
Quantum Mechanical Assessment of Protein–Ligand Hydrogen Bond Strength Patterns: Insights from Semiempirical Tight-Binding and Local Vibrational Mode Theory



Abstract
1. Introduction
2. Results and Discussion
2.1. Hydrogen Bond Database (Database A) and Hydrogen Bond Strength Database (Database B)
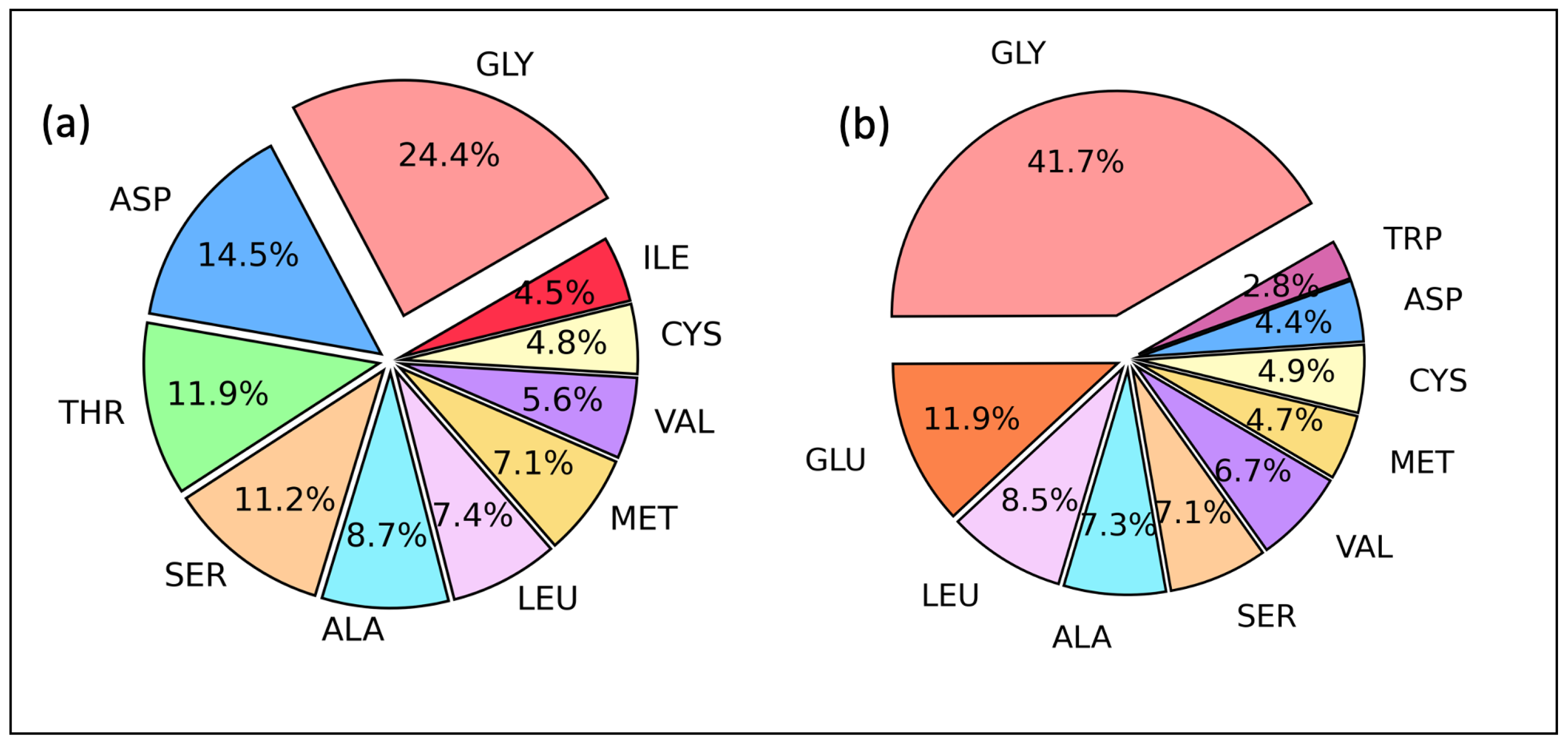
2.2. Amino Acid Residues as Proton Acceptor and Donor
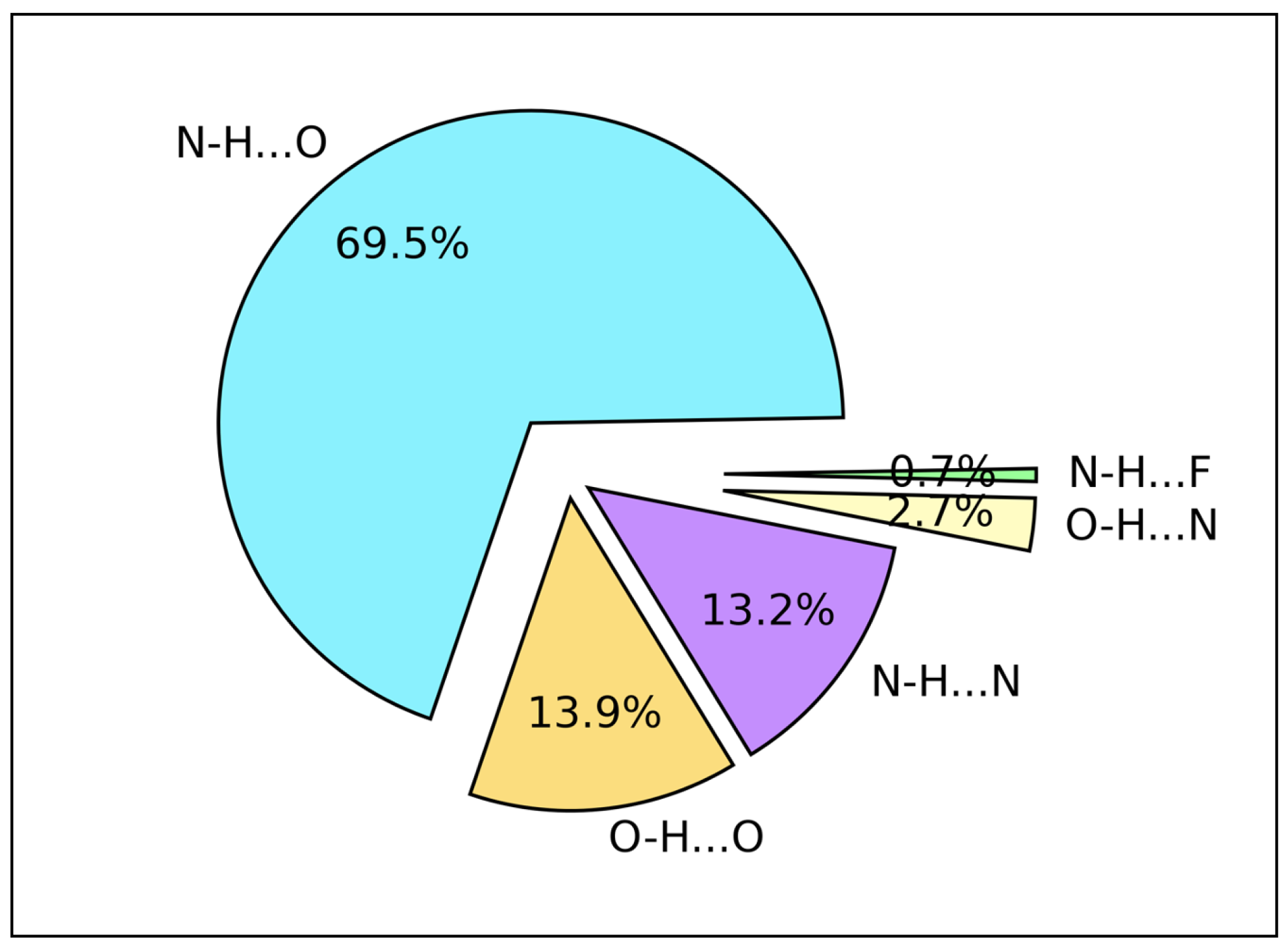
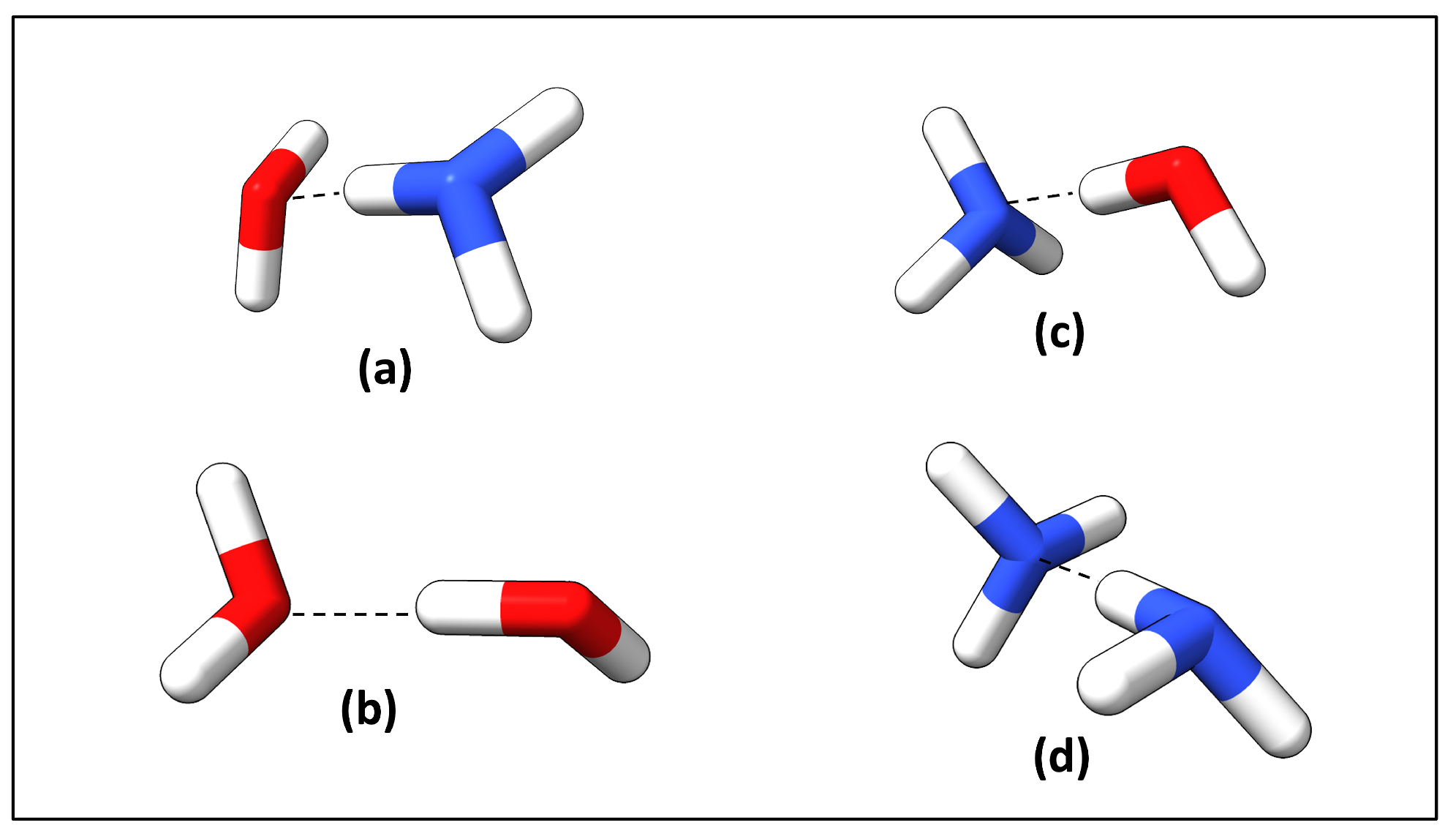
2.3. Hydrogen Bond Types Composition
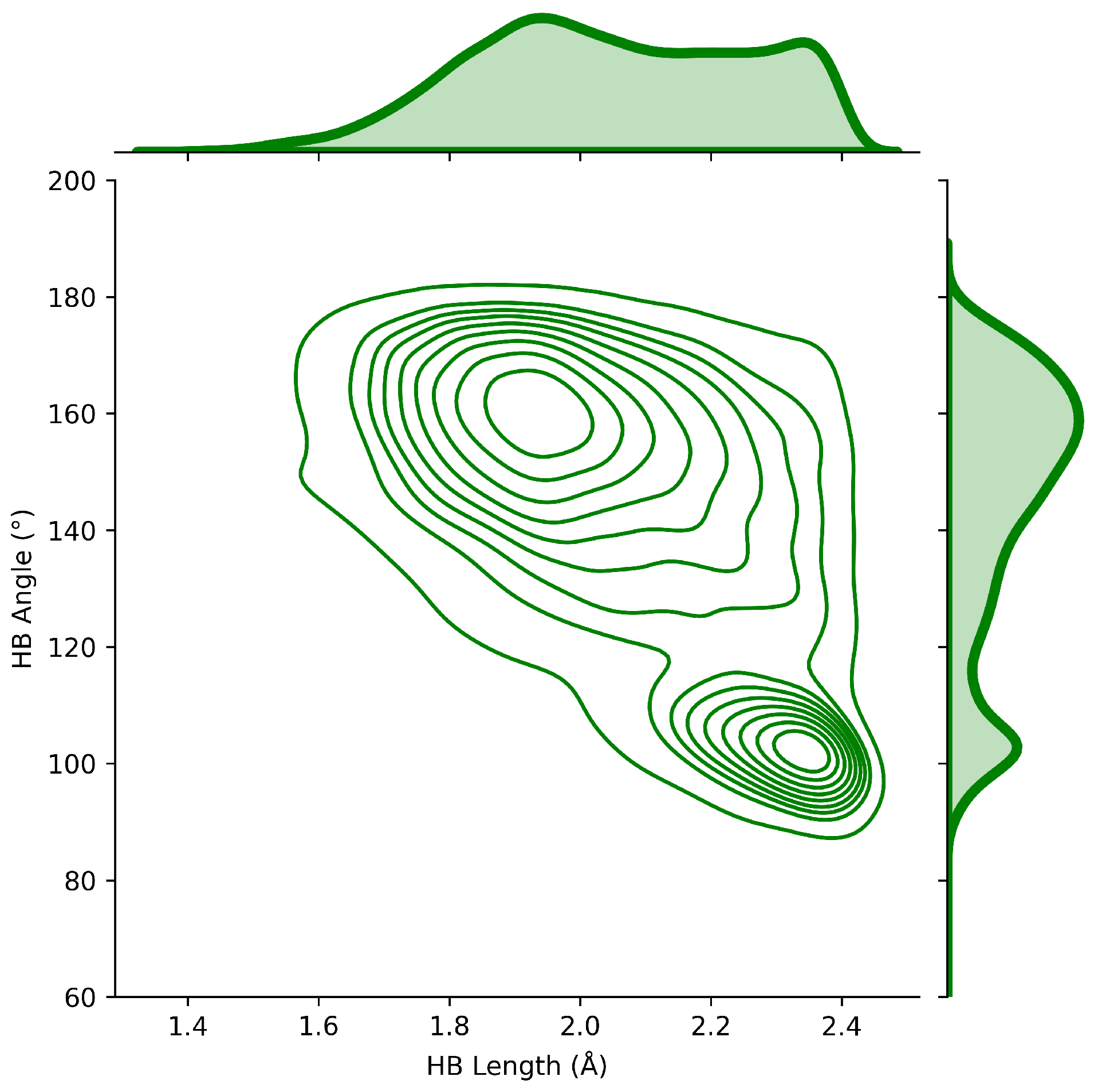
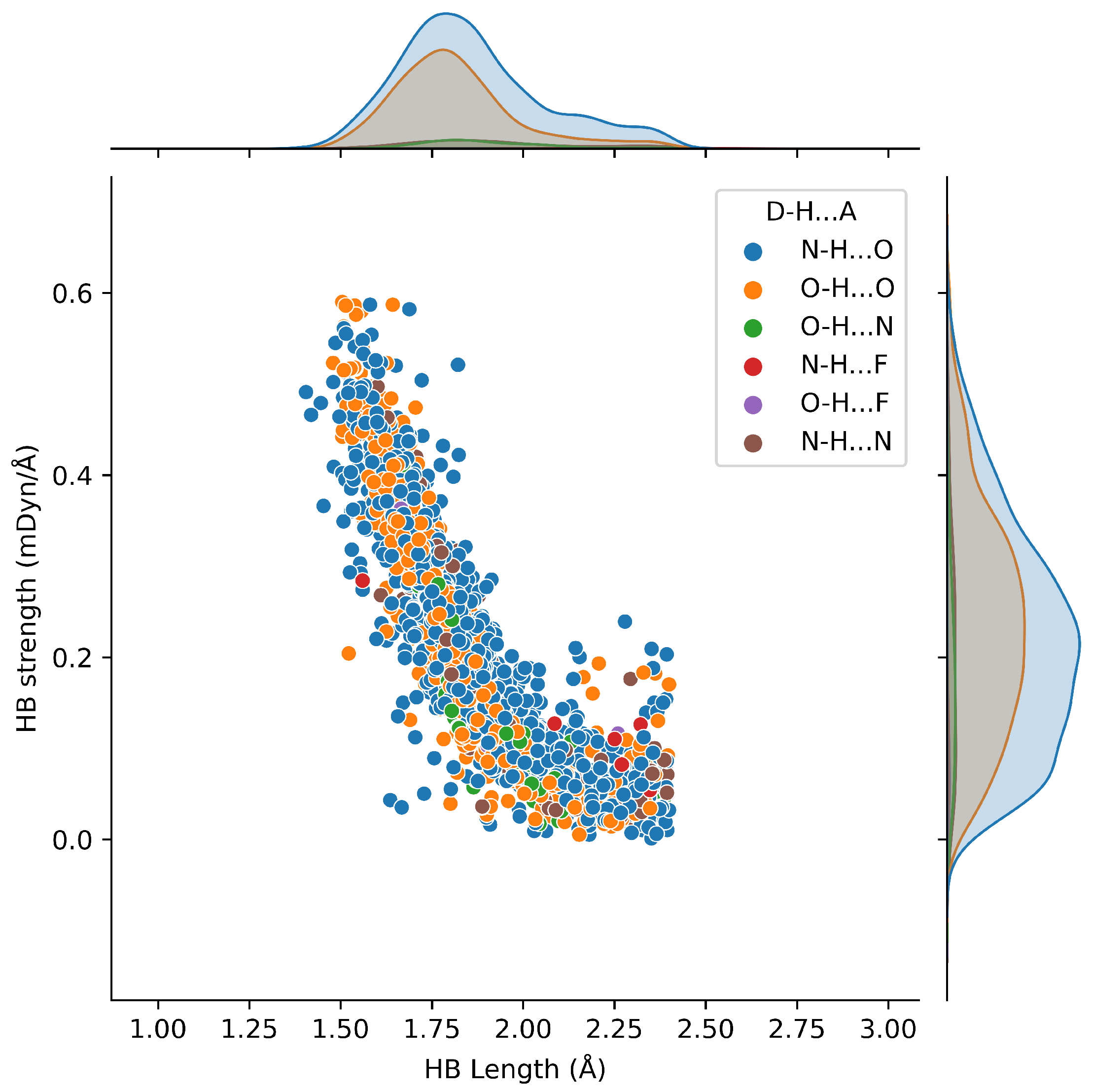
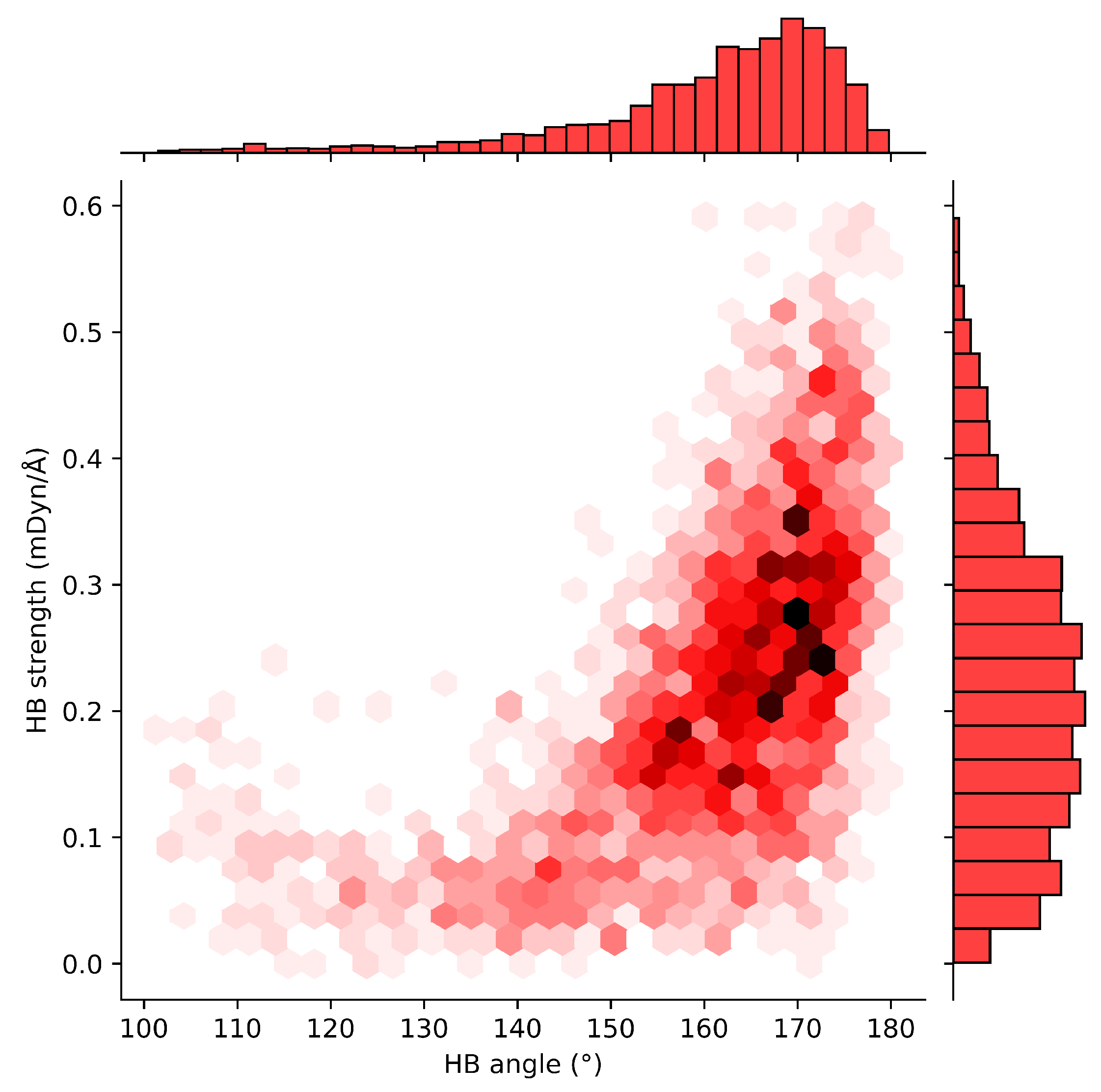
2.4. HB Lengths and Angles Relationship
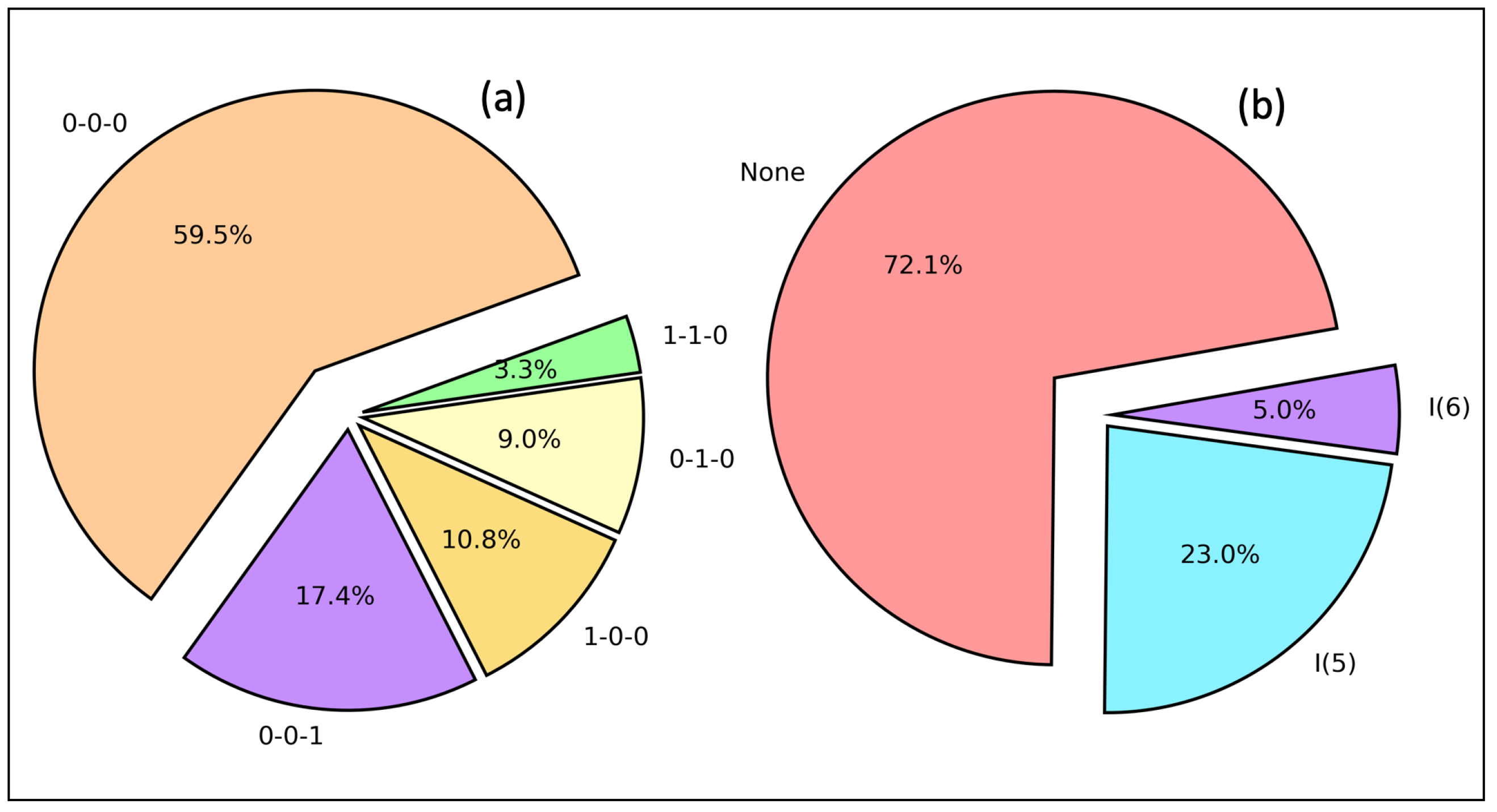
2.5. HB Network and Intramolecular HB
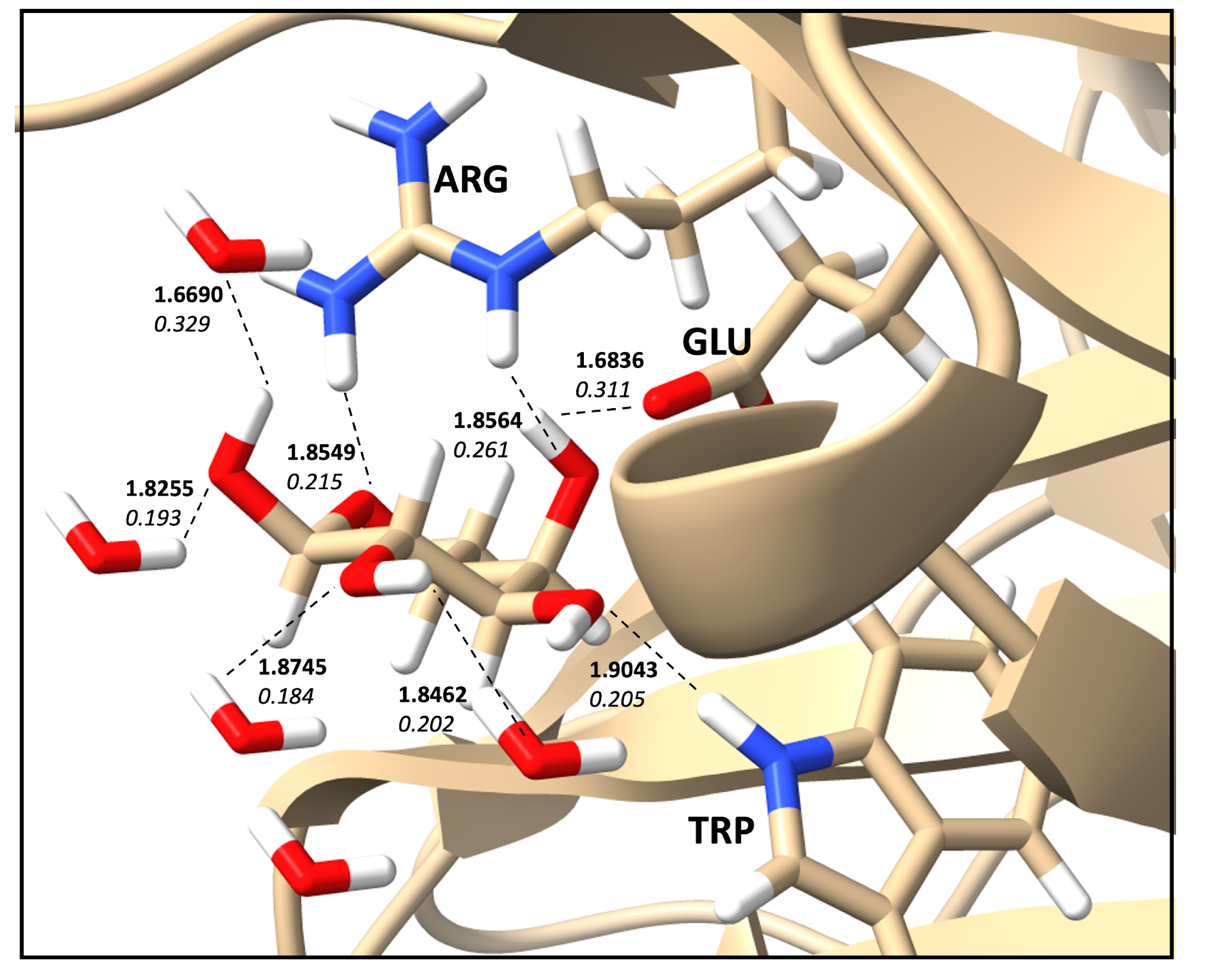
2.6. Protein–Ligand Hydrogen Bonds and Strength Relationships
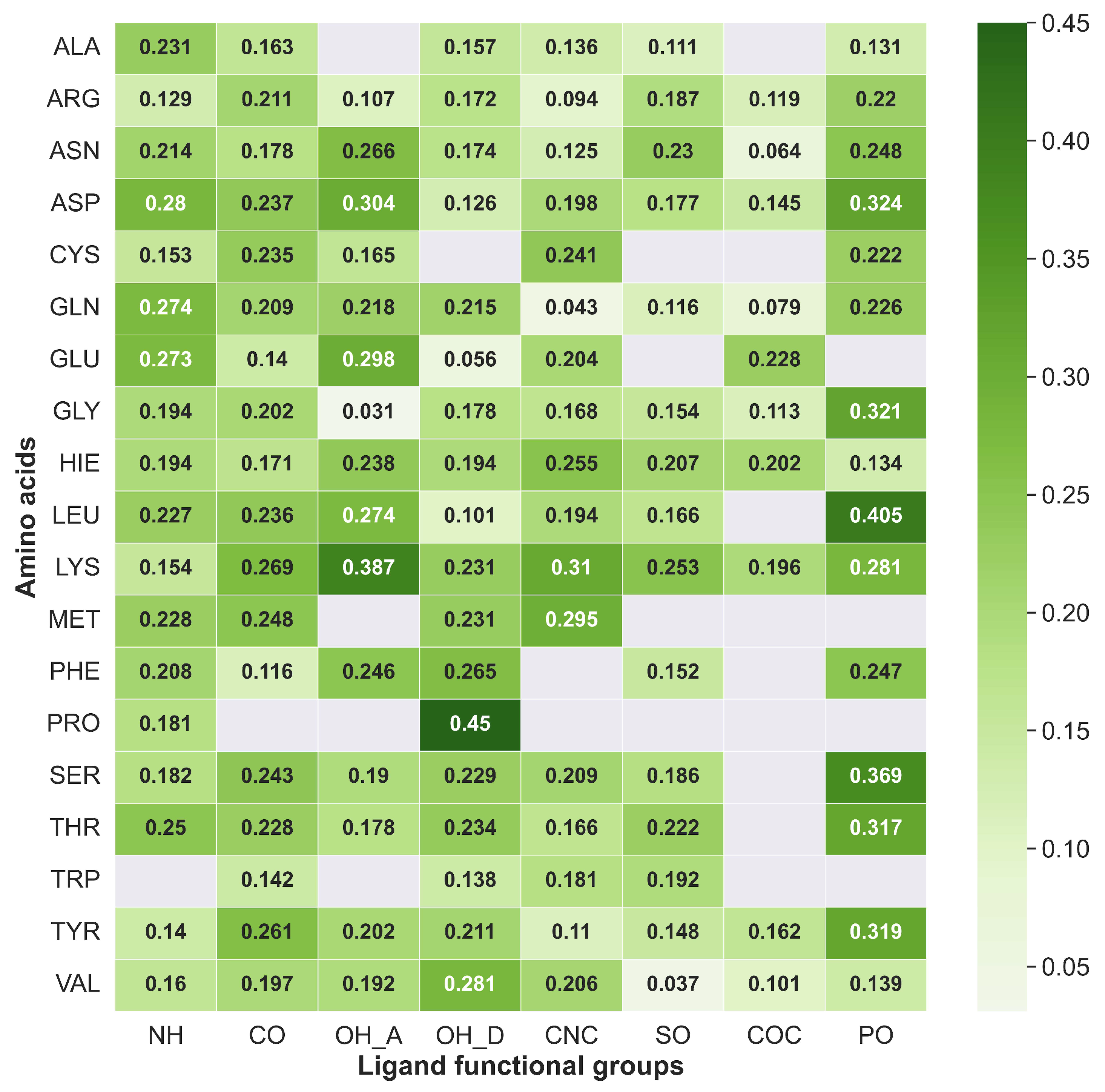
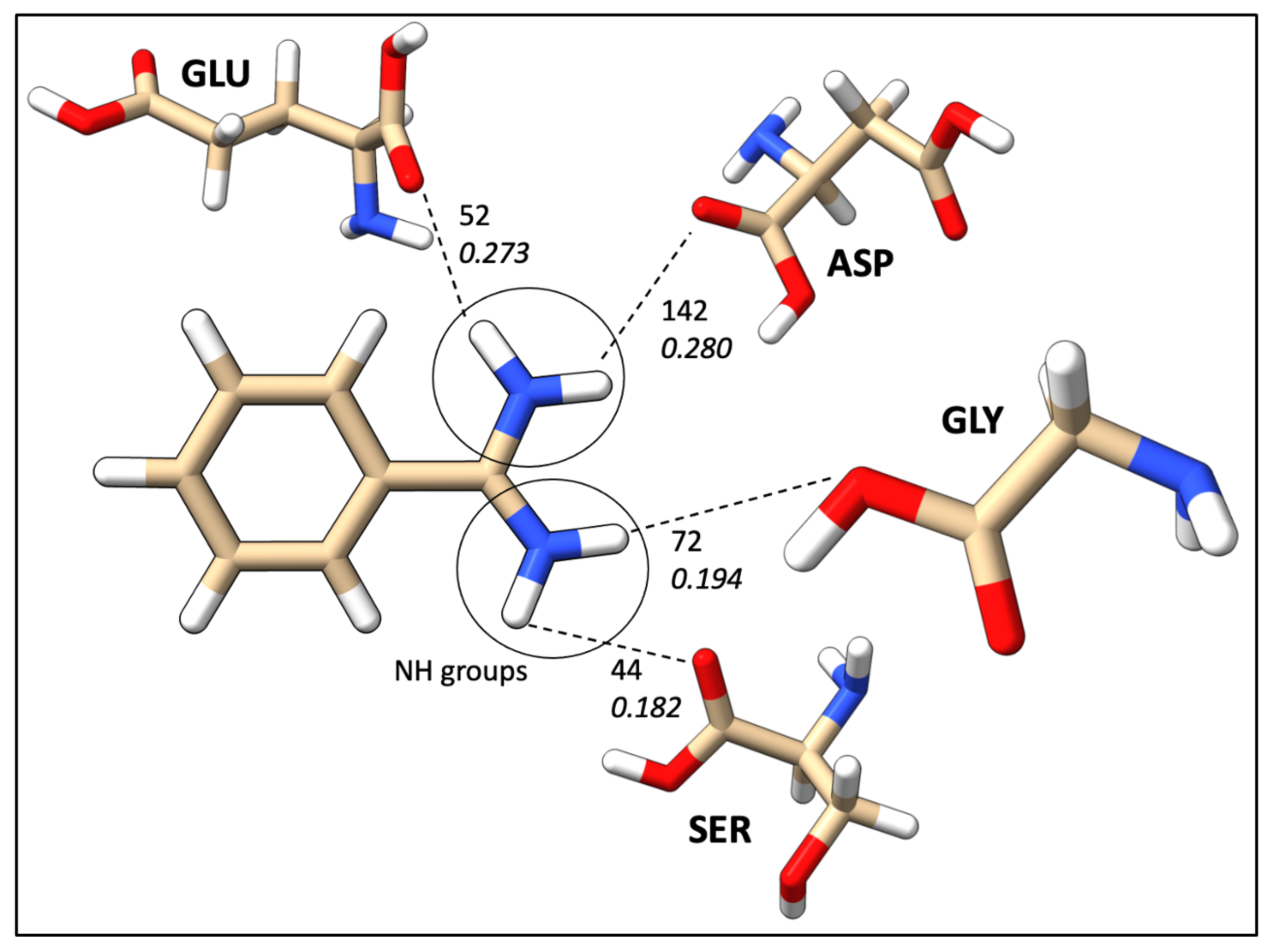
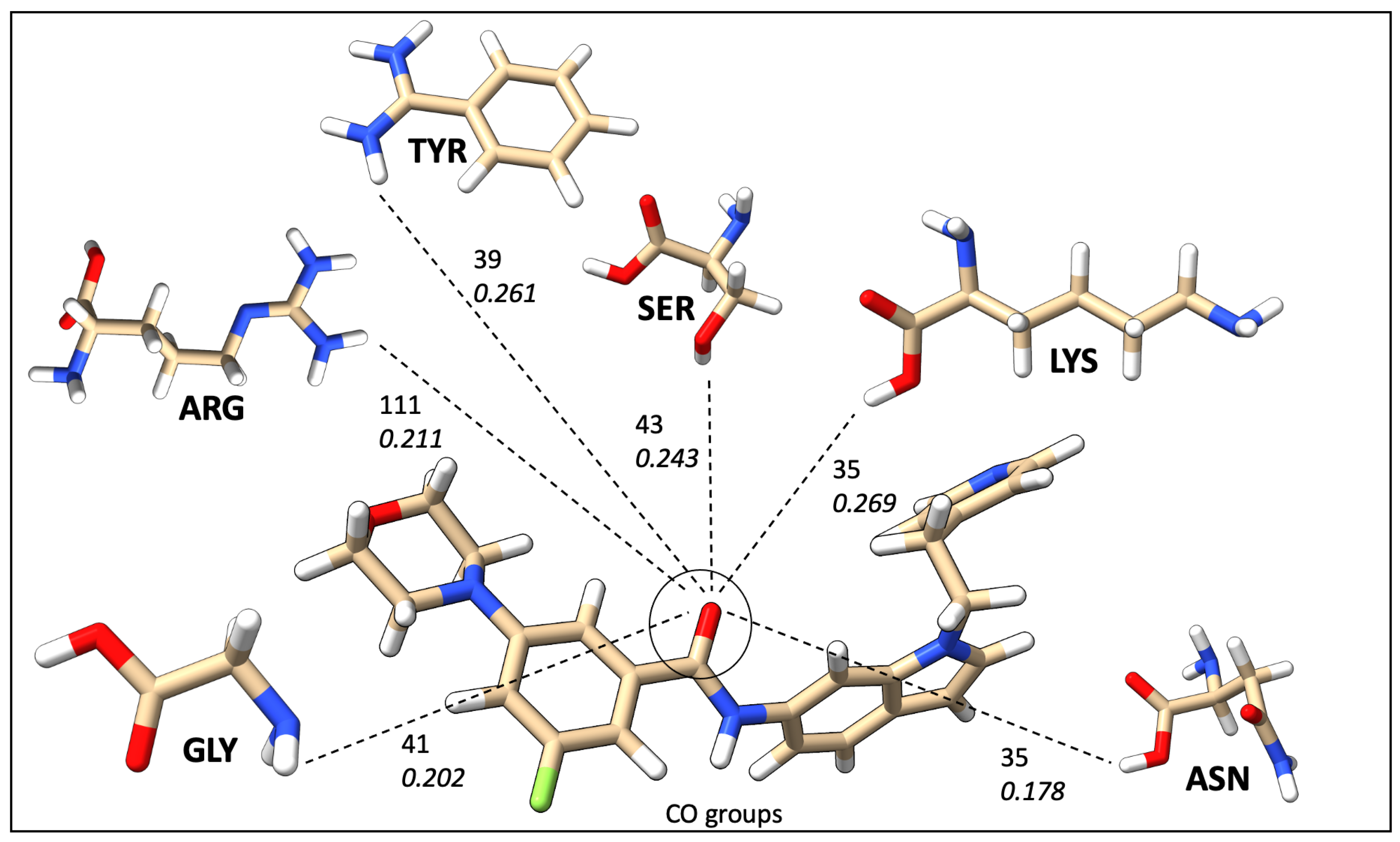
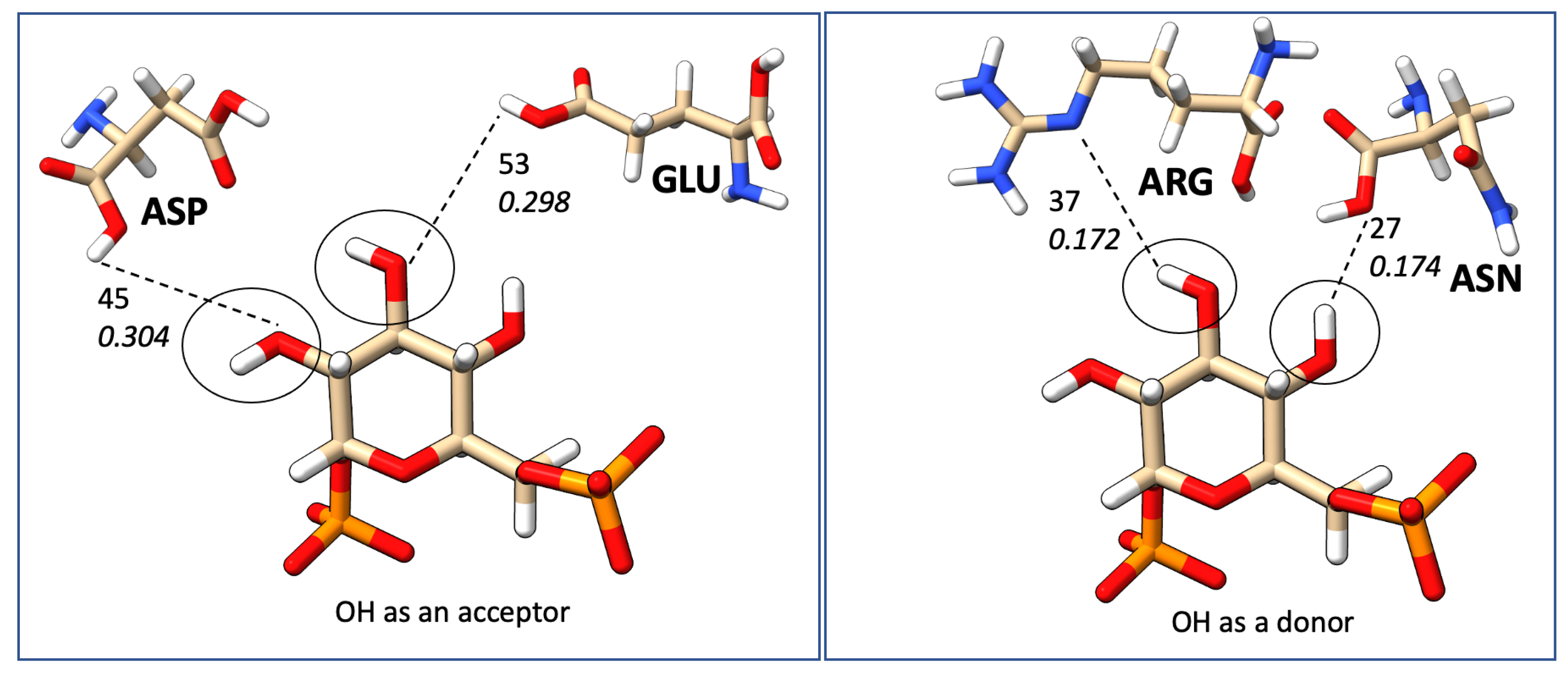
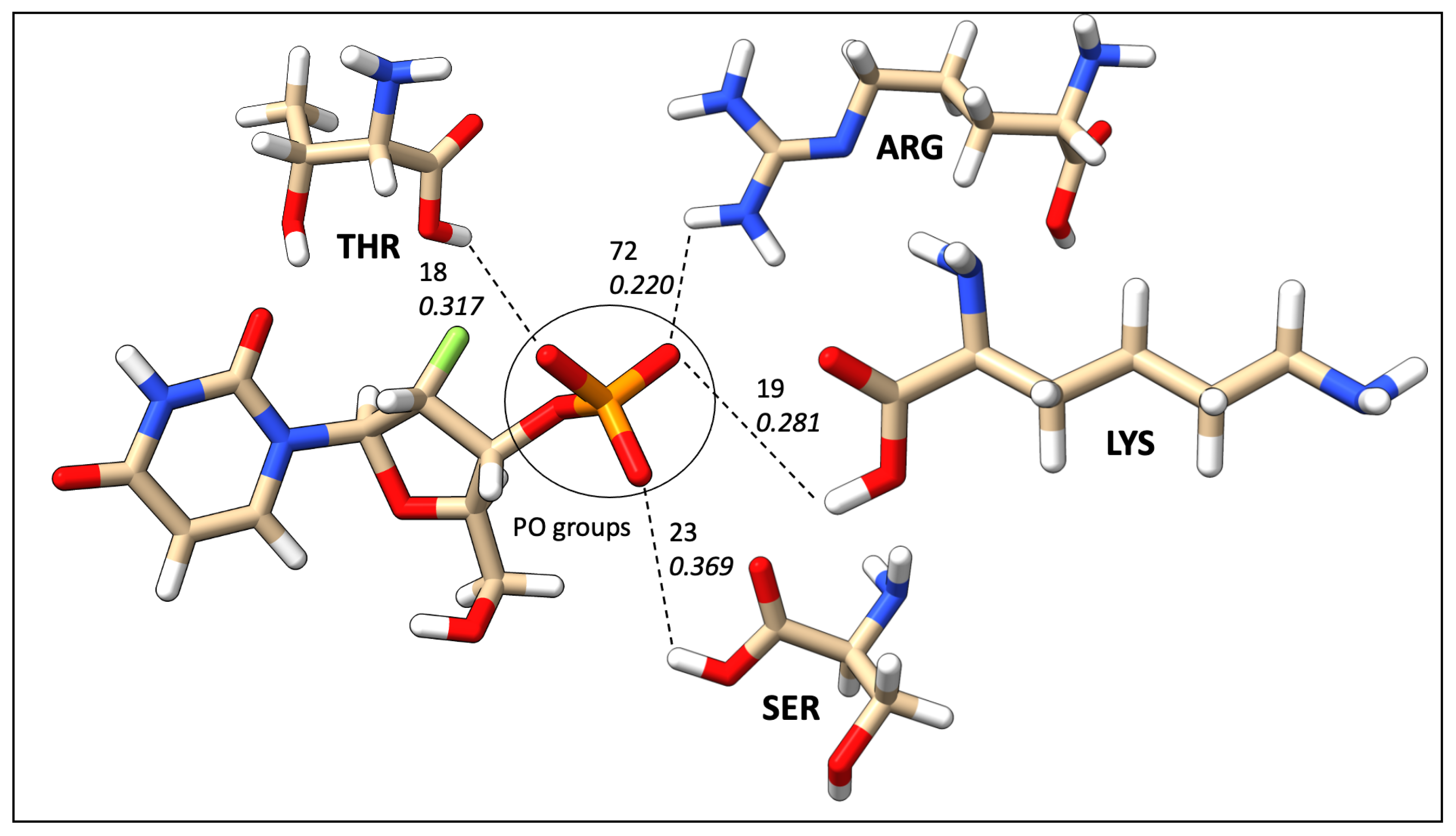
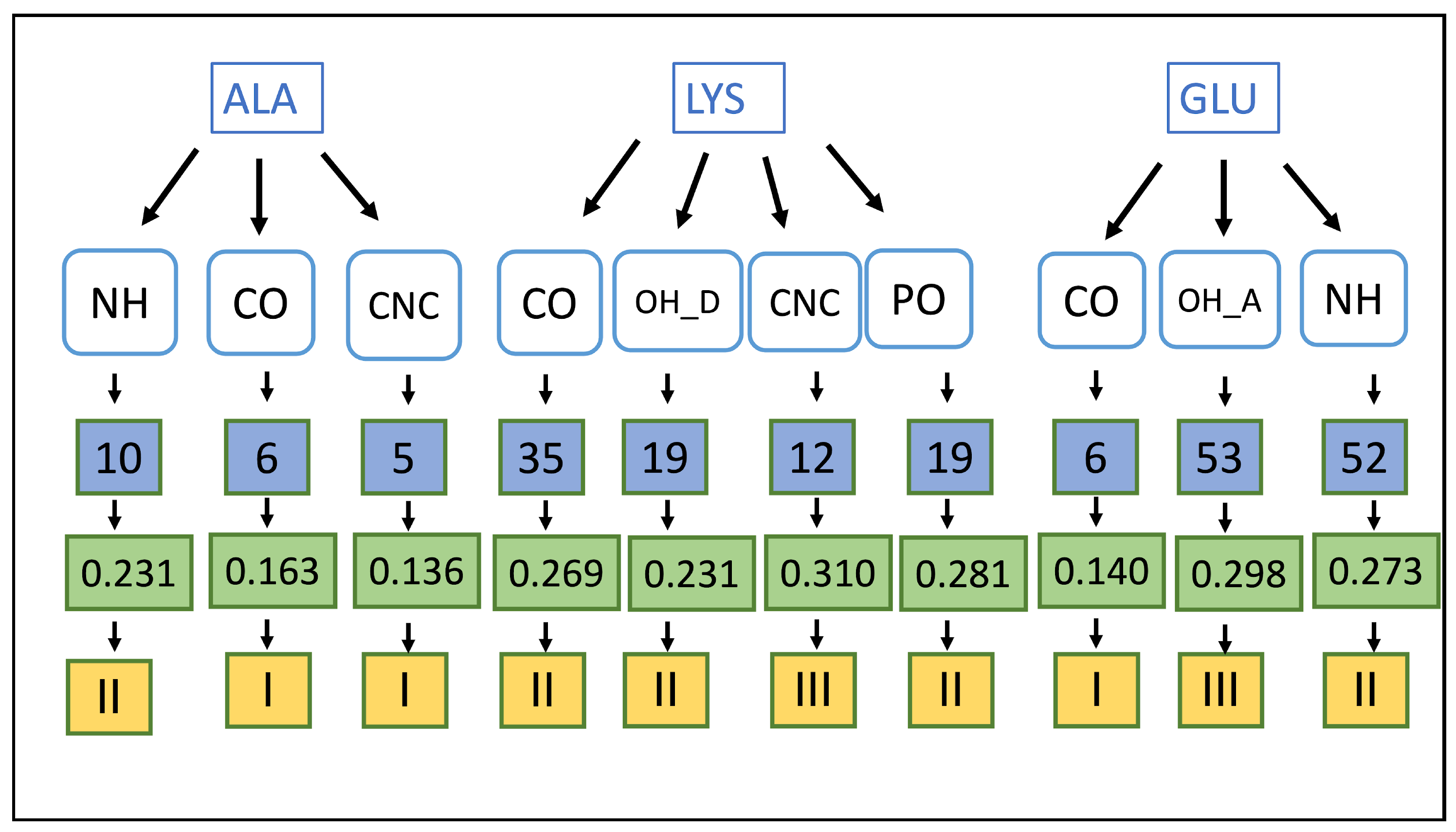
2.7. Mapping HB Strength: Amino Acid and Ligand Functional Group
3. Materials and Methods
3.1. Dataset and Structure Preparation
3.2. EDHB: Efficient Detection of Hydrogen Bonding
3.3. XTB Geometry Optimization and Frequency Calculations
3.4. Local Mode Analysis
3.5. Pattern Analysis
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Wohlert, M.; Benselfelt, T.; Wågberg, L.; Furó, I.; Berglund, L.A.; Wohlert, J. Cellulose and the role of hydrogen bonds: Not in charge of everything. Cellulose 2022, 29, 1–23. [Google Scholar] [CrossRef]
- Mayfield, A.B.; Metternich, J.B.; Trotta, A.H.; Jacobsen, E.N. Stereospecific furanosylations catalyzed by bis-thiourea hydrogen-bond donors. J. Am. Chem. Soc. 2020, 142, 4061–4069. [Google Scholar] [CrossRef]
- Song, P.; Wang, H. High-performance polymeric materials through hydrogen-bond cross-linking. Adv. Mater. 2020, 32, 1901244. [Google Scholar] [CrossRef]
- Pauling, L.; Corey, R.B.; Branson, H.R. The structure of proteins: Two hydrogen-bonded helical configurations of the polypeptide chain. Proc. Natl. Acad. Sci. USA 1951, 37, 205–211. [Google Scholar] [CrossRef] [PubMed]
- Pauling, L.; Corey, R.B. The pleated sheet, a new layer configuration of polypeptide chains. Proc. Natl. Acad. Sci. USA 1951, 37, 251–256. [Google Scholar] [CrossRef] [PubMed]
- Ahmed, I.; Hasan, Z.; Lee, G.; Lee, H.J.; Jhung, S.H. Contribution of hydrogen bonding to liquid-phase adsorptive removal of hazardous organics with metal-organic framework-based materials. J. Chem. Eng. 2022, 430, 132596. [Google Scholar] [CrossRef]
- Chu, Y.; Cui, X.; Kong, W.; Du, K.; Zhen, L.; Wang, L. A robust polymeric binder based on complementary multiple hydrogen bonds in lithium-sulfur batteries. J. Chem. Eng. 2022, 427, 130844. [Google Scholar] [CrossRef]
- Khalid, M.; Ali, A.; Khan, M.U.; Tahir, M.N.; Ahmad, A.; Ashfaq, M.; Hussain, R.; de Alcantara Morais, S.F.; Braga, A.A.C. Non-covalent interactions abetted supramolecular arrangements of N-Substituted benzylidene acetohydrazide to direct its solid-state network. J. Mol. Struct. 2021, 1230, 129827. [Google Scholar] [CrossRef]
- Wallace, A.C.; Laskowski, R.A.; Thornton, J.M. LIGPLOT: A program to generate schematic diagrams of protein-ligand interactions. Protein Eng. Des. Sel. 1995, 8, 127–134. [Google Scholar] [CrossRef]
- Schrödinger, L. Schrödinger, Maestro, NY, USA. Available online: https://www.https://www.schrodinger.com (accessed on 13 January 2023).
- BIOVIA. Pharmacophore and Ligand-Based Design with Biovia Discovery Studio®. Available online: https://www.discngine.com/discovery-studio, (accessed on 17 January 2023).
- Jeffrey, P.D.G.A.; Saenger, P.D.W. Hydrogen Bonding in Biological Structures; Springer: Berlin/Heidelberg, Germany, 1991. [Google Scholar]
- Abraham, M.H.; Duce, P.P.; Prior, D.V.; Barratt, D.G.; Morris, J.J.; Taylor, P.J. Hydrogen bonding. Part 9. Solute proton donor and proton acceptor scales for use in drug design. J. Chem. Soc. Perkin Trans. 1989, 2, 1355–1375. [Google Scholar] [CrossRef]
- Emamian, S.; Lu, T.; Kruse, H.; Emamian, H. Exploring nature and predicting strength of hydrogen bonds: A correlation analysis between atoms-in-molecules descriptors, binding energies, and energy components of symmetry-adapted perturbation theory. J. Comput. Chem. 2019, 40, 2868–2881. [Google Scholar] [CrossRef]
- Jeffrey, G.t.; Maluszynska, H. A survey of hydrogen bond geometries in the crystal structures of amino acids. Int. J. Biol. Macromol. 1982, 4, 173–185. [Google Scholar] [CrossRef]
- Bingham, A.H.; Davenport, R.J.; Gowers, L.; Knight, R.L.; Lowe, C.; Owen, D.A.; Parry, D.M.; Pitt, W.R. A novel series of potent and selective IKK2 inhibitors. Bioorganic Med. Chem. Lett. 2004, 14, 409–412. [Google Scholar] [CrossRef]
- Hao, M.H. Theoretical calculation of hydrogen-bonding strength for drug molecules. J. Chem. Theory Comput. 2006, 2, 863–872. [Google Scholar] [CrossRef]
- Alkorta, I.; Elguero, J.; Frontera, A. Not only hydrogen bonds: Other noncovalent interactions. Crystals 2020, 10, 180. [Google Scholar] [CrossRef]
- Summers, T.J.; Daniel, B.P.; Cheng, Q.; DeYonker, N.J. Quantifying Inter-Residue Contacts through Interaction Energies. J. Chem. Inf. Model. 2019, 59, 5034–5044. [Google Scholar] [CrossRef]
- Lucarini, M.; Pedrielli, P.; Pedulli, G.F.; Cabiddu, S.; Fattuoni, C. Bond dissociation energies of O- H bonds in substituted phenols from equilibration studies. J. Org. Chem. 1996, 61, 9259–9263. [Google Scholar] [CrossRef]
- Bader, R.F.; Nguyen-Dang, T. Quantum theory of atoms in molecules—Dalton revisited. In Advances in Quantum Chemistry; Academic Press: Cambridge, MA, USA, 1981; Volume 14, pp. 63–124. [Google Scholar]
- Grabowski, S.J. Ab initio calculations on conventional and unconventional hydrogen bonds study of the hydrogen bond strength. J. Phys. Chem. A 2001, 105, 10739–10746. [Google Scholar] [CrossRef]
- Rozas, I.; Alkorta, I.; Elguero, J. Monohydride and monofluoride derivatives of B, Al, N and P. Theoretical study of their ability as hydrogen bond acceptors. J. Phys. Chem. A 1999, 103, 8861–8869. [Google Scholar] [CrossRef]
- Reed, A.E.; Curtiss, L.A.; Weinhold, F. Intermolecular interactions from a natural bond orbital, donor-acceptor viewpoint. Chem. Rev. 1988, 88, 899–926. [Google Scholar] [CrossRef]
- Zahedi-Tabrizi, M.; Farahati, R. Calculation of intramolecular hydrogen bonding strength and natural bond orbital (NBO) analysis of naphthazarin with chlorine substitution. Comput. Theor. Chem 2011, 977, 195–200. [Google Scholar] [CrossRef]
- Szatylowicz, H.; Jezierska, A.; Sadlej-Sosnowska, N. Correlations of NBO energies of individual hydrogen bonds in nucleic acid base pairs with some QTAIM parameters. Struct. Chem. 2016, 27, 367–376. [Google Scholar] [CrossRef]
- Kraka, E.; Quintano, M.; Force, H.W.L.; Antonio, J.J.; Freindorf, M. The Local Vibrational Mode Theory and Its Place in the Vibrational Spectroscopy Arena. J. Phys. Chem. A 2022, 126, 8781–8900. [Google Scholar] [CrossRef] [PubMed]
- Kraka, E.; Zou, W.; Tao, Y. Decoding Chemical Information from Vibrational Spectroscopy Data: Local Vibrational Mode Theory. WIREs Comput. Mol. Sci. 2020, 10, 1480. [Google Scholar] [CrossRef]
- Konkoli, Z.; Cremer, D. A New Way of Analyzing Vibrational Spectra. I. Derivation of Adiabatic Internal Modes. Int. J. Quantum Chem. 1998, 67, 1–9. [Google Scholar] [CrossRef]
- Verma, N.; Tao, Y.; Kraka, E. Systematic Detection and Characterization of Hydrogen Bonding in Proteins via Local Vibrational Modes. J. Phys. Chem. B 2021, 125, 2551–2565. [Google Scholar] [CrossRef]
- March, N.; Matthai, C.C. The application of quantum chemistry and condensed matter theory in studying amino-acids, protein folding and anticancer drug technology. Theor. Chem. Acc. 2010, 125, 193–201. [Google Scholar] [CrossRef]
- Shurki, A.; Warshel, A. Structure Function Correlations of Proteins using MM, QM/MM, and Related Approaches: Methods, Concepts, Pitfalls, and Current Progress. Adv. Protein Chem. 2003, 66, 249–313. [Google Scholar]
- Merz Jr, K.M. Using quantum mechanical approaches to study biological systems. Acc. Chem. Res. 2014, 47, 2804–2811. [Google Scholar] [CrossRef]
- Canfield, P.; Dahlbom, M.G.; Hush, N.S.; Reimers, J.R. Density-functional geometry optimization of the 150 000-atom photosystem-I trimer. J. Chem. Phys. 2006, 124, 024301. [Google Scholar] [CrossRef]
- Fanfrlik, J.; Brynda, J.; Rezac, J.; Hobza, P.; Lepsik, M. Interpretation of protein/ligand crystal structure using QM/MM calculations: Case of HIV-1 protease/metallacarborane complex. J. Phys. Chem. B 2008, 112, 15094–15102. [Google Scholar] [CrossRef]
- Sproviero, E.M.; Newcomer, M.B.; Gascón, J.A.; Batista, E.R.; Brudvig, G.W.; Batista, V.S. The MoD-QM/MM methodology for structural refinement of photosystem II and other biological macromolecules. Photosynth. Res. 2009, 102, 455–470. [Google Scholar] [CrossRef]
- Christensen, A.S.; Kubar, T.; Cui, Q.; Elstner, M. Semiempirical quantum mechanical methods for noncovalent interactions for chemical and biochemical applications. Chem. Rev. 2016, 116, 5301–5337. [Google Scholar] [CrossRef] [PubMed]
- Elstner, M. The SCC-DFTB method and its application to biological systems. Theor. Chem. Acc. 2006, 116, 316–325. [Google Scholar] [CrossRef]
- Stewart, J.J. Calculation of the geometry of a small protein using semiempirical methods. J. Mol. Struct. Theochem. 1997, 401, 195–205. [Google Scholar] [CrossRef]
- Stewart, J.J. Application of the PM6 method to modeling proteins. J. Mol. Model. 2009, 15, 765–805. [Google Scholar] [CrossRef]
- Martin, B.P.; Brandon, C.J.; Stewart, J.J.; Braun-Sand, S.B. Accuracy issues involved in modeling in vivo protein structures using PM 7. Proteins: Struct. Funct. Genet. 2015, 83, 1427–1435. [Google Scholar] [CrossRef]
- Su, M.; Yang, Q.; Du, Y.; Feng, G.; Liu, Z.; Li, Y.; Wang, R. Comparative assessment of scoring functions: The CASF-2016 update. J. Chem. Inf. Model 2018, 59, 895–913. [Google Scholar] [CrossRef]
- Li, Y.; Han, L.; Liu, Z.; Wang, R. Comparative assessment of scoring functions on an updated benchmark: 2. Evaluation methods and general results. J. Chem. Inf. Model 2014, 54, 1717–1736. [Google Scholar] [CrossRef]
- Cheng, T.; Li, X.; Li, Y.; Liu, Z.; Wang, R. Comparative assessment of scoring functions on a diverse test set. J. Chem. Inf. Model 2009, 49, 1079–1093. [Google Scholar] [CrossRef]
- García-Nafría, J.; Tate, C.G. Structure determination of GPCRs: Cryo-EM compared with X-ray crystallography. Biochem. Soc. Trans. 2021, 49, 2345–2355. [Google Scholar] [CrossRef]
- Maveyraud, L.; Mourey, L. Protein X-ray crystallography and drug discovery. Molecules 2020, 25, 1030. [Google Scholar] [CrossRef]
- Wimmerova, M.; Mitchell, E.; Sanchez, J.F.; Gautier, C.; Imberty, A. Crystal structure of fungal lectin: Six-bladed β-propeller fold and novel fucose recognition mode for Aleuria aurantia lectin. J. Biol. Chem. 2003, 278, 27059–27067. [Google Scholar] [CrossRef]
- Aurora, R.; Srinivasan, R.; Rose, G.D. Rules for α-helix termination by glycine. Science 1994, 264, 1126–1130. [Google Scholar] [CrossRef]
- Persikov, A.V.; Ramshaw, J.A.; Kirkpatrick, A.; Brodsky, B. Amino acid propensities for the collagen triple-helix. Biochemistry 2000, 39, 14960–14967. [Google Scholar] [CrossRef]
- Goren, S.D. On the deuteron quadrupole coupling constant in hydrogen bonded solids. J. Chem. Phys. 1974, 60, 1892–1893. [Google Scholar] [CrossRef]
- Tan, K.P.; Singh, K.; Hazra, A.; Madhusudhan, M. Peptide bond planarity constrains hydrogen bond geometry and influences secondary structure conformations. Curr. Rese. Struct. Biol. 2021, 3, 1–8. [Google Scholar] [CrossRef] [PubMed]
- Yunta, M.J. It is important to compute intramolecular hydrogen bonding in drug design. Am. J. Model. Optim 2017, 5, 24–57. [Google Scholar]
- Arunan, E.; Desiraju, G.R.; Klein, R.A.; Sadlej, J.; Scheiner, S.; Alkorta, I.; Clary, D.C.; Crabtree, R.H.; Dannenberg, J.J.; Hobza, P.; et al. Definition of the hydrogen bond (IUPAC Recommendations 2011). Pure Appl. Chem. 2011, 83, 1637–1641. [Google Scholar] [CrossRef]
- Judd, E.T.; Stein, N.; Pacheco, A.A.; Elliott, S.J. Hydrogen bonding networks tune proton-coupled redox steps during the enzymatic six-electron conversion of nitrite to ammonia. Biochemistry 2014, 53, 5638–5646. [Google Scholar] [CrossRef]
- Polander, B.C.; Barry, B.A. A hydrogen-bonding network plays a catalytic role in photosynthetic oxygen evolution. Proc. Natl. Acad. Sci. USA 2012, 109, 6112–6117. [Google Scholar] [CrossRef]
- Guo, H.; Salahub, D.R. Cooperative hydrogen bonding and enzyme catalysis. Angew. Chem. Int. Ed. 1998, 37, 2985–2990. [Google Scholar] [CrossRef]
- Redzic, J.S.; Bowler, B.E. Role of hydrogen bond networks and dynamics in positive and negative cooperative stabilization of a protein. Biochemistry 2005, 44, 2900–2908. [Google Scholar] [CrossRef] [PubMed]
- Tian, Z.; Fattahi, A.; Lis, L.; Kass, S.R. Single-centered hydrogen-bonded enhanced acidity (SHEA) acids: A new class of brønsted acids. J. Am. Chem. Soc. 2009, 131, 16984–16988. [Google Scholar] [CrossRef] [PubMed]
- Shokri, A.; Abedin, A.; Fattahi, A.; Kass, S.R. Effect of hydrogen bonds on p K a values: Importance of networking. J. Am. Chem. Soc. 2012, 134, 10646–10650. [Google Scholar] [CrossRef] [PubMed]
- Kawasaki, Y.; Chufan, E.E.; Lafont, V.; Hidaka, K.; Kiso, Y.; Mario Amzel, L.; Freire, E. How much binding affinity can be gained by filling a cavity? Chem. Biol. Drug Des. 2010, 75, 143–151. [Google Scholar] [CrossRef] [PubMed]
- Freire, E. A thermodynamic guide to affinity optimization of drug candidates. In Proteomics and Protein-Protein Interactions; Waksman, G., Ed.; Protein Reviews; Springer: Boston, MA, USA, 2005; pp. 291–307. [Google Scholar] [CrossRef]
- Perrin, L.; Andre, F.; Aninat, C.; Ricoux, R.; Mahy, J.P.; Shangguan, N.; Joullie, M.M.; Delaforge, M. Intramolecular hydrogen bonding as a determinant of the inhibitory potency of N-unsubstituted imidazole derivatives towards mammalian hemoproteins. Metallomics 2009, 1, 148–156. [Google Scholar] [CrossRef]
- Nomura, M.; Kinoshita, S.; Satoh, H.; Maeda, T.; Murakami, K.; Tsunoda, M.; Miyachi, H.; Awano, K. (3-Substituted benzyl) thiazolidine-2, 4-diones as structurally new antihyperglycemic agents. Bioorganic Med. Chem. Lett. 1999, 9, 533–538. [Google Scholar] [CrossRef]
- Harter, W.G.; Albrect, H.; Brady, K.; Caprathe, B.; Dunbar, J.; Gilmore, J.; Hays, S.; Kostlan, C.R.; Lunney, B.; Walker, N. The design and synthesis of sulfonamides as caspase-1 inhibitors. Bioorganic Med. Chem. Lett. 2004, 14, 809–812. [Google Scholar] [CrossRef]
- Kuhn, B.; Mohr, P.; Stahl, M. Intramolecular hydrogen bonding in medicinal chemistry. J. Med. Chem. 2010, 53, 2601–2611. [Google Scholar] [CrossRef]
- Lord, A.M.; Mahon, M.F.; Lloyd, M.D.; Threadgill, M.D. Design, synthesis, and evaluation in vitro of quinoline-8-carboxamides, a new class of poly (adenosine-diphosphate-ribose) polymerase-1 (PARP-1) inhibitor. J. Med. Chem. 2009, 52, 868–877. [Google Scholar] [CrossRef]
- Laurence, C.; Brameld, K.A.; Graton, J.; Le Questel, J.Y.; Renault, E. The p K BHX database: Toward a better understanding of hydrogen-bond basicity for medicinal chemists. J. Med. Chem. 2009, 52, 4073–4086. [Google Scholar] [CrossRef]
- Ferenczy, G.G.; Keseru, G.M. On the enthalpic preference of fragment binding. Med. Chem. Comm. 2016, 7, 332–337. [Google Scholar] [CrossRef]
- Schaller, D.; Šribar, D.; Noonan, T.; Deng, L.; Nguyen, T.N.; Pach, S.; Machalz, D.; Bermudez, M.; Wolber, G. Next generation 3D pharmacophore modeling. Wiley Interdiscip. Rev. Comput. Mol. Sci. 2020, 10, e1468. [Google Scholar] [CrossRef]
- Li, D.; Deng, Y.; Achab, A.; Bharathan, I.; Hopkins, B.A.; Yu, W.; Zhang, H.; Sanyal, S.; Pu, Q.; Zhou, H.; et al. Carbamate and n-pyrimidine mitigate amide hydrolysis: Structure-based drug design of tetrahydroquinoline ido1 inhibitors. ACS Med. Chem. Lett. 2021, 12, 389–396. [Google Scholar] [CrossRef]
- Steiner, T.; Saenger, W. Lengthening of the covalent O–H bond in O–H...O hydrogen bonds re-examined from low-temperature neutron diffraction data of organic compounds. Acta Crystallogr. Sect. Struct. Sci. 1994, 50, 348–357. [Google Scholar] [CrossRef]
- Stromgaard, K.; Krogsgaard-Larsen, P.; Madsen, U. Textbook of Drug Design and Discovery; CRC Press: Boca Raton, FL, USA, 2009. [Google Scholar]
- Velazquez-Campoy, A.; Freire, E. Incorporating target heterogeneity in drug design. J. Cell. Biochem. 2001, 84, 82–88. [Google Scholar] [CrossRef]
- Reynisson, J.; Mcdonald, E. Tuning of hydrogen bond strength using substituents on phenol and aniline: A possible ligand design strategy. J. Comput. Aided Mol. Des. 2004, 18, 421–431. [Google Scholar] [CrossRef]
- Taft, R.W.; Gurka, D.; Joris, L.; Schleyer, P.v.R.; Rakshys, J. Studies of hydrogen-bonded complex formation with p-fluorophenol. V. Linear free energy relationships with OH reference acids. J. Am. Chem. Soc. 1969, 91, 4801–4808. [Google Scholar] [CrossRef]
- Badger, R.M. A relation between internuclear distances and bond force constants. J. Chem. Phys. 1934, 2, 128–131. [Google Scholar] [CrossRef]
- Wilson, E.B.; Decius, J.C.; Cross, P.C. Molecular Vibrations: The Theory of Infrared and Raman Vibrational Spectra; Courier Corporation: Chelmsford, MA, USA, 1980. [Google Scholar]
- Kraka, E.; Larsson, J.A.; Cremer, D. Generalization of the Badger Rule Based on the Use of Adiabatic Vibrational Modes. In Computational Spectroscopy; Grunenberg, J., Ed.; Wiley: New York, NY, USA, 2010; pp. 105–149. [Google Scholar]
- Decius, J. Compliance matrix and molecular vibrations. J. Chem. Phys. 1963, 38, 241–248. [Google Scholar] [CrossRef]
- Jones, L.H.; Swanson, B.I. Interpretation of potential constants: Application to study of bonding forces in metal cyanide complexes and metal carbonyls. Acc. Chem. Res. 1976, 9, 128–134. [Google Scholar] [CrossRef]
- Konkoli, Z.; Larsson, J.A.; Cremer, D. A New Way of Analyzing Vibrational Spectra. II. Comparison of Internal Mode Frequencies. Int. J. Quantum Chem. 1998, 67, 11–27. [Google Scholar] [CrossRef]
- Konkoli, Z.; Cremer, D. A New Way of Analyzing Vibrational Spectra. III. Characterization of Normal Vibrational Modes in terms of Internal Vibrational Modes. Int. J. Quantum Chem. 1998, 67, 29–40. [Google Scholar] [CrossRef]
- Zou, W.; Cremer, D. C2 in a Box: Determining its Intrinsic Bond Strength for the X1Σ+g Ground State. Chem. Eur. J. 2016, 22, 4087–4097. [Google Scholar] [CrossRef] [PubMed]
- Wendler, K.; Thar, J.; Zahn, S.; Kirchner, B. Estimating the hydrogen bond energy. J. Phys. Chem. A 2010, 114, 9529–9536. [Google Scholar] [CrossRef]
- Xing, L.; Blakemore, D.C.; Narayanan, A.; Unwalla, R.; Lovering, F.; Denny, R.A.; Zhou, H.; Bunnage, M.E. Fluorine in drug design: A case study with fluoroanisoles. ChemMedChem 2015, 10, 715–726. [Google Scholar] [CrossRef]
- Hevey, R. The role of fluorine in glycomimetic drug design. Chem. Eur. J. 2021, 27, 2240–2253. [Google Scholar] [CrossRef] [PubMed]
- Juanes, M.; Saragi, R.T.; Pérez, C.; Evangelisti, L.; Enríquez, L.; Jaraíz, M.; Lesarri, A. Hydrogen Bonding in the Dimer and Monohydrate of 2-Adamantanol: A Test Case for Dispersion-Corrected Density Functional Methods. Molecules 2022, 27, 2584. [Google Scholar] [CrossRef]
- Juanes, M.; Usabiaga, I.; León, I.; Evangelisti, L.; Fernández, J.A.; Lesarri, A. The six isomers of the cyclohexanol dimer: A delicate test for dispersion models. Ed. Angew. Chem. Int. Ed. 2020, 59, 14081–14085. [Google Scholar] [CrossRef]
- Van Duijneveldt, F.B.; van Duijneveldt-van de Rijdt, J.G.; van Lenthe, J.H. State of the art in counterpoise theory. Chem. Rev. 1994, 94, 1873–1885. [Google Scholar] [CrossRef]
- Rezac, J. Non-covalent interactions atlas benchmark data sets 2: Hydrogen bonding in an extended chemical space. J. Chem. Theory Comput. 2020, 16, 6305–6316. [Google Scholar] [CrossRef] [PubMed]
- Ferrero, R.; Pantaleone, S.; Delle Piane, M.; Caldera, F.; Corno, M.; Trotta, F.; Brunella, V. On the interactions of melatonin/β-cyclodextrin inclusion complex: A novel approach combining efficient semiempirical extended tight-binding (xTB) results with ab initio methods. Molecules 2021, 26, 5881. [Google Scholar] [CrossRef]
- Ladbury, J.E. Isothermal titration calorimetry: Application to structure-based drug design. Thermochim. Acta 2001, 380, 209–215. [Google Scholar] [CrossRef]
- Wang, S.; Dong, G.; Sheng, C. Structural simplification: An efficient strategy in lead optimization. Acta Pharm. Sin. B 2019, 9, 880–901. [Google Scholar] [CrossRef]
- Manly, C.J.; Chandrasekhar, J.; Ochterski, J.W.; Hammer, J.D.; Warfield, B.B. Strategies and tactics for optimizing the Hit-to-Lead process and beyond—A computational chemistry perspective. Drug Discov. Today 2008, 13, 99–109. [Google Scholar] [CrossRef]
- Case, D.A.; Ben-Shalom, I.Y.; Brozell, S.R.; Cerutti, D.S.; Cheatham, T.E.; Cruzeiro, V.W.D.; Darden, T.A.; Duke, R.E.; Ghoreishi, D.; Gilson, M.K.; et al. AMBER16; University of California: San Francisco, CA, USA, 2016. [Google Scholar]
- Wang, J.; Wang, W.; Kollman, P.A.; Case, D.A. Antechamber: An accessory software package for molecular mechanical calculations. J. Am. Chem. Soc. 2001, 222, U403. [Google Scholar]
- Dennington, R.; Keith, T.A.; Millam, J.M. GaussView Version 6, 2019; Semichem Inc.: Shawnee, Kansas, 2019. [Google Scholar]
- Pettersen, E.F.; Goddard, T.D.; Huang, C.C.; Couch, G.S.; Greenblatt, D.M.; Meng, E.C.; Ferrin, T.E. UCSF Chimera—A visualization system for exploratory research and analysis. J. Comput. Chem. 2004, 25, 1605–1612. [Google Scholar] [CrossRef]
- Thiel, W. Semiempirical quantum–chemical methods. Wiley Interdiscip. Rev. Comput. Mol. Sci. 2014, 4, 145–157. [Google Scholar] [CrossRef]
- Yilmazer, N.D.; Korth, M. Enhanced semiempirical QM methods for biomolecular interactions. Comput. Struct. Biotechnol. J. 2015, 13, 169–175. [Google Scholar] [CrossRef]
- Bannwarth, C.; Ehlert, S.; Grimme, S. GFN2-xTB—An accurate and broadly parametrized self-consistent tight-binding quantum chemical method with multipole electrostatics and density-dependent dispersion contributions. J. Chem. Theory Comput. 2019, 15, 1652–1671. [Google Scholar] [CrossRef] [PubMed]
- Bannwarth, C.; Caldeweyher, E.; Ehlert, S.; Hansen, A.; Pracht, P.; Seibert, J.; Spicher, S.; Grimme, S. Extended tight-binding quantum chemistry methods. Wiley Interdiscip. Rev. Comput. Mol. Sci. 2021, 11, e1493. [Google Scholar] [CrossRef]
- Grimme, S.; Bannwarth, C.; Shushkov, P. A robust and accurate tight-binding quantum chemical method for structures, vibrational frequencies, and noncovalent interactions of large molecular systems parametrized for all spd-block elements (Z = 1–86). J. Chem. Theory Comput. 2017, 13, 1989–2009. [Google Scholar] [CrossRef]
- Xu, X.; Goddard, W.A. Bonding properties of the water dimer: A comparative study of density functional theories. J. Phys. Chem. A 2004, 108, 2305–2313. [Google Scholar] [CrossRef]
- Anderson, J.A.; Tschumper, G.S. Characterizing the potential energy surface of the water dimer with DFT: Failures of some popular functionals for hydrogen bonding. J. Phys. Chem. A 2006, 110, 7268–7271. [Google Scholar] [CrossRef] [PubMed]
- Grimme, S.; Ehrlich, S.; Goerigk, L. Effect of the damping function in dispersion corrected density functional theory. J. Comput. Chem. 2011, 32, 1456–1465. [Google Scholar] [CrossRef] [PubMed]
- Johnson, E.R.; Becke, A.D. A post-Hartree-Fock model of intermolecular interactions: Inclusion of higher-order corrections. J. Chem. Phys. 2006, 124, 174104. [Google Scholar] [PubMed]
- Dunning, T.H., Jr. Gaussian basis sets for use in correlated molecular calculations. I. The atoms boron through neon and hydrogen. J. Chem. Phys. 1989, 90, 1007–1023. [Google Scholar]
- Frisch, M.J.; Trucks, G.W.; Schlegel, H.B.; Scuseria, G.E.; Robb, M.A.; Cheeseman, J.R.; Scalmani, G.; Barone, V.; Petersson, G.A.; Nakatsuji, H.; et al. Gaussian˜16 Revision C.01, 2016; Gaussian Inc.: Wallingford, CT, USA, 2016. [Google Scholar]
- Konkoli, Z.; Larsson, J.A.; Cremer, D. A New Way of Analyzing Vibrational Spectra. IV. Application and Testing of Adiabatic Modes within the Concept of the Characterization of Normal Modes. Int. J. Quantum Chem. 1998, 67, 41–55. [Google Scholar]
- Cremer, D.; Larsson, J.A.; Kraka, E. New Developments in the Analysis of Vibrational Spectra on the Use of Adiabatic Internal Vibrational Modes. In Theoretical and Computational Chemistry; Parkanyi, C., Ed.; Elsevier: Amsterdam, The Netherlands, 1998; pp. 259–327. [Google Scholar]
- Kelley, J.D.; Leventhal, J.J. Problems in Classical and Quantum Mechanics: Normal Modes and Coordinates; Springer: Berlin/Heidelberg, Germany, 2017; pp. 95–117. [Google Scholar]
- Orville-Thomas, W. Vibrational states. J. Mol. Struct. 1977, 39, 155. [Google Scholar] [CrossRef]
- Pitzer, K. The nature of the chemical bond and the structure of molecules and crystals: An introduction to modern structural chemistry. J. Am. Chem. Soc. 1960, 82, 4121. [Google Scholar] [CrossRef]
- Delgado, A.A.A.; Humason, A.; Kraka, E. Pancake bonding seen through the eyes of spectroscopy. In Density Functional Theory—Recent Advances, New Perspectives and Applications; IntechOpen: London, UK, 2021; pp. 1–21. [Google Scholar]
- Delgado, A.A.A.; Humason, A.; Kalescky, R.; Freindorf, M.; Kraka, E. Exceptionally Long Covalent CC Bonds—A Local Vibrational Mode Study. Molecules 2021, 26, 950. [Google Scholar] [CrossRef] [PubMed]
- Kalescky, R.; Kraka, E.; Cremer, D. Identification of the Strongest Bonds in Chemistry. J. Phys. Chem. A 2013, 117, 8981–8995. [Google Scholar] [CrossRef] [PubMed]
- Kraka, E.; Cremer, D. Characterization of CF Bonds with Multiple-Bond Character: Bond Lengths, Stretching Force Constants, and Bond Dissociation Energies. ChemPhysChem 2009, 10, 686–698. [Google Scholar] [CrossRef]
- Kraka, E.; Setiawan, D.; Cremer, D. Re-Evaluation of the Bond Length-Bond Strength Rule: The Stronger Bond Is not Always the Shorter Bond. J. Comp. Chem. 2015, 37, 130–142. [Google Scholar] [CrossRef]
- Setiawan, D.; Sethio, D.; Cremer, D.; Kraka, E. From Strong to Weak NF Bonds: On the Design of a New Class of Fluorinating Agents. Phys. Chem. Chem. Phys. 2018, 20, 23913–23927. [Google Scholar] [CrossRef]
- Freindorf, M.; Yannacone, S.; Oliveira, V.; Verma, N.; Kraka, E. Halogen Bonding Involving I2 and d8 Transition-Metal Pincer Complexes. Crystals 2021, 11, 373. [Google Scholar] [CrossRef]
- Oliveira, V.; Kraka, E.; Cremer, D. The Intrinsic Strength of the Halogen Bond: Electrostatic and Covalent Contributions Described by Coupled Cluster Theory. Phys. Chem. Chem. Phys. 2016, 18, 33031–33046. [Google Scholar] [CrossRef]
- Oliveira, V.; Kraka, E.; Cremer, D. Quantitative Assessment of Halogen Bonding Utilizing Vibrational Spectroscopy. Inorg. Chem. 2016, 56, 488–502. [Google Scholar] [CrossRef]
- Oliveira, V.; Cremer, D. Transition from Metal-Ligand Bonding to Halogen Bonding Involving a Metal as Halogen Acceptor: A Study of Cu, Ag, Au, Pt, and Hg Complexes. Chem. Phys. Lett. 2017, 681, 56–63. [Google Scholar] [CrossRef]
- Yannacone, S.; Oliveira, V.; Verma, N.; Kraka, E. A Continuum from Halogen Bonds to Covalent Bonds: Where Do λ3 Iodanes Fit? Inorganics 2019, 7, 47. [Google Scholar] [CrossRef]
- Oliveira, V.P.; Marcial, B.L.; Machado, F.B.C.; Kraka, E. Metal-Halogen Bonding Seen through the Eyes of Vibrational Spectroscopy. Materials 2020, 13, 55. [Google Scholar] [CrossRef] [PubMed]
- Oliveira, V.; Cremer, D.; Kraka, E. The Many Facets of Chalcogen Bonding: Described by Vibrational Spectroscopy. J. Phys. Chem. A 2017, 121, 6845–6862. [Google Scholar] [CrossRef] [PubMed]
- Oliveira, V.; Kraka, E. Systematic Coupled Cluster Study of Noncovalent Interactions Involving Halogens, Chalcogens, and Pnicogens. J. Phys. Chem. A 2017, 121, 9544–9556. [Google Scholar] [CrossRef]
- Setiawan, D.; Kraka, E.; Cremer, D. Hidden Bond Anomalies: The Peculiar Case of the Fluorinated Amine Chalcogenides. J. Phys. Chem. A 2015, 119, 9541–9556. [Google Scholar] [CrossRef]
- Setiawan, D.; Kraka, E.; Cremer, D. Strength of the Pnicogen Bond in Complexes Involving Group VA Elements N, P, and As. J. Phys. Chem. A 2014, 119, 1642–1656. [Google Scholar] [CrossRef] [PubMed]
- Setiawan, D.; Kraka, E.; Cremer, D. Description of Pnicogen Bonding with the help of Vibrational Spectroscopy-The Missing Link Between Theory and Experiment. Chem. Phys. Lett. 2014, 614, 136–142. [Google Scholar] [CrossRef]
- Setiawan, D.; Cremer, D. Super-Pnicogen Bonding in the Radical Anion of the Fluorophosphine Dimer. Chem. Phys. Lett. 2016, 662, 182–187. [Google Scholar] [CrossRef]
- Sethio, D.; Oliveira, V.; Kraka, E. Quantitative Assessment of Tetrel Bonding Utilizing Vibrational Spectroscopy. Molecules 2018, 23, 2763-1–2763-21. [Google Scholar] [CrossRef]
- Freindorf, M.; Kraka, E.; Cremer, D. A Comprehensive Analysis of Hydrogen Bond Interactions Based on Local Vibrational Modes. Int. J. Quantum Chem. 2012, 112, 3174–3187. [Google Scholar] [CrossRef]
- Kalescky, R.; Zou, W.; Kraka, E.; Cremer, D. Local Vibrational Modes of the Water Dimer—Comparison of Theory and Experiment. Chem. Phys. Lett. 2012, 554, 243–247. [Google Scholar] [CrossRef]
- Kalescky, R.; Kraka, E.; Cremer, D. Local Vibrational Modes of the Formic Acid Dimer—The Strength of the Double H-Bond. Mol. Phys. 2013, 111, 1497–1510. [Google Scholar] [CrossRef]
- Tao, Y.; Zou, W.; Jia, J.; Li, W.; Cremer, D. Different Ways of Hydrogen Bonding in Water—Why Does Warm Water Freeze Faster than Cold Water? J. Chem. Theory Comput. 2017, 13, 55–76. [Google Scholar] [CrossRef] [PubMed]
- Tao, Y.; Zou, W.; Kraka, E. Strengthening of Hydrogen Bonding With the Push-Pull Effect. Chem. Phys. Lett. 2017, 685, 251–258. [Google Scholar] [CrossRef]
- Makoś, M.Z.; Freindorf, M.; Sethio, D.; Kraka, E. New Insights into Fe–H2 and Fe–H- Bonding of a [NiFe] Hydrogenase Mimic—A Local Vibrational Mode Study. Theor. Chem. Acc. 2019, 138, 76. [Google Scholar] [CrossRef]
- Lyu, S.; Beiranvand, N.; Freindorf, M.; Kraka, E. Interplay of Ring Puckering and Hydrogen Bonding in Deoxyribonucleosides. J. Phys. Chem. A 2019, 123, 7087–7103. [Google Scholar] [CrossRef] [PubMed]
- Beiranvand, N.; Freindorf, M.; Kraka, E. Hydrogen Bonding in Natural and Unnatural Base Pairs-Explored with Vibrational Spectroscopy. Molecules 2021, 26, 2268. [Google Scholar] [CrossRef] [PubMed]
- Cremer, D.; Thiel, W. On the Importance of Size-Consistency Corrections in Semiempirical MNDOC Calculations. J. Comput. Chem. 1987, 8, 48–50. [Google Scholar] [CrossRef]
- Moura, R.T.J.; Quintano, M.; Antonio, J.J.; Freindorf, M.; Kraka, E. Automatic Generation of Local Vibrational Mode Parameters: From Small to Large Molecules and QM/MM Systems. J. Phys. Chem. A 2022, 126, 9313–9331. [Google Scholar] [CrossRef]
- Madushanka, A.; Verma, N.; Freindorf, M.; Kraka, E. Papaya Leaf Extracts as Potential Dengue Treatment: An In-Silico Study. Int. J. Mol. Sci. 2022, 23, 12310. [Google Scholar] [CrossRef]
- Freindorf, M.; Delgado, A.A.A.; Kraka, E. CO Bonding in Hexa– and Pentacoordinate Carboxy–Neuroglobin – A QM/MM and Local Vibrational Mode Study. J. Comp. Chem. 2022, 43, 1725–1746. [Google Scholar] [CrossRef]
- Neto, A.N.C.; Jr, R.T.M.; Carlos, L.D.; Malta, O.L.; Sanadar, M.; Melchior, A.; Kraka, E.; Ruggieri, S.; Bettinelli, M.; Piccinelli, F. Dynamics of the Energy Transfer Process in Eu(III) Complexes Containing Polydentate Ligands Based on Pyridine, Quinoline, and Isoquinoline as Chromophoric Antennae. Inorg. Chem. 2022, 61, 16333–16346. [Google Scholar] [CrossRef] [PubMed]
- Moura, R.T.; Quintano, M.; Santos, C.V., Jr.; Albuquerque, V.A.; Aguiar, E.C.; Kraka, E.; Carneiro Neto, A.N. Featuring a new computational protocol for the estimation of intensity and overall quantum yield in lanthanide chelates with applications to Eu(III) mercapto-triazole Schiff base ligands. Opt. Mater. X 2022, 16, 100216. [Google Scholar] [CrossRef]
- Delgado, A.A.A.; Sethio, D.; Matthews, D.; Oliveira, V.; Kraka, E. Substituted hydrocarbon: A CCSD(T) and local vibrational mode investigation. Mol. Phys. 2021, 119, e1970844. [Google Scholar] [CrossRef]
- Kraka, E.; Freindorf, M. Characterizing the Metal Ligand Bond Strength via Vibrational Spectroscopy: The Metal Ligand Electronic Parameter (MLEP). New Dir. Model. Organomet. React. 2020, 67, 1–43. [Google Scholar]
- Cremer, D.; Kraka, E. Generalization of the Tolman Electronic Parameter: The Metal-Ligand Electronic Parameter and the Intrinsic Strength of the Metal-Ligand Bond. Dalton Trans. 2017, 46, 8323–8338. [Google Scholar] [CrossRef] [PubMed]
- Setiawan, D.; Kalescky, R.; Kraka, E.; Cremer, D. Direct Measure of Metal-Ligand Bonding Replacing the Tolman Electronic Parameter. Inorg. Chem. 2016, 55, 2332–2344. [Google Scholar] [CrossRef]
- Peluzo, B.M.T.C.; Kraka, E. Uranium: The Nuclear Fuel Cycle and Beyond. Int. J. Mol. Sci. 2022, 23, 4655. [Google Scholar] [CrossRef]
- Kalescky, R.; Kraka, E.; Cremer, D. New Approach to Tolman’s Electronic Parameter Based on Local Vibrational Modes. Inorg. Chem. 2013, 53, 478–495. [Google Scholar] [CrossRef]
- Quintano, M.; Kraka, E. Theoretical insights into the linear relationship between pKa values and vibrational frequencies. Chem. Phys. Lett. 2022, 803, 139746. [Google Scholar] [CrossRef]
- Verma, N.; Tao, Y.; Zou, W.; Chen, X.; Chen, X.; Freindorf, M.; Kraka, E. A Critical Evaluation of Vibrational Stark Effect (VSE) Probes with the Local Vibrational Mode Theory. Sensors 2020, 20, 2358. [Google Scholar] [CrossRef] [PubMed]
- Quintano, M.; Delgado, A.A.A.; Moura, R.T., Jr.; Freindorf, M.; Kraka, E. Local mode analysis of characteristic vibrational coupling in nucleobases and Watson–Crick base pairs of DNA. Electron. Struct. 2022, 4, 044005. [Google Scholar] [CrossRef]
- Zou, W.; Tao, Y.; Freindorf, M.; Makoś, M.Z.; Verma, N.; Cremer, D.; Kraka, E. Local Vibrational Mode Analysis (LMode A). In Computational and Theoretical Chemistry Group (CATCO); Southern Methodist University: Dallas, TX, USA, 2022. [Google Scholar]
















{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Count | Donor ID | Acceptor ID | HB Type | Length (Å) | Angle (°) | Acceptor Force Constants (mDyn/Å) | Donor Force Constants (mDyn/Å) | Intra. HB | HB Network | Donor Residue | Acceptor Residue | Ligand Functional Group |
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| 1 | O | O | O–H⋯O | 1.6836 | 162.37 | 0.311 | 4.352 | None | 1-0-1 | MOL | GLU | OH |
| 2 | N | O | N–H⋯O | 1.9043 | 154.20 | 0.205 | 6.073 | None | 0-0-0 | TRP | MOL | OH |
| 3 | N | O | N–H⋯O | 1.8549 | 156.11 | 0.215 | 5.387 | None | 0-0-0 | ARG | MOL | OH |
| 4 | N | O | N–H⋯O | 1.8564 | 175.16 | 0.261 | 6.052 | None | 0-0-0 | ARG | MOL | CO |
| 5 | O | O | O–H⋯O | 1.8255 | 163.02 | 0.193 | 6.336 | None | 1-0-2 | WAT | MOL | OH |
| 6 | O | O | O–H⋯O | 1.8745 | 175.04 | 0.184 | 6.506 | None | 1-0-2 | WAT | MOL | OH |
| 7 | O | O | O–H⋯O | 1.8462 | 163.27 | 0.202 | 6.231 | None | 1-0-2 | MOL | WAT | OH |
| 8 | O | O | O–H⋯O | 1.6690 | 172.92 | 0.329 | 4.644 | None | 1-0-2 | MOL | WAT | OH |
| D-H⋯A (HB Type) | HB Count | < Length > (Å) | < Angle > (°) | < k > (mDyn/Å) |
|---|---|---|---|---|
| N–H⋯O | 1304 | 1.8598 | 159 | 0.219 |
| O–H⋯O | 774 | 1.8150 | 163 | 0.234 |
| N–H⋯N | 95 | 1.9310 | 161 | 0.195 |
| O–H⋯N | 72 | 1.8993 | 161 | 0.171 |
| Level of Theory | N–H⋯O | Length | O–H⋯O | Length | N–H⋯N | Length | O–H⋯N | Length |
|---|---|---|---|---|---|---|---|---|
| XTB/GFN2-xTB | 0.069 | 2.1202 | 0.192 | 1.8854 | 0.093 | 2.0980 | 0.206 | 1.8809 |
| B3LYP-D3(BJ)/aug-cc-pVDZ | 0.013 | 3.3389 | 0.166 | 1.9272 | 0.100 | 2.2428 | 0.201 | 1.9334 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Madushanka, A.; Moura, R.T., Jr.; Verma, N.; Kraka, E. Quantum Mechanical Assessment of Protein–Ligand Hydrogen Bond Strength Patterns: Insights from Semiempirical Tight-Binding and Local Vibrational Mode Theory. Int. J. Mol. Sci. 2023, 24, 6311. https://doi.org/10.3390/ijms24076311
Madushanka A, Moura RT Jr., Verma N, Kraka E. Quantum Mechanical Assessment of Protein–Ligand Hydrogen Bond Strength Patterns: Insights from Semiempirical Tight-Binding and Local Vibrational Mode Theory. International Journal of Molecular Sciences. 2023; 24(7):6311. https://doi.org/10.3390/ijms24076311
Chicago/Turabian StyleMadushanka, Ayesh, Renaldo T. Moura, Jr., Niraj Verma, and Elfi Kraka. 2023. "Quantum Mechanical Assessment of Protein–Ligand Hydrogen Bond Strength Patterns: Insights from Semiempirical Tight-Binding and Local Vibrational Mode Theory" International Journal of Molecular Sciences 24, no. 7: 6311. https://doi.org/10.3390/ijms24076311
APA StyleMadushanka, A., Moura, R. T., Jr., Verma, N., & Kraka, E. (2023). Quantum Mechanical Assessment of Protein–Ligand Hydrogen Bond Strength Patterns: Insights from Semiempirical Tight-Binding and Local Vibrational Mode Theory. International Journal of Molecular Sciences, 24(7), 6311. https://doi.org/10.3390/ijms24076311