
Non-Ceruloplasmin Copper Identifies a Subtype of Alzheimer’s Disease (CuAD): Characterization of the Cognitive Profile and Case of a CuAD Patient Carrying an RGS7 Stop-Loss Variant

 ,
,  , , ,
, , ,  and
and
Abstract
1. Introduction
2. Results
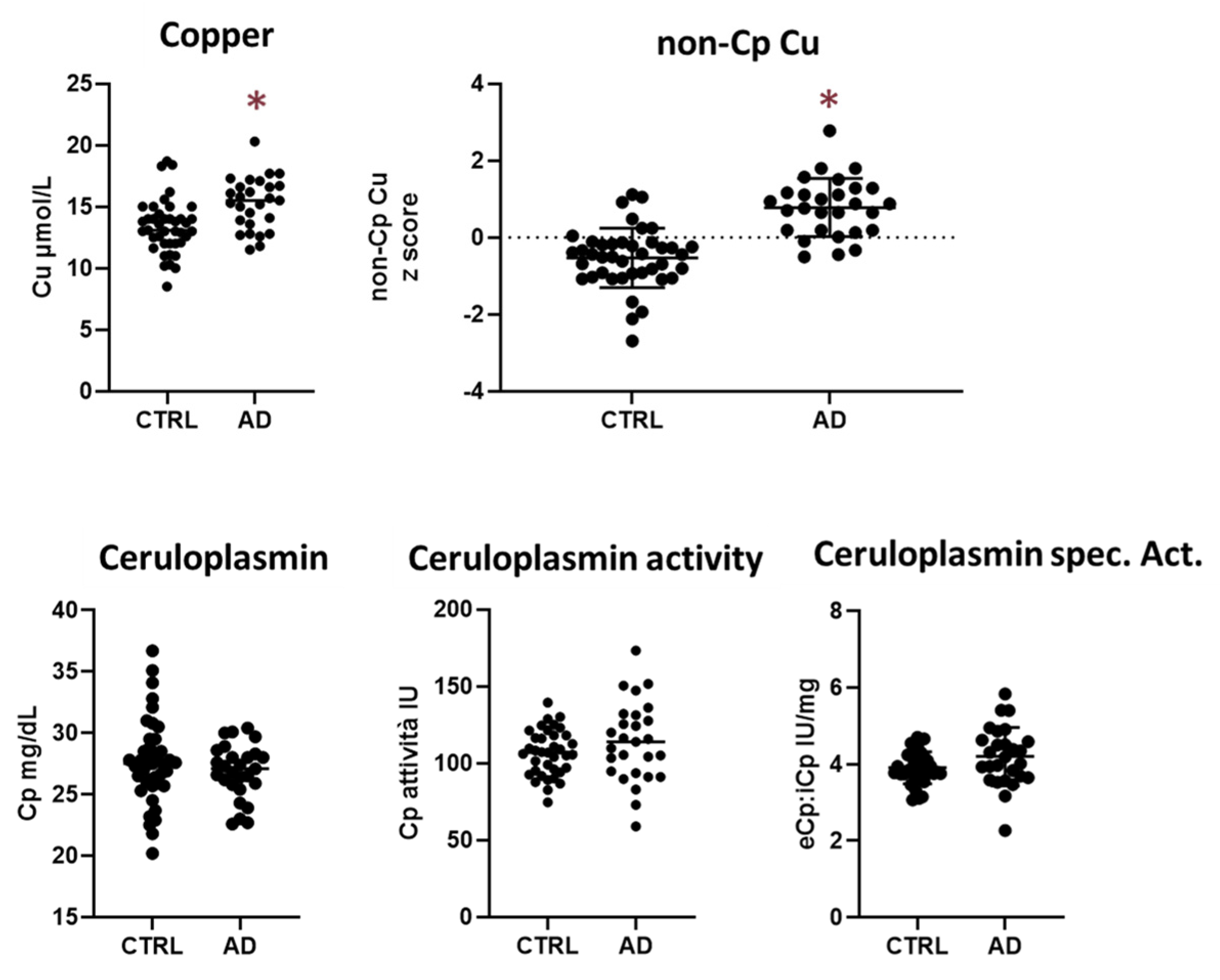
2.1. Alzheimer’s Disease and Healthy Controls Comparison for Cu Panel
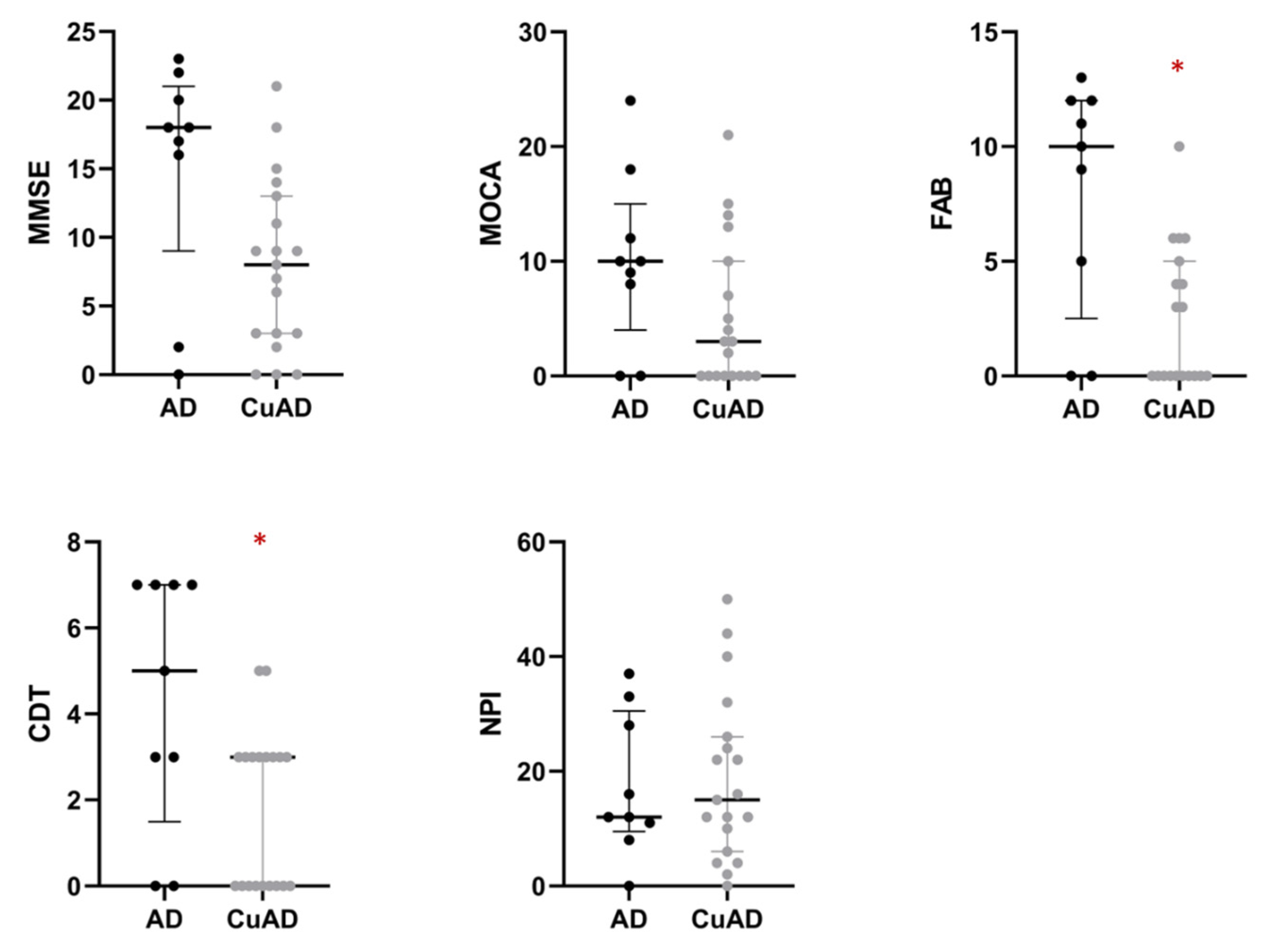
2.2. AD Subgroups Stratification on the Basis of the Add-On Non-Cp Cu Test
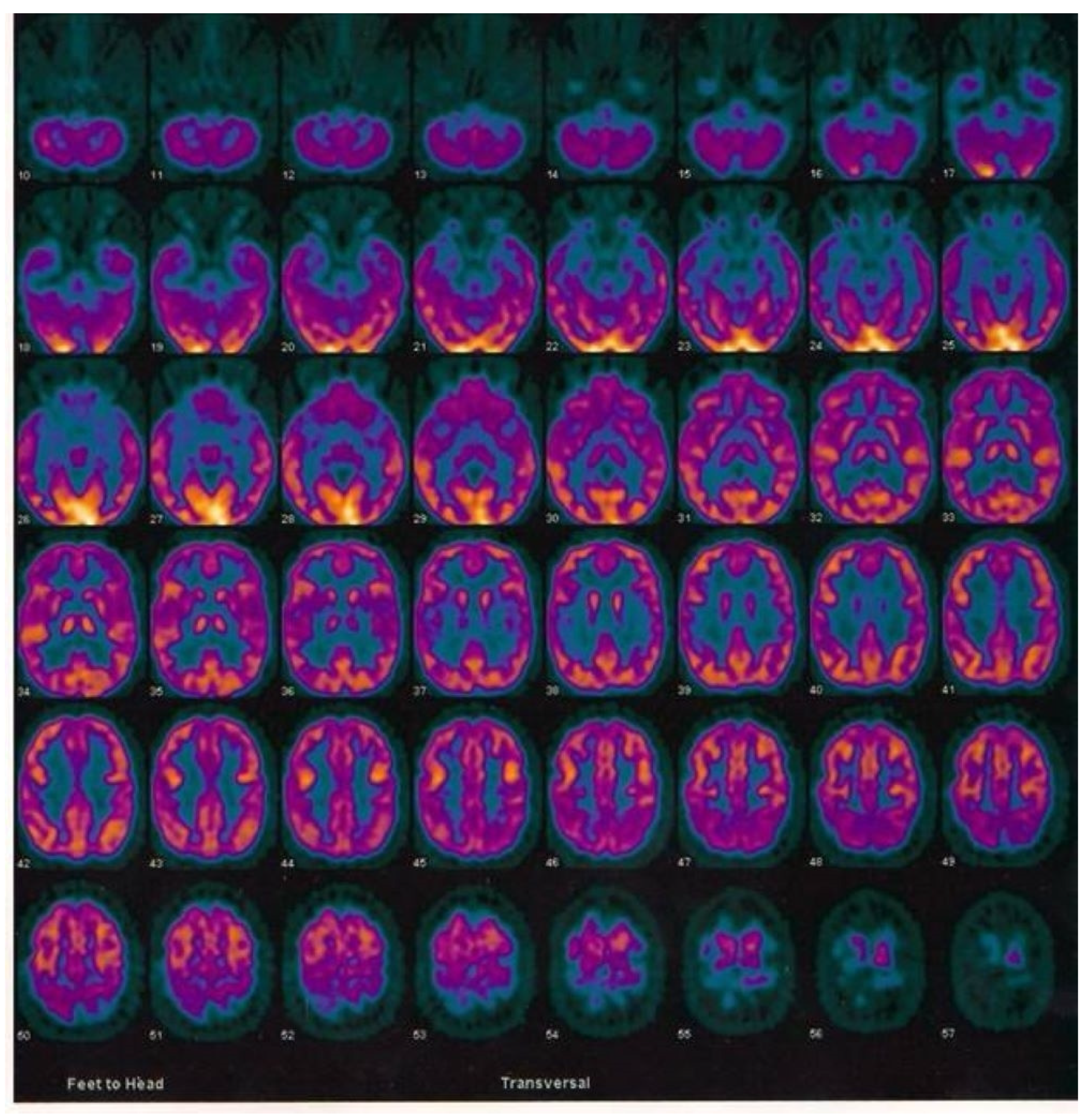
2.3. Case Report of One Patient in the CuAD Subgroup
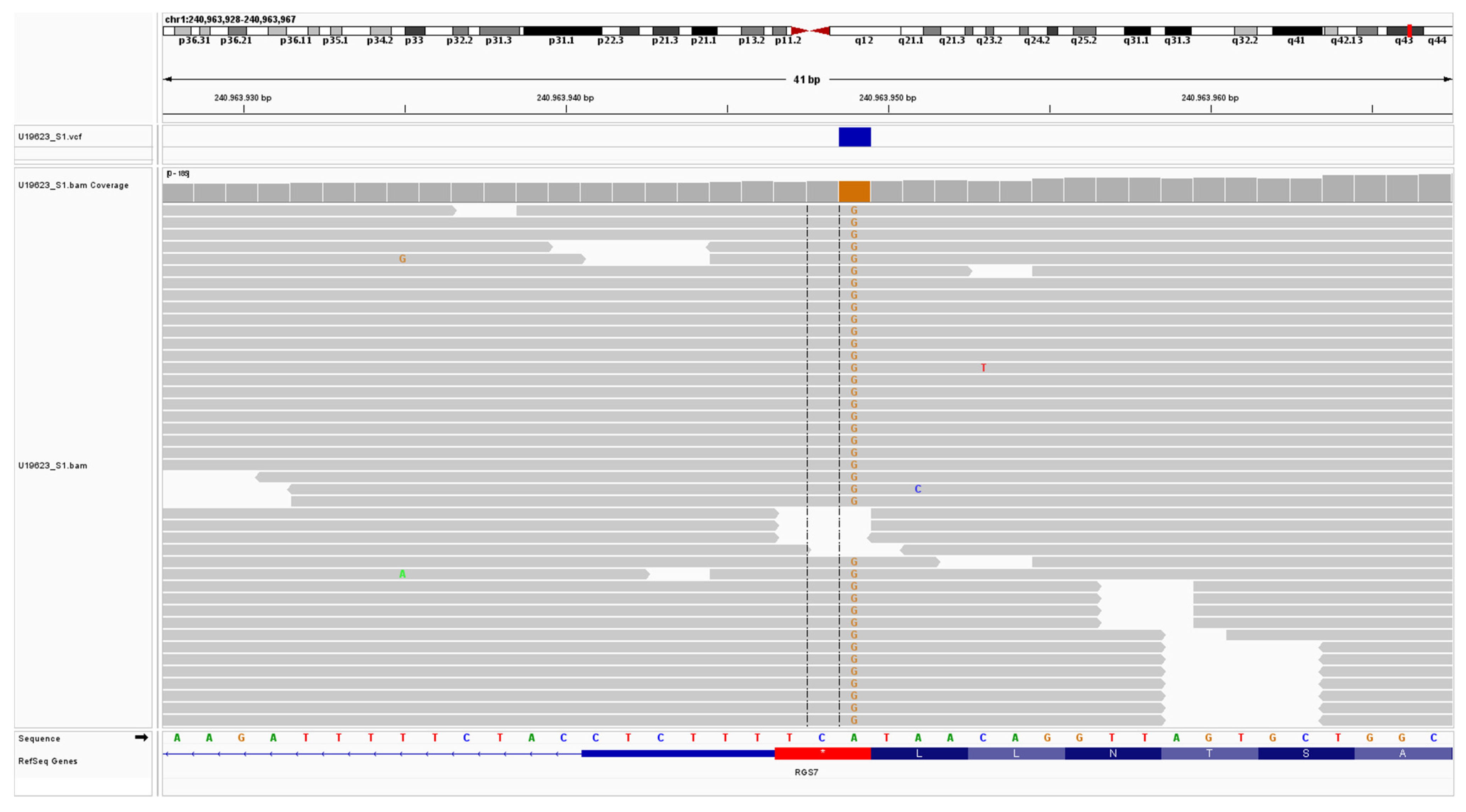
2.4. Analysis and Identification of a Candidate Variant in the CuAD Patient
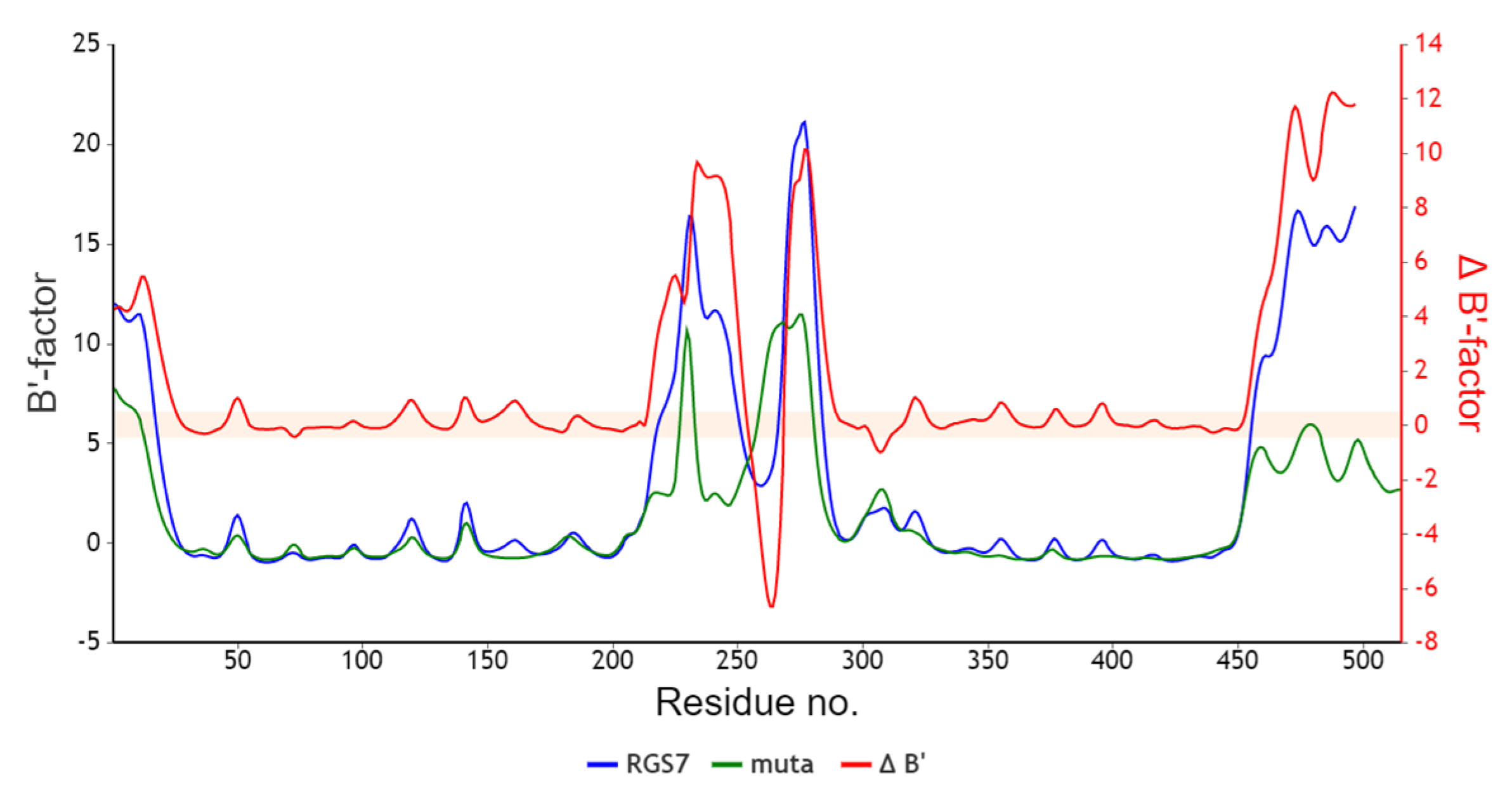
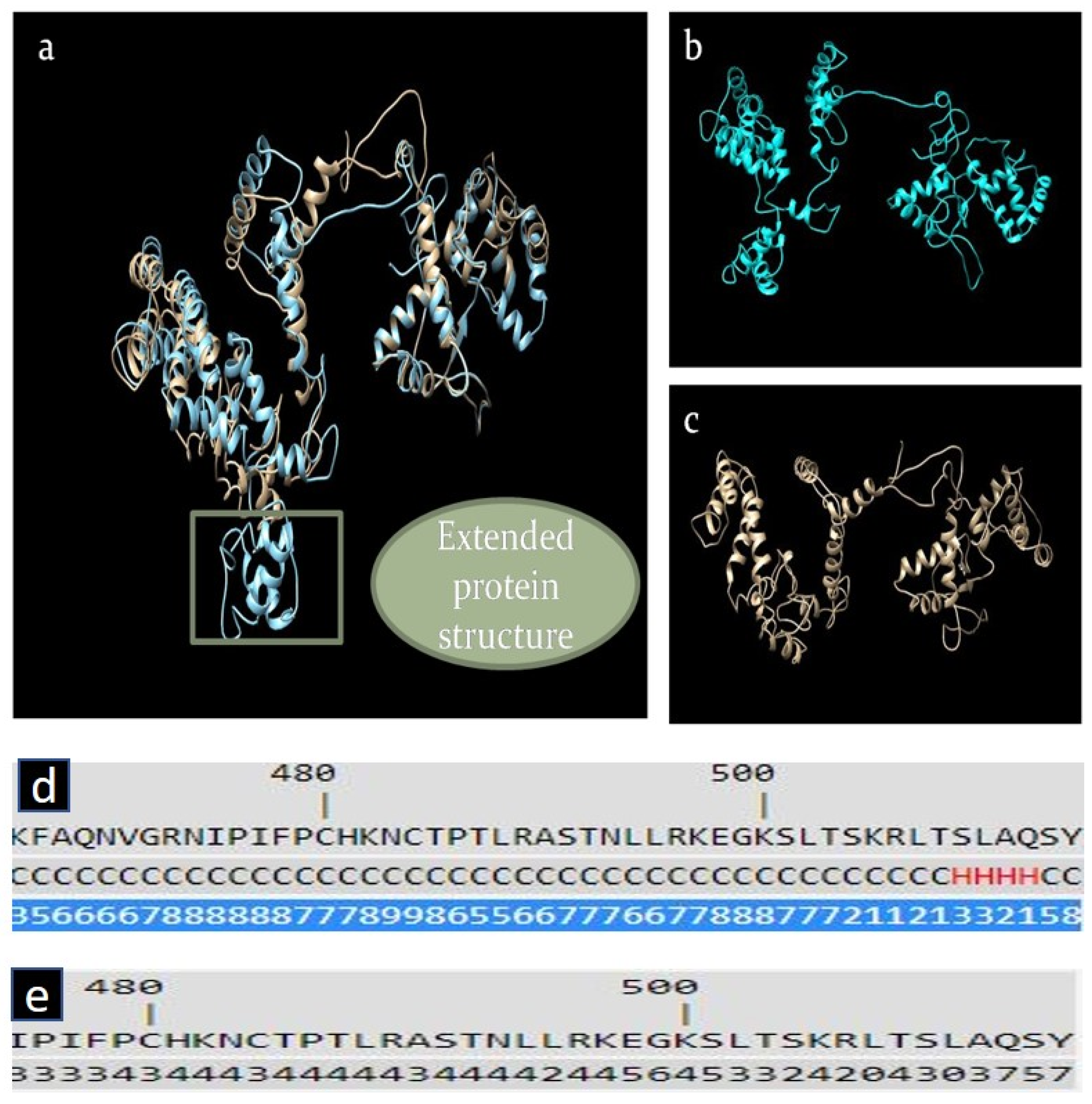
2.5. In Silico Analysis of RGS7 Stop-Loss Mutation
3. Discussion
3.1. Comparison of the AD Subtypes
3.2. Case Report: DNM2 and RGS7 Mutation and Alzheimer’s Disease
3.3. Detection of RGS7 Variants Highlights the Relevance of Alternative Pathways of Intracellular Cu Trafficking in Alzheimer’s Disease
3.4. The Presence of the RGS7 Stop-Loss Variant Highlights the Relevance of Alternative Pathways of Intracellular Fe Trafficking in Alzheimer’s Disease
4. Materials and Methods
- Application of the standard diagnostic pathway for AD according to the IGW2 criteria [28];
- Apply the stratification add-on biomarker to classify the AD population into two subgroups on the basis of the established non-Cp Cu cut-off (1.6 µmol/L) [13];
- Search for genetic variants that might explain the disease-associated metabolic sub-pathways, i.e., searching for mutations that may explain the altered Cu metabolism.
4.1. Subjects
4.2. Biochemical Analyses
4.3. Statistical Analysis
4.4. Case Report and Genetic Analysis
4.5. In Silico Analysis of RGS7 Stop-Loss Mutation
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Hardy, J.A.; Higgins, G.A. Alzheimer’s disease: The amyloid cascade hypothesis. Science 1992, 256, 184–185. [Google Scholar] [CrossRef] [PubMed]
- Brion, J.P. Neurofibrillary tangles and Alzheimer’s disease. Eur. Neurol. 1998, 40, 130–140. [Google Scholar] [CrossRef]
- Rissin, D.M.; Kan, C.W.; Campbell, T.G.; Howes, S.C.; Fournier, D.R.; Song, L.; Piech, T.; Patel, P.P.; Chang, L.; Rivnak, A.J.; et al. Single-molecule enzyme-linked immunosorbent assay detects serum proteins at subfemtomolar concentrations. Nat. Biotechnol. 2010, 28, 595–599. [Google Scholar] [CrossRef] [PubMed]
- Teunissen, C.E.; Verberk, I.M.W.; Thijssen, E.H.; Vermunt, L.; Hansson, O.; Zetterberg, H.; van der Flier, W.M.; Mielke, M.M.; Del Campo, M. Blood-based biomarkers for Alzheimer’s disease: Towards clinical implementation. Lancet Neurol. 2022, 21, 66–77. [Google Scholar] [CrossRef] [PubMed]
- Van Dyck, C.H.; Swanson, C.J.; Aisen, P.; Bateman, R.J.; Chen, C.; Gee, M.; Kanekiyo, M.; Li, D.; Reyderman, L.; Cohen, S.; et al. Lecanemab in Early Alzheimer’s Disease. N. Engl. J. Med. 2023, 388, 9–21. [Google Scholar] [CrossRef]
- Bossuyt, P.M.; Irwig, L.; Craig, J.; Glasziou, P. Comparative accuracy: Assessing new tests against existing diagnostic pathways. BMJ 2006, 332, 1089–1092. [Google Scholar] [CrossRef]
- Schunemann, H.J. Handbook for Grading the Quality of Evidence and the Strength of Recommendations Using the GRADE Approach; Updated October 2013; Schünemann, H., Guyatt, G., Oxman, A., Brożek, J., Eds.; GRADE Working Group: Austin, TX, USA, 2013. [Google Scholar]
- Squitti, R.; Faller, P.; Hureau, C.; Granzotto, A.; White, A.R.; Kepp, K.P. Copper Imbalance in Alzheimer’s Disease and Its Link with the Amyloid Hypothesis: Towards a Combined Clinical, Chemical, and Genetic Etiology. J. Alzheimer’s Dis. 2021, 83, 23–41. [Google Scholar] [CrossRef]
- Mathys, Z.K.; White, A.R. Copper and Alzheimer’s Disease. Adv. Neurobiol. 2017, 18, 199–216. [Google Scholar] [PubMed]
- Roberts, E.A.; Schilsky, M.L.; American Association for Study of Liver, D. Diagnosis and treatment of Wilson disease: An update. Hepatology 2008, 47, 2089–2111. [Google Scholar] [CrossRef] [PubMed]
- Walshe, J.M.; Clinical Investigations Standing Committee of the Association of Clinical, B. Wilson’s disease: The importance of measuring serum caeruloplasmin non-immunologically. Ann. Clin. Biochem. 2003, 40, 115–121. [Google Scholar] [CrossRef] [PubMed]
- Hoogenraad, T. Wilson Disease; Intermed Medical Publishers: Amsterdam, The Netherlands, 2001. [Google Scholar]
- Squitti, R.; Ventriglia, M.; Granzotto, A.; Sensi, S.L.; Rongioletti, M.C.A. Non-Ceruloplasmin Copper as a Stratification Biomarker of Alzheimer’s Disease Patients: How to Measure and Use It. Curr. Alzheimer Res. 2021, 18, 533–545. [Google Scholar] [CrossRef] [PubMed]
- Linder, M.C. Ceruloplasmin and other copper binding components of blood plasma and their functions: An update. Metallomics 2016, 8, 887–905. [Google Scholar] [CrossRef] [PubMed]
- Li, D.-D.; Zhang, W.; Wang, Z.-Y.; Zhao, P. Serum Copper, Zinc, and Iron Levels in Patients with Alzheimer’s Disease: A Meta-Analysis of Case-Control Studies. Front. Aging Neurosci. 2017, 9, 300. [Google Scholar] [CrossRef]
- Gaggelli, E.; Kozlowski, H.; Valensin, D.; Valensin, G. Copper homeostasis and neurodegenerative disorders (Alzheimer’s, prion, and Parkinson’s diseases and amyotrophic lateral sclerosis). Chem. Rev. 2006, 106, 1995–2044. [Google Scholar] [CrossRef] [PubMed]
- Nose, Y.; Wood, L.K.; Kim, B.E.; Prohaska, J.R.; Fry, R.S.; Spears, J.W.; Thiele, D.J. Ctr1 is an apical copper transporter in mammalian intestinal epithelial cells in vivo that is controlled at the level of protein stability. J. Biol. Chem. 2010, 285, 32385–32392. [Google Scholar] [CrossRef]
- Ohrvik, H.; Thiele, D.J. How copper traverses cellular membranes through the mammalian copper transporter 1, Ctr1. Ann. N. Y. Acad. Sci. 2014, 1314, 32–41. [Google Scholar] [CrossRef]
- Christofides, K.; Menon, R.; Jones, C.E. Endocytosis of G Protein-Coupled Receptors and Their Ligands: Is There a Role in Metal Trafficking? Cell Biochem. Biophys. 2018, 76, 329–337. [Google Scholar] [CrossRef] [PubMed]
- Gul, A.S.; Tran, K.K.; Jones, C.E. Neurokinin B and serum albumin limit copper binding to mammalian gonadotropin releasing hormone. Biochem. Biophys. Res. Commun. 2018, 497, 1–6. [Google Scholar] [CrossRef] [PubMed]
- Russino, D.; McDonald, E.; Hejazi, L.; Hanson, G.R.; Jones, C.E. The tachykinin peptide neurokinin B binds copper forming an unusual [CuII(NKB)2] complex and inhibits copper uptake into 1321N1 astrocytoma cells. ACS Chem. Neurosci. 2013, 4, 1371–1381. [Google Scholar] [CrossRef]
- Cabrera-Vera, T.M.; Vanhauwe, J.; Thomas, T.O.; Medkova, M.; Preininger, A.; Mazzoni, M.R.; Hamm, H.E. Insights into G protein structure, function, and regulation. Endocr. Rev. 2003, 24, 765–781. [Google Scholar] [CrossRef] [PubMed]
- Neubig, R.R. RGS-Insensitive G Proteins as In Vivo Probes of RGS Function. Prog. Mol. Biol. Transl. Sci. 2015, 133, 13–30. [Google Scholar]
- Huang, X.; Fu, Y.; Charbeneau, R.A.; Saunders, T.L.; Taylor, D.K.; Hankenson, K.D.; Russell, M.W.; D’Alecy, L.G.; Neubig, R.R. Pleiotropic phenotype of a genomic knock-in of an RGS-insensitive G184S Gnai2 allele. Mol. Cell. Biol. 2006, 26, 6870–6879. [Google Scholar] [CrossRef] [PubMed]
- Tayou, J.; Wang, Q.; Jang, G.F.; Pronin, A.N.; Orlandi, C.; Martemyanov, K.A.; Crabb, J.W.; Slepak, V.Z. Regulator of G Protein Signaling 7 (RGS7) Can Exist in a Homo-oligomeric Form That Is Regulated by Galphao and R7-binding Protein. J. Biol. Chem. 2016, 291, 9133–9147. [Google Scholar] [CrossRef]
- Alqinyah, M.; Hooks, S.B. Regulating the regulators: Epigenetic, transcriptional, and post-translational regulation of RGS proteins. Cell. Signal. 2018, 42, 77–87. [Google Scholar] [CrossRef] [PubMed]
- Ostrovskaya, O.; Xie, K.; Masuho, I.; Fajardo-Serrano, A.; Lujan, R.; Wickman, K.; Martemyanov, K.A. RGS7/Gbeta5/R7BP complex regulates synaptic plasticity and memory by modulating hippocampal GABABR-GIRK signaling. Elife 2014, 3, e02053. [Google Scholar] [CrossRef]
- Dubois, B.; Feldman, H.H.; Jacova, C.; Hampel, H.; Molinuevo, J.L.; Blennow, K.; DeKosky, S.T.; Gauthier, S.; Selkoe, D.; Bateman, R.; et al. Advancing research diagnostic criteria for Alzheimer’s disease: The IWG-2 criteria. Lancet Neurol. 2014, 13, 614–629. [Google Scholar] [CrossRef] [PubMed]
- Folstein, M.F.; Folstein, S.E.; McHugh, P.R. “Mini-mental state”: A practical method for grading the cognitive state of patients for the clinician. J. Psychiatr. Res. 1975, 12, 189–198. [Google Scholar] [CrossRef]
- Santangelo, G.; Siciliano, M.; Pedone, R.; Vitale, C.; Falco, F.; Bisogno, R.; Siano, P.; Barone, P.; Grossi, D.; Santangelo, F.; et al. Normative data for the Montreal Cognitive Assessment in an Italian population sample. Neurol. Sci. 2015, 36, 585–591. [Google Scholar] [CrossRef] [PubMed]
- Dubois, B.; Slachevsky, A.; Litvan, I.; Pillon, B. The FAB: A Frontal Assessment Battery at bedside. Neurology 2000, 55, 1621–1626. [Google Scholar] [CrossRef] [PubMed]
- Caffarra, P.; Gardini, S.; Zonato, F.; Concari, L.; Dieci, F.; Copelli, S.; Freedman, M.; Stracciari, A.; Venneri, A. Italian norms for the Freedman version of the Clock Drawing Test. J. Clin. Exp. Neuropsychol. 2011, 33, 982–988. [Google Scholar] [CrossRef] [PubMed]
- Binetti, G.; Mega, M.S.; Magni, E.; Padovani, A.; Rozzini, L.; Bianchetti, A.; Trabucchi, M.; Cummings, J.L. Behavioral disorders in Alzheimer disease: A transcultural perspective. Arch. Neurol. 1998, 55, 539–544. [Google Scholar] [CrossRef] [PubMed]
- Benjamini, Y.; Hochberg, Y. Controlling the false discovery rate: A practical and powerful approach to multiple hypothesis testing. J. R. Stat. Soc. 1995, 57, 289–300. [Google Scholar] [CrossRef]
- Cohen, J. Statistical Power Analysis in the Behavioral Sciences, 2nd ed.; Lawrence Erlbaum Associates, Inc.: Hillsdale, NJ, USA, 1988. [Google Scholar]
- Squitti, R.; Negrouk, V.; Perera, M.; Llabre, M.M.; Ricordi, C.; Rongioletti, M.C.A.; Mendez, A.J. Serum copper profile in patients with type 1 diabetes in comparison to other metals. J. Trace Elem. Med. Biol. 2019, 56, 156–161. [Google Scholar] [CrossRef]
- Scheinberg, I.H.; Sternlieb, I. Wilson Disease; WB Saunders: Philadelphia, PA, USA, 1984. [Google Scholar]
- Schosinsky, K.H.; Lehmann, H.P.; Beeler, M.F. Measurement of ceruloplasmin from its oxidase activity in serum by use of o-dianisidine dihydrochloride. Clin. Chem. 1974, 20, 1556–1563. [Google Scholar] [CrossRef] [PubMed]
- Rentzsch, P.; Witten, D.; Cooper, G.M.; Shendure, J.; Kircher, M. CADD: Predicting the deleteriousness of variants throughout the human genome. Nucleic. Acids. Res. 2019, 47, D886–D894. [Google Scholar] [CrossRef] [PubMed]
- Robinson, J.T.; Thorvaldsdóttir, H.; Winckler, W.; Guttman, M.; Lander, E.S.; Getz, G.; Mesirov, J.P. Integrative genomics viewer. Nat. Biotechnol. 2011, 29, 24–26. [Google Scholar] [CrossRef]
- Lutsenko, S.; Washington-Hughes, C.; Ralle, M.; Schmidt, K. Copper and the brain noradrenergic system. J. Biol. Inorg. Chem. 2019, 24, 1179–1188. [Google Scholar] [CrossRef] [PubMed]
- Koelle, M.R.; Horvitz, H.R. EGL-10 regulates G protein signaling in the C. elegans nervous system and shares a conserved domain with many mammalian proteins. Cell 1996, 84, 115–125. [Google Scholar] [CrossRef]
- Orlandi, C.; Sutton, L.P.; Muntean, B.S.; Song, C.; Martemyanov, K.A. Homeostatic cAMP regulation by the RGS7 complex controls depression-related behaviors. Neuropsychopharmacology 2019, 44, 642–653. [Google Scholar] [CrossRef]
- Sutton, L.P.; Khalatyan, N.; Savas, J.N.; Martemyanov, K.A. Striatal RGS7 Regulates Depression-Related Behaviors and Stress-Induced Reinstatement of Cocaine Conditioned Place Preference. eNeuro 2021, 8, 1–11. [Google Scholar] [CrossRef]
- Gerber, K.J.; Squires, K.E.; Hepler, J.R. Roles for Regulator of G Protein Signaling Proteins in Synaptic Signaling and Plasticity. Mol. Pharmacol. 2016, 89, 273–286. [Google Scholar] [CrossRef]
- Obsilova, V.; Kopecka, M.; Kosek, D.; Kacirova, M.; Kylarova, S.; Rezabkova, L.; Obsil, T. Mechanisms of the 14-3-3 protein function: Regulation of protein function through conformational modulation. Physiol. Res. 2014, 63 (Suppl. 1), S155–S164. [Google Scholar] [CrossRef] [PubMed]
- Pair, F.S.; Yacoubian, T.A. 14-3-3 Proteins: Novel Pharmacological Targets in Neurodegenerative Diseases. Trends Pharmacol. Sci. 2021, 42, 226–238. [Google Scholar] [CrossRef] [PubMed]
- Foote, M.; Zhou, Y. 14-3-3 proteins in neurological disorders. Int. J. Biochem. Mol. Biol. 2012, 3, 152–164. [Google Scholar]
- Young, J.K. Neurogenesis Makes a Crucial Contribution to the Neuropathology of Alzheimer’s Disease. J. Alzheimers Dis. Rep. 2020, 4, 365–371. [Google Scholar] [CrossRef]
- Dringen, R.; Scheiber, I.F.; Mercer, J.F. Copper metabolism of astrocytes. Front. Aging Neurosci. 2013, 5, 9. [Google Scholar] [CrossRef] [PubMed]
- Murray, M.E.; Graff-Radford, N.R.; Ross, O.A.; Petersen, R.C.; Duara, R.; Dickson, D.W. Neuropathologically defined subtypes of Alzheimer’s disease with distinct clinical characteristics: A retrospective study. Lancet Neurol. 2011, 10, 785–796. [Google Scholar] [CrossRef]
- Fagan, A.M.; Mintun, M.A.; Mach, R.H.; Lee, S.Y.; Dence, C.S.; Shah, A.R.; LaRossa, G.N.; Spinner, M.L.; Klunk, W.E.; Mathis, C.A.; et al. Inverse relation between in vivo amyloid imaging load and cerebrospinal fluid Abeta42 in humans. Ann. Neurol. 2006, 59, 512–519. [Google Scholar] [CrossRef] [PubMed]
- Grimmer, T.; Riemenschneider, M.; Forstl, H.; Henriksen, G.; Klunk, W.E.; Mathis, C.A.; Shiga, T.; Wester, H.J.; Kurz, A.; Drzezga, A. Beta amyloid in Alzheimer’s disease: Increased deposition in brain is reflected in reduced concentration in cerebrospinal fluid. Biol. Psychiatry 2009, 65, 927–934. [Google Scholar] [CrossRef] [PubMed]
- Abe, A.; Yamashita, S.; Noma, A. Sensitive, direct colorimetric assay for copper in serum. Clin. Chem. 1989, 35, 552–554. [Google Scholar] [CrossRef]
- Twomey, P.J.; Viljoen, A.; House, I.M.; Reynolds, T.M.; Wierzbicki, A.S. Copper:caeruloplasmin ratio. J. Clin. Pathol. 2007, 60, 441–442. [Google Scholar] [CrossRef]
- Twomey, P.J.; Viljoen, A.; House, I.M.; Reynolds, T.M.; Wierzbicki, A.S. Adjusting copper concentrations for caeruloplasmin levels in routine clinical practice. J. Clin. Pathol. 2006, 59, 867–869. [Google Scholar] [CrossRef]
- Desvignes, J.P.; Bartoli, M.; Delague, V.; Krahn, M.; Miltgen, M.; Beroud, C.; Salgado, D. VarAFT: A variant annotation and filtration system for human next generation sequencing data. Nucleic. Acids. Res. 2018, 46, W545–W553. [Google Scholar] [CrossRef] [PubMed]
- Sayers, E.W.; Beck, J.; Bolton, E.E.; Bourexis, D.; Brister, J.R.; Canese, K.; Comeau, D.C.; Funk, K.; Kim, S.; Klimke, W.; et al. Database resources of the National Center for Biotechnology Information. Nucleic. Acids. Res. 2021, 49, D10–D17. [Google Scholar] [CrossRef] [PubMed]
- Duvaud, S.; Gabella, C.; Lisacek, F.; Stockinger, H.; Ioannidis, V.; Durinx, C. Expasy, the Swiss Bioinformatics Resource Portal, as designed by its users. Nucleic. Acids. Res. 2021, 49, W216–W227. [Google Scholar] [CrossRef] [PubMed]
- Madeira, F.; Pearce, M.; Tivey, A.R.N.; Basutkar, P.; Lee, J.; Edbali, O.; Madhusoodanan, N.; Kolesnikov, A.; Lopez, R. Search and sequence analysis tools services from EMBL-EBI in 2022. Nucleic. Acids. Res. 2022, 50, W276–W279. [Google Scholar] [CrossRef]
- Yuan, Z.; Bailey, T.L.; Teasdale, R.D. Prediction of protein B-factor profiles. Proteins 2005, 58, 905–912. [Google Scholar] [CrossRef]
- Hekkelman, M.L.; Te Beek, T.A.; Pettifer, S.R.; Thorne, D.; Attwood, T.K.; Vriend, G. WIWS: A protein structure bioinformatics Web service collection. Nucleic. Acids. Res. 2010, 38, W719–W723. [Google Scholar] [CrossRef] [PubMed]
- Baek, M.; DiMaio, F.; Anishchenko, I.; Dauparas, J.; Ovchinnikov, S.; Lee, G.R.; Wang, J.; Cong, Q.; Kinch, L.N.; Schaeffer, R.D.; et al. Accurate prediction of protein structures and interactions using a three-track neural network. Science 2021, 373, 871–876. [Google Scholar] [CrossRef] [PubMed]
- Pettersen, E.F.; Goddard, T.D.; Huang, C.C.; Couch, G.S.; Greenblatt, D.M.; Meng, E.C.; Ferrin, T.E. UCSF Chimera--a visualization system for exploratory research and analysis. J. Comput. Chem. 2004, 25, 1605–1612. [Google Scholar] [CrossRef]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Heathy Controls (CTRL) | Alzheimer’s Disease (AD) | p Value | |
|---|---|---|---|
| n | 41 | 27 | |
| Men, n (%) | 10 (24%) | 13 (48%) | |
| Women, n (%) | 31 (76%) | 14 (52%) | 0.043 |
| Age (±SD) | 70.05 ± 8.8 | 77.78 ± 7.79 | <0.001 |
| Heathy Controls (CTRL) | Alzheimer’s Disease (AD) | p Value | Adjusted p Value b | |
|---|---|---|---|---|
| Cu, µmol/L | 13.37 ± 2.18 | 15.27 ± 2.10 | 0.001 | 0.002 |
| Cp, mg/dL | 27.62 ± 3.45 | 26.87 ± 2.20 | 0.322 | 0.536 |
| Cp-activity, IU | 106.60 ± 14.86 | 113.60 ± 25.88 | 0.212 | 0.222 |
| Cu:Cp ratio | 6.4 ± 0.65 | 7.49 ± 0.60 | <0.001 | <0.001 |
| non-Cp Cu, µmol/L | 0.35 ± 1.34 | 2.58 ± 1.31 | <0.001 a | <0.001 |
| eCp:iCp, IU/mg | 3.91 ± 0.42 | 4.20 ± 0.76 | 0.076 a | 0.142 |
| AD Subtypes | AD | CuAD | p Value |
|---|---|---|---|
| n | 9 | 18 | |
| Men, n (%) | 7 (78%) | 6 (33%) | 0.046 a |
| Women, n (%) | 2 (22%) | 12 (67%) | |
| Age (±SD) | 79.22 ± 8.33 | 77.06 ± 7.65 | 0.507 |
| Disease duration | 9.11 ± 6.83 | 9.83 ± 3.29 | 0.770 |
| AD | CuAD | p Value | Adjusted p Value b | B-H Adjusted p Value c | |
|---|---|---|---|---|---|
| MMSE | 15.11 ± 8.33 | 8.39 ± 6.10 | 0.025 | 0.069 | 0.115 |
| MOCA | 10.11 ± 7.66 | 5.39 ± 6.51 | 0.106 | 0.183 | 0.229 |
| FAB | 8.00 ± 5.10 | 2.61 ± 3.07 | 0.012 a | 0.008 | 0.040 |
| CDT | 4.33 ± 2.96 | 1.89 ± 1.84 | 0.037 a | 0.029 | 0.072 |
| NPI | 17.44 ± 12.41 | 19.39 ± 14.52 | 0.734 | 0.556 | 0.556 |
| Cu | Cp | Cp Activity | Cu:Cp | Non-Cp Cu | eCp:iCp | ||
|---|---|---|---|---|---|---|---|
| MMSE | Pearson Correlation | −0.343 | −0.205 | −0.370 | −0.379 | −0.412 | −0.384 |
| p value | 0.079 | 0.305 | 0.058 | 0.051 | 0.029 | 0.048 | |
| B-H adjusted p a | 0.095 | 0.305 | 0.087 | 0.087 | 0.087 | 0.087 | |
| MOCA | Pearson Correlation | −0.254 | −0.059 | −0.248 | −0.383 | −0.376 | −0.291 |
| p value | 0.256 | 0.770 | 0.256 | 0.147 | 0.147 | 0.256 | |
| B-H adjusted p a | 0.307 | 0.770 | 0.307 | 0.307 | 0.307 | 0.307 | |
| FAB | Pearson Correlation | −0.437 | −0.296 | −0.424 | −0.478 | −0.476 | −0.417 |
| p value | 0.023 | 0.134 | 0.028 | 0.012 | 0.010 | 0.030 | |
| B-H adjusted p a | 0.036 | 0.134 | 0.036 | 0.036 | 0.036 | 0.036 | |
| CDT | Pearson Correlation | −0.328 | −0.245 | −0.363 | −0.330 | −0.352 | −0.360 |
| p value | 0.095 | 0.218 | 0.063 | 0.093 | 0.066 | 0.066 | |
| B-H adjusted p a | 0.114 | 0.218 | 0.114 | 0.114 | 0.114 | 0.114 | |
| NPI | Pearson Correlation | 0.003 | −0.104 | −0.049 | 0.143 | 0.039 | 0.000 |
| p value | 0.990 | 0.605 | 0.810 | 0.478 | 0.843 | 1.000 | |
| B-H adjusted p a | 0.999 | 0.999 | 0.999 | 0.999 | 0.990 | 0.999 | |
| Test | Result | Normal Reference Values |
|---|---|---|
| Ferritin | 230.3 | 24–307 ng/mL |
| γ-GT | 11 | 3–29 U/L |
| AST | 15 | 2–40 U/L |
| ALT | 8 | 2–40 U/L |
| Magnesium | 1.84 | 1.3–2.1 mg/dL |
| Phosphorus | 2.6 | 2.8 to 4.5 mg/dL |
| Lithium | <0.0101 | 0.6 and 1.2 mmol/L |
| Bilirubin total | 0.3 | 0–1.1 mg/dL |
| Iron (Fe) | 31 | 37–164 µg/dL |
| Transferrin | 187 | 200–360 mg/dL |
| TIBC & | 233.8 | 240 to 450 mg/dL |
| Transferrin saturation (%) | 13.3 | 15–50% |
| Copper (Cu) | 5 | 11.0–24.0 µM |
| Ceruloplasmin | 24.23 | 20–60 mg/dl |
| Cp activity | 232 | 62–140 IU |
| Non-Cp Cu * | 4 | 0.05–1.6 µg/dL |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Squitti, R.; Catalli, C.; Gigante, L.; Marianetti, M.; Rosari, M.; Mariani, S.; Bucossi, S.; Mastromoro, G.; Ventriglia, M.; Simonelli, I.; et al. Non-Ceruloplasmin Copper Identifies a Subtype of Alzheimer’s Disease (CuAD): Characterization of the Cognitive Profile and Case of a CuAD Patient Carrying an RGS7 Stop-Loss Variant. Int. J. Mol. Sci. 2023, 24, 6377. https://doi.org/10.3390/ijms24076377
Squitti R, Catalli C, Gigante L, Marianetti M, Rosari M, Mariani S, Bucossi S, Mastromoro G, Ventriglia M, Simonelli I, et al. Non-Ceruloplasmin Copper Identifies a Subtype of Alzheimer’s Disease (CuAD): Characterization of the Cognitive Profile and Case of a CuAD Patient Carrying an RGS7 Stop-Loss Variant. International Journal of Molecular Sciences. 2023; 24(7):6377. https://doi.org/10.3390/ijms24076377
Chicago/Turabian StyleSquitti, Rosanna, Claudio Catalli, Laura Gigante, Massimo Marianetti, Mattia Rosari, Stefania Mariani, Serena Bucossi, Gioia Mastromoro, Mariacarla Ventriglia, Ilaria Simonelli, and et al. 2023. "Non-Ceruloplasmin Copper Identifies a Subtype of Alzheimer’s Disease (CuAD): Characterization of the Cognitive Profile and Case of a CuAD Patient Carrying an RGS7 Stop-Loss Variant" International Journal of Molecular Sciences 24, no. 7: 6377. https://doi.org/10.3390/ijms24076377
APA StyleSquitti, R., Catalli, C., Gigante, L., Marianetti, M., Rosari, M., Mariani, S., Bucossi, S., Mastromoro, G., Ventriglia, M., Simonelli, I., Tondolo, V., Singh, P., Kumar, A., Pal, A., & Rongioletti, M. (2023). Non-Ceruloplasmin Copper Identifies a Subtype of Alzheimer’s Disease (CuAD): Characterization of the Cognitive Profile and Case of a CuAD Patient Carrying an RGS7 Stop-Loss Variant. International Journal of Molecular Sciences, 24(7), 6377. https://doi.org/10.3390/ijms24076377