
Platelets and Cardioprotection: The Role of Nitric Oxide and Carbon Oxide



Abstract
1. Introduction
2. Cardioprotection
3. Some Aspects of Platelet Activation
NO and Platelets
4. Role of NO in Cardioprotection and Platelets
5. The Role of CO in Cardioprotection and Platelet
5.1. CO and Cardioprotection
5.2. CO and Platelets
6. Conclusions
Author Contributions
Funding
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
| ADP | adenosine diphosphate |
| ANP | atrial NP |
| ATP | adenosine triphosphate |
| BNP | brain NP |
| CNP | C-type NP |
| CO | carbon monoxide |
| CORM | CO donors |
| eNOS | endothelial NOS |
| HNO | nitroxyl |
| HO-1 | heme oxygenase 1 |
| iNOS | inducible NOS |
| IP | ischemic preconditioning |
| IR | ischemia reperfusion |
| IRI | ischemia reperfusion injury |
| JAK | Janus kinase |
| KATP | ATP-sensing potassium channels |
| LPS | lipopolysaccharide |
| mitoKATP | mitochondrial KATP channel |
| MOM | mitochondrial outer membrane |
| MPTP | mitochondrial permeability transition pore |
| NAC | N-acetyl-L-cysteine |
| NETs | neutrophil extracellular traps |
| NO | nitric oxide |
| NOS | nitric oxide synthase |
| NPs | natriuretic peptides |
| O2− | superoxide anion |
| ONOO− | peroxynitrite |
| P2Y12 | purinergic Y-type 12 |
| PAF | platelet activating factor phosphoglyceride |
| pGC | particulate GC |
| PKC | protein kinase C |
| PKG | cGMP-dependent protein kinase |
| PMN | polymorphonuclear |
| PMVs | plasma membrane-derived vesicles |
| PostC | Postconditioning |
| RIC | remote ischemic conditioning |
| RIPC | remote ischemic preconditioning |
| RiPerC | remote ischemic perconditioning |
| RiPostC | remote ischemic postconditioning |
| RISK | reperfusion injury salvage kinase |
| RNS | reactive nitrogen species |
| ROS | reactive oxygen species |
| S1P | sphingosine-1-phosphate |
| SAFE | enhancement of survival activating factor |
| sGC | soluble guanylate cyclase |
| SOD | superoxide dismutase |
| STAT3 | signal transducer and activator of transcription 3 |
| TXA2 | thromboxane A2 |
| VEGF | vascular endothelial growth factor |
| ZnPP-IX | zinc protoporphyrin IX |
References
- Virani, S.S.; Alonso, A.; Benjamin, E.J.; Bittencourt, M.S.; Callaway, C.W.; Carson, A.P.; Chamberlain, A.M.; Chang, A.R.; Cheng, S.; Delling, F.N.; et al. Heart Disease and Stroke Statistics-2020 Update: A Report From the American Heart Association. Circulation 2020, 141, e139–e596. [Google Scholar] [CrossRef] [PubMed]
- Yellon, D.M.; Hausenloy, D.J. Myocardial Reperfusion Injury. N. Engl. J. Med. 2007, 357, 1121–1135. [Google Scholar] [CrossRef] [PubMed]
- Davidson, S.M.; Adameová, A.; Barile, L.; Cabrera-Fuentes, H.A.; Lazou, A.; Pagliaro, P.; Stensløkken, K.-O.; Garcia-Dorado, D. EU-Cardioprotection Cost Action (CA16225) Mitochondrial and Mitochondrial-Independent Pathways of Myocardial Cell Death during Ischaemia and Reperfusion Injury. J. Cell. Mol. Med. 2020, 24, 3795–3806. [Google Scholar] [CrossRef] [PubMed]
- Sun, J.; Druhan, L.J.; Zweier, J.L. Reactive Oxygen and Nitrogen Species Regulate Inducible Nitric Oxide Synthase Function Shifting the Balance of Nitric Oxide and Superoxide Production. Arch. Biochem. Biophys. 2010, 494, 130–137. [Google Scholar] [CrossRef]
- Hałucha, K.; Rak-Pasikowska, A.; Bil-Lula, I. Protective Role of Platelets in Myocardial Infarction and Ischemia/Reperfusion Injury. Cardiol. Res. Pract. 2021, 2021, 5545416. [Google Scholar] [CrossRef]
- Valikeserlis, I.; Athanasiou, A.-A.; Stakos, D. Cellular Mechanisms and Pathways in Myocardial Reperfusion Injury. Coron. Artery Dis. 2021, 32, 567–577. [Google Scholar] [CrossRef]
- Ibáñez, B.; Heusch, G.; Ovize, M.; Van de Werf, F. Evolving Therapies for Myocardial Ischemia/Reperfusion Injury. J. Am. Coll. Cardiol. 2015, 65, 1454–1471. [Google Scholar] [CrossRef]
- Penna, C.; Rastaldo, R.; Mancardi, D.; Raimondo, S.; Cappello, S.; Gattullo, D.; Losano, G.; Pagliaro, P. Post-Conditioning Induced Cardioprotection Requires Signaling through a Redox-Sensitive Mechanism, Mitochondrial ATP-Sensitive K+ Channel and Protein Kinase C Activation. Basic Res. Cardiol. 2006, 101, 180–189. [Google Scholar] [CrossRef]
- Downey, J.M.; Cohen, M.V. A Really Radical Observation. Basic Res. Cardiol 2006, 101, 190–191. [Google Scholar] [CrossRef]
- Tsutsumi, Y.M.; Yokoyama, T.; Horikawa, Y.; Roth, D.M.; Patel, H.H. Reactive Oxygen Species Trigger Ischemic and Pharmacological Postconditioning: In Vivo and In Vitro Characterization. Life Sci. 2007, 81, 1223–1227. [Google Scholar] [CrossRef]
- Maslov, L.N.; Popov, S.V.; Mukhomedzyanov, A.V.; Naryzhnaya, N.V.; Voronkov, N.S.; Ryabov, V.V.; Boshchenko, A.A.; Khaliulin, I.; Prasad, N.R.; Fu, F.; et al. Reperfusion Cardiac Injury: Receptors and the Signaling Mechanisms. Curr. Cardiol. Rev. 2022, 18, 63–79. [Google Scholar] [CrossRef] [PubMed]
- Penna, C.; Bassino, E.; Alloatti, G. Platelet Activating Factor: The Good and the Bad in the Ischemic/Reperfused Heart. Exp. Biol. Med. 2011, 236, 390–401. [Google Scholar] [CrossRef] [PubMed]
- Schanze, N.; Bode, C.; Duerschmied, D. Platelet Contributions to Myocardial Ischemia/Reperfusion Injury. Front. Immunol. 2019, 10, 1260. [Google Scholar] [CrossRef] [PubMed]
- Russo, I.; Femminò, S.; Barale, C.; Tullio, F.; Geuna, S.; Cavalot, F.; Pagliaro, P.; Penna, C. Cardioprotective Properties of Human Platelets Are Lost in Uncontrolled Diabetes Mellitus: A Study in Isolated Rat Hearts. Front. Physiol. 2018, 9, 875. [Google Scholar] [CrossRef]
- Lieder, H.R.; Tsoumani, M.; Andreadou, I.; Schrör, K.; Heusch, G.; Kleinbongard, P. Platelet-Mediated Transfer of Cardioprotection by Remote Ischemic Conditioning and Its Abrogation by Aspirin But Not by Ticagrelor. Cardiovasc. Drugs Ther. 2022. [Google Scholar] [CrossRef] [PubMed]
- Penna, C.; Alloatti, G.; Cappello, S.; Gattullo, D.; Berta, G.; Mognetti, B.; Losano, G.; Pagliaro, P. Platelet-Activating Factor Induces Cardioprotection in Isolated Rat Heart Akin to Ischemic Preconditioning: Role of Phosphoinositide 3-Kinase and Protein Kinase C Activation. Am. J. Physiol. Heart Circ. Physiol. 2005, 288, H2512–H2520. [Google Scholar] [CrossRef] [PubMed]
- Vito, C.D.; Hadi, L.A.; Navone, S.E.; Marfia, G.; Campanella, R.; Mancuso, M.E.; Riboni, L. Platelet-Derived Sphingosine-1-Phosphate and Inflammation: From Basic Mechanisms to Clinical Implications. Platelets 2016, 27, 393–401. [Google Scholar] [CrossRef]
- Ziegler, M.; Alt, K.; Paterson, B.M.; Kanellakis, P.; Bobik, A.; Donnelly, P.S.; Hagemeyer, C.E.; Peter, K. Highly Sensitive Detection of Minimal Cardiac Ischemia Using Positron Emission Tomography Imaging of Activated Platelets. Sci. Rep. 2016, 6, 38161. [Google Scholar] [CrossRef]
- Ziegler, M.; Wang, X.; Peter, K. Platelets in Cardiac Ischaemia/Reperfusion Injury: A Promising Therapeutic Target. Cardiovasc. Res. 2019, 115, 1178–1188. [Google Scholar] [CrossRef]
- Maroko, P.R.; Libby, P.; Bloor, C.M.; Sobel, B.E.; Braunwald, E. Reduction by Hyaluronidase of Myocardial Necrosis Following Coronary Artery Occlusion. Circulation 1972, 46, 430–437. [Google Scholar] [CrossRef]
- Maroko, P.R.; Libby, P.; Sobel, B.E.; Bloor, C.M.; Sybers, H.D.; Shell, W.E.; Covell, J.W.; Braunwald, E. Effect of Glucose-Insulin-Potassium Infusion on Myocardial Infarction Following Experimental Coronary Artery Occlusion. Circulation 1972, 45, 1160–1175. [Google Scholar] [CrossRef] [PubMed]
- Murry, C.E.; Jennings, R.B.; Reimer, K.A. Preconditioning with Ischemia: A Delay of Lethal Cell Injury in Ischemic Myocardium. Circulation 1986, 74, 1124–1136. [Google Scholar] [CrossRef]
- Zhao, Z.-Q.; Corvera, J.S.; Halkos, M.E.; Kerendi, F.; Wang, N.-P.; Guyton, R.A.; Vinten-Johansen, J. Inhibition of Myocardial Injury by Ischemic Postconditioning during Reperfusion: Comparison with Ischemic Preconditioning. Am. J. Physiol. Heart Circ. Physiol. 2003, 285, H579–H588. [Google Scholar] [CrossRef] [PubMed]
- Tullio, F.; Angotti, C.; Perrelli, M.-G.; Penna, C.; Pagliaro, P. Redox Balance and Cardioprotection. Basic Res. Cardiol. 2013, 108, 392. [Google Scholar] [CrossRef]
- Heusch, G. Myocardial Ischaemia-Reperfusion Injury and Cardioprotection in Perspective. Nat. Rev. Cardiol. 2020, 17, 773–789. [Google Scholar] [CrossRef]
- Penna, C.; Perrelli, M.-G.; Pagliaro, P. Mitochondrial Pathways, Permeability Transition Pore, and Redox Signaling in Cardioprotection: Therapeutic Implications. Antioxid. Redox Signal 2013, 18, 556–599. [Google Scholar] [CrossRef] [PubMed]
- Boengler, K.; Heusch, G.; Schulz, R. Mitochondria in Postconditioning. Antioxid. Redox Signal 2011, 14, 863–880. [Google Scholar] [CrossRef]
- Rossello, X.; Yellon, D.M. The RISK Pathway and Beyond. Basic Res. Cardiol. 2018, 113, 2. [Google Scholar] [CrossRef]
- Lecour, S. Activation of the Protective Survivor Activating Factor Enhancement (SAFE) Pathway against Reperfusion Injury: Does It Go beyond the RISK Pathway? J. Mol. Cell. Cardiol. 2009, 47, 32–40. [Google Scholar] [CrossRef]
- Hadebe, N.; Cour, M.; Lecour, S. The SAFE pathway for cardioprotection: Is this a promising target? Basic Res. Cardiol. 2018, 113, 9. [Google Scholar] [CrossRef]
- Park, M.; Sandner, P.; Krieg, T. cGMP at the centre of attention: Emerging strategies for activating the cardioprotective PKG pathway. Basic Res. Cardiol. 2018, 113, 24. [Google Scholar] [CrossRef] [PubMed]
- Costa, A.D.; Pierre, S.V.; Cohen, M.V.; Downey, J.M.; Garlid, K.D. cGMP signalling in pre- and post-conditioning: The role of mitochondria. Cardiovasc. Res. 2007, 77, 344–352. [Google Scholar] [CrossRef] [PubMed]
- Le Page, S.; Bejan-Angoulvant, T.; Angoulvant, D.; Prunier, F. Remote ischemic conditioning and cardioprotection: A systematic review and meta-analysis of randomized clinical trials. Basic Res. Cardiol. 2015, 110, 11. [Google Scholar] [CrossRef] [PubMed]
- Battipaglia, I.; Scalone, G.; Milo, M.; Di Franco, A.; Lanza, G.A.; Crea, F. Upper arm intermittent ischaemia reduces exercise-related increase of platelet reactivity in patients with obstructive coronary artery disease. Heart 2011, 97, 1298–1303. [Google Scholar] [CrossRef] [PubMed]
- Lanza, G.A.; Cesarano, M.; De Vita, A.; Villano, A.; Milo, M.; Russo, G.; Crea, F. Effect of Remote Ischemic Preconditioning on Coronary Procedure-Related Impairment of Vascular Dilator Function. J. Am. Coll. Cardiol. 2016, 68, 2490–2492. [Google Scholar] [CrossRef]
- Davidson, S.M.; Andreadou, I.; Barile, L.; Birnbaum, Y.; Cabrera-Fuentes, H.A.; Cohen, M.V.; Downey, J.M.; Girao, H.; Pagliaro, P.; Penna, C.; et al. Circulating blood cells and extracellular vesicles in acute cardioprotection. Cardiovasc. Res. 2019, 115, 1156–1166. [Google Scholar] [CrossRef]
- Li, Z.; Delaney, M.K.; O’Brien, K.A.; Du, X. Signaling During Platelet Adhesion and Activation. Arter. Thromb. Vasc. Biol. 2010, 30, 2341–2349. [Google Scholar] [CrossRef]
- Semple, J.W.; Italiano, J.E.; Freedman, J. Platelets and the immune continuum. Nat. Rev. Immunol. 2011, 11, 264–274. [Google Scholar] [CrossRef]
- Barale, C.; Melchionda, E.; Morotti, A.; Russo, I. Prothrombotic Phenotype in COVID-19: Focus on Platelets. Int. J. Mol. Sci. 2021, 22, 13638. [Google Scholar] [CrossRef] [PubMed]
- Veer, C.V.; Van Der Poll, T.; De Stoppelaar, S.F. The role of platelets in sepsis. Thromb. Haemost. 2014, 112, 666–677. [Google Scholar] [CrossRef]
- Lufrano, M.; Balazy, M. Interactions of peroxynitrite and other nitrating substances with human platelets: The role of glutathione and peroxynitrite permeability. Biochem. Pharmacol. 2002, 65, 515–523. [Google Scholar] [CrossRef]
- Bearer, E.; Prakash, J.; Li, Z. Actin dynamics in platelets. Int. Rev. Cytol. 2002, 217, 137–182. [Google Scholar] [CrossRef] [PubMed]
- Begonja, A.J.; Pluthero, F.G.; Suphamungmee, W.; Giannini, S.; Christensen, H.; Leung, R.; Lo, R.W.; Nakamura, F.; Lehman, W.; Plomann, M.; et al. FlnA binding to PACSIN2 F-BAR domain regulates membrane tubulation in megakaryocytes and platelets. Blood 2015, 126, 80–88. [Google Scholar] [CrossRef] [PubMed]
- Moncada, S.; Higgs, E.A. Nitric Oxide and the Vascular Endothelium. In The Vascular Endothelium I. Handbook of Experimental Pharmacology; Moncada, S., Higgs, A., Eds.; Springer: Berlin/Heidelberg, Germany, 2006; Volume 176/I. [Google Scholar] [CrossRef]
- Smolenski, A. Novel roles of cAMP/cGMP-dependent signaling in platelets. J. Thromb. Haemost. 2012, 10, 167–176. [Google Scholar] [CrossRef] [PubMed]
- Gambaryan, S.; Tsikas, D. A review and discussion of platelet nitric oxide and nitric oxide synthase: Do blood platelets produce nitric oxide from l-arginine or nitrite? Amino Acids 2015, 47, 1779–1793. [Google Scholar] [CrossRef] [PubMed]
- Böhmer, A.; Gambaryan, S.; Tsikas, D. Human blood platelets lack nitric oxide synthase activity. Platelets 2014, 26, 583–588. [Google Scholar] [CrossRef] [PubMed]
- Radziwon-Balicka, A.; Lesyk, G.; Back, V.; Fong, T.; Loredo-Calderon, E.L.; Dong, B.; El-Sikhry, H.; A El-Sherbeni, A.; El-Kadi, A.; Ogg, S.; et al. Differential eNOS-signalling by platelet subpopulations regulates adhesion and aggregation. Cardiovasc. Res. 2017, 113, 1719–1731. [Google Scholar] [CrossRef]
- Preedy, M.E.J.; Baliga, R.S.; Hobbs, A.J. Multiplicity of Nitric Oxide and Natriuretic Peptide Signaling in Heart Failure. J. Cardiovasc. Pharmacol. 2020, 75, 370–384. [Google Scholar] [CrossRef]
- Blanton, R.M. cGMP Signaling and Modulation in Heart Failure. J. Cardiovasc. Pharmacol. 2020, 75, 385–398. [Google Scholar] [CrossRef]
- Dang, T.A.; Schunkert, H.; Kessler, T. cGMP Signaling in Cardiovascular Diseases: Linking Genotype and Phenotype. J. Cardiovasc. Pharmacol. 2020, 75, 516–525. [Google Scholar] [CrossRef]
- Dunkerly-Eyring, B.; Kass, D.A. Myocardial Phosphodiesterases and Their Role in cGMP Regulation: Linking Genotype and Phenotype. J. Cardiovasc. Pharmacol. 2020, 75, 483–493. [Google Scholar] [CrossRef] [PubMed]
- Degjoni, A.; Campolo, F.; Stefanini, L.; Venneri, M.A. The NO/cGMP/PKG pathway in platelets: The therapeutic potential of PDE5 inhibitors in platelet disorders. J. Thromb. Haemost. 2022, 20, 2465–2474. [Google Scholar] [CrossRef] [PubMed]
- Walter, U.; Gambaryan, S. cGMP and cGMP-Dependent Protein Kinase in Platelets and Blood Cells. In cGMP: Generators, Effectors and Therapeutic Implications; Springer: Berlin/Heidelberg, Germany, 2009; pp. 533–548. [Google Scholar] [CrossRef]
- Geiger, J.; Nolte, C.; Walter, U. Regulation of calcium mobilization and entry in human platelets by endothelium-derived factors. Am. J. Physiol. 1994, 267, C236–C244. [Google Scholar] [CrossRef]
- Subramanian, H.; Zahedi, R.P.; Sickmann, A.; Walter, U.; Gambaryan, S. Phosphorylation of CalDAG-GEFI by protein kinase A prevents Rap1b activation. J. Thromb. Haemost. 2013, 11, 1574–1582. [Google Scholar] [CrossRef]
- Tsikas, D.; Ikic, M.; Tewes, K.S.; Raida, M.; Frölich, J.C. Inhibition of platelet aggregation by S-nitroso-cysteine via cGMP-independent mechanisms: Evidence of inhibition of thromboxane A2 synthesis in human blood platelets. FEBS Lett. 1999, 442, 162–166. [Google Scholar] [CrossRef]
- Kobsar, A.; Simonis, S.; Klinker, E.; Koessler, A.; Kuhn, S.; Boeck, M.; Koessler, J. Specific inhibitory effects of the NO donor MAHMA/NONOate on human platelets. Eur. J. Pharmacol. 2014, 735, 169–176. [Google Scholar] [CrossRef] [PubMed]
- Butt, E.; Bernhardt, M.; Smolenski, A.; Kotsonis, P.; Fröhlich, L.G.; Sickmann, A.; Meyer, H.E.; Lohmann, S.M.; Schmidt, H.H.H.W. Endothelial Nitric-oxide Synthase (Type III) Is Activated and Becomes Calcium Independent upon Phosphorylation by Cyclic Nucleotide-dependent Protein Kinases. J. Biol. Chem. 2000, 275, 5179–5187. [Google Scholar] [CrossRef]
- Boo, Y.C.; Sorescu, G.; Boyd, N.; Shiojima, I.; Walsh, K.; Du, J.; Jo, H. Shear Stress Stimulates Phosphorylation of Endothelial Nitric-oxide Synthase at Ser1179 by Akt-independent Mechanisms: Role of Protein Kinase A. J. Biol. Chem. 2002, 277, 3388–3396. [Google Scholar] [CrossRef]
- Queen, L.R.; Xu, B.; Horinouchi, K.; Fisher, I.; Ferro, A. β2 -Adrenoceptors Activate Nitric Oxide Synthase in Human Platelets. Circ. Res. 2000, 87, 39–44. [Google Scholar] [CrossRef]
- Schulz, C.; Fichtlscherer, B.; Kemp, B.E.; Fisslthaler, B.; Busse, R.; Fleming, I. AMP-activated protein kinase (AMPK) regulates the insulin-induced activation of the nitric oxide synthase in human platelets. Thromb. Haemost. 2003, 90, 863–871. [Google Scholar] [CrossRef]
- O’Kane, P.; Xie, L.; Liu, Z.; Queen, L.; Jackson, G.; Ji, Y.; Ferro, A. Aspirin acetylates nitric oxide synthase type 3 in platelets thereby increasing its activity. Cardiovasc. Res. 2009, 83, 123–130. [Google Scholar] [CrossRef] [PubMed]
- Russo, I.; Doronzo, G.; Mattiello, L.; De Salve, A.; Trovati, M.; Anfossi, G. The activity of constitutive nitric oxide synthase is increased by the pathway cAMP/cAMP-activated protein kinase in human platelets. New insights into the antiaggregating effects of cAMP-elevating agents. Thromb. Res. 2004, 114, 265–273. [Google Scholar] [CrossRef] [PubMed]
- Radomski, M.; Palmer, R.; Moncada, S. Characterization of the l-arginine: Nitric oxide pathway in human platelets. Br. J. Pharmacol. 1990, 101, 325–328. [Google Scholar] [CrossRef] [PubMed]
- Radomski, M.W.; Palmer, R.M.; Moncada, S. An L-arginine/nitric oxide pathway present in human platelets regulates aggregation. Proc. Natl. Acad. Sci. USA 1990, 87, 5193–5197. [Google Scholar] [CrossRef] [PubMed]
- Patel, B.; Sharifi, M.; Milward, A.D.; Oberprieler, N.G.; Gibbins, J.M.; Parkin, S.; Naseem, K.M. Platelet nitric oxide synthase is activated by tyrosine dephosphorylation: Possible role for SHP-1 phosphatase. J. Thromb. Haemost. 2006, 4, 2423–2432. [Google Scholar] [CrossRef]
- Mehta, J.L.; Chen, L.Y.; Kone, B.C.; Mehta, P.; Turner, P. Identification of constitutive and inducible forms of nitric oxide synthase in human platelets. J. Lab. Clin. Med. 1995, 125, 370–377. [Google Scholar]
- Radomski, M.W.; Zakar, T.; Salas, E. [9] Nitric oxide in platelets. In Methods in Enzymology; Academic Press: Cambridge, MA, USA, 1996; Volume 269, pp. 88–107. [Google Scholar] [CrossRef]
- Loscalzo, J.; Jin, R.C. Vascular nitric oxide: Formation and function. J. Blood Med. 2010, 1, 147–162. [Google Scholar] [CrossRef]
- Frijhoff, J.; Winyard, P.G.; Zarkovic, N.; Davies, S.S.; Stocker, R.; Cheng, D.; Knight, A.R.; Taylor, E.L.; Oettrich, J.; Ruskovska, T.; et al. Clinical Relevance of Biomarkers of Oxidative Stress. Antioxid. Redox Signal. 2015, 23, 1144–1170. [Google Scholar] [CrossRef]
- Morotti, A.; Barale, C.; Melchionda, E.; Russo, I. Platelet Redox Imbalance in Hypercholesterolemia: A Big Problem for a Small Cell. Int. J. Mol. Sci. 2022, 23, 11446. [Google Scholar] [CrossRef]
- Krötz, F.; Sohn, H.-Y.; Pohl, U. Reactive Oxygen Species: Players in the Platelet Game. Arter. Thromb. Vasc. Biol. 2004, 24, 1988–1996. [Google Scholar] [CrossRef]
- A Moro, M.; Darley-Usmar, V.M.; A Goodwin, D.; Read, N.G.; Zamora-Pino, R.; Feelisch, M.; Radomski, M.W.; Moncada, S. Paradoxical fate and biological action of peroxynitrite on human platelets. Proc. Natl. Acad. Sci. USA 1994, 91, 6702–6706. [Google Scholar] [CrossRef] [PubMed]
- Mondoro, T.H.; Shafer, B.C.; Vostal, J.G. Peroxynitrite-Induced Tyrosine Nitration and Phosphorylation in Human Platelets. Free Radic. Biol. Med. 1997, 22, 1055–1063. [Google Scholar] [CrossRef] [PubMed]
- Bruckdorfer, K. The nitration of proteins in platelets. Comptes Rendus L’académie Sci. Ser. III Sci. Vie 2001, 324, 611–615. [Google Scholar] [CrossRef]
- Sabetkar, M.; Low, S.Y.; Naseem, K.M.; Bruckdorfer, K. The nitration of proteins in platelets: Significance in platelet function1,2. Free Radic. Biol. Med. 2002, 33, 728–736. [Google Scholar] [CrossRef]
- Olas, B.; Nowak, P.; Ponczek, M.; Wachowicz, B. Resveratrol, a natural phenolic compound may reduce carbonylation proteins induced by peroxynitrite in blood platelets. Gen. Physiol. Biophys. 2006, 25, 215–222. [Google Scholar] [PubMed]
- Kabbani, S.S.; Watkins, M.W.; Ashikaga, T.; Terrien, E.F.; Holoch, P.A.; Sobel, B.E.; Schneider, D.J. Platelet Reactivity Characterized Prospectively: A Determinant of Outcome 90 Days after Percutaneous Coronary Intervention. Circulation 2001, 104, 181–186. [Google Scholar] [CrossRef] [PubMed]
- Trip, M.D.; Cats, V.M.; van Capelle, F.J.; Vreeken, J. Platelet Hyperreactivity and Prognosis in Survivors of Myocardial Infarction. N. Engl. J. Med. 1990, 322, 1549–1554. [Google Scholar] [CrossRef] [PubMed]
- Vanschoonbeek, K.; Feijge, M.A.H.; Keuren, J.F.W.; Hemker, H.C.; Lodder, J.J.; Hamulyák, K.; Van Pampus, E.C.M.; Heemskerk, J.W.M. Thrombin-induced hyperactivity of platelets of young stroke patients: Involvement of thrombin receptors in the subject-dependent variability in Ca2+ signal generation. Thromb. Haemost. 2002, 88, 931–937. [Google Scholar] [CrossRef]
- Barale, C.; Cavalot, F.; Frascaroli, C.; Bonomo, K.; Morotti, A.; Guerrasio, A.; Russo, I. Association between High On-Aspirin Platelet Reactivity and Reduced Superoxide Dismutase Activity in Patients Affected by Type 2 Diabetes Mellitus or Primary Hypercholesterolemia. Int. J. Mol. Sci. 2020, 21, 4983. [Google Scholar] [CrossRef]
- Barale, C.; Russo, I. Influence of Cardiometabolic Risk Factors on Platelet Function. Int. J. Mol. Sci. 2020, 21, 623. [Google Scholar] [CrossRef]
- Trovati, M.; Anfossi, G.; Massucco, P.; Mattiello, L.; Costamagna, C.; Piretto, V.; Mularoni, E.; Cavalot, F.; Bosia, A.; Ghigo, D. Insulin Stimulates Nitric Oxide Synthesis in Human Platelets and, Through Nitric Oxide, Increases Platelet Concentrations of Both Guanosine-3′, 5′-Cyclic Monophosphate and Adenosine-3′, 5′-Cyclic Monophosphate. Diabetes 1997, 46, 742–749. [Google Scholar] [CrossRef] [PubMed]
- Anfossi, G.; Russo, I.; Trovati, M. Platelet dysfunction in central obesity. Nutr. Metab. Cardiovasc. Dis. 2009, 19, 440–449. [Google Scholar] [CrossRef] [PubMed]
- Russo, I.; Del Mese, P.; Doronzo, G.; De Salve, A.; Secchi, M.; Trovati, M.; Anfossi, G. Platelet Resistance to the Antiaggregatory Cyclic Nucleotides in Central Obesity Involves Reduced Phosphorylation of Vasodilator-Stimulated Phosphoprotein. Clin. Chem. 2007, 53, 1053–1060. [Google Scholar] [CrossRef]
- Mammadova-Bach, E.; Nagy, M.; Heemskerk, J.W.; Nieswandt, B.; Braun, A. Store-operated calcium entry in thrombosis and thrombo-inflammation. Cell Calcium 2018, 77, 39–48. [Google Scholar] [CrossRef] [PubMed]
- Russo, I.; Traversa, M.; Bonomo, K.; De Salve, A.; Mattiello, L.; Del Mese, P.; Doronzo, G.; Cavalot, F.; Trovati, M.; Anfossi, G. In Central Obesity, Weight Loss Restores Platelet Sensitivity to Nitric Oxide and Prostacyclin. Obesity 2010, 18, 788–797. [Google Scholar] [CrossRef]
- Russo, I.; Penna, C.; Musso, T.; Popara, J.; Alloatti, G.; Cavalot, F.; Pagliaro, P. Platelets, diabetes and myocardial ischemia/reperfusion injury. Cardiovasc. Diabetol. 2017, 16, 71. [Google Scholar] [CrossRef] [PubMed]
- Anfossi, G.; Russo, I.; Massucco, P.; Mattiello, L.; Trovati, M. Platelet resistance to the antiaggregating effect of N-acetyl-l-cysteine in obese, insulin-resistant subjects. Thromb. Res. 2003, 110, 39–46. [Google Scholar] [CrossRef]
- Kinasewitz, G.T.; Yan, S.B.; Basson, B.; Comp, P.; A Russell, J.; Cariou, A.; Um, S.L.; Utterback, B.; Laterre, P.-F.; Dhainaut, J.-F. Universal changes in biomarkers of coagulation and inflammation occur in patients with severe sepsis, regardless of causative micro-organism [ISRCTN74215569]. Crit. Care 2004, 8, R82–R90. [Google Scholar] [CrossRef]
- Mehta, P.; McAuley, D.F.; Brown, M.; Sanchez, E.; Tattersall, R.S.; Manson, J.J. COVID-19: Consider cytokine storm syndromes and immunosuppression. Lancet 2020, 395, 1033–1034. [Google Scholar] [CrossRef]
- Pinheiro, M.B.M.; Rozini, S.V.; Quirino-Teixeira, A.C.; Barbosa-Lima, G.; Lopes, J.F.; Sacramento, C.Q.; Bozza, F.A.; Bozza, P.T.; Hottz, E.D. Dengue induces iNOS expression and nitric oxide synthesis in platelets through IL-1R. Front. Immunol. 2022, 13, 1029213. [Google Scholar] [CrossRef]
- Mancardi, D.; Pagliaro, P.; Ridnour, L.A.; Tocchetti, C.G.; Miranda, K.; Juhaszova, M.; Sollott, S.J.; Wink, D.A.; Paolocci, N. HNO Protects the Myocardium against Reperfusion Injury, Inhibiting the mPTP Opening via PKCε Activation. Antioxidants 2022, 11, 382. [Google Scholar] [CrossRef] [PubMed]
- Pagliaro, P.; Mancardi, D.; Rastaldo, R.; Penna, C.; Gattullo, D.; Miranda, K.M.; Feelisch, M.; A Wink, D.; A Kass, D.; Paolocci, N. Nitroxyl affords thiol-sensitive myocardial protective effects akin to early preconditioning. Free Radic. Biol. Med. 2003, 34, 33–43. [Google Scholar] [CrossRef] [PubMed]
- Penna, C.; Angotti, C.; Pagliaro, P. Protein S-nitrosylation in preconditioning and postconditioning. Exp. Biol. Med. 2014, 239, 647–662. [Google Scholar] [CrossRef] [PubMed]
- Shemarova, I.; Nesterov, V.; Emelyanova, L.; Korotkov, S. Mitochondrial mechanisms by which gasotransmitters (H2S, NO and CO) protect cardiovascular system against hypoxia. Front. Biosci. 2021, 13, 105–130. [Google Scholar] [CrossRef]
- Leary, P.J.; Rajasekaran, S.; Morrison, R.R.; Tuomanen, E.I.; Chin, T.K.; Hofmann, P.A. A cardioprotective role for platelet-activating factor through NOS-dependent S-nitrosylation. Am. J. Physiol. Circ. Physiol. 2008, 294, H2775–H2784. [Google Scholar] [CrossRef] [PubMed]
- Igarashi, J.; Michel, T. S1P and eNOS regulation. Biochim. Biophys. Acta 2008, 1781, 489–495. [Google Scholar] [CrossRef] [PubMed]
- Yokota, T.; Higuma, T.; Endo, T.; Nishizaki, F.; Hanada, K.; Yokoyama, H.; Yamada, M.; Okumura, K.; Tomita, H. Prasugrel versus clopidogrel for residual thrombus burden in patients with ST-segment elevation myocardial infarction: An Optical Coherence Tomog-raphy Study. Coron. Artery Dis. 2018, 29, 663–669. [Google Scholar] [CrossRef]
- Pandit, A.; Aryal, M.R.; Pandit, A.A.; Jalota, L.; Hakim, F.A.; Mookadam, F.; Lee, H.R.; Tleyjeh, I.M. Cangrelor versus clopidogrel in percutaneous coronary intervention: A systematic review and meta-analysis. Eurointervention 2014, 9, 1350–1358. [Google Scholar] [CrossRef]
- Ye, Y.; Birnbaum, G.D.; Perez-Polo, J.R.; Nanhwan, M.K.; Nylander, S.; Birnbaum, Y. Ticagrelor Protects the Heart Against Reperfusion Injury and Improves Remodeling After Myocardial Infarction. Arter. Thromb. Vasc. Biol. 2015, 35, 1805–1814. [Google Scholar] [CrossRef]
- Zhuang, Y.; Yu, M.-L.; Lu, S.-F. Purinergic signaling in myocardial ischemia–reperfusion injury. Purinergic Signal. 2023, 19, 229–243. [Google Scholar] [CrossRef]
- Penna, C.; Aragno, M.; Cento, A.S.; Femminò, S.; Russo, I.; Bello, F.D.; Chiazza, F.; Collotta, D.; Alves, G.F.; Bertinaria, M.; et al. Ticagrelor Conditioning Effects Are Not Additive to Cardioprotection Induced by Direct NLRP3 Inflammasome Inhibition: Role of RISK, NLRP3, and Redox Cascades. Oxidative Med. Cell. Longev. 2020, 2020, 9219825. [Google Scholar] [CrossRef] [PubMed]
- Bell, R.M.; Sivaraman, V.; Kunuthur, S.P.; Cohen, M.V.; Downey, J.M.; Yellon, D.M. Cardioprotective Properties of the Platelet P2Y12 Receptor Inhibitor, Cangrelor: Protective in Diabetics and Reliant Upon the Presence of Blood. Cardiovasc. Drugs Ther. 2015, 29, 415–418. [Google Scholar] [CrossRef] [PubMed]
- Dost, T. Cardioprotective properties of the platelet P2Y12 receptor inhibitor prasugrel on cardiac ischemia/reperfusion injury. Pharmacol. Rep. 2020, 72, 672–679. [Google Scholar] [CrossRef] [PubMed]
- Aungraheeta, R.; Conibear, A.; Butler, M.; Kelly, E.; Nylander, S.; Mumford, A.; Mundell, S.J. Inverse agonism at the P2Y12 receptor and ENT1 transporter blockade contribute to platelet inhibition by ticagrelor. Blood 2016, 128, 2717–2728. [Google Scholar] [CrossRef] [PubMed]
- Gimbel, M.; Qaderdan, K.; Willemsen, L.; Hermanides, R.; Bergmeijer, T.; de Vrey, E.; Heestermans, T.; Gin, M.T.J.; Waalewijn, R.; Hofma, S.; et al. Clopidogrel versus ticagrelor or prasugrel in patients aged 70 years or older with non-ST-elevation acute coronary syndrome (POPular AGE): The randomised, open-label, non-inferiority trial. Lancet 2020, 395, 1374–1381. [Google Scholar] [CrossRef]
- Knauert, M.; Vangala, S.; Haslip, M.; Lee, P.J. Therapeutic Applications of Carbon Monoxide. Oxidative Med. Cell. Longev. 2013, 2013, 360815. [Google Scholar] [CrossRef]
- Ryter, S.W. Therapeutic Potential of Heme Oxygenase-1 and Carbon Monoxide in Acute Organ Injury, Critical Illness, and Inflammatory Disorders. Antioxidants 2020, 9, 1153. [Google Scholar] [CrossRef]
- Brouard, S.; Otterbein, L.E.; Anrather, J.; Tobiasch, E.; Bach, F.H.; Choi, A.M.; Soares, M. Carbon Monoxide Generated by Heme Oxygenase 1 Suppresses Endothelial Cell Apoptosis. J. Exp. Med. 2000, 192, 1015–1026. [Google Scholar] [CrossRef]
- Di Lisa, F.; Canton, M.; Carpi, A.; Kaludercic, N.; Menabò, R.; Menazza, S.; Semenzato, M.; Valls-Lacalle, L.; Barba, I.; Miró-Casas, E.; et al. Mitochondrial Injury and Protection in Ischemic Pre- and Postconditioning. Antioxid. Redox Signal. 2011, 14, 881–891. [Google Scholar] [CrossRef]
- Xu, Z.; Ji, X.; Boysen, P.G. Exogenous nitric oxide generates ROS and induces cardioprotection: Involvement of PKG, mitochondrial KATP channels, and ERK. Am. J. Physiol. Heart Circ. Physiol. 2004, 286, H1433–H1440. [Google Scholar] [CrossRef]
- Chlopicki, S.; Olszanecki, R.; Marcinkiewicz, E.; Lomnicka, M.; Motterlini, R. Carbon monoxide released by CORM-3 inhibits human platelets by a mechanism independent of soluble guanylate cyclase. Cardiovasc. Res. 2006, 71, 393–401. [Google Scholar] [CrossRef] [PubMed]
- Wu, L.; Wang, R. Carbon Monoxide: Endogenous Production, Physiological Functions, and Pharmacological Applications. Pharmacol. Rev. 2005, 57, 585–630. [Google Scholar] [CrossRef] [PubMed]
- Marks, G.S.; Brien, J.F.; Nakatsu, K.; McLaughlin, B.E. Does carbon monoxide have a physiological function? Trends Pharmacol. Sci. 1991, 12, 185–188. [Google Scholar] [CrossRef] [PubMed]
- Brüne, B.; Ullrich, V. Inhibition of platelet aggregation by carbon monoxide is mediated by activation of guanylate cyclase. Mol. Pharmacol. 1987, 32, 497–504. [Google Scholar] [PubMed]
- Brüne, B.; Schmidt, K.-U.; Ullrich, V. Activation of soluble guanylate cyclase by carbon monoxide and inhibition by superoxide anion. JBIC J. Biol. Inorg. Chem. 1990, 192, 683–688. [Google Scholar] [CrossRef]
- Nowell, S.A.; Leakey, J.E.; Warren, J.F.; Lang, N.P.; Frame, L.T. Identification of Enzymes Responsible for the Metabolism of Heme in Human Platelets. J. Biol. Chem. 1998, 273, 33342–33346. [Google Scholar] [CrossRef] [PubMed]
- Morisaki, H.; Katayama, T.; Kotake, Y.; Ito, M.; Tamatani, T.; Sakamoto, S.; Ishimura, Y.; Takeda, J.; Suematsu, M. Roles of Carbon Monoxide in Leukocyte and Platelet Dynamics in Rat Mesenteric during Sevoflurane Anesthesia. Anesthesiology 2001, 95, 192–199. [Google Scholar] [CrossRef]
- Morisaki, H.; Katayama, T.; Kotake, Y.; Ito, M.; Handa, M.; Ikeda, Y.; Takeda, J.; Suematsu, M. Carbon Monoxide Modulates Endotoxin-induced Microvascular Leukocyte Adhesion through Platelet-dependent Mechanisms. Anesthesiology 2002, 97, 701–709. [Google Scholar] [CrossRef]
- Peng, L.; Mundada, L.; Stomel, J.M.; Liu, J.J.; Sun, J.; Yet, S.-F.; Fay, W.P. Induction of Heme Oxygenase-1 Expression Inhibits Platelet-Dependent Thrombosis. Antioxid. Redox Signal. 2004, 6, 729–735. [Google Scholar] [CrossRef]
- True, A.L.; Olive, M.; Boehm, M.; San, H.; Westrick, R.J.; Raghavachari, N.; Xu, X.; Lynn, E.G.; Sack, M.N.; Munson, P.J.; et al. Heme Oxygenase-1 Deficiency Accelerates Formation of Arterial Thrombosis Through Oxidative Damage to the Endothelium, Which Is Rescued by Inhaled Carbon Monoxide. Circ. Res. 2007, 101, 893–901. [Google Scholar] [CrossRef]
- Chen, B.; Guo, L.; Fan, C.; Bolisetty, S.; Joseph, R.; Wright, M.M.; Agarwal, A.; George, J.F. Carbon Monoxide Rescues Heme Oxygenase-1-Deficient Mice from Arterial Thrombosis in Allogeneic Aortic Transplantation. Am. J. Pathol. 2009, 175, 422–429. [Google Scholar] [CrossRef] [PubMed]
- Motterlini, R.; Mann, B.; Johnson, T.; Clark, J.; Foresti, R.; Green, C. Bioactivity and Pharmacological Actions of Carbon Monoxide-Releasing Molecules. Curr. Pharm. Des. 2003, 9, 2525–2539. [Google Scholar] [CrossRef] [PubMed]
- A Gende, O. Carbon monoxide inhibits capacitative calcium entry in human platelets. Thromb. Res. 2004, 114, 113–119. [Google Scholar] [CrossRef] [PubMed]
- Liu, D.; Liang, F.; Wang, X.; Cao, J.; Qin, W.; Sun, B. Suppressive Effect of CORM-2 on LPS-Induced Platelet Activation by Glycoprotein Mediated HS1 Phosphorylation Interference. PLoS ONE 2013, 8, e83112. [Google Scholar] [CrossRef] [PubMed]
- Nielsen, V.G.; Garza, J.I. Comparison of the effects of CORM-2, CORM-3 and CORM-A1 on coagulation in human plasma. Blood Coagul. Fibrinolysis 2014, 25, 801–805. [Google Scholar] [CrossRef]
- Vostal, J.G.; Fratantoni, J.C. Econazole inhibits thapsigargin-induced platelet calcium influx by mechanisms other than cytochrome P-450 inhibition. Biochem. J. 1993, 295, 525–529. [Google Scholar] [CrossRef][Green Version]
- Truss, N.J.; Warner, T.D. Gasotransmitters and platelets. Pharmacol. Ther. 2011, 132, 196–203. [Google Scholar] [CrossRef]
- Zhu, X.-Y.; Yan, X.-H.; Chen, S.-J. H(2)S protects myocardium against ischemia/reperfusion injury and its effect on c-Fos protein expression in rats. Sheng li xue bao Acta Physiol. Sin. 2008, 60, 221–227. [Google Scholar]
- Ananthakrishnan, R.; Li, Q.; O’Shea, K.M.; Quadri, N.; Wang, L.; Abuchowski, A.; Schmidt, A.M.; Ramasamy, R. Carbon monoxide form of PEGylated hemoglobin protects myocardium against ischemia/reperfusion injury in diabetic and normal mice. Artif. Cells Nanomed. Biotechnol. 2013, 41, 428–436. [Google Scholar] [CrossRef]
- Andreadou, I.; Iliodromitis, E.K.; Rassaf, T.; Schulz, R.; Papapetropoulos, A.; Ferdinandy, P. The role of gasotransmitters NO, H2S and CO in myocardial ischaemia/reperfusion injury and cardioprotection by preconditioning, postconditioning and remote conditioning. Br. J. Pharmacol. 2014, 172, 1587–1606. [Google Scholar] [CrossRef]
- Shao, J.; Miao, C.; Geng, Z.; Gu, M.; Wu, Y.; Li, Q. Effect of eNOS on Ischemic Postconditioning-Induced Autophagy against Ischemia/Reperfusion Injury in Mice. BioMed Res. Int. 2019, 2019, 5201014. [Google Scholar] [CrossRef]
- Zhang, J.; Cai, X.; Zhang, Q.; Li, X.; Li, S.; Ma, J.; Zhu, W.; Liu, X.; Wei, M.; Tu, W.; et al. Hydrogen sulfide restores sevoflurane postconditioning mediated cardioprotection in diabetic rats: Role of SIRT1/Nrf2 signaling-modulated mitochondrial dysfunction and oxidative stress: Nitric Oxide, Protein Kinases, and Mitochondria. J. Cell. Physiol. 2020, 236, 5052–5068. [Google Scholar] [CrossRef] [PubMed]
- Heusch, G.; Boengler, K.; Schulz, R. Cardioprotection: Nitric Oxide, Protein Kinases, and Mitochondria. Circulation 2008, 118, 1915–1919. [Google Scholar] [CrossRef] [PubMed]
- Pagliaro, P.; Moro, F.; Tullio, F.; Perrelli, M.-G.; Penna, C. Cardioprotective Pathways During Reperfusion: Focus on Redox Signaling and Other Modalities of Cell Signaling. Antioxid. Redox Signal. 2011, 14, 833–850. [Google Scholar] [CrossRef] [PubMed]



{kind=link}
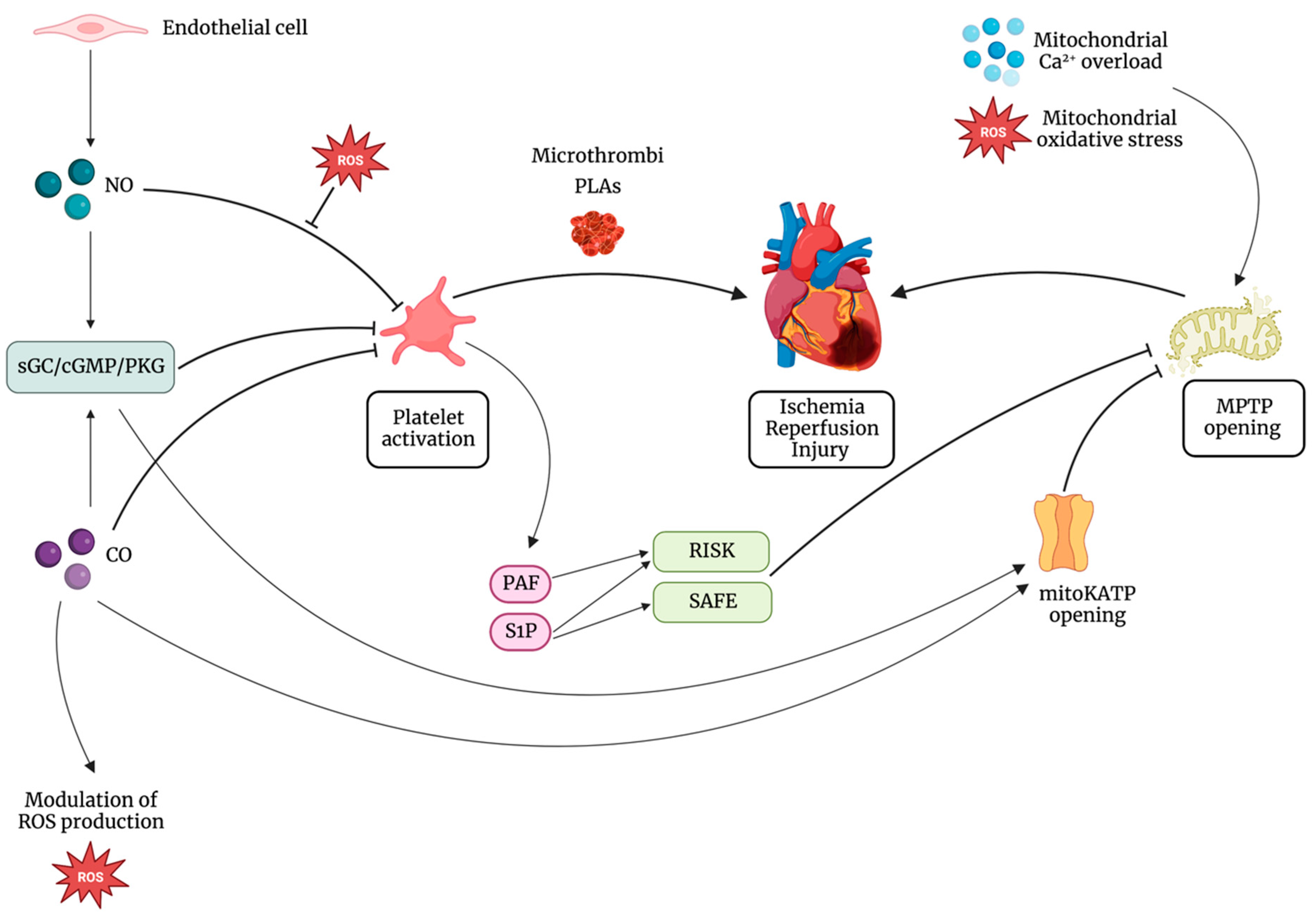{kind=link}
| Gas | Stimuli | Production | Main Pathway | Effect(s) |
|---|---|---|---|---|
| NO | Shear stress, VEGF, insulin | Endothelial cells (eNOS) | NO/cGMP/PKG | Vasodilation |
| [Ca]i increase, interaction protein (HSP70, HSP90, caveolin), insulin, β2 stimulation, acetylsalicylic acid, adenosine, and forskolin | Platelets (eNOS) | NO/cGMP/PKG | Reduction of adhesion, activation, and aggregation | |
| Inflammation | Platelets iNOS | NO/cGMP/PKG | Increased production of NO correlates with IL-1β | |
| Conditioning ischemia | Cardiac cells (eNOS or iNOS) | NO/cGMP/PKG S-nitrosylation | Cardioprotection | |
| HNO | Conditioning ischemia | Cardiac cells (eNOS?) | PKCε translocation to the mitochondria | Cardioprotection |
| ONOO− | Metabolic diseases | NO + O2− | nitration carbonylation and peroxidation | Alteration of haemostatic functions |
| CO | Hemin and sodium arsenite | Platelet HO-1 | cGMP/PKG | Reduction of aggregation and release of ADP and 5-HT |
| Conditioning ischemia | Cardiac cells HO-1 | Opening of KATP channel and closure of the MPTP. | Cardioprotection |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Russo, I.; Barale, C.; Melchionda, E.; Penna, C.; Pagliaro, P. Platelets and Cardioprotection: The Role of Nitric Oxide and Carbon Oxide. Int. J. Mol. Sci. 2023, 24, 6107. https://doi.org/10.3390/ijms24076107
Russo I, Barale C, Melchionda E, Penna C, Pagliaro P. Platelets and Cardioprotection: The Role of Nitric Oxide and Carbon Oxide. International Journal of Molecular Sciences. 2023; 24(7):6107. https://doi.org/10.3390/ijms24076107
Chicago/Turabian StyleRusso, Isabella, Cristina Barale, Elena Melchionda, Claudia Penna, and Pasquale Pagliaro. 2023. "Platelets and Cardioprotection: The Role of Nitric Oxide and Carbon Oxide" International Journal of Molecular Sciences 24, no. 7: 6107. https://doi.org/10.3390/ijms24076107
APA StyleRusso, I., Barale, C., Melchionda, E., Penna, C., & Pagliaro, P. (2023). Platelets and Cardioprotection: The Role of Nitric Oxide and Carbon Oxide. International Journal of Molecular Sciences, 24(7), 6107. https://doi.org/10.3390/ijms24076107