
Clinical and Mechanistic Implications of R-Loops in Human Leukemias

Abstract
1. Introduction
2. R-Loop-Mediated DNA Damage and Genomic Instability
2.1. How Do R-Loops Generate DNA Breaks?
2.2. R-Loops and Human Cancers
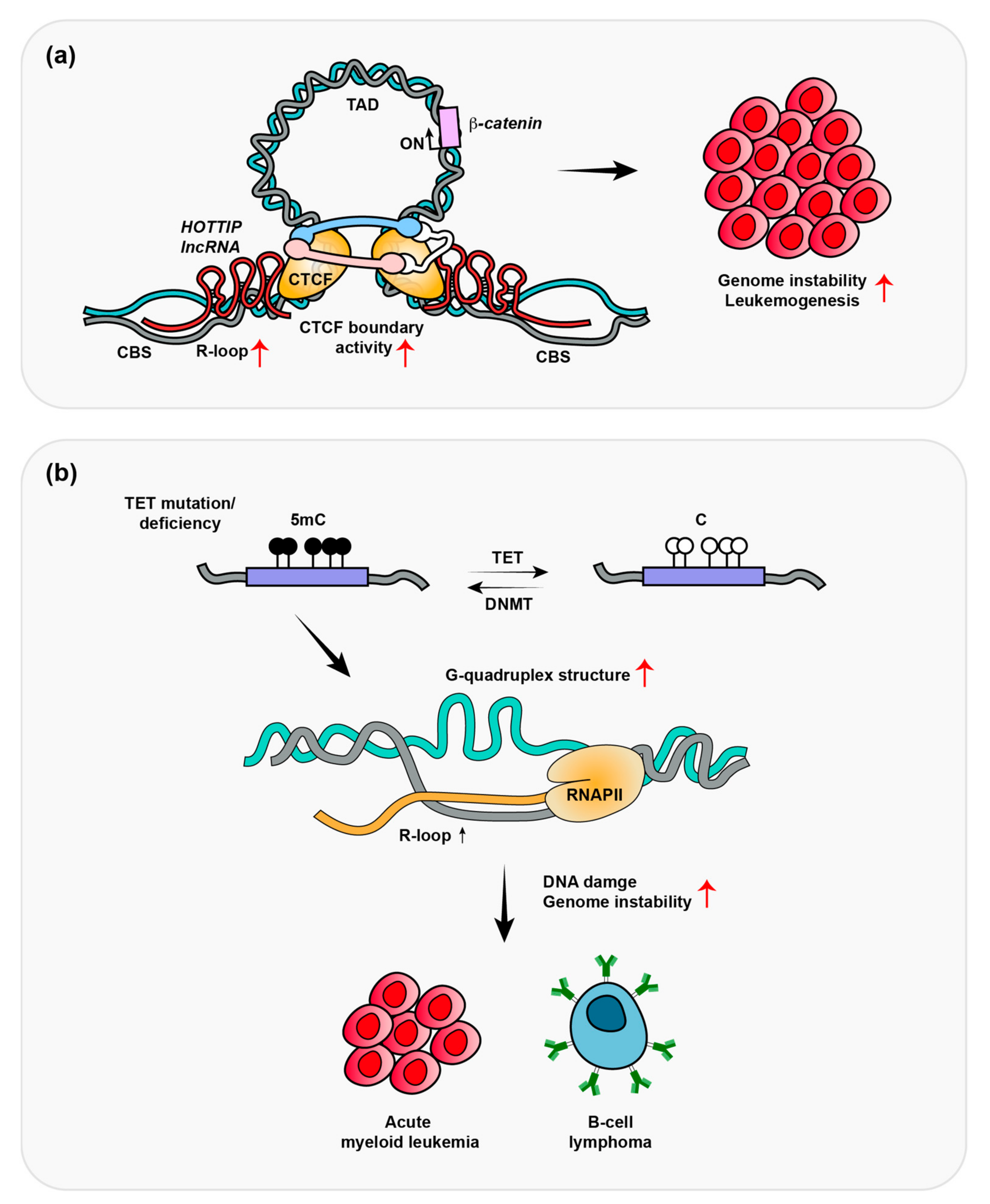
3. HOTTIP-Dependent R-Loop Formation Induces Leukemogenesis
4. Depletion of TET Promotes R-Loop Formation-Induced B-Cell Lymphomas
5. R-Loops Regulate Transcription through DNA Demethylation
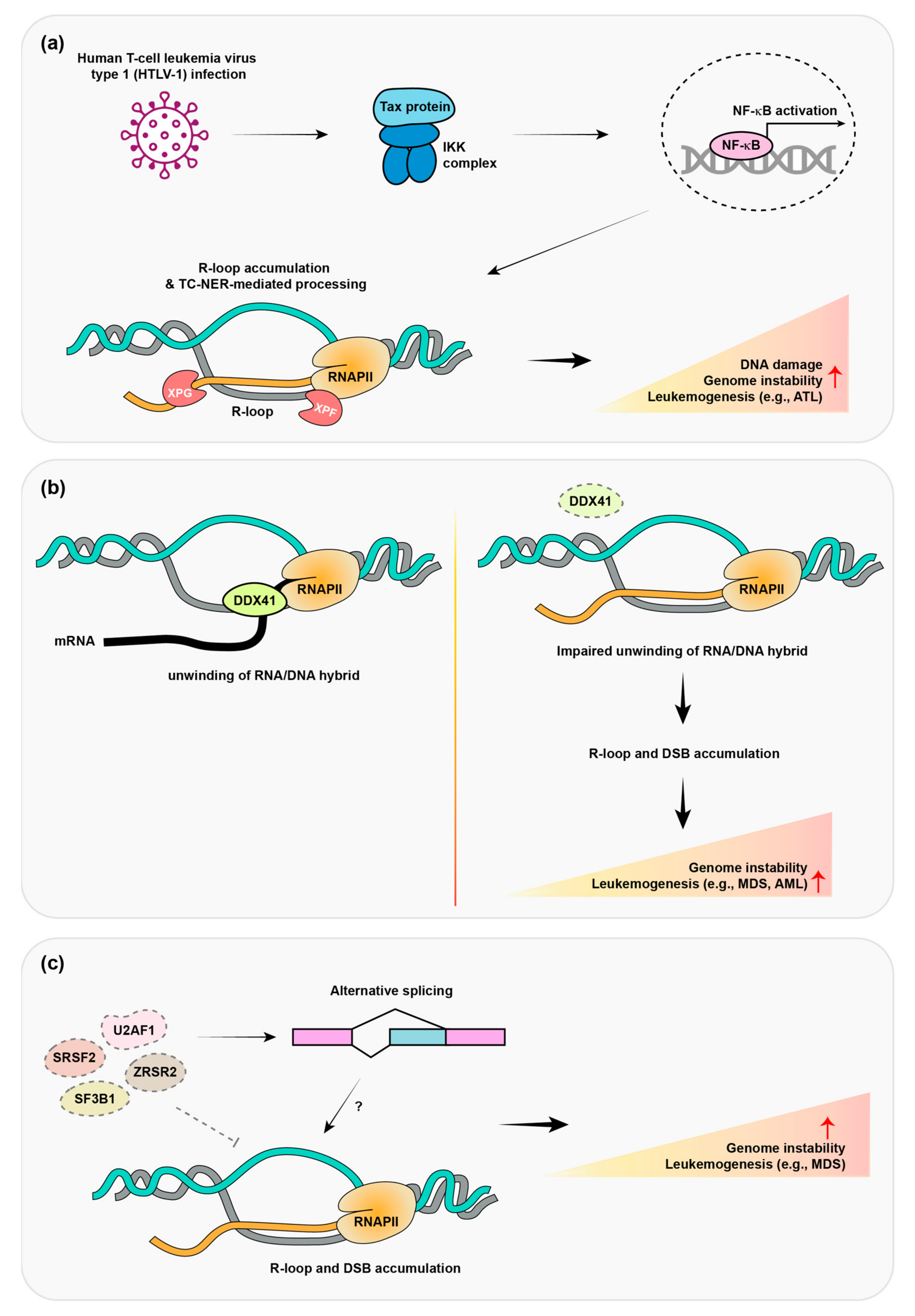
6. Human T-Cell Leukemia Virus Type 1 (HTLV-1) Promotes R-Loop Formation and Adult T-Cell Lymphomas
7. Mutation of a Dead Box Helicase Protein Promotes Adult Myelodysplastic Syndrome (MDS)/Acute Myeloid Leukemia (AML)
8. Mutations in Pre-mRNA Splicing Protein Promote MDS and Leukemia
9. Mutation of mRNA Cleavage and Polyadenylation Proteins Associated with Eosinophilic Leukemia
10. Concluding Remarks
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Hutter, J.J. Childhood leukemia. Pediatr. Rev. 2010, 31, 234–241. [Google Scholar] [CrossRef]
- Ilhan, G.; Karakus, S.; Andic, N. Risk factors and primary prevention of acute leukemia. Asian Pac. J. Cancer Prev. 2006, 7, 515–517. [Google Scholar]
- Mizutani, S. Recent advances in the study of the hereditary and environmental basis of childhood leukemia. Int. J. Hematol. 1998, 68, 131–143. [Google Scholar] [CrossRef]
- Severson, R.K.; Ross, J.A. The causes of acute leukemia. Curr. Opin. Oncol. 1999, 11, 20–24. [Google Scholar] [CrossRef] [PubMed]
- Lindahl, T.; Barnes, D.E. Repair of endogenous DNA damage. Cold Spring Harb. Symp. Quant. Biol. 2000, 65, 127–133. [Google Scholar] [CrossRef]
- Yousefzadeh, M.; Henpita, C.; Vyas, R.; Soto-Palma, C.; Robbins, P.; Niedernhofer, L. DNA damage-how and why we age? Elife 2021, 10, e62852. [Google Scholar] [CrossRef]
- Tomasetti, C.; Vogelstein, B. Cancer etiology. Variation in cancer risk among tissues can be explained by the number of stem cell divisions. Science 2015, 347, 78–81. [Google Scholar] [CrossRef] [PubMed]
- Wu, S.; Powers, S.; Zhu, W.; Hannun, Y.A. Substantial contribution of extrinsic risk factors to cancer development. Nature 2016, 529, 43–47. [Google Scholar] [CrossRef] [PubMed]
- Alexandrov, L.B.; Kim, J.; Haradhvala, N.J.; Huang, M.N.; Tian Ng, A.W.; Wu, Y.; Boot, A.; Covington, K.R.; Gordenin, D.A.; Bergstrom, E.N.; et al. The repertoire of mutational signatures in human cancer. Nature 2020, 578, 94–101. [Google Scholar] [CrossRef] [PubMed]
- Tubbs, A.; Nussenzweig, A. Endogenous DNA Damage as a Source of Genomic Instability in Cancer. Cell 2017, 168, 644–656. [Google Scholar] [CrossRef]
- Petermann, E.; Lan, L.; Zou, L. Sources, resolution and physiological relevance of R-loops and RNA-DNA hybrids. Nat. Rev. Mol. Cell. Biol. 2022, 23, 521–540. [Google Scholar] [CrossRef] [PubMed]
- Crossley, M.P.; Bocek, M.; Cimprich, K.A. R-Loops as Cellular Regulators and Genomic Threats. Mol. Cell. 2019, 73, 398–411. [Google Scholar] [CrossRef]
- Chen, L.; Chen, J.Y.; Zhang, X.; Gu, Y.; Xiao, R.; Shao, C.; Tang, P.; Qian, H.; Luo, D.; Li, H.; et al. R-ChIP Using Inactive RNase H Reveals Dynamic Coupling of R-loops with Transcriptional Pausing at Gene Promoters. Mol. Cell 2017, 68, 745–757.e5. [Google Scholar] [CrossRef] [PubMed]
- Dumelie, J.G.; Jaffrey, S.R. Defining the location of promoter-associated R-loops at near-nucleotide resolution using bisDRIP-seq. Elife 2017, 6, e28306. [Google Scholar] [CrossRef] [PubMed]
- Nadel, J.; Athanasiadou, R.; Lemetre, C.; Wijetunga, N.A.; Broin, P.Ó.; Sato, H.; Zhang, Z.; Jeddeloh, J.; Montagna, C.; Golden, A.; et al. RNA:DNA hybrids in the human genome have distinctive nucleotide characteristics, chromatin composition, and transcriptional relationships. Epigenet. Chromatin 2015, 8, 46. [Google Scholar] [CrossRef]
- Ginno, P.A.; Lott, P.L.; Christensen, H.C.; Korf, I.; Chedin, F. R-loop formation is a distinctive characteristic of unmethylated human CpG island promoters. Mol. Cell 2012, 45, 814–825. [Google Scholar] [CrossRef]
- Grunseich, C.; Wang, I.X.; Watts, J.A.; Burdick, J.T.; Guber, R.D.; Zhu, Z.; Bruzel, A.; Lanman, T.; Chen, K.; Schindler, A.B.; et al. Senataxin Mutation Reveals How R-Loops Promote Transcription by Blocking DNA Methylation at Gene Promoters. Mol. Cell 2018, 69, 426–437.e7. [Google Scholar] [CrossRef]
- Chen, P.B.; Chen, H.V.; Acharya, D.; Rando, O.J.; Fazzio, T.G. R loops regulate promoter-proximal chromatin architecture and cellular differentiation. Nat. Struct. Mol. Biol. 2015, 22, 999–1007. [Google Scholar] [CrossRef]
- Kim, A.; Wang, G.G. R-loop and its functions at the regulatory interfaces between transcription and (epi)genome. Biochim. Biophys. Acta Gene Regul. Mech. 2021, 1864, 194750. [Google Scholar] [CrossRef] [PubMed]
- Wells, J.P.; White, J.; Stirling, P.C. R Loops and Their Composite Cancer Connections. Trends Cancer 2019, 5, 619–631. [Google Scholar] [CrossRef]
- Khan, E.S.; Danckwardt, S. Pathophysiological Role and Diagnostic Potential of R-Loops in Cancer and Beyond. Genes 2022, 13, 2181. [Google Scholar] [CrossRef] [PubMed]
- Garcia-Muse, T.; Aguilera, A. R Loops: From Physiological to Pathological Roles. Cell 2019, 179, 604–618. [Google Scholar] [CrossRef] [PubMed]
- Yu, K.; Chedin, F.; Hsieh, C.L.; Wilson, T.E.; Lieber, M.R. R-loops at immunoglobulin class switch regions in the chromosomes of stimulated B cells. Nat. Immunol. 2003, 4, 442–451. [Google Scholar] [CrossRef] [PubMed]
- Richard, P.; Manley, J.L. R Loops and Links to Human Disease. J. Mol. Biol. 2017, 429, 3168–3180. [Google Scholar] [CrossRef]
- Bianchi, J.; Rudd, S.G.; Jozwiakowski, S.K.; Bailey, L.J.; Soura, V.; Taylor, E.; Stevanovic, I.; Green, A.J.; Stracker, T.H.; Lindsay, H.D.; et al. PrimPol bypasses UV photoproducts during eukaryotic chromosomal DNA replication. Mol. Cell 2013, 52, 566–573. [Google Scholar] [CrossRef]
- Garcia-Gomez, S.; Reyes, A.; Martinez-Jimenez, M.I.; Chocron, E.S.; Mouron, S.; Terrados, G.; Powell, C.; Salido, E.; Mendez, J.; Holt, I.J.; et al. PrimPol, an archaic primase/polymerase operating in human cells. Mol. Cell 2013, 52, 541–553. [Google Scholar] [CrossRef] [PubMed]
- Gloor, J.W.; Balakrishnan, L.; Campbell, J.L.; Bambara, R.A. Biochemical analyses indicate that binding and cleavage specificities define the ordered processing of human Okazaki fragments by Dna2 and FEN1. Nucleic Acids Res. 2012, 40, 6774–6786. [Google Scholar] [CrossRef] [PubMed]
- McCann, J.L.; Cristini, A.; Law, E.K.; Lee, S.Y.; Tellier, M.; Carpenter, M.A.; Beghè, C.; Kim, J.J.; Jarvis, M.C.; Stefanovska, B.; et al. R-loop homeostasis and cancer mutagenesis promoted by the DNA cytosine deaminase APOBEC3B. bioRxiv 2021. [Google Scholar] [CrossRef]
- Sollier, J.; Cimprich, K.A. Breaking bad: R-loops and genome integrity. Trends Cell. Biol. 2015, 25, 514–522. [Google Scholar] [CrossRef]
- Heinaniemi, M.; Vuorenmaa, T.; Teppo, S.; Kaikkonen, M.U.; Bouvy-Liivrand, M.; Mehtonen, J.; Niskanen, H.; Zachariadis, V.; Laukkanen, S.; Liuksiala, T.; et al. Transcription-coupled genetic instability marks acute lymphoblastic leukemia structural variation hotspots. Elife 2016, 5, e13087. [Google Scholar] [CrossRef]
- Stavnezer, J.; Schrader, C.E. Mismatch repair converts AID-instigated nicks to double-strand breaks for antibody class-switch recombination. Trends Genet. 2006, 22, 23–28. [Google Scholar] [CrossRef] [PubMed]
- Rinaldi, C.; Pizzul, P.; Longhese, M.P.; Bonetti, D. Sensing R-Loop-Associated DNA Damage to Safeguard Genome Stability. Front. Cell. Dev. Biol. 2020, 8, 618157. [Google Scholar] [CrossRef] [PubMed]
- Schrader, C.E.; Guikema, J.E.; Linehan, E.K.; Selsing, E.; Stavnezer, J. Activation-induced cytidine deaminase-dependent DNA breaks in class switch recombination occur during G1 phase of the cell cycle and depend upon mismatch repair. J. Immunol. 2007, 179, 6064–6071. [Google Scholar] [CrossRef] [PubMed]
- Tian, M.; Alt, F.W. Transcription-induced cleavage of immunoglobulin switch regions by nucleotide excision repair nucleases in vitro. J. Biol. Chem. 2000, 275, 24163–24172. [Google Scholar] [CrossRef]
- Hegazy, Y.A.; Fernando, C.M.; Tran, E.J. The balancing act of R-loop biology: The good, the bad, and the ugly. J. Biol. Chem. 2020, 295, 905–913. [Google Scholar] [CrossRef]
- Rhodes, D.; Lipps, H.J. G-quadruplexes and their regulatory roles in biology. Nucleic Acids Res. 2015, 43, 8627–8637. [Google Scholar] [CrossRef]
- De Magis, A.; Manzo, S.G.; Russo, M.; Marinello, J.; Morigi, R.; Sordet, O.; Capranico, G. DNA damage and genome instability by G-quadruplex ligands are mediated by R loops in human cancer cells. Proc. Natl. Acad. Sci. USA 2019, 116, 816–825. [Google Scholar] [CrossRef]
- Gregersen, L.H.; Svejstrup, J.Q. The Cellular Response to Transcription-Blocking DNA Damage. Trends Biochem. Sci. 2018, 43, 327–341. [Google Scholar] [CrossRef]
- Sollier, J.; Stork, C.T.; Garcia-Rubio, M.L.; Paulsen, R.D.; Aguilera, A.; Cimprich, K.A. Transcription-coupled nucleotide excision repair factors promote R-loop-induced genome instability. Mol. Cell 2014, 56, 777–785. [Google Scholar] [CrossRef]
- Hamperl, S.; Bocek, M.J.; Saldivar, J.C.; Swigut, T.; Cimprich, K.A. Transcription-Replication Conflict Orientation Modulates R-Loop Levels and Activates Distinct DNA Damage Responses. Cell 2017, 170, 774–786.e19. [Google Scholar] [CrossRef]
- Matos, D.A.; Zhang, J.M.; Ouyang, J.; Nguyen, H.D.; Genois, M.M.; Zou, L. ATR Protects the Genome against R Loops through a MUS81-Triggered Feedback Loop. Mol. Cell 2020, 77, 514–527.e4. [Google Scholar] [CrossRef] [PubMed]
- Bhatia, V.; Barroso, S.I.; Garcia-Rubio, M.L.; Tumini, E.; Herrera-Moyano, E.; Aguilera, A. BRCA2 prevents R-loop accumulation and associates with TREX-2 mRNA export factor PCID2. Nature 2014, 511, 362–365. [Google Scholar] [CrossRef]
- Hamperl, S.; Cimprich, K.A. The contribution of co-transcriptional RNA:DNA hybrid structures to DNA damage and genome instability. DNA Repair 2014, 19, 84–94. [Google Scholar] [CrossRef] [PubMed]
- San Martin Alonso, M.; Noordermeer, S.M. Untangling the crosstalk between BRCA1 and R-loops during DNA repair. Nucleic Acids Res. 2021, 49, 4848–4863. [Google Scholar] [CrossRef] [PubMed]
- Shivji, M.K.K.; Renaudin, X.; Williams, C.H.; Venkitaraman, A.R. BRCA2 Regulates Transcription Elongation by RNA Polymerase II to Prevent R-Loop Accumulation. Cell Rep. 2018, 22, 1031–1039. [Google Scholar] [CrossRef] [PubMed]
- Royfman, R.; Whiteley, E.; Noe, O.; Morand, S.; Creeden, J.; Stanbery, L.; Hamouda, D.; Nemunaitis, J. BRCA1/2 signaling and homologous recombination deficiency in breast and ovarian cancer. Future Oncol. 2021, 17, 2817–2830. [Google Scholar] [CrossRef] [PubMed]
- Garcia-Rubio, M.L.; Perez-Calero, C.; Barroso, S.I.; Tumini, E.; Herrera-Moyano, E.; Rosado, I.V.; Aguilera, A. The Fanconi Anemia Pathway Protects Genome Integrity from R-loops. PLoS Genet. 2015, 11, e1005674. [Google Scholar] [CrossRef]
- Chang, E.Y.; Tsai, S.; Aristizabal, M.J.; Wells, J.P.; Coulombe, Y.; Busatto, F.F.; Chan, Y.A.; Kumar, A.; Dan Zhu, Y.; Wang, A.Y.; et al. MRE11-RAD50-NBS1 promotes Fanconi Anemia R-loop suppression at transcription-replication conflicts. Nat. Commun. 2019, 10, 4265. [Google Scholar] [CrossRef]
- Liang, Z.; Liang, F.; Teng, Y.; Chen, X.; Liu, J.; Longerich, S.; Rao, T.; Green, A.M.; Collins, N.B.; Xiong, Y.; et al. Binding of FANCI-FANCD2 Complex to RNA and R-Loops Stimulates Robust FANCD2 Monoubiquitination. Cell Rep. 2019, 26, 564–572.e5. [Google Scholar] [CrossRef]
- Liu, W.; Palovcak, A.; Li, F.; Zafar, A.; Yuan, F.; Zhang, Y. Fanconi anemia pathway as a prospective target for cancer intervention. Cell Biosci. 2020, 10, 39. [Google Scholar] [CrossRef]
- Niraj, J.; Farkkila, A.; D’Andrea, A.D. The Fanconi Anemia Pathway in Cancer. Annu. Rev. Cancer Biol. 2019, 3, 457–478. [Google Scholar] [CrossRef] [PubMed]
- Kennedy, R.D.; D’Andrea, A.D. The Fanconi Anemia/BRCA pathway: New faces in the crowd. Genes Dev. 2005, 19, 2925–2940. [Google Scholar] [CrossRef] [PubMed]
- Chen, L.; Chen, J.Y.; Huang, Y.J.; Gu, Y.; Qiu, J.; Qian, H.; Shao, C.; Zhang, X.; Hu, J.; Li, H.; et al. The Augmented R-Loop Is a Unifying Mechanism for Myelodysplastic Syndromes Induced by High-Risk Splicing Factor Mutations. Mol. Cell 2018, 69, 412–425.e6. [Google Scholar] [CrossRef] [PubMed]
- Luo, H.; Zhu, G.; Eshelman, M.A.; Fung, T.K.; Lai, Q.; Wang, F.; Zeisig, B.B.; Lesperance, J.; Ma, X.; Chen, S.; et al. HOTTIP-dependent R-loop formation regulates CTCF boundary activity and TAD integrity in leukemia. Mol. Cell 2022, 82, 833–851.e11. [Google Scholar] [CrossRef]
- Shukla, V.; Samaniego-Castruita, D.; Dong, Z.; Gonzalez-Avalos, E.; Yan, Q.; Sarma, K.; Rao, A. TET deficiency perturbs mature B cell homeostasis and promotes oncogenesis associated with accumulation of G-quadruplex and R-loop structures. Nat. Immunol. 2022, 23, 99–108. [Google Scholar] [CrossRef]
- He, Y.; Pasupala, N.; Zhi, H.; Dorjbal, B.; Hussain, I.; Shih, H.M.; Bhattacharyya, S.; Biswas, R.; Miljkovic, M.; Semmes, O.J.; et al. NF-kappaB-induced R-loop accumulation and DNA damage select for nucleotide excision repair deficiencies in adult T cell leukemia. Proc. Natl. Acad. Sci. USA 2021, 118, e2005568118. [Google Scholar] [CrossRef]
- Mosler, T.; Conte, F.; Longo, G.M.C.; Mikicic, I.; Kreim, N.; Mockel, M.M.; Petrosino, G.; Flach, J.; Barau, J.; Luke, B.; et al. R-loop proximity proteomics identifies a role of DDX41 in transcription-associated genomic instability. Nat. Commun. 2021, 12, 7314. [Google Scholar] [CrossRef]
- Li, X.; Manley, J.L. Inactivation of the SR protein splicing factor ASF/SF2 results in genomic instability. Cell 2005, 122, 365–378. [Google Scholar] [CrossRef]
- Nguyen, H.D.; Leong, W.Y.; Li, W.; Reddy, P.N.G.; Sullivan, J.D.; Walter, M.J.; Zou, L.; Graubert, T.A. Spliceosome Mutations Induce R Loop-Associated Sensitivity to ATR Inhibition in Myelodysplastic Syndromes. Cancer Res. 2018, 78, 5363–5374. [Google Scholar] [CrossRef]
- Singh, S.; Ahmed, D.; Dolatshad, H.; Tatwavedi, D.; Schulze, U.; Sanchi, A.; Ryley, S.; Dhir, A.; Carpenter, L.; Watt, S.M.; et al. SF3B1 mutations induce R-loop accumulation and DNA damage in MDS and leukemia cells with therapeutic implications. Leukemia 2020, 34, 2525–2530. [Google Scholar] [CrossRef]
- Stirling, P.C.; Chan, Y.A.; Minaker, S.W.; Aristizabal, M.J.; Barrett, I.; Sipahimalani, P.; Kobor, M.S.; Hieter, P. R-loop-mediated genome instability in mRNA cleavage and polyadenylation mutants. Genes Dev. 2012, 26, 163–175. [Google Scholar] [CrossRef] [PubMed]
- Wang, K.C.; Yang, Y.W.; Liu, B.; Sanyal, A.; Corces-Zimmerman, R.; Chen, Y.; Lajoie, B.R.; Protacio, A.; Flynn, R.A.; Gupta, R.A.; et al. A long noncoding RNA maintains active chromatin to coordinate homeotic gene expression. Nature 2011, 472, 120–124. [Google Scholar] [CrossRef] [PubMed]
- Luo, H.; Zhu, G.; Xu, J.; Lai, Q.; Yan, B.; Guo, Y.; Fung, T.K.; Zeisig, B.B.; Cui, Y.; Zha, J.; et al. HOTTIP lncRNA Promotes Hematopoietic Stem Cell Self-Renewal Leading to AML-like Disease in Mice. Cancer Cell. 2019, 36, 645–659 e8. [Google Scholar] [CrossRef] [PubMed]
- Deng, C.; Li, Y.; Zhou, L.; Cho, J.; Patel, B.; Terada, N.; Li, Y.; Bungert, J.; Qiu, Y.; Huang, S. HoxBlinc RNA Recruits Set1/MLL Complexes to Activate Hox Gene Expression Patterns and Mesoderm Lineage Development. Cell Rep. 2016, 14, 103–114. [Google Scholar] [CrossRef]
- Deschamps, J.; van Nes, J. Developmental regulation of the Hox genes during axial morphogenesis in the mouse. Development 2005, 132, 2931–2942. [Google Scholar] [CrossRef] [PubMed]
- Forlani, S.; Lawson, K.A.; Deschamps, J. Acquisition of Hox codes during gastrulation and axial elongation in the mouse embryo. Development 2003, 130, 3807–3819. [Google Scholar] [CrossRef]
- Shah, N.; Sukumar, S. The Hox genes and their roles in oncogenesis. Nat. Rev. Cancer 2010, 10, 361–371. [Google Scholar] [CrossRef]
- Ferrando, A.A.; Armstrong, S.A.; Neuberg, D.S.; Sallan, S.E.; Silverman, L.B.; Korsmeyer, S.J.; Look, A.T. Gene expression signatures in MLL-rearranged T-lineage and B-precursor acute leukemias: Dominance of HOX dysregulation. Blood 2003, 102, 262–268. [Google Scholar] [CrossRef]
- Armstrong, S.A.; Golub, T.R.; Korsmeyer, S.J. MLL-rearranged leukemias: Insights from gene expression profiling. Semin. Hematol. 2003, 40, 268–273. [Google Scholar] [CrossRef]
- Faber, J.; Krivtsov, A.V.; Stubbs, M.C.; Wright, R.; Davis, T.N.; van den Heuvel-Eibrink, M.; Zwaan, C.M.; Kung, A.L.; Armstrong, S.A. HOXA9 is required for survival in human MLL-rearranged acute leukemias. Blood 2009, 113, 2375–2385. [Google Scholar] [CrossRef]
- Golub, T.R.; Slonim, D.K.; Tamayo, P.; Huard, C.; Gaasenbeek, M.; Mesirov, J.P.; Coller, H.; Loh, M.L.; Downing, J.R.; Caligiuri, M.A.; et al. Molecular classification of cancer: Class discovery and class prediction by gene expression monitoring. Science 1999, 286, 531–537. [Google Scholar] [CrossRef] [PubMed]
- Collins, C.T.; Hess, J.L. Role of HOXA9 in leukemia: Dysregulation, cofactors and essential targets. Oncogene 2016, 35, 1090–1098. [Google Scholar] [CrossRef] [PubMed]
- Costantino, L.; Koshland, D. The Yin and Yang of R-loop biology. Curr. Opin. Cell. Biol. 2015, 34, 39–45. [Google Scholar] [CrossRef] [PubMed]
- Niehrs, C.; Luke, B. Regulatory R-loops as facilitators of gene expression and genome stability. Nat. Rev. Mol. Cell Biol. 2020, 21, 167–178. [Google Scholar] [CrossRef] [PubMed]
- Kosmider, O.; Delabesse, E.; de Mas, V.M.; Cornillet-Lefebvre, P.; Blanchet, O.; Delmer, A.; Recher, C.; Raynaud, S.; Bouscary, D.; Viguie, F.; et al. TET2 mutations in secondary acute myeloid leukemias: A French retrospective study. Haematologica 2011, 96, 1059–1063. [Google Scholar] [CrossRef]
- Weissmann, S.; Alpermann, T.; Grossmann, V.; Kowarsch, A.; Nadarajah, N.; Eder, C.; Dicker, F.; Fasan, A.; Haferlach, C.; Haferlach, T.; et al. Landscape of TET2 mutations in acute myeloid leukemia. Leukemia 2012, 26, 934–942. [Google Scholar] [CrossRef]
- Shaikh, A.R.K.; Ujjan, I.; Irfan, M.; Naz, A.; Shamsi, T.; Khan, M.T.M.; Shakeel, M. TET2 mutations in acute myeloid leukemia: A comprehensive study in patients of Sindh, Pakistan. PeerJ 2021, 9, e10678. [Google Scholar] [CrossRef]
- Delhommeau, F.; Dupont, S.; Della Valle, V.; James, C.; Trannoy, S.; Masse, A.; Kosmider, O.; Le Couedic, J.P.; Robert, F.; Alberdi, A.; et al. Mutation in TET2 in myeloid cancers. N. Engl. J. Med. 2009, 360, 2289–2301. [Google Scholar] [CrossRef]
- Kubuki, Y.; Yamaji, T.; Hidaka, T.; Kameda, T.; Shide, K.; Sekine, M.; Kamiunten, A.; Akizuki, K.; Shimoda, H.; Tahira, Y.; et al. TET2 mutation in diffuse large B-cell lymphoma. J. Clin. Exp. Hematop. 2017, 56, 145–149. [Google Scholar] [CrossRef]
- Dominguez, P.M.; Ghamlouch, H.; Rosikiewicz, W.; Kumar, P.; Beguelin, W.; Fontan, L.; Rivas, M.A.; Pawlikowska, P.; Armand, M.; Mouly, E.; et al. TET2 Deficiency Causes Germinal Center Hyperplasia, Impairs Plasma Cell Differentiation, and Promotes B-cell Lymphomagenesis. Cancer Discov. 2018, 8, 1632–1653. [Google Scholar] [CrossRef]
- Chou, W.C.; Chou, S.C.; Liu, C.Y.; Chen, C.Y.; Hou, H.A.; Kuo, Y.Y.; Lee, M.C.; Ko, B.S.; Tang, J.L.; Yao, M.; et al. TET2 mutation is an unfavorable prognostic factor in acute myeloid leukemia patients with intermediate-risk cytogenetics. Blood 2011, 118, 3803–3810. [Google Scholar] [CrossRef] [PubMed]
- Morin, R.D.; Mendez-Lago, M.; Mungall, A.J.; Goya, R.; Mungall, K.L.; Corbett, R.D.; Johnson, N.A.; Severson, T.M.; Chiu, R.; Field, M.; et al. Frequent mutation of histone-modifying genes in non-Hodgkin lymphoma. Nature 2011, 476, 298–303. [Google Scholar] [CrossRef] [PubMed]
- Asmar, F.; Punj, V.; Christensen, J.; Pedersen, M.T.; Pedersen, A.; Nielsen, A.B.; Hother, C.; Ralfkiaer, U.; Brown, P.; Ralfkiaer, E.; et al. Genome-wide profiling identifies a DNA methylation signature that associates with TET2 mutations in diffuse large B-cell lymphoma. Haematologica 2013, 98, 1912–1920. [Google Scholar] [CrossRef] [PubMed]
- Ley, T.J.; Ding, L.; Walter, M.J.; McLellan, M.D.; Lamprecht, T.; Larson, D.E.; Kandoth, C.; Payton, J.E.; Baty, J.; Welch, J.; et al. DNMT3A mutations in acute myeloid leukemia. N. Engl. J. Med. 2010, 363, 2424–2433. [Google Scholar] [CrossRef]
- Kim, H.D.; Choe, J.; Seo, Y.S. The sen1(+) gene of Schizosaccharomyces pombe, a homologue of budding yeast SEN1, encodes an RNA and DNA helicase. Biochemistry 1999, 38, 14697–14710. [Google Scholar] [CrossRef]
- Martin-Tumasz, S.; Brow, D.A. Saccharomyces cerevisiae Sen1 Helicase Domain Exhibits 5’- to 3’-Helicase Activity with a Preference for Translocation on DNA Rather than RNA. J. Biol. Chem. 2015, 290, 22880–22889. [Google Scholar] [CrossRef]
- Leonaite, B.; Han, Z.; Basquin, J.; Bonneau, F.; Libri, D.; Porrua, O.; Conti, E. Sen1 has unique structural features grafted on the architecture of the Upf1-like helicase family. EMBO J. 2017, 36, 1590–1604. [Google Scholar] [CrossRef]
- Chen, Y.Z.; Bennett, C.L.; Huynh, H.M.; Blair, I.P.; Puls, I.; Irobi, J.; Dierick, I.; Abel, A.; Kennerson, M.L.; Rabin, B.A.; et al. DNA/RNA helicase gene mutations in a form of juvenile amyotrophic lateral sclerosis (ALS4). Am. J. Hum. Genet. 2004, 74, 1128–1135. [Google Scholar] [CrossRef]
- Kang, G.H.; Shim, Y.H.; Ro, J.Y. Correlation of methylation of the hMLH1 promoter with lack of expression of hMLH1 in sporadic gastric carcinomas with replication error. Lab. Investig. 1999, 79, 903–909. [Google Scholar]
- Kulis, M.; Esteller, M. DNA methylation and cancer. Adv. Genet. 2010, 70, 27–56. [Google Scholar]
- Gnyszka, A.; Jastrzebski, Z.; Flis, S. DNA methyltransferase inhibitors and their emerging role in epigenetic therapy of cancer. Anticancer. Res. 2013, 33, 2989–2996. [Google Scholar]
- Ganetsky, A. The role of decitabine for the treatment of acute myeloid leukemia. Ann. Pharmacother. 2012, 46, 1511–1517. [Google Scholar] [CrossRef] [PubMed]
- Sriraman, A.; Debnath, T.K.; Xhemalce, B.; Miller, K.M. Making it or breaking it: DNA methylation and genome integrity. Essays Biochem. 2020, 64, 687–703. [Google Scholar] [PubMed]
- Forlani, G.; Shallak, M.; Accolla, R.S.; Romanelli, M.G. HTLV-1 Infection and Pathogenesis: New Insights from Cellular and Animal Models. Int. J. Mol. Sci. 2021, 22, 8001. [Google Scholar] [CrossRef] [PubMed]
- Bangham, C.R.; Araujo, A.; Yamano, Y.; Taylor, G.P. HTLV-1-associated myelopathy/tropical spastic paraparesis. Nat. Rev. Dis. Primers 2015, 1, 15012. [Google Scholar] [CrossRef]
- Zuo, X.; Zhou, R.; Yang, S.; Ma, G. HTLV-1 persistent infection and ATLL oncogenesis. J. Med. Virol. 2023, 95, e28424. [Google Scholar] [CrossRef]
- Sun, S.C.; Yamaoka, S. Activation of NF-kappaB by HTLV-I and implications for cell transformation. Oncogene 2005, 24, 5952–5964. [Google Scholar] [CrossRef]
- Xiao, G. NF-kappaB activation: Tax sumoylation is out, but what about Tax ubiquitination? Retrovirology 2012, 9, 78. [Google Scholar] [CrossRef]
- Yang, L.; Kotomura, N.; Ho, Y.K.; Zhi, H.; Bixler, S.; Schell, M.J.; Giam, C.Z. Complex cell cycle abnormalities caused by human T-lymphotropic virus type 1 Tax. J. Virol. 2011, 85, 3001–3009. [Google Scholar] [CrossRef]
- Liu, M.; Yang, L.; Zhang, L.; Liu, B.; Merling, R.; Xia, Z.; Giam, C.Z. Human T-cell leukemia virus type 1 infection leads to arrest in the G1 phase of the cell cycle. J. Virol. 2008, 82, 8442–8455. [Google Scholar] [CrossRef]
- Shudofsky, A.M.D.; Giam, C.Z. Cells of adult T-cell leukemia evade HTLV-1 Tax/NF-kappaB hyperactivation-induced senescence. Blood Adv. 2019, 3, 564–569. [Google Scholar] [CrossRef] [PubMed]
- Frame, J.M.; North, T.E. Ddx41 loss R-loops in cGAS to fuel inflammatory HSPC production. Dev. Cell 2021, 56, 571–572. [Google Scholar] [CrossRef] [PubMed]
- Weinreb, J.T.; Ghazale, N.; Pradhan, K.; Gupta, V.; Potts, K.S.; Tricomi, B.; Daniels, N.J.; Padgett, R.A.; De Oliveira, S.; Verma, A.; et al. Excessive R-loops trigger an inflammatory cascade leading to increased HSPC production. Dev. Cell 2021, 56, 627–640.e5. [Google Scholar] [CrossRef]
- Kurre, P. Hematopoietic development: A gap in our understanding of inherited bone marrow failure. Exp. Hematol. 2018, 59, 1–8. [Google Scholar] [CrossRef] [PubMed]
- Sperling, A.S.; Gibson, C.J.; Ebert, B.L. The genetics of myelodysplastic syndrome: From clonal haematopoiesis to secondary leukaemia. Nat. Rev. Cancer 2017, 17, 5–19. [Google Scholar] [CrossRef]
- Espin-Palazon, R.; Weijts, B.; Mulero, V.; Traver, D. Proinflammatory Signals as Fuel for the Fire of Hematopoietic Stem Cell Emergence. Trends Cell. Biol. 2018, 28, 58–66. [Google Scholar] [CrossRef]
- Pietras, E.M. Inflammation: A key regulator of hematopoietic stem cell fate in health and disease. Blood 2017, 130, 1693–1698. [Google Scholar] [CrossRef]
- Papaemmanuil, E.; Gerstung, M.; Malcovati, L.; Tauro, S.; Gundem, G.; Van Loo, P.; Yoon, C.J.; Ellis, P.; Wedge, D.C.; Pellagatti, A.; et al. Clinical and biological implications of driver mutations in myelodysplastic syndromes. Blood 2013, 122, 3616–3627. [Google Scholar] [CrossRef] [PubMed]
- Haferlach, T.; Nagata, Y.; Grossmann, V.; Okuno, Y.; Bacher, U.; Nagae, G.; Schnittger, S.; Sanada, M.; Kon, A.; Alpermann, T.; et al. Landscape of genetic lesions in 944 patients with myelodysplastic syndromes. Leukemia 2014, 28, 241–247. [Google Scholar] [CrossRef] [PubMed]
- Yip, B.H.; Dolatshad, H.; Roy, S.; Pellagatti, A.; Boultwood, J. Impact of Splicing Factor Mutations on Pre-mRNA Splicing in the Myelodysplastic Syndromes. Curr. Pharm. Des. 2016, 22, 2333–2344. [Google Scholar] [CrossRef]
- Kim, E.; Ilagan, J.O.; Liang, Y.; Daubner, G.M.; Lee, S.C.; Ramakrishnan, A.; Li, Y.; Chung, Y.R.; Micol, J.B.; Murphy, M.E.; et al. SRSF2 Mutations Contribute to Myelodysplasia by Mutant-Specific Effects on Exon Recognition. Cancer Cell. 2015, 27, 617–630. [Google Scholar] [CrossRef] [PubMed]
- Kon, A.; Yamazaki, S.; Nannya, Y.; Kataoka, K.; Ota, Y.; Nakagawa, M.M.; Yoshida, K.; Shiozawa, Y.; Morita, M.; Yoshizato, T.; et al. Physiological Srsf2 P95H expression causes impaired hematopoietic stem cell functions and aberrant RNA splicing in mice. Blood 2018, 131, 621–635. [Google Scholar] [CrossRef] [PubMed]
- Shirai, C.L.; Ley, J.N.; White, B.S.; Kim, S.; Tibbitts, J.; Shao, J.; Ndonwi, M.; Wadugu, B.; Duncavage, E.J.; Okeyo-Owuor, T.; et al. Mutant U2AF1 Expression Alters Hematopoiesis and Pre-mRNA Splicing In Vivo. Cancer Cell. 2015, 27, 631–643. [Google Scholar] [CrossRef] [PubMed]
- Obeng, E.A.; Chappell, R.J.; Seiler, M.; Chen, M.C.; Campagna, D.R.; Schmidt, P.J.; Schneider, R.K.; Lord, A.M.; Wang, L.; Gambe, R.G.; et al. Physiologic Expression of Sf3b1(K700E) Causes Impaired Erythropoiesis, Aberrant Splicing, and Sensitivity to Therapeutic Spliceosome Modulation. Cancer Cell. 2016, 30, 404–417. [Google Scholar] [CrossRef] [PubMed]
- Paulsen, R.D.; Soni, D.V.; Wollman, R.; Hahn, A.T.; Yee, M.C.; Guan, A.; Hesley, J.A.; Miller, S.C.; Cromwell, E.F.; Solow-Cordero, D.E.; et al. A genome-wide siRNA screen reveals diverse cellular processes and pathways that mediate genome stability. Mol. Cell 2009, 35, 228–239. [Google Scholar] [CrossRef]
- Chan, Y.A.; Aristizabal, M.J.; Lu, P.Y.; Luo, Z.; Hamza, A.; Kobor, M.S.; Stirling, P.C.; Hieter, P. Genome-wide profiling of yeast DNA:RNA hybrid prone sites with DRIP-chip. PLoS Genet. 2014, 10, e1004288. [Google Scholar] [CrossRef]
- Alhmoud, J.F.; Mustafa, A.G.; Malki, M.I. Targeting DNA Repair Pathways in Hematological Malignancies. Int. J. Mol. Sci. 2020, 21, 7365. [Google Scholar] [CrossRef]
- Baldwin, E.L.; Osheroff, N. Etoposide, topoisomerase II and cancer. Curr. Med. Chem. Anticancer Agents 2005, 5, 363–372. [Google Scholar] [CrossRef]
- Walsby, E.J.; Coles, S.J.; Knapper, S.; Burnett, A.K. The topoisomerase II inhibitor voreloxin causes cell cycle arrest and apoptosis in myeloid leukemia cells and acts in synergy with cytarabine. Haematologica 2011, 96, 393–399. [Google Scholar] [CrossRef]
- Kim, J.J.; Lee, S.Y.; Gong, F.; Battenhouse, A.M.; Boutz, D.R.; Bashyal, A.; Refvik, S.T.; Chiang, C.M.; Xhemalce, B.; Paull, T.T.; et al. Systematic bromodomain protein screens identify homologous recombination and R-loop suppression pathways involved in genome integrity. Genes. Dev. 2019, 33, 1751–1774. [Google Scholar] [CrossRef]
- Ezoe, S. Secondary leukemia associated with the anti-cancer agent, etoposide, a topoisomerase II inhibitor. Int. J. Environ. Res. Public. Health 2012, 9, 2444–2453. [Google Scholar] [CrossRef] [PubMed]
- Das, M. Venetoclax with decitabine or azacitidine for AML. Lancet Oncol. 2018, 19, e672. [Google Scholar] [CrossRef] [PubMed]




{kind=link}
{kind=link}
{kind=link}
| Types | R-Loop Regulators | Status | Description | Reference |
|---|---|---|---|---|
| lncRNA | HOTTIP | Upregulated in MDS | Forms an R-loop structure at CBS sites for TAD formation and oncogene expression | [54] |
| Protein | TET2, TET3 | Mutation | Increases DNA methylation to promote R-loop formation | [55] |
| Virus | HTLV-1 | Infection | Constitutively activates NF-kB to promote R-loop formation | [56] |
| Protein | DDX41 | Mutation | Unwinds RNA–DNA hybrids | [57] |
| Protein | SRSF1 | Mutation | Splicing factor that binds to Pol II CTD and suppresses R-loop formation | [58] |
| Protein | U2AF1 | Mutation | Splicing factor that suppresses R-loop formation, but the mechanism is unknown | [59] |
| Protein | SF3B1 | Mutation | Splicing factor that suppresses R-loop formation, but the mechanism is unknown | [60] |
| Protein | FIP1L1 | Mutation | mRNA cleavage and polyadenylation(mCP) protein suppresses R-loop formation | [61] |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Lee, S.-Y.; Miller, K.M.; Kim, J.-J. Clinical and Mechanistic Implications of R-Loops in Human Leukemias. Int. J. Mol. Sci. 2023, 24, 5966. https://doi.org/10.3390/ijms24065966
Lee S-Y, Miller KM, Kim J-J. Clinical and Mechanistic Implications of R-Loops in Human Leukemias. International Journal of Molecular Sciences. 2023; 24(6):5966. https://doi.org/10.3390/ijms24065966
Chicago/Turabian StyleLee, Seo-Yun, Kyle M. Miller, and Jae-Jin Kim. 2023. "Clinical and Mechanistic Implications of R-Loops in Human Leukemias" International Journal of Molecular Sciences 24, no. 6: 5966. https://doi.org/10.3390/ijms24065966
APA StyleLee, S.-Y., Miller, K. M., & Kim, J.-J. (2023). Clinical and Mechanistic Implications of R-Loops in Human Leukemias. International Journal of Molecular Sciences, 24(6), 5966. https://doi.org/10.3390/ijms24065966