánchez-López, A.L.; Alatorre-Jiménez, M.A. Role of the blood-brain barrier in multiple sclerosis. Arch. Med. Res. 2014, 45, 687–697. [Google Scholar] [CrossRef]
- Sheikh, M.H.; Henson, S.M.; Loiola, R.A.; Mercurio, S.; Colamatteo, A.; Maniscalco, G.T.; De Rosa, V.; McArthur, S.; Solito, E. Immuno-metabolic impact of the multiple sclerosis patients’ sera on endothelial cells of the blood-brain barrier. J. Neuroinflammation 2020, 17, 153. [Google Scholar] [CrossRef]
- Lassmann, H. Pathogenic Mechanisms Associated With Different Clinical Courses of Multiple Sclerosis. Front. Immunol. 2019, 9, 3116. [Google Scholar] [CrossRef]
- Mezzaroba, L.; Alfieri, D.F.; Colado Simão, A.N.; Vissoci Reiche, E.M. The role of zinc, copper, manganese and iron in neurodegenerative diseases. Neurotoxicology 2019, 74, 230–241. [Google Scholar] [CrossRef] [PubMed]
- Machado-Santos, J.; Saji, E.; Tröscher, A.R.; Paunovic, M.; Liblau, R.; Gabriely, G.; Bien, C.G.; Bauer, J.; Lassmann, H. The compartmentalized inflammatory response in the multiple sclerosis brain is composed of tissue-resident CD8+ T lymphocytes and B cells. Brain 2018, 141, 2066–2082. [Google Scholar] [CrossRef] [PubMed]
- Jin, W.C.; Jeon, J.H.; Yoon, S.R.; Choi, I. Vitamin D3 upregulated protein 1 (VDUP1) is a regulator for redox signaling and stress-mediated diseases. J. Dermatol. 2006, 33, 662–669. [Google Scholar]
- Sang, Y.K.; Suh, H.W.; Jin, W.C.; Yoon, S.R.; Choi, I. Diverse functions of VDUP1 in cell proliferation, differentiation, and diseases. Cell. Mol. Immunol. 2007, 4, 345–351. [Google Scholar]
- Nishiyama, A.; Matsui, M.; Iwata, S.; Hirota, K.; Masutani, H.; Nakamura, H.; Takagi, Y.; Sono, H.; Gon, Y.; Yodoi, J. Identification of thioredoxin-binding protein-2/vitamin D(3) up-regulated protein 1 as a negative regulator of thioredoxin function and expression. J. Biol. Chem. 1999, 274, 21645–21650. [Google Scholar] [CrossRef] [PubMed]
- Saxena, G.; Chen, J.; Shalev, A. Intracellular Shuttling and Mitochondrial Function of Thioredoxin-interacting Protein. J. Biol. Chem. 2010, 285, 3997–4005. [Google Scholar] [CrossRef]
- Tsubaki, H.; Tooyama, I.; Walker, D.G. Thioredoxin-Interacting Protein (TXNIP) with Focus on Brain and Neurodegenerative Diseases. Int. J. Mol. Sci. 2020, 21, 9357. [Google Scholar] [CrossRef]
- Yu, Y.; Wu, D.M.; Li, J.; Deng, S.H.; Xu, Y. Bixin Attenuates Experimental Autoimmune Encephalomyelitis by Suppressing TXNIP/NLRP3 inflammasome Activity and Activating Nrf2 Signaling. Front. Media SA 2020, 11, 593368. [Google Scholar] [CrossRef] [PubMed]
- Mahmoudian, E.; Khalilnezhad, A.; Gharagozli, K.; Amani, D. Thioredoxin-1, redox factor-1 and thioredoxin-interacting protein, mRNAs are differentially expressed in Multiple Sclerosis patients exposed and non-exposed to interferon and immunosuppressive treatments. Gene 2017, 634, 29–36. [Google Scholar] [CrossRef]
- Raivich, G. c-Jun expression, activation and function in neural cell death, inflammation and repair. J. Neurochem. 2010, 107, 898–906. [Google Scholar] [CrossRef]
- Behrens, A.; Sibilia, M.; Wagner, E.F. Amino-terminal phosphorylation of c-Jun regulates stress-induced apoptosis and cellular proliferation. Nat. Genet. 1999, 21, 326–329. [Google Scholar] [CrossRef] [PubMed]
- Martin-Villalba, A.; Hahne, M.; Kleber, S.; Vogel, J.; Falk, W.; Schenkel, J.; Krammer, P.H. Therapeutic neutralization of CD95-ligand and TNF attenuates brain damage in stroke. Cell Death Differ. 2001, 8, 679–686. [Google Scholar] [CrossRef] [PubMed]
- Winter, C.; Weiss, C.; Martin-Villalba, A.; Zimmermann, M.; Schenkel, J. JunB and Bcl-2 overexpression results in protection against cell death of nigral neurons following axotomy. Mol. Brain Res. 2002, 104, 194–202. [Google Scholar] [CrossRef]
- Mancias, J.D.; Wang, X.; Gygi, S.P.; Harper, J.W.; Kimmelman, A.C. Quantitative proteomics identifies NCOA4 as the cargo receptor mediating ferritinophagy. Nature 2014, 509, 105–109. [Google Scholar] [CrossRef]
- Rey, M.; Mancias, J.D. NCOA4-Mediated Ferritinophagy: A Potential Link to Neurodegeneration. Front. Neurosci. 2019, 13, 238. [Google Scholar]
- Orsini, H.; Araujo, L.P.; Maricato, J.T.; Guereschi, M.G.; Mariano, M.; Castilho, B.A.; Basso, A.S. GCN2 kinase plays an important role triggering the remission phase of experimental autoimmune encephalomyelitis (EAE) in mice. Brain Behav. Immun. 2014, 37, 177–186. [Google Scholar] [CrossRef]
- Haro, C.D.; Méndez, R.; Santoyo, J. The eIF-2α kinases and the control of protein synthesis. Faseb J. 1996, 10, 1378–1387. [Google Scholar] [CrossRef]
- Kang, Y.; Tiziani, S.; Park, G.; Kaul, M.; Paternostro, G. Cellular Protection using Flt3 and PI3Kα inhibitors demonstrates multiple mechanisms of oxidative glutamate toxicity. Nat. Publ. Group 2014, 5, 3672. [Google Scholar] [CrossRef] [PubMed]
- Jha, H.C.; Mehta, D.; Lu, J.; El-Naccache, D.; Shukla, S.K.; Kovacsics, C.; Kolson, D.; Robertson, E.S. Gammaherpesvirus Infection of Human Neuronal Cells. Mbio 2015, 6, e01844-15. [Google Scholar] [CrossRef]
- Enbom, M. Multiple sclerosis and Kaposi’s sarcoma—Chronic diseases associated with new human herpesviruses? Scand. J. Infect. Dis. 2001, 33, 648–658. [Google Scholar] [CrossRef] [PubMed]
- Tully, T.; Barkley, A.; Silber, E. Kaposi sarcoma in a patient with relapsing-remitting multiple sclerosis receiving fingolimod. Neurology 2015, 84, 1999–2001. [Google Scholar] [CrossRef]
- Walker, S.; Brew, B. Kaposi sarcoma in a fingolimod-treated patient with multiple sclerosis. J. Clin. Neurosci. 2016, 31, 217–218. [Google Scholar] [CrossRef]
- Veillet-Lemay, G.M.; Sawchuk, M.A.; Kanigsberg, N.D. Primary Cutaneous Histoplasma capsulatum Infection in a Patient Treated With Fingolimod: A Case Report. J. Cutan. Med. Surg. 2017, 21, 553–555. [Google Scholar] [CrossRef]
- Foppoli, C.; Marco, F.D.; Cini, C.; Perluigi, M. Redox control of viral carcinogenesis: The human papillomavirus paradigm. Biochim. Biophys. Acta 2015, 1850, 1622–1632. [Google Scholar] [CrossRef] [PubMed]
- Gruhne, B.; Sompallae, R.; Marescotti, D.; Kamranvar, S.A.; Gastaldello, S.; Masucci, M.G. The Epstein-Barr virus nuclear antigen-1 promotes genomic instability via induction of reactive oxygen species. Proc. Natl. Acad. Sci. USA 2009, 106, 2313–2318. [Google Scholar] [CrossRef]
- Jiang, L.; Xie, C.; Lung, H.L.; Lo, K.W.; Law, G.L.; Mak, N.K.; Wong, K.L. EBNA1-targeted inhibitors: Novel approaches for the treatment of Epstein-Barr virus-associated cancers. Theranostics 2018, 8, 5307–5319. [Google Scholar] [CrossRef]
- Lin, Z.; Liu, J.; Kang, R.; Yang, M.; Tang, D. Lipid Metabolism in Ferroptosis. Adv. Biol. 2021, 5, 2100396. [Google Scholar] [CrossRef]
- Mathur, D.; Mishra, B.K.; Rout, S.; Lopez-Iranzo, F.J.; Lopez-Rodas, G.A.-O.X.; Vallamkondu, J.; Kandimalla, R.A.-O.; Casanova, B. Potential Biomarkers Associated with Multiple Sclerosis Pathology. Int. J. Mol. Sci. 2021, 22, 10323. [Google Scholar] [CrossRef] [PubMed]
- Brownlee, W.J.; Hardy, T.A.; Fazekas, F.; Miller, D.H. Diagnosis of multiple sclerosis: Progress and challenges. Lancet 2016, 389, 1336–1346. [Google Scholar] [CrossRef] [PubMed]








Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Song, X.; Wang, Z.; Tian, Z.; Wu, M.; Zhou, Y.; Zhang, J. Identification of Key Ferroptosis-Related Genes in the Peripheral Blood of Patients with Relapsing-Remitting Multiple Sclerosis and Its Diagnostic Value. Int. J. Mol. Sci. 2023, 24, 6399. https://doi.org/10.3390/ijms24076399
Song X, Wang Z, Tian Z, Wu M, Zhou Y, Zhang J. Identification of Key Ferroptosis-Related Genes in the Peripheral Blood of Patients with Relapsing-Remitting Multiple Sclerosis and Its Diagnostic Value. International Journal of Molecular Sciences. 2023; 24(7):6399. https://doi.org/10.3390/ijms24076399
Chicago/Turabian StyleSong, Xi, Zixuan Wang, Zixin Tian, Meihuan Wu, Yitao Zhou, and Jun Zhang. 2023. "Identification of Key Ferroptosis-Related Genes in the Peripheral Blood of Patients with Relapsing-Remitting Multiple Sclerosis and Its Diagnostic Value" International Journal of Molecular Sciences 24, no. 7: 6399. https://doi.org/10.3390/ijms24076399
APA StyleSong, X., Wang, Z., Tian, Z., Wu, M., Zhou, Y., & Zhang, J. (2023). Identification of Key Ferroptosis-Related Genes in the Peripheral Blood of Patients with Relapsing-Remitting Multiple Sclerosis and Its Diagnostic Value. International Journal of Molecular Sciences, 24(7), 6399. https://doi.org/10.3390/ijms24076399