
Hypoxia-Inducible Factor 1-Alpha and Glucose Metabolism during Cardiac Remodeling Progression from Hypertrophy to Heart Failure
 ,
,
Abstract
1. Introduction
2. Results
2.1. Clinical-Pathological Signs of Heart Failure
2.2. Body and Lung Weights and Post Mortem Cardiac Morphology
2.3. Heart Rate, Structural and Functional Echocardiographic Data
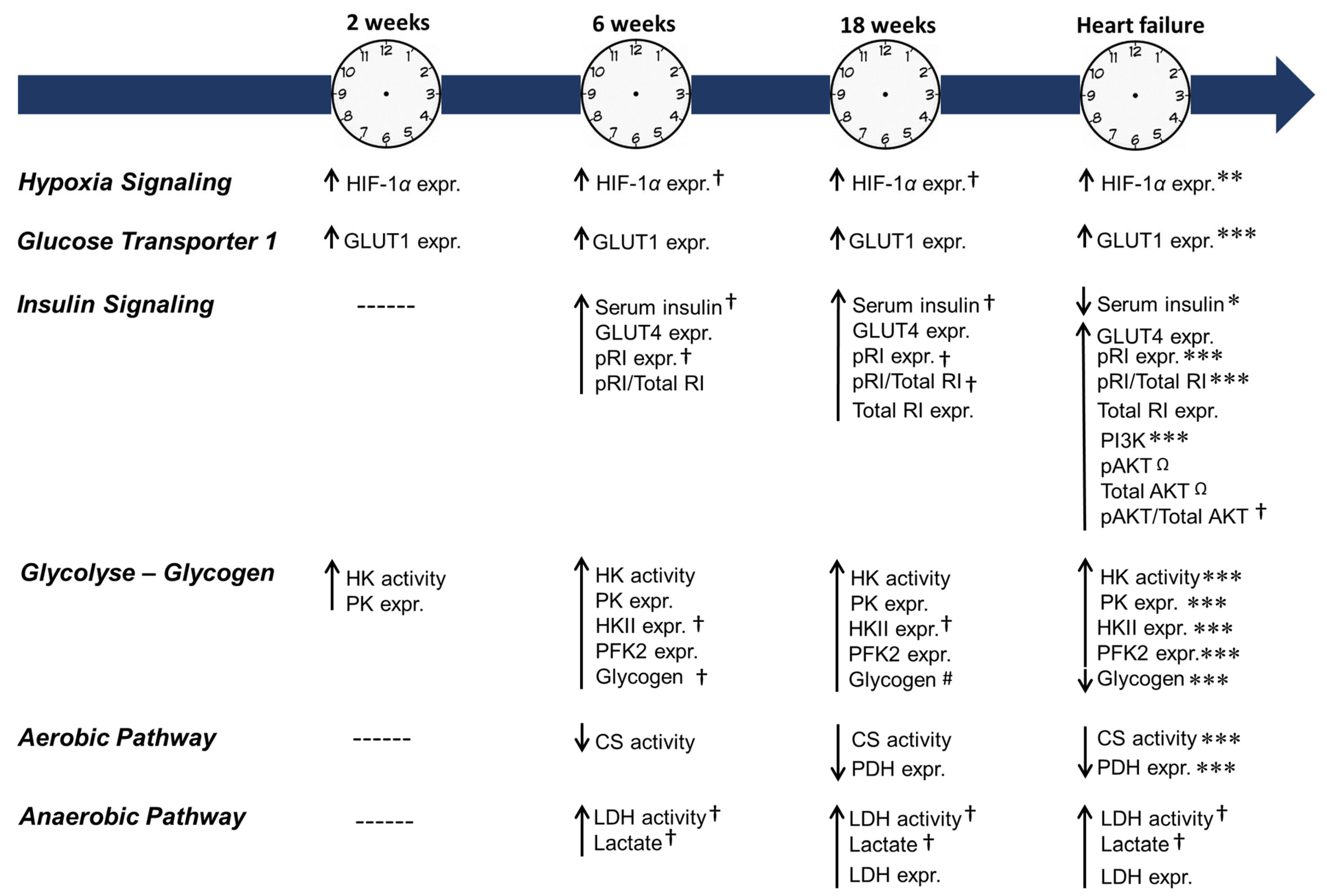
2.4. Cardiac HIF-1α Expression
2.5. Blood Glucose and Serum Insulin
2.6. Cardiac Insulin Signaling and Glucose Transporters 1 and 4
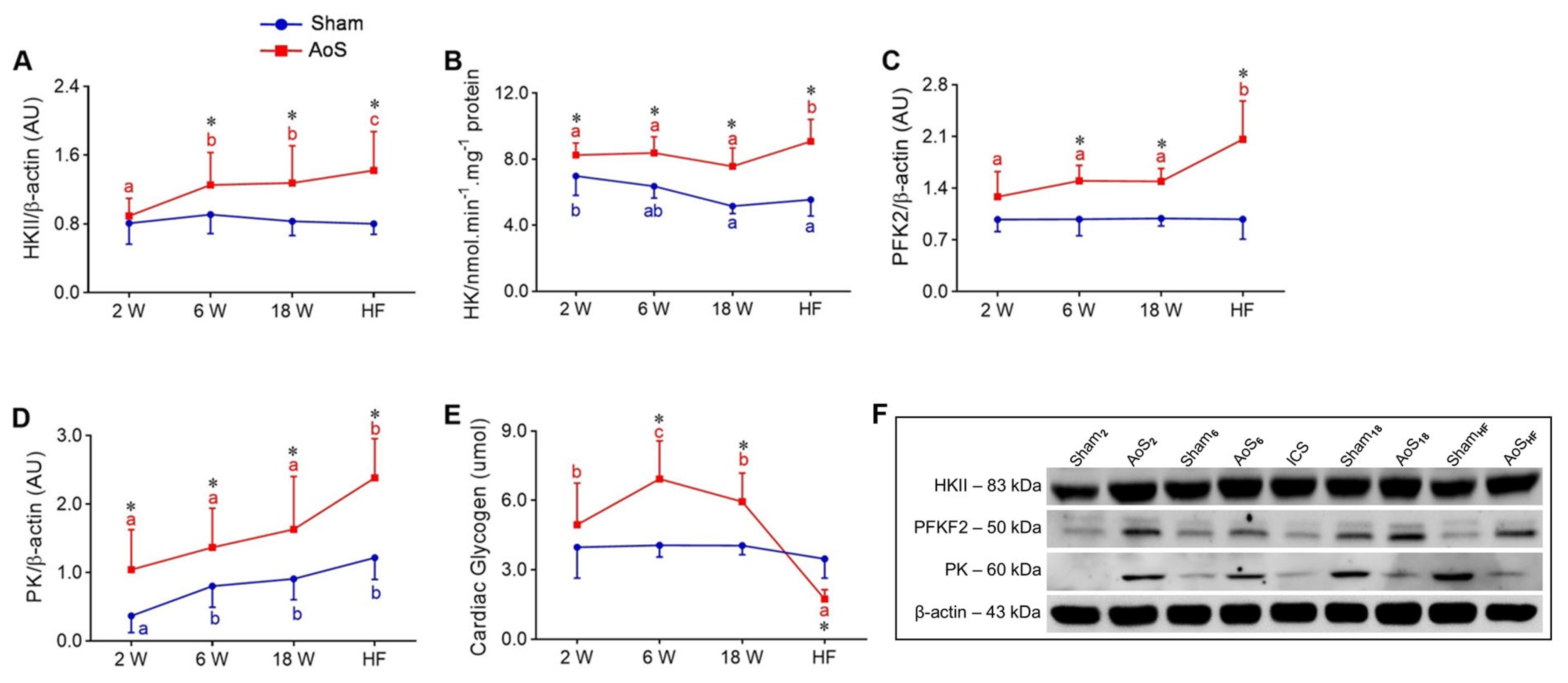
2.7. Cardiac Glycolysis and Glycogen
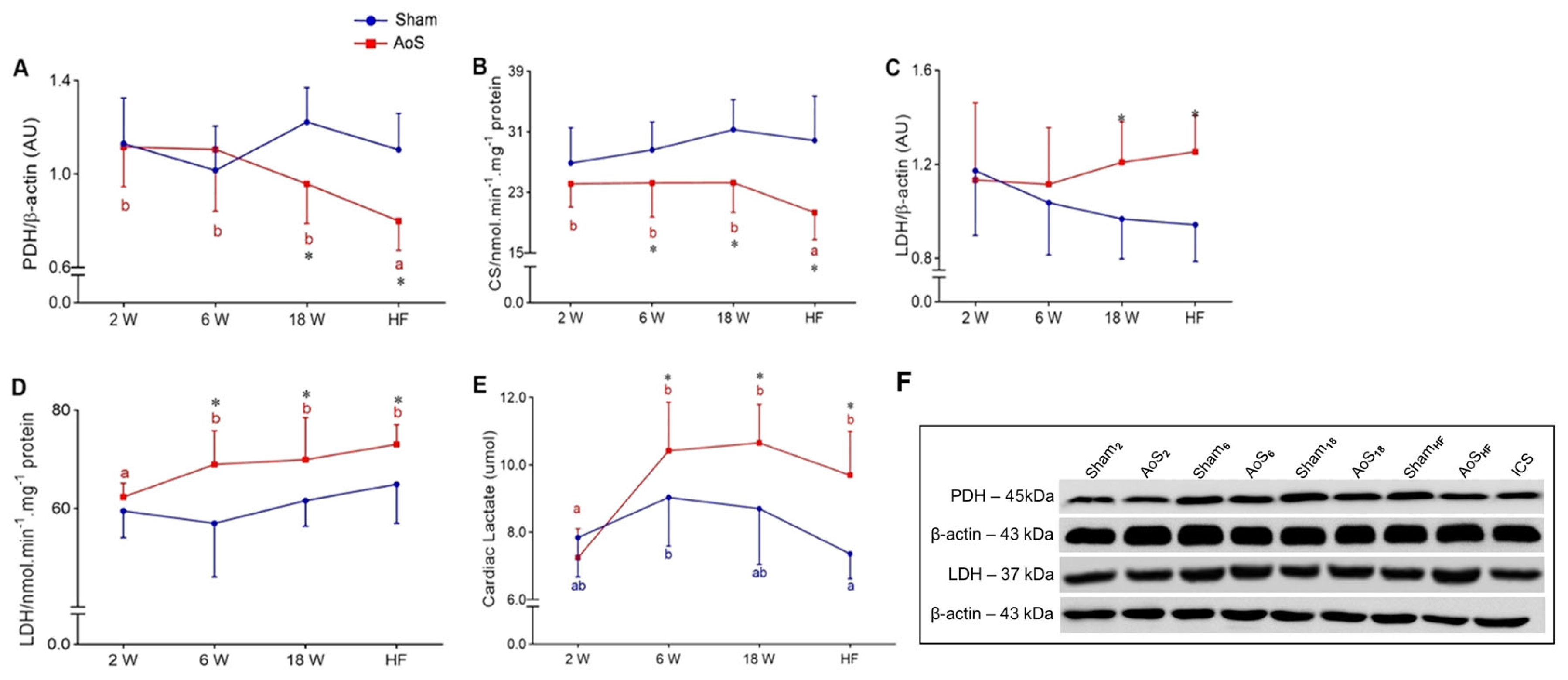
2.8. Cardiac Aerobic and Anaerobic Pathway
2.9. Canonical Correlation Analysis (CCA)
3. Discussion
4. Materials and Methods
4.1. Animals, Supravalvar Surgery, and Experimental Groups
4.2. Body and Lung Weights and Post Mortem Cardiac Morphology
4.3. Structural and Functional Echocardiographic Data
4.4. Serum Glucose and Insulin Levels
4.5. Cardiac HIF-1α and Glucose Metabolism
4.6. Enzymatic Activity of Cardiac Glucose Metabolism
4.7. Cardiac Glycogen
4.8. Statistical Analysis
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Ritterhoff, J.; Tian, R. Metabolism in Cardiomyopathy: Every Substrate Matters. Cardiovasc. Res. 2017, 113, 411–421. [Google Scholar] [CrossRef] [PubMed]
- Gibb, A.A.; Hill, B.G. Metabolic Coordination of Physiological and Pathological Cardiac Remodeling. Circ. Res. 2018, 123, 107–128. [Google Scholar] [CrossRef]
- Karwi, Q.G.; Uddin, G.M.; Ho, K.L.; Lopaschuk, G.D. Loss of Metabolic Flexibility in the Failing Heart. Front. Cardiovasc. Med. 2018, 5, 68. [Google Scholar] [CrossRef]
- Felker, G.M.; Mann, D.L. Heart Failure: A Companion to Braunwald’s Heart Disease, 3rd ed.; Elsevier-Saunders: Amsterdam, The Netherlands, 2015. [Google Scholar]
- Rocha, S. Gene Regulation under Low Oxygen: Holding Your Breath for Transcription. Trends Biochem. Sci. 2007, 32, 389–397. [Google Scholar] [CrossRef] [PubMed]
- Semenza, G.L. Hypoxia-Inducible Factor 1: Oxygen Homeostasis and Disease Pathophysiology. Trends Mol. Med. 2001, 7, 345–350. [Google Scholar] [CrossRef] [PubMed]
- Sutton, M.G.S.J.; Plappert, T.; Hilpisch, K.E.; Abraham, W.T.; Hayes, D.L.; Chinchoy, E. Sustained Reverse Left Ventricular Structural Remodeling with Cardiac Resynchronization at One Year Is a Function of Etiology: Quantitative Doppler Echocardiographic Evidence from the Multicenter InSync Randomized Clinical Evaluation (MIRACLE). Circulation 2006, 113, 266–272. [Google Scholar] [CrossRef] [PubMed]
- Ye, J.; Gao, Z.; Yin, J.; He, Q. Hypoxia Is a Potential Risk Factor for Chronic Inflammation and Adiponectin Reduction in Adipose Tissue of Ob/Ob and Dietary Obese Mice. Am. J. Physiol. Endocrinol. Metab. 2007, 293, E1118–E1128. [Google Scholar] [CrossRef]
- Lim, C.S.; Kiriakidis, S.; Sandison, A.; Paleolog, E.M.; Davies, A.H. Hypoxia-Inducible Factor Pathway and Diseases of the Vascular Wall. J. Vasc. Surg. 2013, 58, 219–230. [Google Scholar] [CrossRef]
- Semenza, G.L. Involvement of Oxygen-Sensing Pathways in Physiologic and Pathologic Erythropoiesis. Blood 2009, 114, 2015–2019. [Google Scholar] [CrossRef]
- Li, X.; Zhang, Q.; Nasser, M.; Xu, L.; Zhang, X.; Zhu, P.; He, Q.; Zhao, M. Oxygen Homeostasis and Cardiovascular Disease: A Role for HIF? Biomed. Pharmacother. 2020, 128, 110338. [Google Scholar] [CrossRef]
- Mirtschink, P.; Krek, W. Hypoxia-Driven Glycolytic and Fructolytic Metabolic Programs: Pivotal to Hypertrophic Heart Disease. Biochim. Biophys. Acta 2016, 1863 Pt B, 1822–1828. [Google Scholar] [CrossRef]
- Kolwicz, S.C.; Tian, R. Glucose Metabolism and Cardiac Hypertrophy. Cardiovasc. Res. 2011, 90, 194–201. [Google Scholar] [CrossRef] [PubMed]
- Doenst, T.; Nguyen, T.D.; Abel, E.D. Cardiac Metabolism in Heart Failure: Implications beyond ATP Production. Circ. Res. 2013, 113, 709–724. [Google Scholar] [CrossRef] [PubMed]
- Zhang, L.; Jaswal, J.S.; Ussher, J.R.; Sankaralingam, S.; Wagg, C.; Zaugg, M.; Lopaschuk, G.D. Cardiac Insulin-Resistance and Decreased Mitochondrial Energy Production Precede the Development of Systolic Heart Failure after Pressure-Overload Hypertrophy. Circ. Heart Fail. 2013, 6, 1039–1048. [Google Scholar] [CrossRef] [PubMed]
- Doenst, T.; Pytel, G.; Schrepper, A.; Amorim, P.; Färber, G.; Shingu, Y.; Mohr, F.W.; Schwarzer, M. Decreased Rates of Substrate Oxidation Ex Vivo Predict the Onset of Heart Failure and Contractile Dysfunction in Rats with Pressure Overload. Cardiovasc. Res. 2010, 86, 461–470. [Google Scholar] [CrossRef] [PubMed]
- Peterzan, M.A.; Lygate, C.A.; Neubauer, S.; Rider, O.J. Metabolic Remodeling in Hypertrophied and Failing Myocardium: A Review. Am. J. Physiol. Heart Circ. Physiol. 2017, 313, H597–H616. [Google Scholar] [CrossRef]
- Lazzeroni, D.; Rimoldi, O.; Camici, P.G. From Left Ventricular Hypertrophy to Dysfunction and Failure. Circ. J. 2016, 80, 555–564. [Google Scholar] [CrossRef]
- Friehs, I.; Moran, A.M.; Stamm, C.; Colan, S.D.; Takeuchi, K.; Cao-Danh, H.; Rader, C.M.; McGowan, F.X.; del Nido, P.J. Impaired Glucose Transporter Activity in Pressure-Overload Hypertrophy Is an Early Indicator of Progression to Failure. Circulation 1999, 100 (Suppl. S19), II-187–II-193. [Google Scholar] [CrossRef]
- Wigerup, C.; Påhlman, S.; Bexell, D. Therapeutic Targeting of Hypoxia and Hypoxia-Inducible Factors in Cancer. Pharmacol. Ther. 2016, 164, 152–169. [Google Scholar] [CrossRef]
- Semenza, G.L. Hypoxia-Inducible Factors: Mediators of Cancer Progression and Targets for Cancer Therapy. Trends Pharmacol. Sci. 2012, 33, 207–214. [Google Scholar] [CrossRef]
- Masoud, G.N.; Li, W. HIF-1α Pathway: Role, Regulation and Intervention for Cancer Therapy. Acta Pharm. Sin. B 2015, 5, 378–389. [Google Scholar] [CrossRef] [PubMed]
- Sant’Ana, P.G.; Batah, S.S.; Leão, P.S.; Teodoro, W.R.; de Souza, S.L.B.; Ferreira Mota, G.A.; Vileigas, D.F.; da Silva, V.L.; de Campos, D.H.S.; Okoshi, K.; et al. Heart Remodeling Produced by Aortic Stenosis Promotes Cardiomyocyte Apoptosis Mediated by Collagen V Imbalance. Pathophysiology 2018, 25, 373–379. [Google Scholar] [CrossRef] [PubMed]
- Tromp, J.; Khan, M.A.F.; Klip, I.T.; Meyer, S.; de Boer, R.A.; Jaarsma, T.; Hillege, H.; van Veldhuisen, D.J.; van der Meer, P.; Voors, A.A. Biomarker Profiles in Heart Failure Patients with Preserved and Reduced Ejection Fraction. J. Am. Heart Assoc. 2017, 6, e003989. [Google Scholar] [CrossRef] [PubMed]
- Mohammed, S.F.; Hussain, S.; Mirzoyev, S.A.; Edwards, W.D.; Maleszewski, J.J.; Redfield, M.M. Coronary Microvascular Rarefaction and Myocardial Fibrosis in Heart Failure with Preserved Ejection Fraction. Circulation 2015, 131, 550–559. [Google Scholar] [CrossRef]
- Oldfield, C.J.; Duhamel, T.A.; Dhalla, N.S. Mechanisms for the Transition from Physiological to Pathological Cardiac Hypertrophy. Can. J. Physiol. Pharmacol. 2020, 98, 74–84. [Google Scholar] [CrossRef]
- Nakamura, M.; Sadoshima, J. Mechanisms of Physiological and Pathological Cardiac Hypertrophy. Nat. Rev. Cardiol. 2018, 15, 387–407. [Google Scholar] [CrossRef]
- Hurst, J.H. William Kaelin, Peter Ratcliffe, and Gregg Semenza Receive the 2016 Albert Lasker Basic Medical Research Award. J. Clin. Investig. 2016, 126, 3628–3638. [Google Scholar] [CrossRef]
- Anzai, T. Inflammatory Mechanisms of Cardiovascular Remodeling. Circ. J. 2018, 82, 629–635. [Google Scholar] [CrossRef]
- Pitoulis, F.G.; Terracciano, C.M. Heart Plasticity in Response to Pressure- and Volume-Overload: A Review of Findings in Compensated and Decompensated Phenotypes. Front. Physiol. 2020, 11, 92. [Google Scholar] [CrossRef]
- Riehle, C.; Abel, E.D. Insulin Signaling and Heart Failure. Circ. Res. 2016, 118, 1151–1169. [Google Scholar] [CrossRef]
- Cohn, J.N.; Ferrari, R.; Sharpe, N. Cardiac Remodeling--Concepts and Clinical Implications: A Consensus Paper from an International Forum on Cardiac Remodeling. Behalf of an International Forum on Cardiac Remodeling. J. Am. Coll. Cardiol. 2000, 35, 569–582. [Google Scholar] [CrossRef] [PubMed]
- Bekeredjian, R.; Walton, C.B.; MacCannell, K.A.; Ecker, J.; Kruse, F.; Outten, J.T.; Sutcliffe, D.; Gerard, R.D.; Bruick, R.K.; Shohet, R.V. Conditional HIF-1alpha Expression Produces a Reversible Cardiomyopathy. PLoS ONE 2010, 5, e11693. [Google Scholar] [CrossRef] [PubMed]
- Ronkainen, V.-P.; Skoumal, R.; Tavi, P. Hypoxia and HIF-1 Suppress SERCA2a Expression in Embryonic Cardiac Myocytes through Two Interdependent Hypoxia Response Elements. J. Mol. Cell. Cardiol. 2011, 50, 1008–1016. [Google Scholar] [CrossRef]
- Bourdier, G.; Détrait, M.; Bouyon, S.; Lemarié, E.; Brasseur, S.; Doutreleau, S.; Pépin, J.-L.; Godin-Ribuot, D.; Belaidi, E.; Arnaud, C. Intermittent Hypoxia Triggers Early Cardiac Remodeling and Contractile Dysfunction in the Time-Course of Ischemic Cardiomyopathy in Rats. J. Am. Heart Assoc. 2020, 9, e016369. [Google Scholar] [CrossRef] [PubMed]
- Ruthenborg, R.J.; Ban, J.-J.; Wazir, A.; Takeda, N.; Kim, J.-W. Regulation of Wound Healing and Fibrosis by Hypoxia and Hypoxia-Inducible Factor-1. Mol. Cells 2014, 37, 637–643. [Google Scholar] [CrossRef]
- Shimizu, I.; Minamino, T.; Toko, H.; Okada, S.; Ikeda, H.; Yasuda, N.; Tateno, K.; Moriya, J.; Yokoyama, M.; Nojima, A.; et al. Excessive Cardiac Insulin Signaling Exacerbates Systolic Dysfunction Induced by Pressure Overload in Rodents. J. Clin. Investig. 2010, 120, 1506–1514. [Google Scholar] [CrossRef]
- Shao, D.; Tian, R. Glucose Transporters in Cardiac Metabolism and Hypertrophy. Compr. Physiol. 2015, 6, 331–351. [Google Scholar] [CrossRef] [PubMed]
- Pereira, R.O.; Wende, A.R.; Olsen, C.; Soto, J.; Rawlings, T.; Zhu, Y.; Anderson, S.M.; Abel, E.D. Inducible Overexpression of GLUT1 Prevents Mitochondrial Dysfunction and Attenuates Structural Remodeling in Pressure Overload but Does Not Prevent Left Ventricular Dysfunction. J. Am. Heart Assoc. 2013, 2, e000301. [Google Scholar] [CrossRef] [PubMed]
- Semenza, G. Signal Transduction to Hypoxia-Inducible Factor 1. Biochem. Pharmacol. 2002, 64, 993–998. [Google Scholar] [CrossRef]
- Montessuit, C.; Thorburn, A. Transcriptional Activation of the Glucose Transporter GLUT1 in Ventricular Cardiac Myocytes by Hypertrophic Agonists. J. Biol. Chem. 1999, 274, 9006–9012. [Google Scholar] [CrossRef]
- Tham, Y.K.; Bernardo, B.C.; Ooi, J.Y.Y.; Weeks, K.L.; McMullen, J.R. Pathophysiology of Cardiac Hypertrophy and Heart Failure: Signaling Pathways and Novel Therapeutic Targets. Arch. Toxicol. 2015, 89, 1401–1438. [Google Scholar] [CrossRef] [PubMed]
- Ascaso, J.F.; Real, J.T.; Priego, A.; Carmena, R.; Romero, P.; Valdecabres, C. Cuantificación de Insulinorresistencia Con Los Valores de Insulina Basal e Índice HOMA En Una Población No Diabética. Med. Clin. 2001, 117, 530–533. [Google Scholar] [CrossRef] [PubMed]
- DeBoer, M.D. Animal Models of Anorexia and Cachexia. Expert Opin. Drug Discov. 2009, 4, 1145–1155. [Google Scholar] [CrossRef] [PubMed]
- Cook, S.A.; Varela-Carver, A.; Mongillo, M.; Kleinert, C.; Khan, M.T.; Leccisotti, L.; Strickland, N.; Matsui, T.; Das, S.; Rosenzweig, A.; et al. Abnormal Myocardial Insulin Signalling in Type 2 Diabetes and Left-Ventricular Dysfunction. Eur. Heart J. 2010, 31, 100–111. [Google Scholar] [CrossRef]
- Katz, A.M. Physiology of the Heart, 5th ed.; Wolters Kluwer Health/Lippincott Williams & Wilkins Health: Philadelphia, PA, USA, 2011. [Google Scholar]
- Li, X.-B.; Gu, J.-D.; Zhou, Q.-H. Review of Aerobic Glycolysis and Its Key Enzymes—New Targets for Lung Cancer Therapy. Thorac. Cancer 2015, 6, 17–24. [Google Scholar] [CrossRef]
- Alves-Filho, J.C.; Pålsson-McDermott, E.M. Pyruvate Kinase M2: A Potential Target for Regulating Inflammation. Front. Immunol. 2016, 7, 145. [Google Scholar] [CrossRef]
- Wilson, J.E. Isozymes of Mammalian Hexokinase: Structure, Subcellular Localization and Metabolic Function. J. Exp. Biol. 2003, 206 Pt 12, 2049–2057. [Google Scholar] [CrossRef]
- Mor, I.; Cheung, E.C.; Vousden, K.H. Control of Glycolysis through Regulation of PFK1: Old Friends and Recent Additions. Cold Spring Harb. Symp. Quant. Biol. 2011, 76, 211–216. [Google Scholar] [CrossRef]
- Tian, R.; Musi, N.; D’Agostino, J.; Hirshman, M.F.; Goodyear, L.J. Increased Adenosine Monophosphate-Activated Protein Kinase Activity in Rat Hearts with Pressure-Overload Hypertrophy. Circulation 2001, 104, 1664–1669. [Google Scholar] [CrossRef]
- Schaefer, S.; Ramasamy, R. Glycogen Utilization and Ischemic Injury in the Isolated Rat Heart. Cardiovasc. Res. 1997, 35, 90–98. [Google Scholar] [CrossRef]
- Tran, D.H.; Wang, Z.V. Glucose Metabolism in Cardiac Hypertrophy and Heart Failure. J. Am. Heart Assoc. 2019, 8, e012673. [Google Scholar] [CrossRef] [PubMed]
- Rabinowitz, J.D.; Enerbäck, S. Lactate: The Ugly Duckling of Energy Metabolism. Nat. Metab. 2020, 2, 566–571. [Google Scholar] [CrossRef]
- Urbańska, K.; Orzechowski, A. Unappreciated Role of LDHA and LDHB to Control Apoptosis and Autophagy in Tumor Cells. Int. J. Mol. Sci. 2019, 20, 2085. [Google Scholar] [CrossRef]
- Taegtmeyer, H.; Overturf, M.L. Effects of Moderate Hypertension on Cardiac Function and Metabolism in the Rabbit. Hypertension 1988, 11, 416–426. [Google Scholar] [CrossRef] [PubMed]
- Sousa Fialho, M.D.L.; Abd Jamil, A.H.; Stannard, G.A.; Heather, L.C. Hypoxia-Inducible Factor 1 Signalling, Metabolism and Its Therapeutic Potential in Cardiovascular Disease. Biochim. Biophys. Acta Mol. Basis. Dis. 2019, 1865, 831–843. [Google Scholar] [CrossRef] [PubMed]
- Mota, G.A.F.; de Souza, S.L.B.; da Silva, V.L.; Gatto, M.; de Campos, D.H.S.; Sant’Ana, P.G.; Vileigas, D.F.; Padovani, C.R.; Casarini, D.E.; de Oliveira, E.M.; et al. Cardioprotection Generated by Aerobic Exercise Training Is Not Related to the Proliferation of Cardiomyocytes and Angiotensin-(1-7) Levels in the Hearts of Rats with Supravalvar Aortic Stenosis. Cell. Physiol. Biochem. 2020, 54, 719–735. [Google Scholar] [CrossRef]
- De Souza, S.L.B.; Mota, G.A.F.; da Silva, V.L.; Sant’Ana, P.G.; Vileigas, D.F.; de Campos, D.H.S.; Padovani, C.R.; Rodrigues, M.A.M.; do Nascimento, A.F.; Sugizaki, M.M.; et al. Adjustments in β-Adrenergic Signaling Contribute to the Amelioration of Cardiac Dysfunction by Exercise Training in Supravalvular Aortic Stenosis. Cell. Physiol. Biochem. 2020, 54, 665–681. [Google Scholar] [CrossRef]
- Mazeto, I.F.S.; Okoshi, K.; Silveira, C.F.S.M.P.; Sant’Ana, P.G.; da Silva, V.L.; Mota, G.A.F.; de Souza, S.L.B.; Vileigas, D.F.; Padovani, C.R.; Cicogna, A.C. Calcium Homeostasis Behavior and Cardiac Function on Left Ventricular Remodeling by Pressure Overload. Braz. J. Med. Biol. Res. 2021, 54, e10138. [Google Scholar] [CrossRef]
- Casquel De Tomasi, L.; Salomé Campos, D.H.; Grippa Sant’Ana, P.; Okoshi, K.; Padovani, C.R.; Masahiro Murata, G.; Nguyen, S.; Kolwicz, S.C.; Cicogna, A.C. Pathological Hypertrophy and Cardiac Dysfunction Are Linked to Aberrant Endogenous Unsaturated Fatty Acid Metabolism. PLoS ONE 2018, 13, e0193553. [Google Scholar] [CrossRef]
- Souza, R.W.A.; Piedade, W.P.; Soares, L.C.; Souza, P.A.T.; Aguiar, A.F.; Vechetti-Júnior, I.J.; Campos, D.H.S.; Fernandes, A.A.H.; Okoshi, K.; Carvalho, R.F.; et al. Aerobic Exercise Training Prevents Heart Failure-Induced Skeletal Muscle Atrophy by Anti-Catabolic, but Not Anabolic Actions. PLoS ONE 2014, 9, e110020. [Google Scholar] [CrossRef]
- Momken, I.; Kahapip, J.; Bahi, L.; Badoual, T.; Hittinger, L.; Ventura-Clapier, R.; Veksler, V. Does Angiotensin-Converting Enzyme Inhibition Improve the Energetic Status of Cardiac and Skeletal Muscles in Heart Failure Induced by Aortic Stenosis in Rats? J. Mol. Cell. Cardiol. 2003, 35, 399–407. [Google Scholar] [CrossRef]
- Moreira, V.O.; de Castro, A.V.B.; Yaegaschi, M.Y.; Cicogna, A.C.; Okoshi, M.P.; Pereira, C.A.; Aragon, F.F.; Bruno, M.B.; Padovani, C.R.; Okoshi, K. Echocardiographic Criteria for the Definition of Ventricular Dysfunction Severity in Aortic Banded Rats. Arq. Bras. Cardiol. 2006, 86, 432–438. [Google Scholar] [CrossRef] [PubMed]
- Crabtree, B.; Newsholme, E.A. The Activities of Phosphorylase, Hexokinase, Phosphofructokinase, Lactate Dehydrogenase and the Glycerol 3-Phosphate Dehydrogenases in Muscles from Vertebrates and Invertebrates. Biochem. J. 1972, 126, 49–58. [Google Scholar] [CrossRef] [PubMed]
- Alp, P.R.; Newsholme, E.A.; Zammit, V.A. Activities of Citrate Synthase and NAD+-Linked and NADP+-Linked Isocitrate Dehydrogenase in Muscle from Vertebrates and Invertebrates. Biochem. J. 1976, 154, 689–700. [Google Scholar] [CrossRef]
- Trinder, P. Determination of Blood Glucose Using an Oxidase-Peroxidase System with a Non-Carcinogenic Chromogen. J. Clin. Pathol. 1969, 22, 158–161. [Google Scholar] [CrossRef] [PubMed]
- Zar, J.H. Bioestatical Analysis, 5th ed.; Pearson Prentice Hall: Hoboken, NJ, USA, 2010. [Google Scholar]
- Mingoti, S.M. Análise de Dados Através de Métodos de Estatística Multivariada: Uma Abordagem Aplicada, 1st ed.; UFMG: Belo Horizonte, Brazil, 2005. [Google Scholar]











{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Signs of HF | Grups | Intensity and RF |
|---|---|---|
| Respiratory pattern change | ShamHF | (−) |
| AoSHF | (+++) 100% | |
| Ascites | ShamHF | (−) |
| AoSHF | (+++) 100% | |
| Pleural effusion | ShamHF | (−) |
| AoSHF | (+++) 100% | |
| Thrombus in LA | ShamHF | (−) |
| AoSHF | (++) 20%; (+++) 80% | |
| Hypertrophy of RV | ShamHF | (−) |
| AoSHF | (++) 10%; (+++) 90% |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Sant’Ana, P.G.; Tomasi, L.C.d.; Murata, G.M.; Vileigas, D.F.; Mota, G.A.F.; Souza, S.L.B.d.; Silva, V.L.; Campos, L.P.d.; Okoshi, K.; Padovani, C.R.; et al. Hypoxia-Inducible Factor 1-Alpha and Glucose Metabolism during Cardiac Remodeling Progression from Hypertrophy to Heart Failure. Int. J. Mol. Sci. 2023, 24, 6201. https://doi.org/10.3390/ijms24076201
Sant’Ana PG, Tomasi LCd, Murata GM, Vileigas DF, Mota GAF, Souza SLBd, Silva VL, Campos LPd, Okoshi K, Padovani CR, et al. Hypoxia-Inducible Factor 1-Alpha and Glucose Metabolism during Cardiac Remodeling Progression from Hypertrophy to Heart Failure. International Journal of Molecular Sciences. 2023; 24(7):6201. https://doi.org/10.3390/ijms24076201
Chicago/Turabian StyleSant’Ana, Paula Grippa, Loreta Casquel de Tomasi, Gilson Masahiro Murata, Danielle Fernandes Vileigas, Gustavo Augusto Ferreira Mota, Sérgio Luiz Borges de Souza, Vitor Loureiro Silva, Livia Paschoalino de Campos, Katashi Okoshi, Carlos Roberto Padovani, and et al. 2023. "Hypoxia-Inducible Factor 1-Alpha and Glucose Metabolism during Cardiac Remodeling Progression from Hypertrophy to Heart Failure" International Journal of Molecular Sciences 24, no. 7: 6201. https://doi.org/10.3390/ijms24076201
APA StyleSant’Ana, P. G., Tomasi, L. C. d., Murata, G. M., Vileigas, D. F., Mota, G. A. F., Souza, S. L. B. d., Silva, V. L., Campos, L. P. d., Okoshi, K., Padovani, C. R., & Cicogna, A. C. (2023). Hypoxia-Inducible Factor 1-Alpha and Glucose Metabolism during Cardiac Remodeling Progression from Hypertrophy to Heart Failure. International Journal of Molecular Sciences, 24(7), 6201. https://doi.org/10.3390/ijms24076201