
Interactions between Malassezia and New Therapeutic Agents in Atopic Dermatitis Affecting Skin Barrier and Inflammation in Recombinant Human Epidermis Model

 , , and
, , and 
Abstract
1. Introduction
2. Results
2.1. Variation of Tissue Morphology of the AD-RHE Stimulated with Malassezia Restricta (MR)
2.2. Expression of Epidermal Skin Barrier and Antimicrobial Peptide-Related Molecules in AD-RHE Stimulated with Malassezia Restricta (MR)
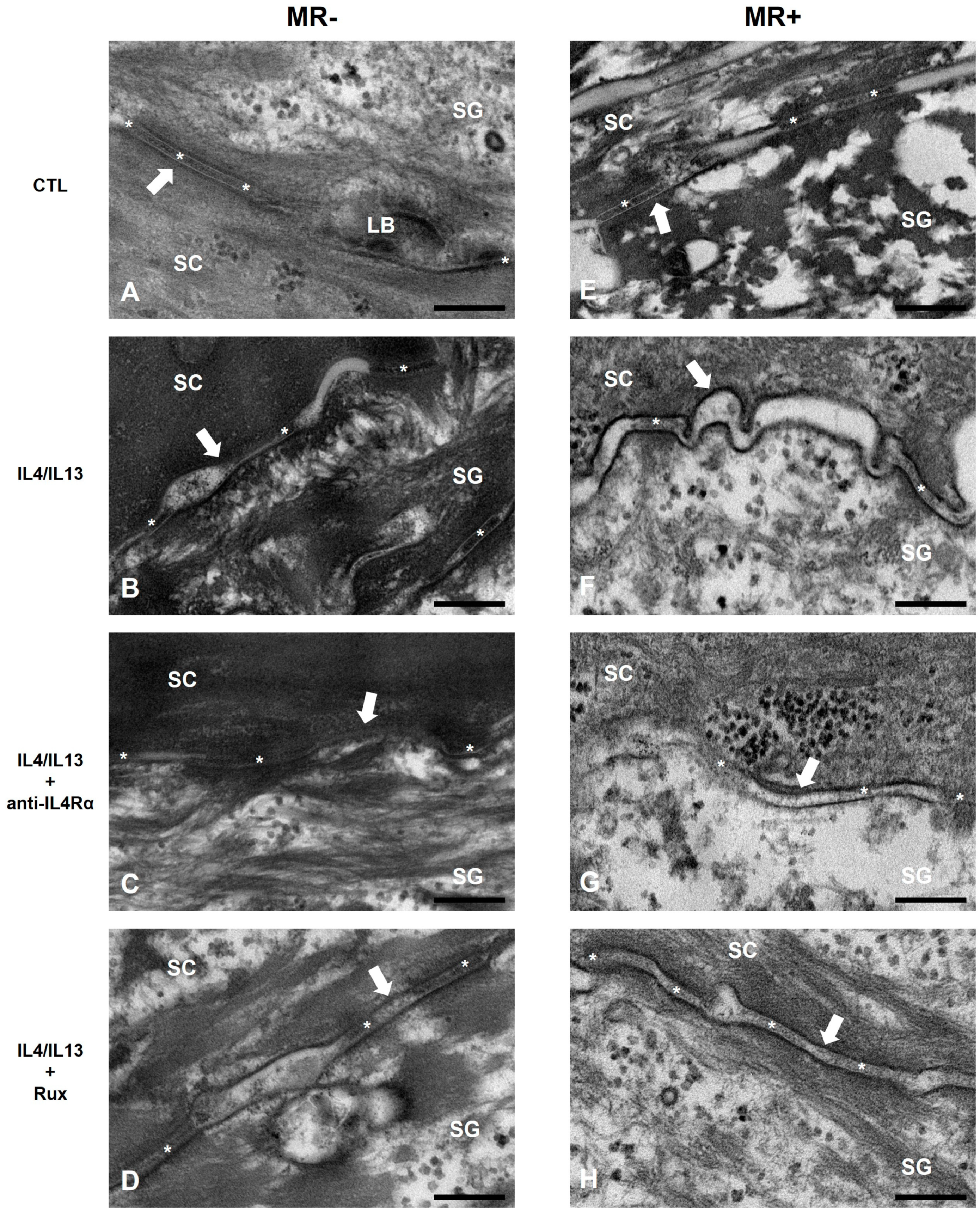
2.3. Epidermal Permeability Barrier of the AD-RHE Stimulated with Malassezia Restricta (MR)
2.4. Expression of Genes Related to Th1, Th2, Th17 Inflammation in AD-RHE Stimulated with Malassezia Restricta (MR)
2.5. Protein Expression of Th2- and Th17-Related Molecules in AD-RHE Stimulated with Malassezia Restricta (MR)
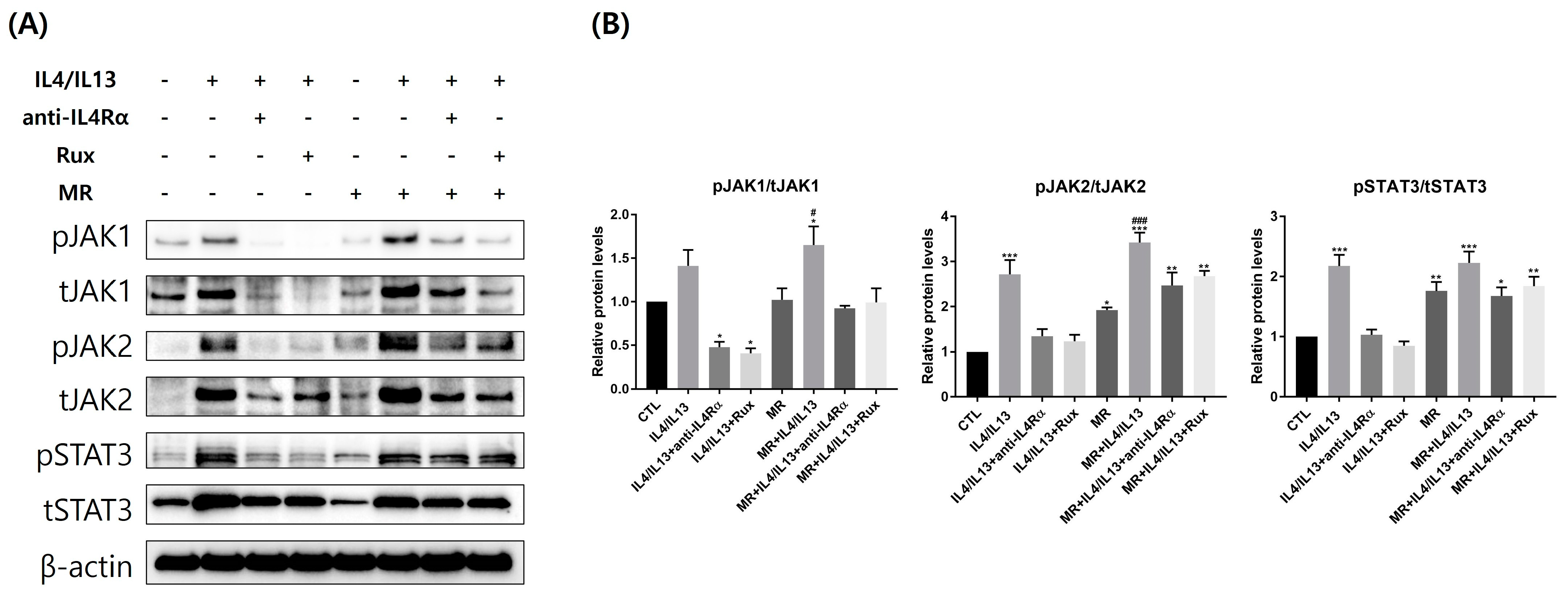
2.6. Protein Expression of JAK/STAT pathway-Related Molecules in AD-RHE Stimulated with Malassezia Restricta (MR)
3. Discussion
4. Materials and Methods
4.1. Malassezia Culture
4.2. HaCaT Cell Culture
4.3. Setting Up an In Vitro 2D AD Model
4.4. 3D Skin AD Modeling
4.5. Cell Viability Assay
4.6. Real-Time PCR
4.7. Western Blot Analysis
4.8. H&E Staining and Immunofluorescent Staining
4.9. Transmission Electron Microscopy (TEM)
4.10. Statistical Analysis
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Lee, H.-J.; Lee, S.-H. Epidermal permeability barrier defects and barrier repair therapy in atopic dermatitis. Allergy Asthma Immunol. Res. 2014, 6, 276–287. [Google Scholar] [CrossRef]
- Wickett, R.R.; Visscher, M.O. Structure and function of the epidermal barrier. Am. J. Infect. Control 2006, 34, S98–S110. [Google Scholar] [CrossRef]
- Elias, P.M.; Choi, E.H. Interactions among stratum corneum defensive functions. Exp. Dermatol. 2005, 14, 719–726. [Google Scholar] [CrossRef]
- Kim, J.E.; Kim, J.S.; Cho, D.H.; Park, H.J. Molecular mechanisms of cutaneous inflammatory disorder: Atopic dermatitis. Int. J. Mol. Sci. 2016, 17, 1234. [Google Scholar] [CrossRef]
- Mao-Qiang, M.; Fowler, A.J.; Schmuth, M.; Lau, P.; Chang, S.; Brown, B.E.; Moser, A.H.; Michalik, L.; Desvergne, B.; Wahli, W. Peroxisome-proliferator-activated receptor (PPAR)-γ activation stimulates keratinocyte differentiation. J. Investig. Dermatol. 2004, 123, 305–312. [Google Scholar]
- Cork, M.J.; Danby, S.; Vasilopoulos, Y.; Moustafa, M.; MacGowan, A.; Varghese, J.; Duff, G.W.; Tazi-Ahnini, R.; Ward, S.J. Epidermal barrier dysfunction in atopic dermatitis. In Textbook of Atopic Dermatitis; CRC Press: Boca Raton, FL, USA, 2008; pp. 47–70. [Google Scholar]
- Haftek, M. Epidermal barrier disorders and corneodesmosome defects. Cell Tissue Res. 2015, 360, 483–490. [Google Scholar] [CrossRef]
- Huan, Y.; Kong, Q.; Mou, H.; Yi, H. Antimicrobial peptides: Classification, design, application and research progress in multiple fields. Front. Microbiol. 2020, 11, 582779. [Google Scholar] [CrossRef]
- Nguyen, H.L.T.; Trujillo-Paez, J.V.; Umehara, Y.; Yue, H.; Peng, G.; Kiatsurayanon, C.; Chieosilapatham, P.; Song, P.; Okumura, K.; Ogawa, H. Role of antimicrobial peptides in skin barrier repair in individuals with atopic dermatitis. Int. J. Mol. Sci. 2020, 21, 7607. [Google Scholar] [CrossRef]
- Glatz, M.; Bosshard, P.P.; Hoetzenecker, W.; Schmid-Grendelmeier, P. The role of Malassezia spp. in atopic dermatitis. J. Clin. Med. 2015, 4, 1217–1228. [Google Scholar]
- Hobi, S.; Cafarchia, C.; Romano, V.; Barrs, V.R. Malassezia: Zoonotic Implications, Parallels and Differences in Colonization and Disease in Humans and Animals. J. Fungi 2022, 8, 708. [Google Scholar] [CrossRef]
- Gupta, A.K.; Batra, R.; Bluhm, R.; Boekhout, T.; Dawson, T.L., Jr. Skin diseases associated with Malassezia species. J. Am. Acad. Dermatol. 2004, 51, 785–798. [Google Scholar] [CrossRef]
- White, T.C.; Findley, K.; Dawson, T.L.; Scheynius, A.; Boekhout, T.; Cuomo, C.A.; Xu, J.; Saunders, C.W. Fungi on the skin: Dermatophytes and Malassezia. Cold Spring Harb. Perspect. Med. 2014, 4, a019802. [Google Scholar] [CrossRef]
- Park, H.R.; Oh, J.H.; Lee, Y.J.; Park, S.H.; Lee, Y.W.; Lee, S.; Kang, H.; Kim, J.E. Inflammasome-mediated Inflammation by Malassezia in human keratinocytes: A comparative analysis with different strains. Mycoses 2021, 64, 292–299. [Google Scholar] [CrossRef]
- Sparber, F.; LeibundGut-Landmann, S. Host responses to Malassezia spp. in the mammalian skin. Front. Immunol. 2017, 8, 1614. [Google Scholar] [CrossRef]
- Später, S.; Hipler, U.-C.; Haustein, U.-F.; Nenoff, P. Generation of reactive oxygen species in vitro by Malassezia yeasts. Der Hautarzt 2009, 60, 122–127. [Google Scholar] [CrossRef] [PubMed]
- Sparber, F.; Ruchti, F.; LeibundGut-Landmann, S. Host immunity to Malassezia in health and disease. Front. Cell. Infect. Microbiol. 2020, 10, 198. [Google Scholar] [CrossRef]
- Saunte, D.M.; Gaitanis, G.; Hay, R.J. Malassezia-associated skin diseases, the use of diagnostics and treatment. Front. Cell. Infect. Microbiol. 2020, 10, 112. [Google Scholar] [CrossRef] [PubMed]
- Prohic, A.; Jovovic Sadikovic, T.; Krupalija-Fazlic, M.; Kuskunovic-Vlahovljak, S. Malassezia species in healthy skin and in dermatological conditions. Int. J. Dermatol. 2016, 55, 494–504. [Google Scholar] [CrossRef]
- Brandt, E.B.; Sivaprasad, U. Th2 cytokines and atopic dermatitis. J. Clin. Cell. Immunol. 2011, 2, 110. [Google Scholar] [CrossRef]
- Yamanaka, K.-i.; Mizutani, H. The role of cytokines/chemokines in the pathogenesis of atopic dermatitis. Pathog. Manag. Atopic Dermat. 2011, 41, 80–92. [Google Scholar]
- Bao, L.; Zhang, H.; Chan, L.S. The involvement of the JAK-STAT signaling pathway in chronic inflammatory skin disease atopic dermatitis. Jak-Stat 2013, 2, e24137. [Google Scholar] [CrossRef]
- Welsch, K.; Holstein, J.; Laurence, A.; Ghoreschi, K. Targeting JAK/STAT signalling in inflammatory skin diseases with small molecule inhibitors. Eur. J. Immunol. 2017, 47, 1096–1107. [Google Scholar] [CrossRef]
- Huang, I.-H.; Chung, W.-H.; Wu, P.-C.; Chen, C.-B. JAK–STAT signaling pathway in the pathogenesis of atopic dermatitis: An updated review. Front. Immunol. 2022, 13, 1068260. [Google Scholar] [CrossRef]
- Lees, J.R. Interferon gamma in autoimmunity: A complicated player on a complex stage. Cytokine 2015, 74, 18–26. [Google Scholar] [CrossRef] [PubMed]
- Kim, J.; Kim, B.E.; Leung, D.Y. Pathophysiology of atopic dermatitis: Clinical implications. Allergy Asthma 2019, 40, 84–92. [Google Scholar] [CrossRef]
- Cua, D.J.; Tato, C.M. Innate IL-17-producing cells: The sentinels of the immune system. Nat. Rev. Immunol. 2010, 10, 479–489. [Google Scholar] [CrossRef] [PubMed]
- Conti, H.R.; Shen, F.; Nayyar, N.; Stocum, E.; Sun, J.N.; Lindemann, M.J.; Ho, A.W.; Hai, J.H.; Yu, J.J.; Jung, J.W. Th17 cells and IL-17 receptor signaling are essential for mucosal host defense against oral candidiasis. J. Exp. Med. 2009, 206, 299–311. [Google Scholar] [CrossRef] [PubMed]
- Sugaya, M. The role of Th17-related cytokines in atopic dermatitis. Int. J. Mol. Sci. 2020, 21, 1314. [Google Scholar] [CrossRef] [PubMed]
- Junttila, I.S. Tuning the cytokine responses: An update on interleukin (IL)-4 and IL-13 receptor complexes. Front. Immunol. 2018, 9, 888. [Google Scholar] [CrossRef]
- Le Floc’h, A.; Allinne, J.; Nagashima, K.; Scott, G.; Birchard, D.; Asrat, S.; Bai, Y.; Lim, W.K.; Martin, J.; Huang, T. Dual blockade of IL-4 and IL-13 with dupilumab, an IL-4Rα antibody, is required to broadly inhibit type 2 inflammation. Allergy 2020, 75, 1188–1204. [Google Scholar] [CrossRef]
- Nguyen, H.L.; Anderson, K.R.; Tollefson, M.M. New and emerging therapies for pediatric atopic dermatitis. Pediatr. Drugs 2019, 21, 239–260. [Google Scholar] [CrossRef] [PubMed]
- Soria, A.; Du-Thanh, A.; Seneschal, J.; Jachiet, M.; Staumont-Sallé, D.; Barbarot, S.; GREAT Research Group. Development or exacerbation of head and neck dermatitis in patients treated for atopic dermatitis with dupilumab. JAMA Dermatol. 2019, 155, 1312–1315. [Google Scholar] [CrossRef]
- Chen, L.; Shao, J.; Jiang, F.; Liu, J.; Zhiming, L. Dupilumab with Concomitant JAK Inhibitor: A Novel Strategy for Atopic Dermatitis with Poor Response to Dupilumab. Br. J. Dermatol. 2022, 187, 828–830. [Google Scholar]
- De Vuyst, E.; Salmon, M.; Evrard, C.; Lambert de Rouvroit, C.; Poumay, Y. Atopic dermatitis studies through in vitro models. Front. Med. 2017, 4, 119. [Google Scholar] [CrossRef]
- Bieber, T. Atopic Dermatitis. N. Engl. J. Med. 2008, 358, 1483–1494. [Google Scholar] [CrossRef]
- Girolomoni, G.; de Bruin-Weller, M.; Aoki, V.; Kabashima, K.; Deleuran, M.; Puig, L.; Bansal, A.; Rossi, A.B. Nomenclature and clinical phenotypes of atopic dermatitis. Ther. Adv. Chronic Dis. 2021, 12, 20406223211002979. [Google Scholar] [CrossRef] [PubMed]
- Kim, B.E.; Leung, D.Y. Significance of skin barrier dysfunction in atopic dermatitis. Allergy Asthma Immunol. Res. 2018, 10, 207–215. [Google Scholar] [CrossRef]
- Furue, M. Regulation of filaggrin, loricrin, and involucrin by IL-4, IL-13, IL-17A, IL-22, AHR, and NRF2: Pathogenic implications in atopic dermatitis. Int. J. Mol. Sci. 2020, 21, 5382. [Google Scholar] [CrossRef]
- Petrova, A.; Celli, A.; Jacquet, L.; Dafou, D.; Crumrine, D.; Hupe, M.; Arno, M.; Hobbs, C.; Cvoro, A.; Karagiannis, P. 3D in vitro model of a functional epidermal permeability barrier from human embryonic stem cells and induced pluripotent stem cells. Stem Cell Rep. 2014, 2, 675–689. [Google Scholar] [CrossRef]
- Van Smeden, J.; Bouwstra, J.A. Stratum corneum lipids: Their role for the skin barrier function in healthy subjects and atopic dermatitis patients. Ski. Barrier Funct. 2016, 49, 8–26. [Google Scholar]
- Roger, M.; Fullard, N.; Costello, L.; Bradbury, S.; Markiewicz, E.; O’Reilly, S.; Darling, N.; Ritchie, P.; Määttä, A.; Karakesisoglou, I. Bioengineering the microanatomy of human skin. J. Anat. 2019, 234, 438–455. [Google Scholar] [CrossRef]
- Clausen, M.-L.; Slotved, H.-C.; Krogfelt, K.A.; Agner, T. Measurements of AMPs in stratum corneum of atopic dermatitis and healthy skin–tape stripping technique. Sci. Rep. 2018, 8, 1666. [Google Scholar] [CrossRef]
- Ballardini, N.; Johansson, C.; Lilja, G.; Lindh, M.; Linde, Y.; Scheynius, A.; Agerberth, B. Enhanced expression of the antimicrobial peptide LL-37 in lesional skin of adults with atopic eczema. Br. J. Dermatol. 2009, 161, 40–47. [Google Scholar] [CrossRef]
- Harder, J.; Dressel, S.; Wittersheim, M.; Cordes, J.; Meyer-Hoffert, U.; Mrowietz, U.; Fölster-Holst, R.; Proksch, E.; Schröder, J.-M.; Schwarz, T. Enhanced expression and secretion of antimicrobial peptides in atopic dermatitis and after superficial skin injury. J. Investig. Dermatol. 2010, 130, 1355–1364. [Google Scholar] [CrossRef] [PubMed]
- Rademacher, F.; Gläser, R.; Harder, J. Antimicrobial peptides and proteins: Interaction with the skin microbiota. Exp. Dermatol. 2021, 30, 1496–1508. [Google Scholar] [CrossRef]
- Kubicek-Sutherland, J.Z.; Lofton, H.; Vestergaard, M.; Hjort, K.; Ingmer, H.; Andersson, D.I. Antimicrobial peptide exposure selects for Staphylococcus aureus resistance to human defence peptides. J. Antimicrob. Chemother. 2016, 72, 115–127. [Google Scholar] [CrossRef]
- Steinstraesser, L.; Kraneburg, U.; Jacobsen, F.; Al-Benna, S. Host defense peptides and their antimicrobial-immunomodulatory duality. Immunobiology 2011, 216, 322–333. [Google Scholar] [CrossRef] [PubMed]
- Pfalzgraff, A.; Brandenburg, K.; Weindl, G. Antimicrobial peptides and their therapeutic potential for bacterial skin infections and wounds. Front. Pharmacol. 2018, 9, 281. [Google Scholar] [CrossRef] [PubMed]
- Lee, H.J.; Hong, Y.J.; Kim, M. Angiogenesis in chronic inflammatory skin disorders. Int. J. Mol. Sci. 2021, 22, 12035. [Google Scholar] [CrossRef]
- Kortekaas Krohn, I.; Aerts, J.L.; Breckpot, K.; Goyvaerts, C.; Knol, E.; Van Wijk, F.; Gutermuth, J. T-cell subsets in the skin and their role in inflammatory skin disorders. Allergy 2022, 77, 827–842. [Google Scholar] [CrossRef]
- Chang, K.-T.; Lin, H.Y.-H.; Kuo, C.-H.; Hung, C.-H. Tacrolimus suppresses atopic dermatitis-associated cytokines and chemokines in monocytes. J. Microbiol. Immunol. Infect. 2016, 49, 409–416. [Google Scholar] [CrossRef]
- Jovanovic, D.V.; Di Battista, J.A.; Martel-Pelletier, J.; Jolicoeur, F.C.; He, Y.; Zhang, M.; Mineau, F.; Pelletier, J.-P. IL-17 stimulates the production and expression of proinflammatory cytokines, IL-β and TNF-α, by human macrophages. J. Immunol. 1998, 160, 3513–3521. [Google Scholar] [CrossRef]
- Glatz, M.; Jo, J.-H.; Kennedy, E.A.; Polley, E.C.; Segre, J.A.; Simpson, E.L.; Kong, H.H. Emollient use alters skin barrier and microbes in infants at risk for developing atopic dermatitis. PloS ONE 2018, 13, e0192443. [Google Scholar] [CrossRef]
- Sparber, F.; De Gregorio, C.; Steckholzer, S.; Ferreira, F.M.; Dolowschiak, T.; Ruchti, F.; Kirchner, F.R.; Mertens, S.; Prinz, I.; Joller, N. The skin commensal yeast Malassezia triggers a type 17 response that coordinates anti-fungal immunity and exacerbates skin inflammation. Cell Host Microbe 2019, 25, 389–403.e386. [Google Scholar] [CrossRef]
- Tajima, M.; Sugita, T.; Nishikawa, A.; Tsuboi, R. Molecular analysis of Malassezia microflora in seborrheic dermatitis patients: Comparison with other diseases and healthy subjects. J. Investig. Dermatol. 2008, 128, 345–351. [Google Scholar] [CrossRef] [PubMed]
- Darabi, K.; Hostetler, S.G.; Bechtel, M.A.; Zirwas, M. The role of Malassezia in atopic dermatitis affecting the head and neck of adults. J. Am. Acad. Dermatol. 2009, 60, 125–136. [Google Scholar] [CrossRef] [PubMed]
- Jain, C.; Das, S.; Ramachandran, V.; Saha, R.; Bhattacharya, S.; Dar, S. Malassezia yeast and cytokine gene polymorphism in atopic dermatitis. J. Clin. Diagn. Res. JCDR 2017, 11, DC01. [Google Scholar] [CrossRef]
- Furue, M. Regulation of skin barrier function via competition between AHR axis versus IL-13/IL-4-JAK-STAT6/STAT3 axis: Pathogenic and therapeutic implications in atopic dermatitis. J. Clin. Med. 2020, 9, 3741. [Google Scholar] [CrossRef] [PubMed]
- Clarysse, K.; Pfaff, C.; Marquardt, Y.; Huth, L.; Kortekaas Krohn, I.; Kluwig, D.; Lüscher, B.; Gutermuth, J.; Baron, J. JAK1/3 inhibition preserves epidermal morphology in full-thickness 3D skin models of atopic dermatitis and psoriasis. J. Eur. Acad. Dermatol. Venereol. 2019, 33, 367–375. [Google Scholar] [CrossRef] [PubMed]
- Park, M.; Cho, Y.J.; Lee, Y.W.; Jung, W.H. Whole genome sequencing analysis of the cutaneous pathogenic yeast Malassezia restricta and identification of the major lipase expressed on the scalp of patients with dandruff. Mycoses 2017, 60, 188–197. [Google Scholar] [CrossRef]
- Leeming, J.P.; Notman, F.H. Improved methods for isolation and enumeration of Malassezia furfur from human skin. J. Clin. Microbiol. 1987, 25, 2017–2019. [Google Scholar] [CrossRef] [PubMed]
- Mukundan, A.; Tsao, Y.-M.; Artemkina, S.B.; Fedorov, V.E.; Wang, H.-C. Growth mechanism of periodic-structured MoS2 by transmission electron microscopy. Nanomaterials 2021, 12, 135. [Google Scholar] [CrossRef] [PubMed]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Normal Skin IL-4/IL-13 (-) | Barrier Disrupted Skin IL-4/IL-13 (+) | Barrier Recovered Skin IL-4/IL-13 (+) with anti-IL4Rα or Ruxolitinib | ||||
|---|---|---|---|---|---|---|
| With M. restricta vs. Without M. restricta | MR (-) | MR (+) | MR (-) # | MR (+) | MR (-) † | MR (+) |
| Skin barrier molecules | FLG, LOR, IVL | LOR↓ | FLG↓ LOR, IVL↓↓ | ↓ * | FLG, LOR↑ IVL↑↑ | LOR, IVL↓ |
| Lipid synthesis | CERS3, ELOVL1 | CERS3↓ | ↓↓ | ↓ * | ↑↑ | CERS3↓↓, ELOVL1↓ |
| Vascular | VEGF | ↑ | ↑↑ | ↑ | ↓↓ | ↑ |
| Innate immunity | IL-1β IL-17, IL-22 LL-37, β-D2 | IL-1β↑ IL-17↑ LL-37, β-D2↑↑ | IL-1β↑↑ IL-17, IL-22↑↑ β-D2↑ | IL-1β↑ * IL-17↑ * LL-37, β-D2↑ * | IL-1β↓ IL-17, IL-22↓↓ LL-37↑ | IL-1β↑ IL-17↑ LL-37, β-D2↑ |
| Proinflammatory cytokines and chemokines | IL-4, TSLP IL-1α, TNF-α CCL17, CCL22 | IL-4, TSLP↑↑ IL-1α↑↑, TNF-α↑ CCL17↑, CCL22↑↑ | ↑↑ | IL-4, TSLP↑ * CCL17, CCL22↑ * TNF-α↑ | IL-4, TSLP↓↓ IL-1α↓↓, TNF-α↓ CCL17↓, CCL22↓↓ | IL-4↑, TSLP↑↑ |
| JAK/STAT pathway | JAK1, JAK2, STAT3 | JAK2, STAT3↑ | JAK2, STAT3↑↑ | ↑ * | JAK1, JAK2↓ STAT3↓↓ | ↑ |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Lee, Y.-J.; Yassa, C.; Park, S.-H.; Song, S.W.; Jung, W.H.; Lee, Y.W.; Kang, H.; Kim, J.-E. Interactions between Malassezia and New Therapeutic Agents in Atopic Dermatitis Affecting Skin Barrier and Inflammation in Recombinant Human Epidermis Model. Int. J. Mol. Sci. 2023, 24, 6171. https://doi.org/10.3390/ijms24076171
Lee Y-J, Yassa C, Park S-H, Song SW, Jung WH, Lee YW, Kang H, Kim J-E. Interactions between Malassezia and New Therapeutic Agents in Atopic Dermatitis Affecting Skin Barrier and Inflammation in Recombinant Human Epidermis Model. International Journal of Molecular Sciences. 2023; 24(7):6171. https://doi.org/10.3390/ijms24076171
Chicago/Turabian StyleLee, Yu-Jin, Caren Yassa, Song-Hee Park, Seo Won Song, Won Hee Jung, Yang Won Lee, Hoon Kang, and Jung-Eun Kim. 2023. "Interactions between Malassezia and New Therapeutic Agents in Atopic Dermatitis Affecting Skin Barrier and Inflammation in Recombinant Human Epidermis Model" International Journal of Molecular Sciences 24, no. 7: 6171. https://doi.org/10.3390/ijms24076171
APA StyleLee, Y.-J., Yassa, C., Park, S.-H., Song, S. W., Jung, W. H., Lee, Y. W., Kang, H., & Kim, J.-E. (2023). Interactions between Malassezia and New Therapeutic Agents in Atopic Dermatitis Affecting Skin Barrier and Inflammation in Recombinant Human Epidermis Model. International Journal of Molecular Sciences, 24(7), 6171. https://doi.org/10.3390/ijms24076171