

Structural Characterization of Neisseria gonorrhoeae Bacterial Peroxidase—Insights into the Catalytic Cycle of Bacterial Peroxidases



Abstract
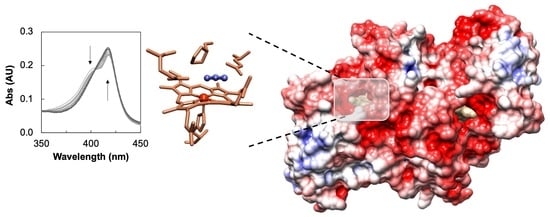
1. Introduction
2. Results
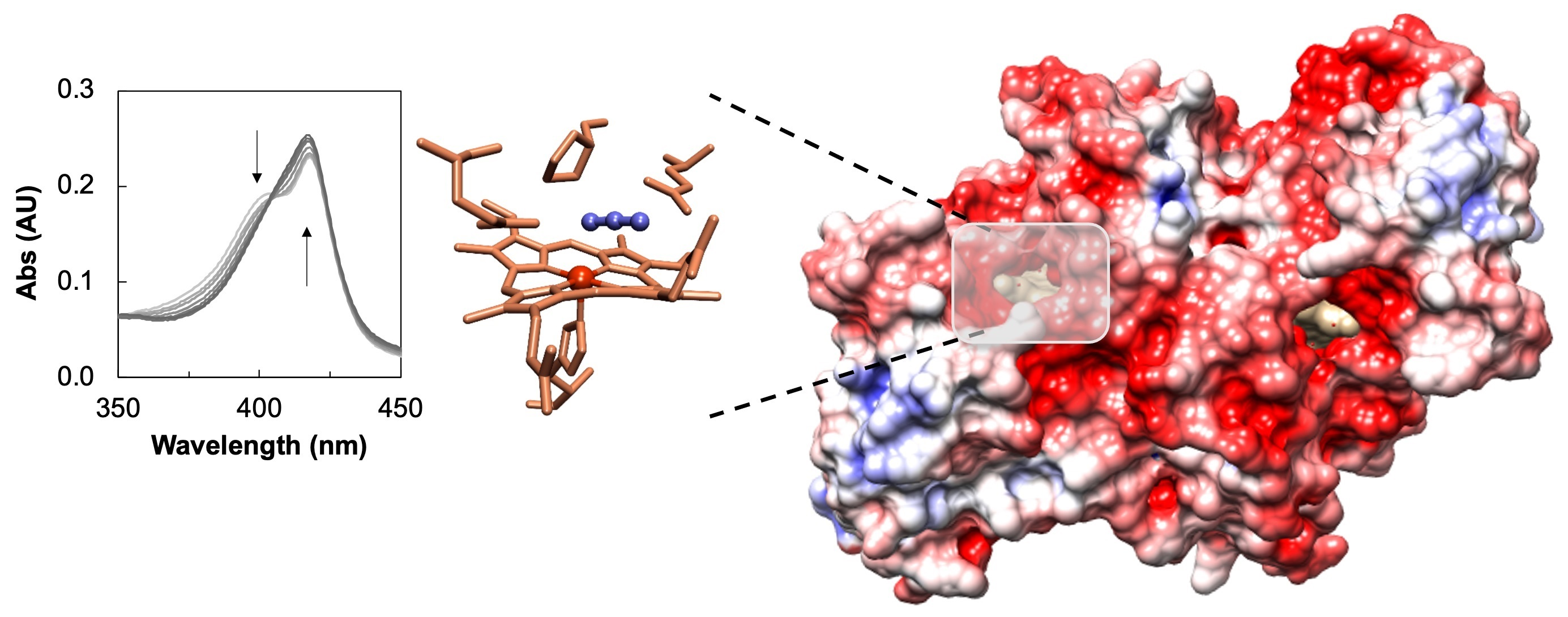
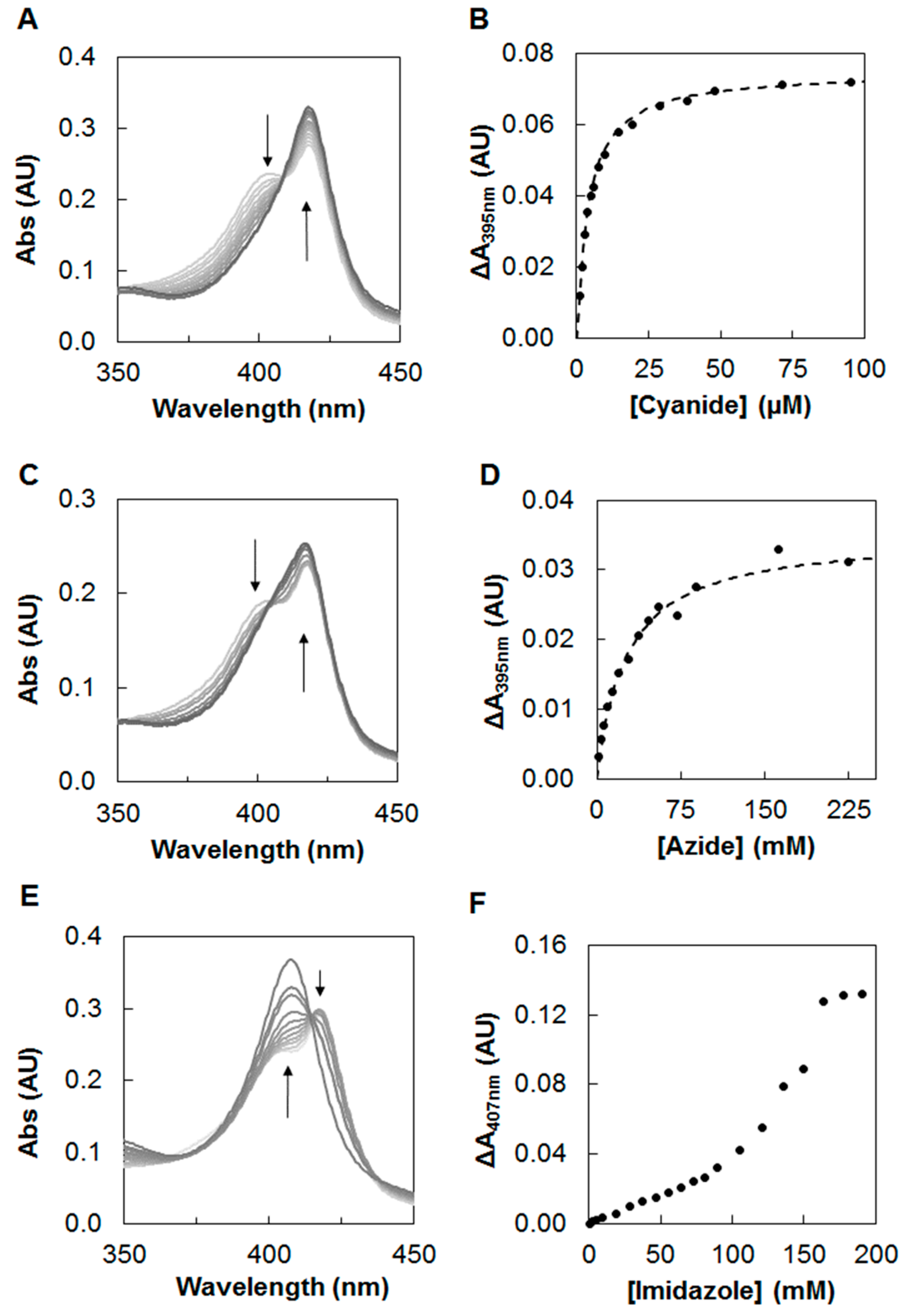
2.1. Inhibition Studies—Binding and Steady-State Kinetics
2.2. Crystallization of the Active and Azide Inhibited NgBCCP
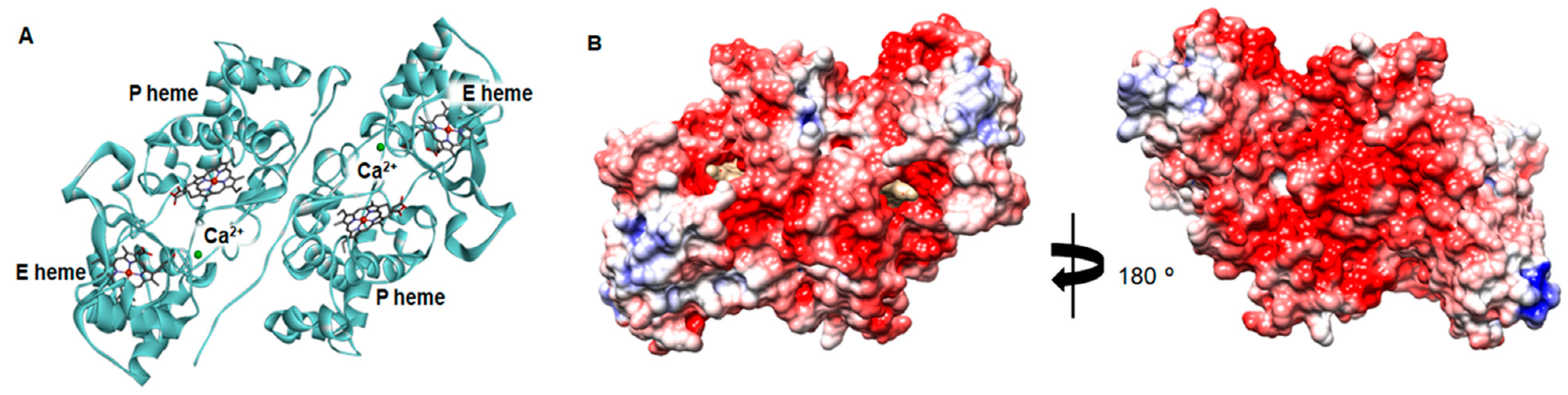
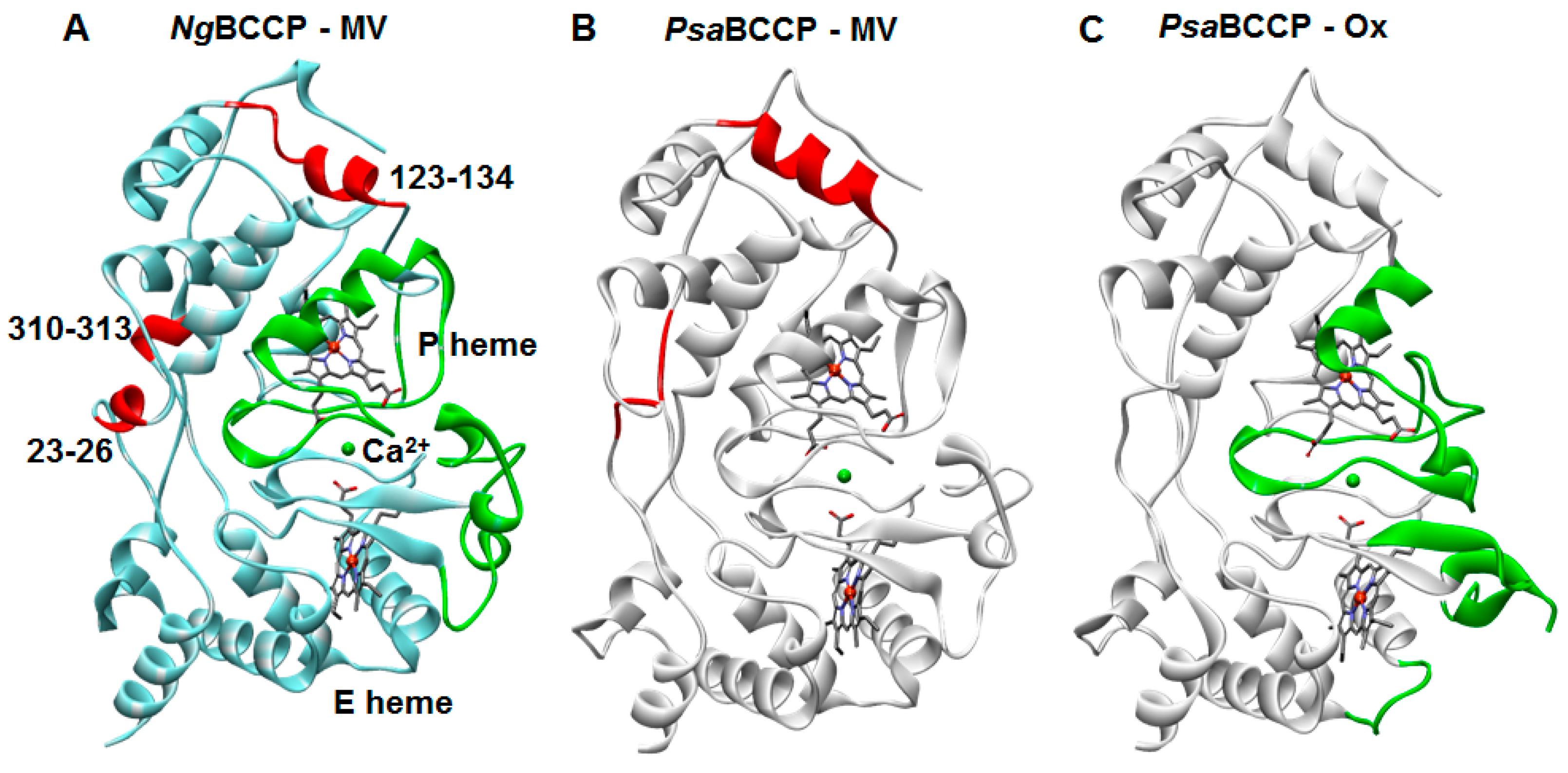
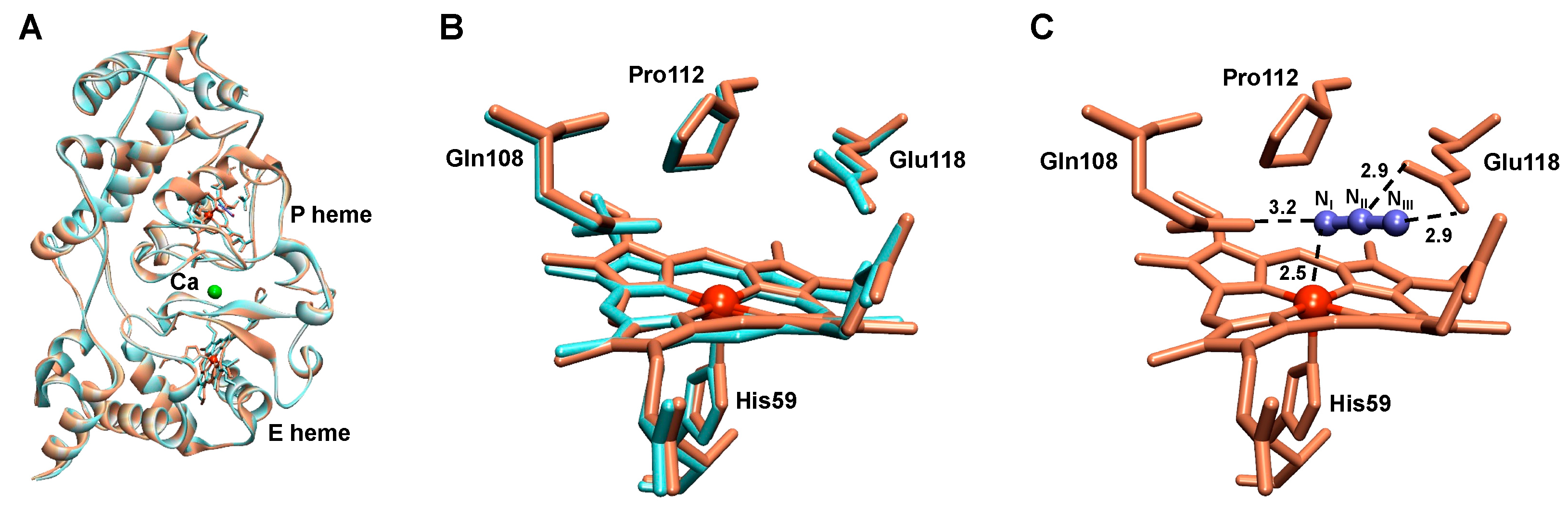
2.3. Structural Analysis of the Active NgBCCP
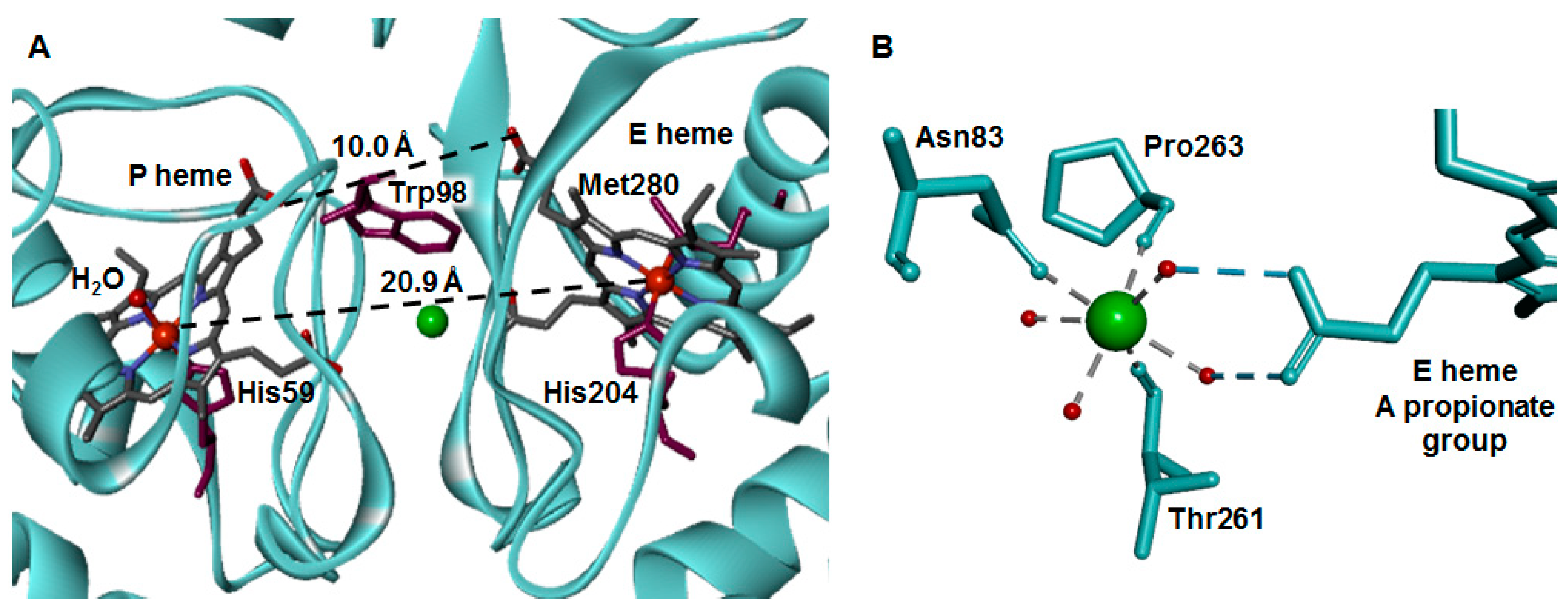
2.4. Dimer Interface
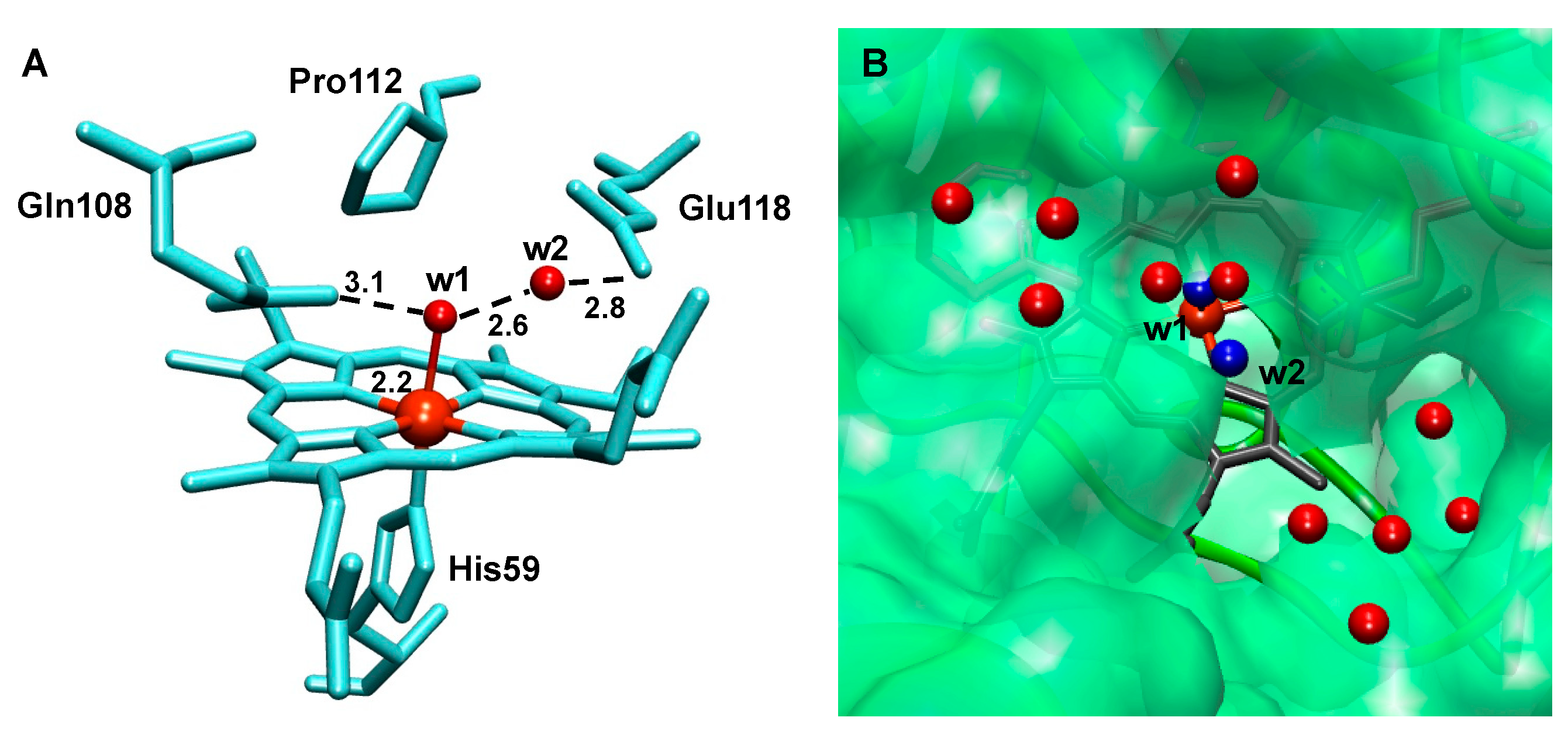
2.5. The Active Site
2.6. The Azide-Inhibited NgBCCP
3. Discussion
3.1. NgBCCP Inhibiting Compounds
3.2. NgBCCP Crystal Structure
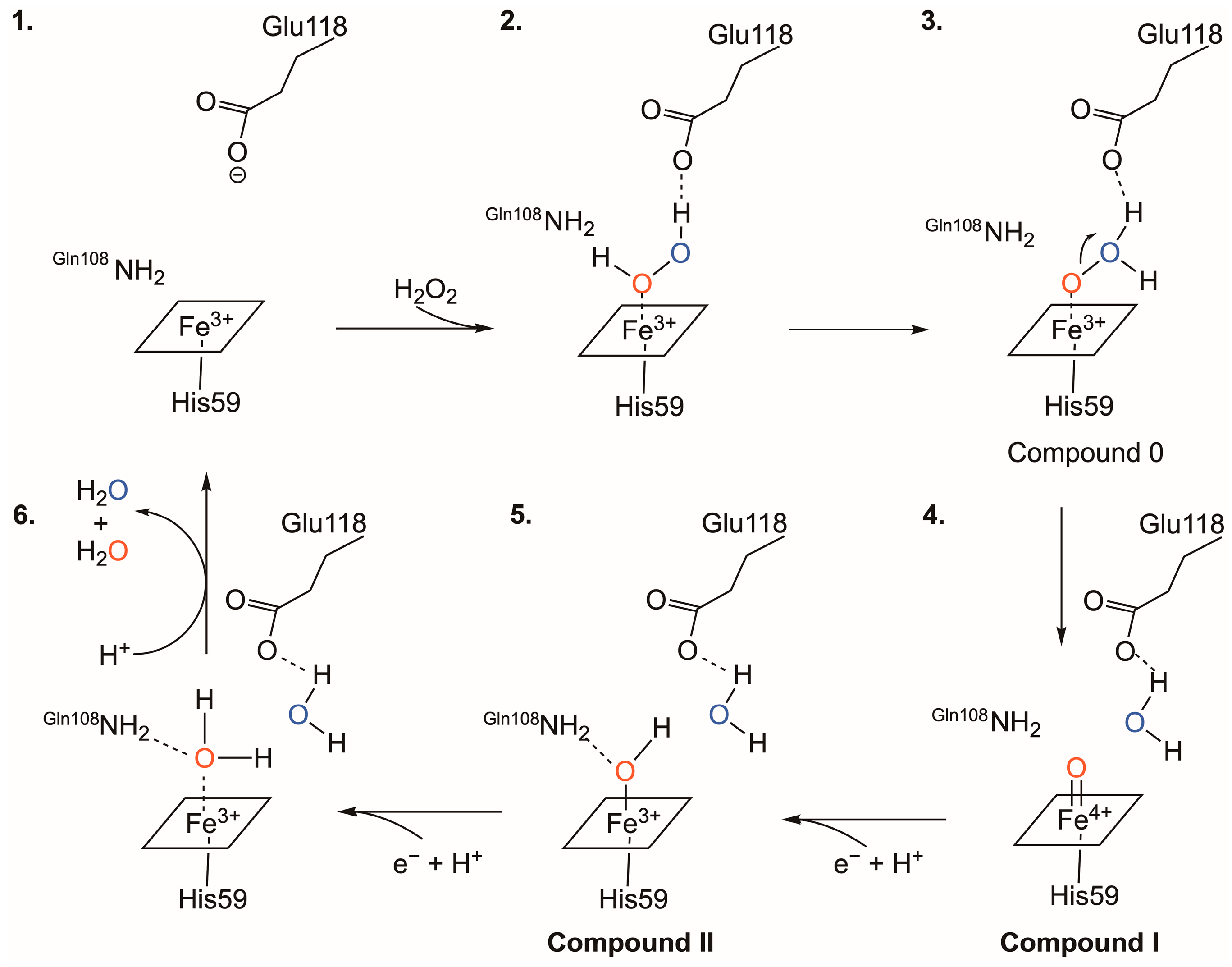
3.3. The Catalytic Mechanism
4. Materials and Methods
4.1. NgBCCP Production and Purification
4.2. UV-Visible Spectroscopy
4.3. Steady-State Kinetics with Inhibitors
4.4. Crystallization of NgBCCP in the Mixed-Valence State
4.5. Data Collection and Processing
4.6. Structure Solution and Refinement of Preliminary Model
4.7. Protein Structure and Surface Analysis
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Seib, K.L.; Wu, H.-J.; Kidd, S.P.; Apicella, M.A.; Jennings, M.P.; McEwan, A.G. Defenses against oxidative stress in Neisseria gonorrhoeae: A system tailored for a challenging environment. Microbiol. Mol. Biol. Rev. 2006, 70, 344–361. [Google Scholar] [CrossRef] [PubMed]
- Johnson, S.R.; Steiner, B.M.; Cruce, D.D.; Perkins, G.H.; Arko, R.J. Characterization of a catalase-deficient strain of Neisseria gonorrhoeae: Evidence for the significance of catalase in the biology of N. gonorrhoeae. Infect. Immun. 1993, 61, 1232–1238. [Google Scholar] [CrossRef] [PubMed]
- Turner, S.; Reid, E.; Smith, H.; Cole, J. A novel cytochrome c peroxidase from Neisseria gonorrhoeae: A lipoprotein from a Gram-negative bacterium. Biochem. J. 2003, 373, 865–873. [Google Scholar] [CrossRef] [PubMed]
- Bingham-Ramos, L.K.; Hendrixson, D.R. Characterization of two putative cytochrome c peroxidases of Campylobacter jejuni involved in promoting commensal colonization of poultry. Infect. Immun. 2008, 76, 1105–1114. [Google Scholar] [CrossRef]
- Nóbrega, C.S.; Raposo, M.; Van Driessche, G.; Devreese, B.; Pauleta, S.R. Biochemical characterization of the bacterial peroxidase from the human pathogen Neisseria gonorrhoeae. J. Inorg. Biochem. 2017, 171, 108–119. [Google Scholar] [CrossRef]
- Li, X.; Parker, S.; Deeudom, M.; Moir, J.W. Tied down: Tethering redox proteins to the outer membrane in Neisseria and other genera. Biochem. Soc. Trans. 2011, 39, 1895–1899. [Google Scholar] [CrossRef]
- Nóbrega, C.S.; Saraiva, I.H.; Carreira, C.; Devreese, B.; Matzapetakis, M.; Pauleta, S.R. The solution structure of the soluble form of the lipid-modified azurin from Neisseria gonorrhoeae, the electron donor of cytochrome c peroxidase. Biochim. Biophys. Acta 2016, 1857, 169–176. [Google Scholar] [CrossRef]
- Nóbrega, C.S.; Pauleta, S.R. Interaction between Neisseria gonorrhoeae bacterial peroxidase and its electron donor, the lipid-modified azurin. FEBS Lett. 2018, 592, 1473–1483. [Google Scholar] [CrossRef]
- Pettigrew, G.W.; Echalier, A.; Pauleta, S.R. Structure and mechanism in the bacterial dihaem cytochrome c peroxidases. J. Inorg. Biochem. 2006, 100, 551–567. [Google Scholar] [CrossRef]
- Nóbrega, C.S.; Pauleta, S.R. Reduction of hydrogen peroxide in gram-negative bacteria—Bacterial peroxidases. Adv. Microb. Physiol. 2019, 74, 415–464. [Google Scholar]
- Echalier, A.; Brittain, T.; Wright, J.; Boycheva, S.; Mortuza, G.B.; Fülöp, V.; Watmough, N.J. Redox-linked structural changes associated with the formation of a catalytically competent form of the diheme cytochrome c peroxidase from Pseudomonas aeruginosa. Biochemistry 2008, 47, 1947–1956. [Google Scholar] [CrossRef] [PubMed]
- Echalier, A.; Goodhew, C.F.; Pettigrew, G.W.; Fülöp, V. Activation and catalysis of the di-heme cytochrome c peroxidase from Paracoccus pantotrophus. Structure 2006, 14, 107–117. [Google Scholar] [CrossRef] [PubMed]
- Dias, J.M.; Alves, T.; Bonifacio, C.; Pereira, A.S.; Trincao, J.; Bourgeois, D.; Moura, I.; Romao, M.J. Structural basis for the mechanism of Ca(2+) activation of the di-heme cytochrome c peroxidase from Pseudomonas nautica 617. Structure 2004, 12, 961–973. [Google Scholar] [CrossRef] [PubMed]
- Nobrega, C.S.; Devreese, B.; Pauleta, S.R. YhjA—An Escherichia coli trihemic enzyme with quinol peroxidase activity. Biochim. Biophys. Acta Bioenerg. 2018, 1859, 411–422. [Google Scholar] [CrossRef] [PubMed]
- Dumortier, C.; Meyer, T.E.; Cusanovich, M.A. Protein dynamics: Imidazole binding to class I c-type cytochromes. Arch. Biochem. Biophys. 1999, 371, 142–148. [Google Scholar] [CrossRef] [PubMed]
- Viola, F.; Aime, S.; Coletta, M.; Desideri, A.; Fasano, M.; Paoletti, S.; Tarricone, C.; Ascenzi, P. Azide, cyanide, fluoride, imidazole and pyridine binding to ferric and ferrous native horse heart cytochrome c and to its carboxymethylated derivative: A comparative study. J. Inorg. Biochem. 1996, 62, 213–222. [Google Scholar] [CrossRef]
- Ikeda-Saito, M.; Iizuka, T. Studies on the heme environment of horse heart ferric cytochrome c. Azide and imidazole complexes of ferric cytochrome c. Biochim. Biophys. Acta 1975, 393, 335–342. [Google Scholar] [CrossRef]
- Soininen, R.; Ellfolk, N. Pseudomonas cytochrome c peroxidase. V. Absorption spectra of the enzyme and of its compounds with ligands. Inhibition of the enzyme by cyanide and azide. Acta Chem. Scand. 1973, 27, 35–46. [Google Scholar] [CrossRef]
- Gilmour, R.; Goodhew, C.F.; Pettigrew, G.W.; Prazeres, S.; Moura, I.; Moura, J.J. Spectroscopic characterization of cytochrome c peroxidase from Paracoccus denitrificans. Biochem. J. 1993, 294 Pt 3, 745–752. [Google Scholar] [CrossRef]
- De Smet, L.; Savvides, S.N.; Van Horen, E.; Pettigrew, G.; Van Beeumen, J.J. Structural and mutagenesis studies on the cytochrome c peroxidase from Rhodobacter capsulatus provide new insights into structure-function relationships of bacterial di-heme peroxidases. J. Biol. Chem. 2006, 281, 4371–4379. [Google Scholar] [CrossRef]
- Arciero, D.M.; Hooper, A.B. A di-heme cytochrome c peroxidase from Nitrosomonas europaea catalytically active in both the oxidized and half-reduced states. J. Biol. Chem. 1994, 269, 11878–11886. [Google Scholar] [CrossRef]
- Elliott, S.J.; Bradley, A.L.; Arciero, D.M.; Hooper, A.B. Protonation and inhibition of Nitrosomonas europaea cytochrome c peroxidase observed with protein film voltammetry. J. Inorg. Biochem. 2007, 101, 173–179. [Google Scholar] [CrossRef] [PubMed]
- Dixon, M. The determination of enzyme inhibitor constants. Biochem. J. 1953, 55, 170–171. [Google Scholar] [CrossRef]
- Cornish-Bowden, A. A simple graphical method for determining the inhibition constants of mixed, uncompetitive and non-competitive inhibitors. Biochem. J. 1974, 137, 143–144. [Google Scholar] [CrossRef] [PubMed]
- Fülöp, V.; Ridout, C.J.; Greenwood, C.; Hajdu, J. Crystal structure of the di-haem cytochrome c peroxidase from Pseudomonas aeruginosa. Structure 1995, 3, 1225–1233. [Google Scholar] [CrossRef]
- Shimizu, H.; Schuller, D.J.; Lanzilotta, W.N.; Sundaramoorthy, M.; Arciero, D.M.; Hooper, A.B.; Poulos, T.L. Crystal structure of Nitrosomonas europaea cytochrome c peroxidase and the structural basis for ligand switching in bacterial di-heme peroxidases. Biochemistry 2001, 40, 13483–13490. [Google Scholar] [CrossRef]
- Ellfolk, N.; Rönnberg, M.; Aasa, R.; Andréasson, L.E.; Vänngård, T. Anion binding to resting and half-reduced Pseudomonas cytochrome c peroxidase. Biochim. Biophys. Acta 1984, 784, 62–67. [Google Scholar] [CrossRef]
- Kokhan, O.; Shinkarev, V.P.; Wraight, C.A. Binding of imidazole to the heme of cytochrome c1 and inhibition of the bc1 complex from Rhodobacter sphaeroides: II. Kinetics and mechanism of binding. J. Biol. Chem. 2010, 285, 22522–22531. [Google Scholar] [CrossRef] [PubMed]
- Gonzaga de França Lopes, L.; Gouveia Júnior, F.S.; Karine Medeiros Holanda, A.; Maria Moreira de Carvalho, I.; Longhinotti, E.; Paulo, T.F.; Abreu, D.S.; Bernhardt, P.V.; Gilles-Gonzalez, M.-A.; Cirino Nogueira Diógenes, I.; et al. Bioinorganic systems responsive to the diatomic gases O2, NO, and CO: From biological sensors to therapy. Coord. Chem. Rev. 2021, 445, 214096. [Google Scholar] [CrossRef]
- Chen, Y.R.; Deterding, L.J.; Tomer, K.B.; Mason, R.P. Nature of the inhibition of horseradish peroxidase and mitochondrial cytochrome c oxidase by cyanyl radical. Biochemistry 2000, 39, 4415–4422. [Google Scholar] [CrossRef]
- Jacobson, T.; Williamson, J.; Wasilewski, A.; Felesik, J.; Vitello, L.B.; Erman, J.E. Azide binding to yeast cytochrome c peroxidase and horse metmyoglobin: Comparative thermodynamic investigation using isothermal titration calorimetry. Arch. Biochem. Biophys. 2004, 422, 125–136. [Google Scholar] [CrossRef]
- Hadarovich, A.; Chakravarty, D.; Tuzikov, A.V.; Ben-Tal, N.; Kundrotas, P.J.; Vakser, I.A. Structural motifs in protein cores and at protein-protein interfaces are different. Protein Sci. 2021, 30, 381–390. [Google Scholar] [CrossRef] [PubMed]
- Singer, A. The uterine cervix from adolescence to the menopause. Br. J. Obstet. Gynaecol. 1975, 82, 81–99. [Google Scholar] [CrossRef] [PubMed]
- Hsiao, H.C.; Boycheva, S.; Watmough, N.J.; Brittain, T. Activation of the cytochrome c peroxidase of Pseudomonas aeruginosa. The role of a heme-linked protein loop: A mutagenesis study. J. Inorg. Biochem. 2007, 101, 1133–1139. [Google Scholar] [CrossRef] [PubMed]
- Abu Tarboush, N.; Yukl, E.T.; Shin, S.; Feng, M.; Wilmot, C.M.; Davidson, V.L. Carboxyl group of Glu113 is required for stabilization of the diferrous and bis-Fe(IV) states of MauG. Biochemistry 2013, 52, 6358–6367. [Google Scholar] [CrossRef] [PubMed]
- Geng, J.; Huo, L.; Liu, A. Heterolytic OO bond cleavage: Functional role of Glu113 during bis-Fe(IV) formation in MauG. J. Inorg. Biochem. 2017, 167, 60–67. [Google Scholar] [CrossRef]
- Shin, S.; Yukl, E.T.; Sehanobish, E.; Wilmot, C.M.; Davidson, V.L. Site-directed mutagenesis of Gln103 reveals the influence of this residue on the redox properties and stability of MauG. Biochemistry 2014, 53, 1342–1349. [Google Scholar] [CrossRef]
- Kwon, H.; Basran, J.; Pathak, C.; Hussain, M.; Freeman, S.L.; Fielding, A.J.; Bailey, A.J.; Stefanou, N.; Sparkes, H.A.; Tosha, T.; et al. XFEL crystal structures of peroxidase compound II. Angew. Chem. Int. Ed. Engl. 2021, 60, 14578–14585. [Google Scholar] [CrossRef]
- Bradley, A.L.; Chobot, S.E.; Arciero, D.M.; Hooper, A.B.; Elliott, S.J. A distinctive electrocatalytic response from the cytochrome c peroxidase of Nitrosomonas europaea. J. Biol. Chem. 2004, 279, 13297–13300. [Google Scholar] [CrossRef]
- Frato, K.E.; Walsh, K.A.; Elliott, S.J. Functionally distinct bacterial cytochrome c peroxidases proceed through a common (electro)catalytic intermediate. Biochemistry 2016, 55, 125–132. [Google Scholar] [CrossRef]
- Gumiero, A.; Metcalfe, C.L.; Pearson, A.R.; Raven, E.L.; Moody, P.C. Nature of the ferryl heme in compounds I and II. J. Biol. Chem. 2011, 286, 1260–1268. [Google Scholar] [CrossRef]
- Vidossich, P.; Magistrato, A. QM/MM molecular dynamics studies of metal binding proteins. Biomolecules 2014, 4, 616–645. [Google Scholar] [CrossRef] [PubMed]
- Casadei, C.M.; Gumiero, A.; Metcalfe, C.L.; Murphy, E.J.; Basran, J.; Concilio, M.G.; Teixeira, S.C.; Schrader, T.E.; Fielding, A.J.; Ostermann, A.; et al. Heme enzymes. Neutron cryo-crystallography captures the protonation state of ferryl heme in a peroxidase. Science 2014, 345, 193–197. [Google Scholar] [CrossRef] [PubMed]
- Pulcu, G.S.; Frato, K.E.; Gupta, R.; Hsu, H.R.; Levine, G.A.; Hendrich, M.P.; Elliott, S.J. The diheme cytochrome c peroxidase from Shewanella oneidensis requires reductive activation. Biochemistry 2012, 51, 974–985. [Google Scholar] [CrossRef]
- Childs, R.E.; Bardsley, W.G. The steady-state kinetics of peroxidase with 2,2′-azino-di-(3-ethyl-benzthiazoline-6-sulphonic acid) as chromogen. Biochem. J. 1975, 145, 93–103. [Google Scholar] [CrossRef] [PubMed]
- Winn, M.D.; Ballard, C.C.; Cowtan, K.D.; Dodson, E.J.; Emsley, P.; Evans, P.R.; Keegan, R.M.; Krissinel, E.B.; Leslie, A.G.; McCoy, A.; et al. Overview of the CCP4 suite and current developments. Acta Crystallogr. D Biol. Crystallogr. 2011, 67 Pt 4, 235–242. [Google Scholar] [CrossRef]
- Battye, T.G.; Kontogiannis, L.; Johnson, O.; Powell, H.R.; Leslie, A.G. iMOSFLM: A new graphical interface for diffraction-image processing with MOSFLM. Acta Crystallogr. D Biol. Crystallogr. 2011, 67 Pt 4, 271–281. [Google Scholar] [CrossRef]
- Evans, P.R.; Murshudov, G.N. How good are my data and what is the resolution? Acta Crystallogr. D Biol. Crystallogr. 2013, 69 Pt 7, 1204–1214. [Google Scholar] [CrossRef]
- McCoy, A.J.; Grosse-Kunstleve, R.W.; Adams, P.D.; Winn, M.D.; Storoni, L.C.; Read, R.J. Phaser crystallographic software. J. Appl. Crystallogr. 2007, 40 Pt 4, 658–674. [Google Scholar] [CrossRef]
- Adams, P.D.; Afonine, P.V.; Bunkoczi, G.; Chen, V.B.; Davis, I.W.; Echols, N.; Headd, J.J.; Hung, L.W.; Kapral, G.J.; Grosse-Kunstleve, R.W.; et al. PHENIX: A comprehensive Python-based system for macromolecular structure solution. Acta Crystallogr. D Biol. Crystallogr. 2010, 66 Pt 2, 213–221. [Google Scholar] [CrossRef]
- Emsley, P.; Lohkamp, B.; Scott, W.G.; Cowtan, K. Features and development of Coot. Acta Crystallogr. D Biol. Crystallogr. 2010, 66 Pt 4, 486–501. [Google Scholar] [CrossRef] [PubMed]
- Murshudov, G.N.; Skubak, P.; Lebedev, A.A.; Pannu, N.S.; Steiner, R.A.; Nicholls, R.A.; Winn, M.D.; Long, F.; Vagin, A.A. REFMAC5 for the refinement of macromolecular crystal structures. Acta Crystallogr. D Biol. Crystallogr. 2011, 67 Pt 4, 355–367. [Google Scholar] [CrossRef] [PubMed]
- Joosten, R.P.; Long, F.; Murshudov, G.N.; Perrakis, A. The PDB_REDO server for macromolecular structure model optimization. IUCrJ 2014, 1 Pt 4, 213–220. [Google Scholar] [CrossRef]
- Krissinel, E.; Henrick, K. Secondary-structure matching (SSM), a new tool for fast protein structure alignment in three dimensions. Acta Crystallogr. D Biol. Crystallogr. 2004, 60 Pt 12, 2256–2268. [Google Scholar] [CrossRef] [PubMed]
- Krissinel, E.; Henrick, K. Inference of macromolecular assemblies from crystalline state. J. Mol. Biol. 2007, 372, 774–797. [Google Scholar] [CrossRef]
- Baker, N.A.; Sept, D.; Joseph, S.; Holst, M.J.; McCammon, J.A. Electrostatics of nanosystems: Application to microtubules and the ribosome. Proc. Natl. Acad. Sci. USA 2001, 98, 10037–10041. [Google Scholar] [CrossRef]
- Lotan, I.; Head-Gordon, T. An analytical electrostatic model for salt screened interactions between multiple proteins. J. Chem. Theory Comput. 2006, 2, 541–555. [Google Scholar] [CrossRef]
- Dolinsky, T.J.; Nielsen, J.E.; McCammon, J.A.; Baker, N.A. PDB2PQR: An automated pipeline for the setup of Poisson-Boltzmann electrostatics calculations. Nucleic Acids Res. 2004, 32, W665–W667. [Google Scholar] [CrossRef]
- Olsson, M.H.; Sondergaard, C.R.; Rostkowski, M.; Jensen, J.H. PROPKA3: Consistent treatment of internal and surface residues in empirical pKa Predictions. J. Chem. Theory Comput. 2011, 7, 525–537. [Google Scholar] [CrossRef]










{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
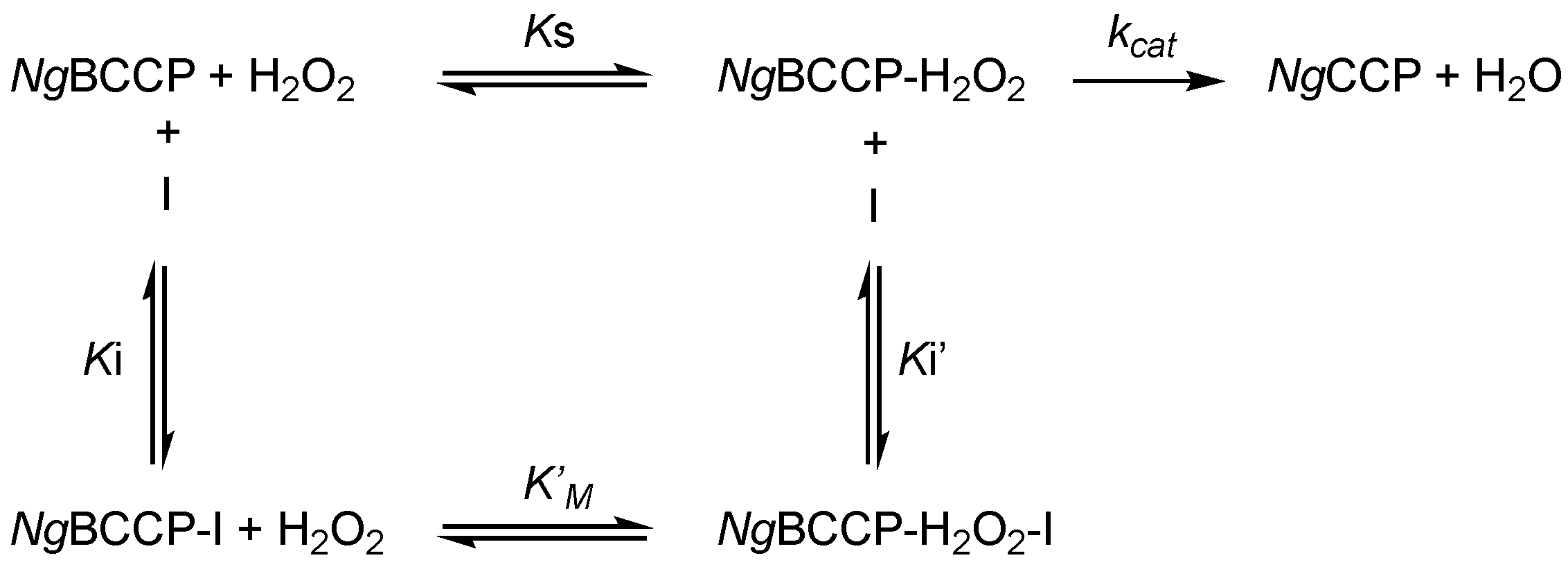{kind=link}
{kind=link}
| NgBCCP (Active, 1.8 Å) | NgBCCP (Active, 1.4 Å) | NgBCCP (Azide-Inhibited, 2.3 Å) | |
|---|---|---|---|
| Data Collection and Processing | |||
| Source | IμS 3.0 | PXIII (X06DA), SLS | BM30, ESRF |
| Detector | CMOS Photon 100 | PILATUS 2M-F | ADSC Q315r CCD |
| Wavelength (Å) | 1.5418 | 1.0 | 0.979 |
| a (Å) | 78.9 | 78.8 | 79.1 |
| b (Å) | 88.8 | 89.1 | 89.1 |
| c (Å) | 93.1 | 89.7 | 94.8 |
| Space group | P212121 | P212121 | P212121 |
| Molecules per ASU | 2 | 2 | 2 |
| Matthews coefficient (Å3.Da−1) | 2.17 | 2.10 | 2.23 |
| Resolution range (Å) | 23.81–1.80 (1.85–1.80) | 39.90–1.40 (1.42–1.40) | 65.00–2.30 (2.38–2.30) |
| <I/σI> | 10.3 (2.31) | 11.4 (2.1) | 9.2 (2.2) |
| Wilson B-factor | 14.5 | 12.7 | 27.4 |
| Rmerge (%) * | 15.7 (62.6) | 6.8 (65.6) | 11.7 (72.3) |
| Rpim (%) † | - | 4.2 (44.9) | 8.0 (48.7) |
| Half-dataset correlation CC1/2 | - | 0.999 (0.813) | 0.995 (0.762) |
| Multiplicity | 10 (7.8) | 6.4 (5.6) | 5.6 (5.6) |
| No. of observed reflections | 588,364 | 797,508 (33,769) | 169,921 (16,295) |
| No. of unique reflections | 61,589 | 124,619 (6083) | 30,270 (2907) |
| Completeness (%) | 98.8 (99.9) | 99.9 (99.3) | 97.7 (94.9) |
| Model Building and Refinement | |||
| No. of protein atoms | 5080 | 5096 | 5032 |
| No. of water molecules | 535 | 577 | 191 |
| Rwork ‡ (%) | 21.9 | 19.6 | 18.0 |
| Rfree § (%) | 25.5 | 22.1 | 23.3 |
| R.m.s.d. bond lengths (Å) | 0.029 | 0.012 | 0.015 |
| R.m.s.d. bond angles (⁰) | 2.810 | 1.972 | 2.124 |
| Average B-factor (Å2) | |||
| Protein | |||
| Main-chain (A, B) | 20.1, 19.2 | 17.3, 16.3 | 37.8, 30.5 |
| Side-chain (A, B) | 22.5, 21.4 | 20.6, 19.5 | 40.3, 33.5 |
| Heme groups P heme_A, E heme_A P heme_B, E heme_B | 12.1, 14.9, 13.0, 13.7 | 11.0, 15.0 11.0, 12.0 | 25.2, 36.8 22.9, 21.1 |
| Iron atoms P heme_A, E heme_A P heme_B, E heme_B | 13.0, 16.5, 13.9, 15.1 | 11.5, 14.8 11.2, 12.4 | 24.2, 36.7 21.9, 24.3 |
| Calcium ions (A, B) | 11.7, 10.5 | 12.8, 10.4 | 28.3, 22.1 |
| Water molecules | 25.9 (535) | 26.6 (577) | 30.1 (191) |
| Azide ions (A, B) | - | - | 39.8, 51.3 |
| Ramachandran plot | |||
| Residues in favored regions (%) | 96.1 | 96.8 | 96.1 |
| Residues in allowed regions (%) | 2.9 | 2.3 | 2.8 |
| Residues outliers (%) | 0.9 | 0.9 | 1.1 |
| PDB accession codes | 6FU3 | 7ZS8 | 6QKN |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Nóbrega, C.S.; Carvalho, A.L.; Romão, M.J.; Pauleta, S.R. Structural Characterization of Neisseria gonorrhoeae Bacterial Peroxidase—Insights into the Catalytic Cycle of Bacterial Peroxidases. Int. J. Mol. Sci. 2023, 24, 6246. https://doi.org/10.3390/ijms24076246
Nóbrega CS, Carvalho AL, Romão MJ, Pauleta SR. Structural Characterization of Neisseria gonorrhoeae Bacterial Peroxidase—Insights into the Catalytic Cycle of Bacterial Peroxidases. International Journal of Molecular Sciences. 2023; 24(7):6246. https://doi.org/10.3390/ijms24076246
Chicago/Turabian StyleNóbrega, Cláudia S., Ana Luísa Carvalho, Maria João Romão, and Sofia R. Pauleta. 2023. "Structural Characterization of Neisseria gonorrhoeae Bacterial Peroxidase—Insights into the Catalytic Cycle of Bacterial Peroxidases" International Journal of Molecular Sciences 24, no. 7: 6246. https://doi.org/10.3390/ijms24076246
APA StyleNóbrega, C. S., Carvalho, A. L., Romão, M. J., & Pauleta, S. R. (2023). Structural Characterization of Neisseria gonorrhoeae Bacterial Peroxidase—Insights into the Catalytic Cycle of Bacterial Peroxidases. International Journal of Molecular Sciences, 24(7), 6246. https://doi.org/10.3390/ijms24076246