
N-Type Ca Channel in Epileptic Syndromes and Epilepsy: A Systematic Review of Its Genetic Variants

 , , ,
, , ,
Abstract
1. Introduction
1.1. Voltage-Dependent Calcium Channels


{kind=link}
{kind=link}
{kind=link}
| Channel Type | Channel Name | Gene (Pore-Forming Subunit) | Locus | LVA/HVA |
|---|---|---|---|---|
| P/Q | Cav2.1 | CACNA1A | 19p13.13 | HVA |
| N | Cav2.2 | CACNA1B | 9q34.3 | HVA |
| L | Cav1.2 | CACNA1C | 12p13.33 | HVA |
| L | Cav1.3 | CACNA1D | 3p21.1 | HVA |
| R | Cav2.3 | CACNA1E | 1q25.3 | HVA |
| L | Cav1.4 | CACNA1F | Xp11.23 | HVA |
| T | Cav3.1 | CACNA1G | 17q21.33 | LVA |
| T | Cav3.2 | CACNA1H | 16p13.3 | LVA |
| T | Cav3.3 | CACNA1I | 22q13.1 | LVA |
| L | Cav1.1 | CACNA1S | 1q32.1 | HVA |
| Channel Type | Conductance (pS) | Activation Potential (mV) | Inactivation Kinetics (ms) | Specific Blocker |
|---|---|---|---|---|
| L | 25 | ~10–50 | 150–2000 (slow) | dihydropyridines |
| N | 11–20 | ~20 | 100–200 (intermediate) | ω-conotoxin |
| P/Q | 15–16 | ~50 | 500–1000 (intermediate–slow) | ω-Agatoxin |
| R | 15–20 | ~25–40 | 50–100 (intermediate–fast) | SNX-482 |
| T | 8 | ~70 | 10–70 (fast) | Mibefradil |
| Gene | Subunit Name | Locus | OMIM * |
|---|---|---|---|
| CACNB1 | β1 | 17q12 | 114207 |
| CACNB2 | β2 | 10p12.33-p12.31 | 600003 |
| CACNB3 | β3 | 12q13.12 | 601958 |
| CACNB4 | β4 | 2q23.3 | 601949 |
| CACNA2D1 | α2δ1 | 7q21.11 | 114204 |
| CACNA2D2 | α2δ2 | 3p21.31 | 607082 |
| CACNA2D3 | α2δ3 | 3p21.1-p14.3 | 606399 |
| CACNA2D4 | α2δ4 | 12p13.33 | 608171 |
| CACNG1 | γ1 | 17q24.2 | 114209 |
| CACNG2 | γ2 | 22q12.3 | 602911 |
| CACNG3 | γ3 | 16p12.1 | 606403 |
| CACNG4 | γ4 | 17q24.2 | 606404 |
| CACNG5 | γ5 | 17q24.2 | 606405 |
| CACNG6 | γ6 | 19q13.42 | 606898 |
| CACNG7 | γ7 | 19q13.42 | 606899 |
| CACNG8 | γ8 | 19q13.42 | 606900 |
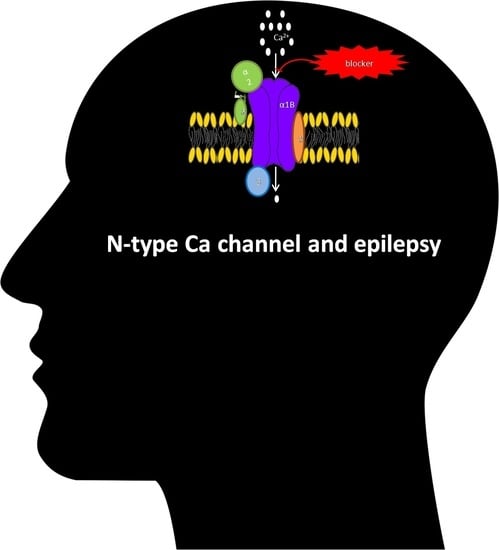
1.2. N-Type Ca Channel
2. Methods
3. Results and Discussion
3.1. N-Type Ca Channel and Epilepsy: Clinical Evidence
3.1.1. α1 Subunit: CACNA1B
3.1.2. α2δ Subunits: CACNA2D1 and CACNA2D2
CACNA2D1
| Ref. | Gene | Nº Patients | Epilepsy | Cognitive Involvement | MRI | Prenatal/ Birth | Other |
|---|---|---|---|---|---|---|---|
| [24] | CACNA1B | 6 | DEE | Yes (severe/regression) | Unknown | Normal | Hyperkinetic movement disorder, postnatal microcephaly, hypotonia. Death in childhood (5/6) |
| [25] | CACNA2D1 | 2 | DEE | Yes (severe) | Atrophy, thin corpus callosum | Normal | Facial dysmorphism, hyperkinetic movement disorder, insensibility to pain |
| [28] | CACNA2D1 | 3 | Focal epilepsy (2/3 refractory epilepsy) | Yes (mild to moderate) | 1 normal, 1 atrophy in the follow-up,1 frontotemporal polymicrogyria | Normal | 2 facial dysmorphism, 1 hyperinsulinism and obesity |
| [26] | CACNA2D1 | 1 | Epilepsy (no additional data) | Yes | Unknown | Unknown | |
| [27] | CACNA2D1 | 1 | DEE, infantile spasms | No | Normal | Unknown | Facial dysmorphism |
| [30] | CACNA2D2 | 3 | DEE | Yes (severe) | 2/3 paucity of white matter and cerebellar atrophy | Normal | Hyperkinetic movement disorder, hypotonia |
| [31] | CACNA2D2 | 1 | DEE | Yes | Cerebellar atrophy | Normal | Estrabismus, ocular apraxia |
| [29] | CACNA2D2 | 1 | DEE | Yes | Cerebellar atrophy | Fetal distress | Ataxia, hypotonia, atypical eye movements |
| [44] | CACNA2D2 | 1 | 1 febrile seizure | No (only motor delay) | Cerebellar atrophy | Ataxia, neonatal hypotonia, myoclonus | |
| [33] | CACNA2D2 | 3 | DEE | Yes (severe) | Cerebellar and brain atrophy | Normal | 1 Facial dysmorphism, 1 ataxia. Death in childhood (1/3) |
| [35] | CACNB4 | 10 | 3/10 generalized idiopathic epilepsy | No | Unknown | Unknown | 5/10 episodic ataxia, 2/10 asymptomatic |
| [36] | CACNB4 | 1 | Severe myoclonic epilepsy in infancy | Yes | Normal | Unknown | Ataxia. Death in childhood |
| [37] | CACNB4 | 1 | NA | NA | NA | NA | NA |
| [38] | CACNB4 | 2 | 1 DEE/1 focal epilepsy | Yes (severe) | Cerebellar atrophy | Normal | Hyperkinetic movement disorder, hypotonia |
| [40] | CACNG2 | 1 | Benign rolandic epilepsy | No | Unknown | Unknown |
CACNA2D2
3.1.3. β Subunit: CACNB4
3.1.4. γ Subunit: CACNG2
3.2. N-Type Ca Channel and Epilepsy: Evidences in Animal Models
| Animal | Tissue/Seizure Type | Effect | Ref. |
|---|---|---|---|
| rat | neocortex | Epileptiform activity in the rat neocortex may occur, at least partially, via the activation of the N-type VGCC. | [57] |
| mouse | dentate gyrus/CA1 hippocampus | Somatostatin (SST) reduction of dendritic Ca2+ through N-type VGCC may contribute to the modulation of synaptic plasticity at long-term potentiation (LTP) synapses. Loss of SST postseizure could contribute to epileptogenesis. | [64] |
| rat | pyramidal (Pyr) neurons of the sensorimotor cortical | Decreased calcium influx via N-type VGCC in presynaptic GABAergic terminals is a mechanism contributing to decreased inhibitory input onto layer V Pyr cells in this model of cortical posttraumatic epileptogenesis. | [55] |
| rat | frontal and occipital cortical/absence epilepsy | T and N-type VGCCs play activator roles in spike-wave discharges (SWDs) and have positive effects on the frequency and duration of these discharges. | [56] |
| rat | inferior colliculus neurons (IC)/ethanol withdrawal seizures | Ethanol withdrawal significantly decreased the protein levels of α1B subunit, specific for the N-Type VGCC, and secondary enhanced the susceptibility to seizures. | [63] |
| Xenopus | oocytes | Loss of neuronal protein stargazin is associated with recurrent epileptic seizures and ataxia in mice. Stargazin modulates neuronal N-type VGCC by a Gβγ-dependent mechanism. | [54] |
| rat | cerebral cortex synaptosomes | Crotoxin (Crtx) acts in the CNS, causing chronic seizure effects and other cytotoxic effects. Crtx induces calcium-dependent glutamate release via N and P/Q VGCC. | [65] |
| rat | hippocampus | Amygdala kindling induced an increase of N-type VGCC expression in the hippocampus. | [58] |
| rat | hippocampus | N-type VGCC trafficking mechanisms to cause a persistent, local, remodeling of their distributions in CA1 dendrites. | [59] |
| mouse | neurons and astrocytes/pilocarpine-induced status epilepticus (PISE) | N-type VGCC translocation occurs at acute stages during and after pilocarpine-induced status epilepticus (PISE). The increased expression of this channel in the strata granulosum and dentate gyrus at the chronic stage may be involved in the occurrence of spontaneously recurrent seizures. | [60] |
| gerbil | hippocampus | The elevated expressions of VGCC subtypes, including N-type VGCC, may increase Ca2+-dependent excitatory transmission in the hippocampus of the seizure-sensitive gerbil. | [61] |
| rat | hippocampus, entorhinal cortex, and neocortex | No overall age-related change in the number of L-type and N-type VGCC in brain areas frequently involved in seizure activity suggests that age-related changes in brain Ca2+ physiology may be associated with VGCC function rather than a channel number. | [66] |
| mouse | brain/audiogenic seizure | Development of N-Type VGCC in the brain is different in epileptic mice (generalized seizures when exposed to auditory stimulation) from nonepileptic mice. | [62] |
3.3. Anti-Epileptic-Drug Response and Therapeutic Target
4. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Catterall, W.A. Voltage-Gated Calcium Channels. Cold Spring Harb. Perspect. Biol. 2011, 3, a003947. [Google Scholar] [CrossRef] [PubMed]
- Catterall, W.A.; Perez-Reyes, E.; Snutch, T.P.; Striessnig, J. International Union of Pharmacology. XLVIII. Nomenclature and Structure-Function Relationships of Voltage-Gated Calcium Channels. Pharmacol. Rev. 2005, 57, 411–425. [Google Scholar] [CrossRef] [PubMed]
- Hille, B. Ion Channels of Excitable Membranes, 3rd ed.; Sinauer Associates: Sunderland, MA, USA, 2001. [Google Scholar]
- Meriney, S.D.; Fanselow, E.E. (Eds.) Calcium Homeostasis, Calcium Channels, and Transmitter Release. In Synaptic Transmission; Academic Press: Cambridge, MA, USA, 2019; pp. 121–153. ISBN 9780128153208. [Google Scholar]
- Klugbauer, N.; Marais, E.; Hofmann, F. Calcium Channel α2δ Subunits: Differential Expression, Function, and Drug Binding. J. Bioenerg. Biomembr. 2003, 35, 639–647. [Google Scholar] [CrossRef] [PubMed]
- Black, J.L., 3rd. The Voltage-Gated Calcium Channel Gamma Subunits: A Review of the Literature. J. Bioenerg. Biomembr. 2003, 35, 649–660. [Google Scholar] [CrossRef]
- Heron, S.E.; Phillips, H.A.; Mulley, J.C.; Mazarib, A.; Neufeld, M.Y.; Berkovic, S.F.; Scheffer, I.E. Genetic Variation of CACNA1H in Idiopathic Generalized Epilepsy. Ann. Neurol. 2004, 55, 595–596. [Google Scholar] [CrossRef]
- Heron, S.E.; Khosravani, H.; Varela, D.; Bladen, C.; Williams, T.C.; Newman, M.R.; Scheffer, I.E.; Berkovic, S.F.; Mulley, J.C.; Zamponi, G.W. Extended Spectrum of Idiopathic Generalized Epilepsies Associated with CACNA1H Functional Variants. Ann. Neurol. 2007, 62, 560–568. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.; Lu, J.; Pan, H.; Zhang, Y.; Wu, H.; Xu, K.; Liu, X.; Jiang, Y.; Bao, X.; Yao, Z.; et al. Association between Genetic Variation of CACNA1H and Childhood Absence Epilepsy. Ann. Neurol. 2003, 54, 239–243. [Google Scholar] [CrossRef]
- Calhoun, J.D.; Huffman, A.M.; Bellinski, I.; Kinsley, L.; Bachman, E.; Gerard, E.; Kearney, J.A.; Carvill, G.L. CACNA1H Variants Are Not a Cause of Monogenic Epilepsy. Hum. Mutat. 2020, 41, 1138–1144. [Google Scholar] [CrossRef]
- Gurkoff, G.; Shahlaie, K.; Lyeth, B.; Berman, R. Voltage-Gated Calcium Channel Antagonists and Traumatic Brain Injury. Pharmaceuticals 2013, 6, 788–812. [Google Scholar] [CrossRef]
- Jurkovicova-Tarabova, B.; Lacinova, L. Structure, Function and Regulation of Ca(V) 2.2 N-Type Calcium Channels. Gen. Physiol. Biophys. 2019, 38, 101–110. [Google Scholar] [CrossRef]
- Reynolds, I.J.; Wagner, J.A.; Snyder, S.H.; Thayer, S.A.; Olivera, B.M.; Miller, R.J. Brain Voltage-Sensitive Calcium Channel Subtypes Differentiated by Omega-Conotoxin Fraction GVIA. Proc. Natl. Acad. Sci. USA 1986, 83, 8804–8807. [Google Scholar] [CrossRef]
- Schroeder, C.I.; Doering, C.J.; Zamponi, G.W.; Lewis, R.J. N-Type Calcium Channel Blockers: Novel Therapeutics for the Treatment of Pain. Med. Chem. 2006, 2, 535–543. [Google Scholar] [CrossRef] [PubMed]
- De Jongh, K.S.; Warner, C.; Catterall, W.A. Subunits of Purified Calcium Channels. Alpha 2 and Delta Are Encoded by the Same Gene. J. Biol. Chem. 1990, 265, 14738–14741. [Google Scholar] [CrossRef]
- Davies, A.; Kadurin, I.; Alvarez-Laviada, A.; Douglas, L.; Nieto-Rostro, M.; Bauer, C.S.; Pratt, W.S.; Dolphin, A.C. The Alpha2delta Subunits of Voltage-Gated Calcium Channels Form GPI-Anchored Proteins, a Posttranslational Modification Essential for Function. Proc. Natl. Acad. Sci. USA 2010, 107, 1654–1659. [Google Scholar] [CrossRef] [PubMed]
- Cassidy, J.S.; Ferron, L.; Kadurin, I.; Pratt, W.S.; Dolphin, A.C. Functional Exofacially Tagged N-Type Calcium Channels Elucidate the Interaction with Auxiliary A2δ-1 Subunits. Proc. Natl. Acad. Sci. USA 2014, 111, 8979–8984. [Google Scholar] [CrossRef]
- Kadurin, I.; Ferron, L.; Rothwell, S.W.; Meyer, J.O.; Douglas, L.R.; Bauer, C.S.; Lana, B.; Margas, W.; Alexopoulos, O.; Nieto-Rostro, M.; et al. Proteolytic Maturation of α(2)δ Represents a Checkpoint for Activation and Neuronal Trafficking of Latent Calcium Channels. eLife 2016, 5, e21143. [Google Scholar] [CrossRef] [PubMed]
- Nieto-Rostro, M.; Ramgoolam, K.; Pratt, W.S.; Kulik, A.; Dolphin, A.C. Ablation of α2δ-1 Inhibits Cell-Surface Trafficking of Endogenous N-Type Calcium Channels in the Pain Pathway in Vivo. Proc. Natl. Acad. Sci. USA 2018, 115, E12043–E12052. [Google Scholar] [CrossRef]
- Buraei, Z.; Yang, J. The ß Subunit of Voltage-Gated Ca2+ Channels. Physiol. Rev. 2010, 90, 1461–1506. [Google Scholar] [CrossRef]
- Müller, C.S.; Haupt, A.; Bildl, W.; Schindler, J.; Knaus, H.-G.; Meissner, M.; Rammner, B.; Striessnig, J.; Flockerzi, V.; Fakler, B.; et al. Quantitative Proteomics of the Cav2 Channel Nano-Environments in the Mammalian Brain. Proc. Natl. Acad. Sci. USA 2010, 107, 14950–14957. [Google Scholar] [CrossRef]
- Murakami, M.; Fleischmann, B.; De Felipe, C.; Freichel, M.; Trost, C.; Ludwig, A.; Wissenbach, U.; Schwegler, H.; Hofmann, F.; Hescheler, J.; et al. Pain Perception in Mice Lacking the Beta3 Subunit of Voltage-Activated Calcium Channels. J. Biol. Chem. 2002, 277, 40342–40351. [Google Scholar] [CrossRef]
- Page, M.J.; McKenzie, J.E.; Bossuyt, P.M.; Boutron, I.; Hoffmann, T.C.; Mulrow, C.D.; Shamseer, L.; Tetzlaff, J.M.; Akl, E.A.; Brennan, S.E.; et al. The PRISMA 2020 Statement: An Updated Guideline for Reporting Systematic Reviews. BMJ 2021, 372, n71. [Google Scholar] [CrossRef] [PubMed]
- Gorman, K.M.; Meyer, E.; Grozeva, D.; Spinelli, E.; McTague, A.; Sanchis-Juan, A.; Carss, K.J.; Bryant, E.; Reich, A.; Schneider, A.L.; et al. Bi-Allelic Loss-of-Function CACNA1B Mutations in Progressive Epilepsy-Dyskinesia. Am. J. Hum. Genet. 2019, 104, 948–956. [Google Scholar] [CrossRef] [PubMed]
- Dahimene, S.; von Elsner, L.; Holling, T.; Mattas, L.S.; Pickard, J.; Lessel, D.; Pilch, K.S.; Kadurin, I.; Pratt, W.S.; Zhulin, I.B.; et al. Biallelic CACNA2D1 Loss-of-Function Variants Cause Early-Onset Developmental Epileptic Encephalopathy. Brain 2022, 145, 2721–2729. [Google Scholar] [CrossRef]
- Valentino, F.; Bruno, L.P.; Doddato, G.; Giliberti, A.; Tita, R.; Resciniti, S.; Fallerini, C.; Bruttini, M.; Lo Rizzo, C.; Mencarelli, M.A.; et al. Exome Sequencing in 200 Intellectual Disability/Autistic Patients: New Candidates and Atypical Presentations. Brain Sci. 2021, 11, 936. [Google Scholar] [CrossRef]
- Hino-Fukuyo, N.; Kikuchi, A.; Arai-Ichinoi, N.; Niihori, T.; Sato, R.; Suzuki, T.; Kudo, H.; Sato, Y.; Nakayama, T.; Kakisaka, Y.; et al. Genomic Analysis Identifies Candidate Pathogenic Variants in 9 of 18 Patients with Unexplained West Syndrome. Hum. Genet. 2015, 134, 649–658. [Google Scholar] [CrossRef]
- Vergult, S.; Dheedene, A.; Meurs, A.; Faes, F.; Isidor, B.; Janssens, S.; Gautier, A.; Le Caignec, C.; Menten, B. Genomic Aberrations of the CACNA2D1 Gene in Three Patients with Epilepsy and Intellectual Disability. Eur. J. Hum. Genet. 2015, 23, 628–632. [Google Scholar] [CrossRef]
- Butler, K.M.; Holt, P.J.; Milla, S.S.; da Silva, C.; Alexander, J.J.; Escayg, A. Epileptic Encephalopathy and Cerebellar Atrophy Resulting from Compound Heterozygous CACNA2D2 Variants. Case Rep. Genet. 2018, 2018, 6308283. [Google Scholar] [CrossRef]
- Edvardson, S.; Oz, S.; Abulhijaa, F.A.; Taher, F.B.; Shaag, A.; Zenvirt, S.; Dascal, N.; Elpeleg, O. Early Infantile Epileptic Encephalopathy Associated with a High Voltage Gated Calcium Channelopathy. J. Med. Genet. 2013, 50, 118–123. [Google Scholar] [CrossRef]
- Pippucci, T.; Parmeggiani, A.; Palombo, F.; Maresca, A.; Angius, A.; Crisponi, L.; Cucca, F.; Liguori, R.; Valentino, M.L.; Seri, M.; et al. A Novel Null Homozygous Mutation Confirms CACNA2D2 as a Gene Mutated in Epileptic Encephalopathy. PLoS ONE 2013, 8, e82154. [Google Scholar] [CrossRef]
- Courage, C.; Oliver, K.L.; Park, E.J.; Cameron, J.M.; Grabińska, K.A.; Muona, M.; Canafoglia, L.; Gambardella, A.; Said, E.; Afawi, Z.; et al. Progressive Myoclonus Epilepsies-Residual Unsolved Cases Have Marked Genetic Heterogeneity Including Dolichol-Dependent Protein Glycosylation Pathway Genes. Am. J. Hum. Genet. 2021, 108, 722–738. [Google Scholar] [CrossRef] [PubMed]
- Punetha, J.; Karaca, E.; Gezdirici, A.; Lamont, R.E.; Pehlivan, D.; Marafi, D.; Appendino, J.P.; Hunter, J.V.; Akdemir, Z.C.; Fatih, J.M.; et al. Biallelic CACNA2D2 Variants in Epileptic Encephalopathy and Cerebellar Atrophy. Ann. Clin. Transl. Neurol. 2019, 6, 1395–1406. [Google Scholar] [CrossRef]
- Truty, R.; Patil, N.; Sankar, R.; Sullivan, J.; Millichap, J.; Carvill, G.; Entezam, A.; Esplin, E.D.; Fuller, A.; Hogue, M.; et al. Possible Precision Medicine Implications from Genetic Testing Using Combined Detection of Sequence and Intragenic Copy Number Variants in a Large Cohort with Childhood Epilepsy. Epilepsia Open 2019, 4, 397–408. [Google Scholar] [CrossRef]
- Escayg, A.; De Waard, M.; Lee, D.D.; Bichet, D.; Wolf, P.; Mayer, T.; Johnston, J.; Baloh, R.; Sander, T.; Meisler, M.H. Coding and Noncoding Variation of the Human Calcium-Channel Beta4-Subunit Gene CACNB4 in Patients with Idiopathic Generalized Epilepsy and Episodic Ataxia. Am. J. Hum. Genet. 2000, 66, 1531–1539. [Google Scholar] [CrossRef] [PubMed]
- Ohmori, I.; Ouchida, M.; Miki, T.; Mimaki, N.; Kiyonaka, S.; Nishiki, T.; Tomizawa, K.; Mori, Y.; Matsui, H. A CACNB4 Mutation Shows That Altered Ca(v)2.1 Function May Be a Genetic Modifier of Severe Myoclonic Epilepsy in Infancy. Neurobiol. Dis. 2008, 32, 349–354. [Google Scholar] [CrossRef] [PubMed]
- Ko, A.; Youn, S.E.; Kim, S.H.; Lee, J.S.; Kim, S.; Choi, J.R.; Kim, H.D.; Lee, S.T.; Kang, H.C. Targeted Gene Panel and Genotype-Phenotype Correlation in Children with Developmental and Epileptic Encephalopathy. Epilepsy Res. 2018, 141, 48–55. [Google Scholar] [CrossRef]
- Coste de Bagneaux, P.; von Elsner, L.; Bierhals, T.; Campiglio, M.; Johannsen, J.; Obermair, G.J.; Hempel, M.; Flucher, B.E.; Kutsche, K. A Homozygous Missense Variant in CACNB4 Encoding the Auxiliary Calcium Channel Beta4 Subunit Causes a Severe Neurodevelopmental Disorder and Impairs Channel and Non-Channel Functions. PLoS Genet. 2020, 16, e1008625. [Google Scholar] [CrossRef] [PubMed]
- Klee, E.W.; Cousin, M.A.; e Vairo, F.P.; Morales-Rosado, J.A.; Macke, E.L.; Jenkinson, W.G.; Ferrer, A.; Schultz-Rogers, L.E.; Olson, R.J.; Oliver, G.R.; et al. Impact of Integrated Translational Research on Clinical Exome Sequencing. Genet. Med. Off. J. Am. Coll. Med. Genet. 2021, 23, 498–507. [Google Scholar] [CrossRef]
- Rudolf, G.; de Bellescize, J.; de Saint Martin, A.; Arzimanoglou, A.; Valenti Hirsch, M.P.; Labalme, A.; Boulay, C.; Simonet, T.; Boland, A.; Deleuze, J.F.; et al. Exome Sequencing in 57 Patients with Self-Limited Focal Epilepsies of Childhood with Typical or Atypical Presentations Suggests Novel Candidate Genes. Eur. J. Paediatr. Neurol. EJPN Off. J. Eur. Paediatr. Neurol. Soc. 2020, 27, 104–110. [Google Scholar] [CrossRef]
- Karczewski, K.J.; Francioli, L.C.; Tiao, G.; Cummings, B.B.; Alföldi, J.; Wang, Q.; Collins, R.L.; Laricchia, K.M.; Ganna, A.; Birnbaum, D.P.; et al. The Mutational Constraint Spectrum Quantified from Variation in 141,456 Humans. Nature 2020, 581, 434–443. [Google Scholar] [CrossRef]
- Itsara, A.; Cooper, G.M.; Baker, C.; Girirajan, S.; Li, J.; Absher, D.; Krauss, R.M.; Myers, R.M.; Ridker, P.M.; Chasman, D.I.; et al. Population Analysis of Large Copy Number Variants and Hotspots of Human Genetic Disease. Am. J. Hum. Genet. 2009, 84, 148–161. [Google Scholar] [CrossRef]
- Xu, H.; Poh, W.-T.; Sim, X.; Ong, R.T.-H.; Suo, C.; Tay, W.-T.; Khor, C.-C.; Seielstad, M.; Liu, J.; Aung, T.; et al. SgD-CNV, a Database for Common and Rare Copy Number Variants in Three Asian Populations. Hum. Mutat. 2011, 32, 1341–1349. [Google Scholar] [CrossRef] [PubMed]
- Valence, S.; Cochet, E.; Rougeot, C.; Garel, C.; Chantot-Bastaraud, S.; Lainey, E.; Afenjar, A.; Barthez, M.-A.; Bednarek, N.; Doummar, D.; et al. Exome Sequencing in Congenital Ataxia Identifies Two New Candidate Genes and Highlights a Pathophysiological Link between Some Congenital Ataxias and Early Infantile Epileptic Encephalopathies. Genet. Med. Off. J. Am. Coll. Med. Genet. 2019, 21, 553–563. [Google Scholar] [CrossRef]
- Brodbeck, J.; Davies, A.; Courtney, J.-M.; Meir, A.; Balaguero, N.; Canti, C.; Moss, F.J.; Page, K.M.; Pratt, W.S.; Hunt, S.P.; et al. The Ducky Mutation in Cacna2d2 Results in Altered Purkinje Cell Morphology and Is Associated with the Expression of a Truncated Alpha 2 Delta-2 Protein with Abnormal Function. J. Biol. Chem. 2002, 277, 7684–7693. [Google Scholar] [CrossRef]
- Burgess, D.L.; Jones, J.M.; Meisler, M.H.; Noebels, J.L. Mutation of the Ca2+ Channel Beta Subunit Gene Cchb4 Is Associated with Ataxia and Seizures in the Lethargic (Lh) Mouse. Cell 1997, 88, 385–392. [Google Scholar] [CrossRef]
- Tadmouri, A.; Kiyonaka, S.; Barbado, M.; Rousset, M.; Fablet, K.; Sawamura, S.; Bahembera, E.; Pernet-Gallay, K.; Arnoult, C.; Miki, T.; et al. Cacnb4 Directly Couples Electrical Activity to Gene Expression, a Process Defective in Juvenile Epilepsy. EMBO J. 2012, 31, 3730–3744. [Google Scholar] [CrossRef]
- Sandoval, A.; Andrade, A.; Beedle, A.M.; Campbell, K.P.; Felix, R. Inhibition of Recombinant N-Type Ca(V) Channels by the Gamma 2 Subunit Involves Unfolded Protein Response (UPR)-Dependent and UPR-Independent Mechanisms. J. Neurosci. Off. J. Soc. Neurosci. 2007, 27, 3317–3327. [Google Scholar] [CrossRef]
- Hamdan, F.F.; Gauthier, J.; Araki, Y.; Lin, D.-T.; Yoshizawa, Y.; Higashi, K.; Park, A.-R.; Spiegelman, D.; Dobrzeniecka, S.; Piton, A.; et al. Excess of de Novo Deleterious Mutations in Genes Associated with Glutamatergic Systems in Nonsyndromic Intellectual Disability. Am. J. Hum. Genet. 2011, 88, 306–316. [Google Scholar] [CrossRef]
- Noebels, J.L.; Qiao, X.; Bronson, R.T.; Spencer, C.; Davisson, M.T. Stargazer: A New Neurological Mutant on Chromosome 15 in the Mouse with Prolonged Cortical Seizures. Epilepsy Res. 1990, 7, 129–135. [Google Scholar] [CrossRef]
- Letts, V.A. Stargazer—A Mouse to Seize! Epilepsy Curr. 2005, 5, 161–165. [Google Scholar] [CrossRef] [PubMed]
- Miyamoto, N.; Namiki, K.; Tokuhara, N.; Uesugi, M.; Takahashi, E.; Kuromitsu, J.; Kasuya, Y. The Utilization of Gene Targeting Models during in Preclinical Study of Drug Discovery Process--Example of Phenotypic and Functional Analysis of Cacna1beta Gene Product. Curr. Pharm. Biotechnol. 2009, 10, 261–267. [Google Scholar] [CrossRef] [PubMed]
- Jeon, D.; Kim, C.; Yang, Y.-M.; Rhim, H.; Yim, E.; Oh, U.; Shin, H.-S. Impaired Long-Term Memory and Long-Term Potentiation in N-Type Ca2+ Channel-Deficient Mice. Genes Brain Behav. 2007, 6, 375–388. [Google Scholar] [CrossRef] [PubMed]
- Tselnicker, I.; Tsemakhovich, V.A.; Dessauer, C.W.; Dascal, N. Stargazin Modulates Neuronal Voltage-Dependent Ca2+ Channel Ca(v)2.2 by a Gbetagamma-Dependent Mechanism. J. Biol. Chem. 2010, 285, 20462–20471. [Google Scholar] [CrossRef] [PubMed]
- Faria, L.C.; Parada, I.; Prince, D.A. Interneuronal Calcium Channel Abnormalities in Posttraumatic Epileptogenic Neocortex. Neurobiol. Dis. 2012, 45, 821–828. [Google Scholar] [CrossRef]
- Durmus, N.; Gültürk, S.; Kaya, T.; Demir, T.; Parlak, M.; Altun, A. Evaluation of Effects of T and N Type Calcium Channel Blockers on the Electroencephalogram Recordings in Wistar Albino Glaxo/Rij Rats, an Absence Epilepsy Model. Indian J. Pharmacol. 2015, 47, 34–38. [Google Scholar] [CrossRef]
- Boulton, C.L.; O’Shaughnessy, C.T. The Effect of Calcium Channel Antagonists on Spontaneous and Evoked Epileptiform Activity in the Rat Neocortex In Vitro. Eur. J. Neurosci. 1991, 3, 992–1000. [Google Scholar] [CrossRef]
- Bernstein, G.M.; Mendonça, A.; Wadia, J.; McIntyre Burnham, W.; Jones, O.T. Kindling Induces an Asymmetric Enhancement of N-Type Ca2+ Channel Density in the Dendritic Fields of the Rat Hippocampus. Neurosci. Lett. 1999, 268, 155–158. [Google Scholar] [CrossRef]
- Bernstein, G.M.; Mendonça, A.; Wadia, J.; Burnham, W.M.; Jones, O.T. Kindling Induces a Long-Term Enhancement in the Density of N-Type Calcium Channels in the Rat Hippocampus. Neuroscience 1999, 94, 1083–1095. [Google Scholar] [CrossRef] [PubMed]
- Xu, J.H.; Long, L.; Wang, J.; Tang, Y.C.; Hu, H.T.; Soong, T.W.; Tang, F.R. Nuclear Localization of Ca(v)2.2 and Its Distribution in the Mouse Central Nervous System, and Changes in the Hippocampus during and after Pilocarpine-Induced Status Epilepticus. Neuropathol. Appl. Neurobiol. 2010, 36, 71–85. [Google Scholar] [CrossRef]
- Kang, T.C.; Kim, D.S.; Yoo, K.Y.; Hwang, I.K.; Kwak, S.E.; Kim, J.E.; Jung, J.Y.; Won, M.H.; Suh, J.G.; Oh, Y.S. Elevated Voltage-Gated Ca2+ Channel Immunoreactivities in the Hippocampus of Seizure-Prone Gerbil. Brain Res. 2004, 1029, 168–178. [Google Scholar] [CrossRef]
- Esplin, M.S.; Abbott, J.R.; Smart, M.L.; Burroughs, A.F.; Frandsen, T.C.; Litzinger, M.J. Voltage-Sensitive Calcium Channel Development in Epileptic DBA/2J Mice Suggests Altered Presynaptic Function. Epilepsia 1994, 35, 911–914. [Google Scholar] [CrossRef]
- N’Gouemo, P.; Yasuda, R.P.; Morad, M. Ethanol Withdrawal Is Accompanied by Downregulation of Calcium Channel Alpha 1B Subunit in Rat Inferior Colliculus Neurons. Brain Res. 2006, 1108, 216–220. [Google Scholar] [CrossRef] [PubMed]
- Baratta, M.V.; Lamp, T.; Tallent, M.K. Somatostatin Depresses Long-Term Potentiation and Ca2+ Signaling in Mouse Dentate Gyrus. J. Neurophysiol. 2002, 88, 3078–3086. [Google Scholar] [CrossRef] [PubMed]
- Lomeo, R.D.S.; de Faria Gonçalves, A.P.; da Silva, C.N.; de Paula, A.T.; Costa Santos, D.O.; Fortes-Dias, C.L.; Gomes, D.A.; de Lima, M.E. Crotoxin from Crotalus Durissus Terrificus Snake Venom Induces the Release of Glutamate from Cerebrocortical Synaptosomes via N and P/Q Calcium Channels. Toxicon 2014, 85, 5–16. [Google Scholar] [CrossRef] [PubMed]
- Kelly, K.M.; Kume, A.; Albin, R.L.; Macdonald, R.L. Autoradiography of L-Type and N-Type Calcium Channels in Aged Rat Hippocampus, Entorhinal Cortex, and Neocortex. Neurobiol. Aging 2001, 22, 17–23. [Google Scholar] [CrossRef]
- Zamani, M.; Budde, T.; Bozorgi, H. Intracerebroventricular Administration of N-Type Calcium Channel Blocker Ziconotide Displays Anticonvulsant, Anxiolytic, and Sedative Effects in Rats: A Preclinical and Pilot Study. Epilepsy Behav. 2020, 111, 107251. [Google Scholar] [CrossRef]
- Mattson, R.H.; Cramer, J.A.; Collins, J.F.; Smith, D.B.; Delgado-Escueta, A.V.; Browne, T.R.; Williamson, P.D.; Treiman, D.M.; McNamara, J.O.; McCutchen, C.B. Comparison of Carbamazepine, Phenobarbital, Phenytoin, and Primidone in Partial and Secondarily Generalized Tonic-Clonic Seizures. N. Engl. J. Med. 1985, 313, 145–151. [Google Scholar] [CrossRef]
- Perucca, E.; Gram, L.; Avanzini, G.; Dulac, O. Antiepileptic Drugs as a Cause of Worsening Seizures. Epilepsia 1998, 39, 5–17. [Google Scholar] [CrossRef]
- Abou-Khalil, B.W. Update on Antiseizure Medications 2022. Continuum 2022, 28, 500–535. [Google Scholar] [CrossRef]
- Privitera, M. Efficacy of Levetiracetam: A Review of Three Pivotal Clinical Trials. Epilepsia 2001, 42 (Suppl. S4), 31–35. [Google Scholar] [CrossRef]
- Vossler, D.G.; Knake, S.; O’Brien, T.J.; Watanabe, M.; Brock, M.; Steiniger-Brach, B.; Williams, P.; Roebling, R. Efficacy and Safety of Adjunctive Lacosamide in the Treatment of Primary Generalised Tonic-Clonic Seizures: A Double-Blind, Randomised, Placebo-Controlled Trial. J. Neurol. Neurosurg. Psychiatry 2020, 91, 1067–1075. [Google Scholar] [CrossRef]
- Wang, S.J.; Huang, C.C.; Hsu, K.S.; Tsai, J.J.; Gean, P.W. Inhibition of N-Type Calcium Currents by Lamotrigine in Rat Amygdalar Neurones. Neuroreport 1996, 7, 3037–3040. [Google Scholar] [CrossRef] [PubMed]
- Pisani, A.; Bonsi, P.; Martella, G.; De Persis, C.; Costa, C.; Pisani, F.; Bernardi, G.; Calabresi, P. Intracellular Calcium Increase in Epileptiform Activity: Modulation by Levetiracetam and Lamotrigine. Epilepsia 2004, 45, 719–728. [Google Scholar] [CrossRef] [PubMed]
- Stefani, A.; Spadoni, F.; Siniscalchi, A.; Bernardi, G. Lamotrigine Inhibits Ca2+ Currents in Cortical Neurons: Functional Implications. Eur. J. Pharmacol. 1996, 307, 113–116. [Google Scholar] [CrossRef] [PubMed]
- Zhu, G.; Okada, M.; Murakami, T.; Kawata, Y.; Kamata, A.; Kaneko, S. Interaction between Carbamazepine, Zonisamide and Voltage-Sensitive Ca2+ Channel on Acetylcholine Release in Rat Frontal Cortex. Epilepsy Res. 2002, 49, 49–60. [Google Scholar] [CrossRef]
- Schumacher, T.B.; Beck, H.; Steffens, R.; Blümcke, I.; Schramm, J.; Elger, C.E.; Steinhäuser, C. Modulation of Calcium Channels by Group I and Group II Metabotropic Glutamate Receptors in Dentate Gyrus Neurons from Patients with Temporal Lobe Epilepsy. Epilepsia 2000, 41, 1249–1258. [Google Scholar] [CrossRef]
- Vega-Hernández, A.; Felix, R. Down-Regulation of N-Type Voltage-Activated Ca2+ Channels by Gabapentin. Cell. Mol. Neurobiol. 2002, 22, 185–190. [Google Scholar] [CrossRef]
- Bertrand, S.; Nouel, D.; Morin, F.; Nagy, F.; Lacaille, J.-C. Gabapentin Actions on Kir3 Currents and N-Type Ca2+ Channels via GABAB Receptors in Hippocampal Pyramidal Cells. Synapse 2003, 50, 95–109. [Google Scholar] [CrossRef]
- Lukyanetz, E.A.; Shkryl, V.M.; Kostyuk, P.G. Selective Blockade of N-Type Calcium Channels by Levetiracetam. Epilepsia 2002, 43, 9–18. [Google Scholar] [CrossRef]
- Martella, G.; Bonsi, P.; Sciamanna, G.; Platania, P.; Madeo, G.; Tassone, A.; Cuomo, D.; Pisani, A. Seletracetam (Ucb 44212) Inhibits High-Voltage-Activated Ca2+ Currents and Intracellular Ca2+ Increase in Rat Cortical Neurons In Vitro. Epilepsia 2009, 50, 702–710. [Google Scholar] [CrossRef]
- Moutal, A.; François-Moutal, L.; Perez-Miller, S.; Cottier, K.; Chew, L.A.; Yeon, S.K.; Dai, J.; Park, K.D.; Khanna, M.; Khanna, R. (S)-Lacosamide Binding to Collapsin Response Mediator Protein 2 (CRMP2) Regulates CaV2.2 Activity by Subverting Its Phosphorylation by Cdk5. Mol. Neurobiol. 2016, 53, 1959–1976. [Google Scholar] [CrossRef]

| Drug | Effect | Model | Tissue/Seizure | Ref. |
|---|---|---|---|---|
| Lamotrigine (LAG) 1 | Inhibition of N-type and P-type VGCC | Rat | amygdalar and cortical neurons | [73,74,75] |
| Carbamazepine (CBZ) 1 | Inhibitory effects on VGCC activity | Rat | frontal cortex | [76] |
| (S)-4-Carboxy-3-hydroxyphenylglycine ((S)-4C3HPG) 2 | Induced HVA current inhibition was mediated through the inhibition of group I and activation of group II mGluRs. Group II mGluRs affected N-type channels | Human cell | dentate gyms neurons/pharmacoresistant TLE | [77] |
| Gabapentin (GBP) 1 | Induces a significant reduction in the number of N-type functional VGCC in the plasma membrane. Involve GABAB receptor coupling to G-proteins and modulation of potassium channels and N-type VGCC | Cell line Rat | - hippocampal slices | [78,79] |
| Levetiracetam (LEV) 1 | Selective blockers of N-type VGCC | Rat | CA1 hippocampal neurons cortical neurons | [74,80] |
| Seletracetam (SEL) 1 | Selective blockers of N-type VGCC | Rat | pyramidal neurons (paroxysmal depolarization shifts) | [81] |
| (S)-lacosamide (LCM) 1 | Inhibits CRMP2 phosphorylation culminating in a reduction of calcium influx via N-type VGCC | - | - | [82] |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Mayo, S.; Gómez-Manjón, I.; Marco-Hernández, A.V.; Fernández-Martínez, F.J.; Camacho, A.; Martínez, F. N-Type Ca Channel in Epileptic Syndromes and Epilepsy: A Systematic Review of Its Genetic Variants. Int. J. Mol. Sci. 2023, 24, 6100. https://doi.org/10.3390/ijms24076100
Mayo S, Gómez-Manjón I, Marco-Hernández AV, Fernández-Martínez FJ, Camacho A, Martínez F. N-Type Ca Channel in Epileptic Syndromes and Epilepsy: A Systematic Review of Its Genetic Variants. International Journal of Molecular Sciences. 2023; 24(7):6100. https://doi.org/10.3390/ijms24076100
Chicago/Turabian StyleMayo, Sonia, Irene Gómez-Manjón, Ana Victoria Marco-Hernández, Francisco Javier Fernández-Martínez, Ana Camacho, and Francisco Martínez. 2023. "N-Type Ca Channel in Epileptic Syndromes and Epilepsy: A Systematic Review of Its Genetic Variants" International Journal of Molecular Sciences 24, no. 7: 6100. https://doi.org/10.3390/ijms24076100
APA StyleMayo, S., Gómez-Manjón, I., Marco-Hernández, A. V., Fernández-Martínez, F. J., Camacho, A., & Martínez, F. (2023). N-Type Ca Channel in Epileptic Syndromes and Epilepsy: A Systematic Review of Its Genetic Variants. International Journal of Molecular Sciences, 24(7), 6100. https://doi.org/10.3390/ijms24076100