
Inherited Disorders of Coenzyme A Biosynthesis: Models, Mechanisms, and Treatments

Abstract
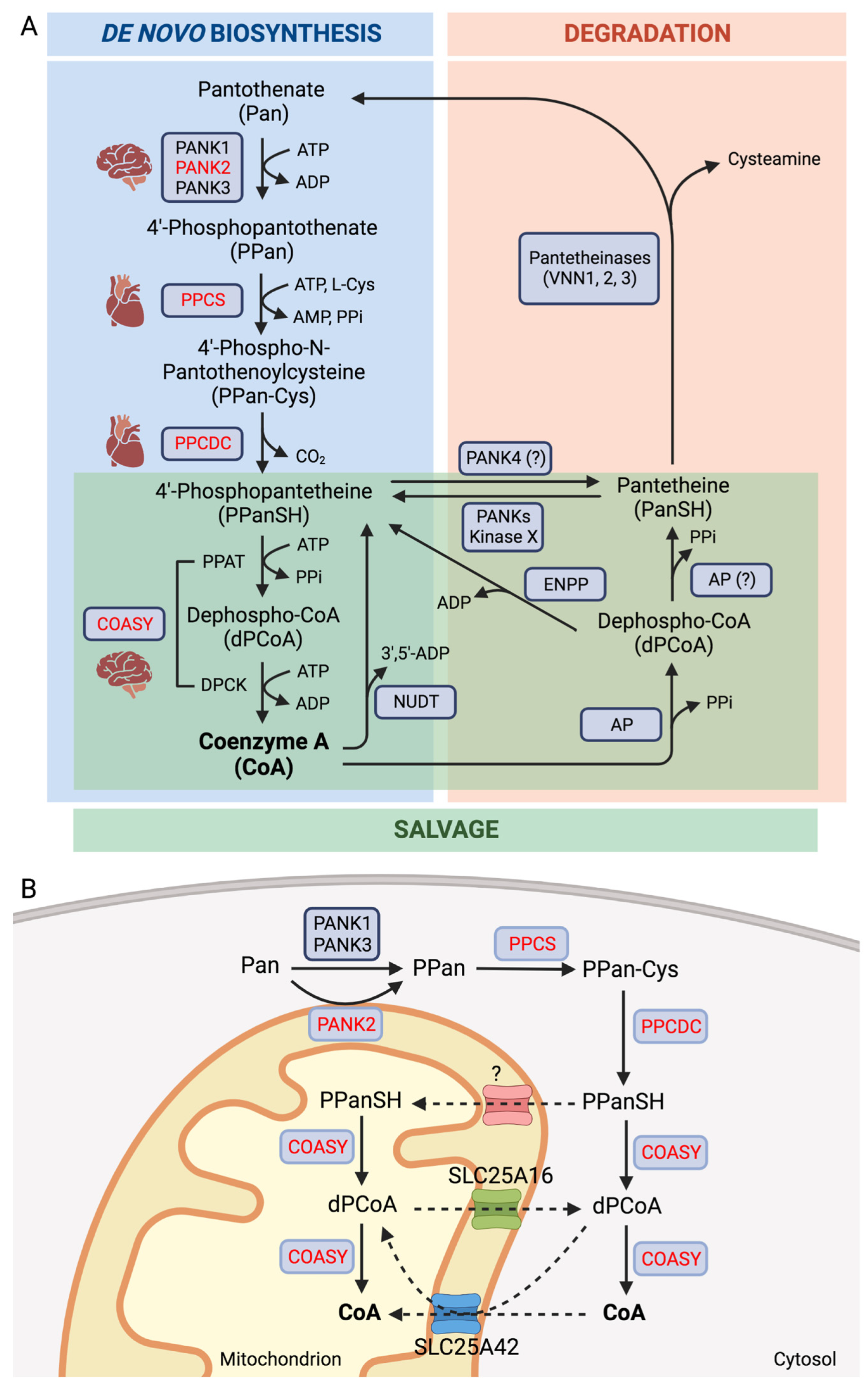
1. Introduction
2. Coenzyme A Homeostasis and Functions
3. Inborn Errors of CoA Biosynthesis
3.1. PANK2
3.2. COASY
3.3. PPCS
3.4. PPCDC
4. Proposed Therapeutic Options
4.1. Metabolic Supplementation
4.1.1. Pantothenate
4.1.2. Pantethine
4.1.3. Fosmetpantotenate
4.1.4. Phosphopantetheine and Acetyl-Phosphopantetheine
4.1.5. Coenzyme A
4.2. Allosteric Activators of PANKs Isoforms
4.3. Improvement of Mitochondrial Function
4.4. Iron Chelators
5. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Davaapil, H.; Tsuchiya, Y.; Gout, I. Signalling Functions of Coenzyme A and Its Derivatives in Mammalian Cells. Biochem. Soc. Trans. 2014, 42, 1056–1062. [Google Scholar] [CrossRef] [PubMed]
- Zhou, B.; Westaway, S.K.; Levinson, B.; Johnson, M.A.; Gitschier, J.; Hayflick, S.J. A Novel Pantothenate Kinase Gene (PANK2) Is Defective in Hallervorden-Spatz Syndrome. Nat. Genet. 2001, 28, 345–349. [Google Scholar] [CrossRef]
- Dusi, S.; Valletta, L.; Haack, T.B.; Tsuchiya, Y.; Venco, P.; Pasqualato, S.; Goffrini, P.; Tigano, M.; Demchenko, N.; Wieland, T.; et al. Exome Sequence Reveals Mutations in CoA Synthase as a Cause of Neurodegeneration with Brain Iron Accumulation. Am. J. Hum. Genet. 2014, 94, 11–22. [Google Scholar] [CrossRef] [PubMed]
- Kurian, M.A.; McNeill, A.; Lin, J.-P.; Maher, E.R. Childhood Disorders of Neurodegeneration with Brain Iron Accumulation (NBIA). Dev. Med. Child Neurol. 2011, 53, 394–404. [Google Scholar] [CrossRef]
- Spaull, R.V.V.; Soo, A.K.S.; Hogarth, P.; Hayflick, S.J.; Kurian, M.A. Towards Precision Therapies for Inherited Disorders of Neurodegeneration with Brain Iron Accumulation. Tremor Other Hyperkinetic Mov. 2021, 11, 51. [Google Scholar] [CrossRef]
- Iuso, A.; Wiersma, M.; Schüller, H.-J.; Pode-Shakked, B.; Marek-Yagel, D.; Grigat, M.; Schwarzmayr, T.; Berutti, R.; Alhaddad, B.; Kanon, B.; et al. Mutations in PPCS, Encoding Phosphopantothenoylcysteine Synthetase, Cause Autosomal-Recessive Dilated Cardiomyopathy. Am. J. Hum. Genet. 2018, 102, 1018–1030. [Google Scholar] [CrossRef]
- Lok, A.; Fernandez-Garcia, M.A.; Taylor, R.W.; French, C.; MacFarland, R.; Bodi, I.; Champion, M.; Josifova, D.; Raymond, F.L.; Iuso, A.; et al. Novel Phosphopantothenoylcysteine Synthetase (PPCS) Mutations with Prominent Neuromuscular Features: Expanding the Phenotypical Spectrum of PPCS-Related Disorders. Am. J. Med. Genet. Part A 2022, 188, 2783–2789. [Google Scholar] [CrossRef] [PubMed]
- Bravo-Alonso, I.; Morin, M.; Arribas-Carreira, L.; Álvarez, M.; Pedrón-Giner, C.; Soletto, L.; Santolaria, C.; Ramón-Maiques, S.; Ugarte, M.; Rodríguez-Pombo, P.; et al. Pathogenic Variants of the Coenzyme A Biosynthesis-Associated Enzyme Phosphopantothenoylcysteine Decarboxylase Cause Autosomal-Recessive Dilated Cardiomyopathy. J. Inherit. Metab. Dis. 2023, 46, 261–272. [Google Scholar] [CrossRef]
- Wang, Y.; Yang, H.; Geerts, C.; Furtos, A.; Waters, P.; Cyr, D.; Wang, S.; Mitchell, G.A. The Multiple Facets of Acetyl-CoA Metabolism: Energetics, Biosynthesis, Regulation, Acylation and Inborn Errors. Mol. Genet. Metab. 2023, 138, 106966. [Google Scholar] [CrossRef]
- Srinivasan, B.; Sibon, O.C.M. Coenzyme A, More than “just” a Metabolic Cofactor. Biochem. Soc. Trans. 2014, 42, 1075–1079. [Google Scholar] [CrossRef]
- Lipmann, F.; Kaplan, N.O. Coenzyme for Acetylation, a Pantothenic Acid Derivative. J. Biol. Chem. 1947, 167, 869. [Google Scholar]
- Robishaw, J.D.; Neely, J.R. Coenzyme A Metabolism. Am. J. Physiol. Endocrinol. Metab. 1985, 248, E1–E9. [Google Scholar] [CrossRef]
- Alfonso-Pecchio, A.; Garcia, M.; Leonardi, R.; Jackowski, S. Compartmentalization of Mammalian Pantothenate Kinases. PLoS ONE 2012, 7, e49509. [Google Scholar] [CrossRef]
- Dansie, L.E.; Reeves, S.; Miller, K.; Zano, S.P.; Frank, M.; Pate, C.; Wang, J.; Jackowski, S. Physiological Roles of the Pantothenate Kinases. Biochem. Soc. Trans. 2014, 42, 1033–1036. [Google Scholar] [CrossRef] [PubMed]
- Yao, J.; Subramanian, C.; Rock, C.O.; Jackowski, S. Human Pantothenate Kinase 4 Is a Pseudo-Pantothenate Kinase. Protein Sci. 2019, 28, 1031–1047. [Google Scholar] [CrossRef] [PubMed]
- Dibble, C.C.; Barritt, S.A.; Perry, G.E.; Lien, E.C.; Geck, R.C.; DuBois-Coyne, S.E.; Bartee, D.; Zengeya, T.T.; Cohen, E.B.; Yuan, M.; et al. PI3K Drives the de Novo Synthesis of Coenzyme A from Vitamin B5. Nature 2022, 608, 192–198. [Google Scholar] [CrossRef]
- Zano, S.P.; Pate, C.; Frank, M.; Rock, C.O.; Jackowski, S. Correction of a Genetic Deficiency in Pantothenate Kinase 1 Using Phosphopantothenate Replacement Therapy. Mol. Genet. Metab. 2015, 116, 281–288. [Google Scholar] [CrossRef]
- Czumaj, A.; Szrok-Jurga, S.; Hebanowska, A.; Turyn, J.; Swierczynski, J.; Sledzinski, T.; Stelmanska, E. The Pathophysiological Role of CoA. Int. J. Mol. Sci. 2020, 21, 9057. [Google Scholar] [CrossRef]
- Naquet, P.; Kerr, E.W.; Vickers, S.D.; Leonardi, R. Regulation of Coenzyme A Levels by Degradation: The “Ins and Outs”. Prog. Lipid Res. 2020, 78, 101028. [Google Scholar] [CrossRef]
- Zhang, Y.-M.; Rock, C.O.; Jackowski, S. Feedback Regulation of Murine Pantothenate Kinase 3 by Coenzyme A and Coenzyme A Thioesters*. J. Biol. Chem. 2005, 280, 32594–32601. [Google Scholar] [CrossRef]
- Leonardi, R.; Zhang, Y.-M.; Rock, C.O.; Jackowski, S. Coenzyme A: Back in Action. Prog. Lipid Res. 2005, 44, 125–153. [Google Scholar] [CrossRef] [PubMed]
- Srinivasan, B.; Baratashvili, M.; van der Zwaag, M.; Kanon, B.; Colombelli, C.; Lambrechts, R.A.; Schaap, O.; Nollen, E.A.; Podgoršek, A.; Kosec, G.; et al. Extracellular 4’-Phosphopantetheine Is a Source for Intracellular Coenzyme A Synthesis. Nat. Chem. Biol. 2015, 11, 784–792. [Google Scholar] [CrossRef] [PubMed]
- Rana, A.; Seinen, E.; Siudeja, K.; Muntendam, R.; Srinivasan, B.; van der Want, J.J.; Hayflick, S.; Reijngoud, D.-J.; Kayser, O.; Sibon, O.C.M. Pantethine Rescues a Drosophila Model for Pantothenate Kinase-Associated Neurodegeneration. Proc. Natl. Acad. Sci. USA 2010, 107, 6988–6993. [Google Scholar] [CrossRef] [PubMed]
- Zhyvoloup, A.; Nemazanyy, I.; Panasyuk, G.; Valovka, T.; Fenton, T.; Rebholz, H.; Wang, M.-L.; Foxon, R.; Lyzogubov, V.; Usenko, V.; et al. Subcellular Localization and Regulation of Coenzyme A Synthase*. J. Biol. Chem. 2003, 278, 50316–50321. [Google Scholar] [CrossRef]
- Rhee, H.-W.; Zou, P.; Udeshi, N.D.; Martell, J.D.; Mootha, V.K.; Carr, S.A.; Ting, A.Y. Proteomic Mapping of Mitochondria in Living Cells via Spatially Restricted Enzymatic Tagging. Science 2013, 339, 1328–1331. [Google Scholar] [CrossRef] [PubMed]
- Xue, L.; Schnacke, P.; Frei, M.S.; Koch, B.; Hiblot, J.; Wombacher, R.; Fabritz, S.; Johnsson, K. Probing Coenzyme A Homeostasis with Semisynthetic Biosensors. Nat. Chem. Biol. 2022, 19, 346–355. [Google Scholar] [CrossRef] [PubMed]
- Fiermonte, G.; Paradies, E.; Todisco, S.; Marobbio, C.M.T.; Palmieri, F. A Novel Member of Solute Carrier Family 25 (SLC25A42) Is a Transporter of Coenzyme A and Adenosine 3′,5′-Diphosphate in Human Mitochondria. J. Biol. Chem. 2009, 284, 18152–18159. [Google Scholar] [CrossRef] [PubMed]
- Vozza, A.; De Leonardis, F.; Paradies, E.; De Grassi, A.; Pierri, C.L.; Parisi, G.; Marobbio, C.M.T.; Lasorsa, F.M.; Muto, L.; Capobianco, L.; et al. Biochemical Characterization of a New Mitochondrial Transporter of Dephosphocoenzyme A in Drosophila Melanogaster. Biochim. Biophys. Acta (BBA)-Bioenerg. 2017, 1858, 137–146. [Google Scholar] [CrossRef]
- Prohl, C.; Pelzer, W.; Diekert, K.; Kmita, H.; Bedekovics, T.; Kispal, G.; Lill, R. The Yeast Mitochondrial Carrier Leu5p and Its Human Homologue Graves’ Disease Protein Are Required for Accumulation of Coenzyme A in the Matrix. Mol. Cell. Biol. 2001, 21, 1089–1097. [Google Scholar] [CrossRef]
- Khan, S.; Ansar, M.; Khan, A.K.; Shah, K.; Muhammad, N.; Shahzad, S.; Nickerson, D.A.; Bamshad, M.J.; Santos-Cortez, R.L.P.; Leal, S.M.; et al. A Homozygous Missense Mutation in SLC25A16 Is Associated with Autosomal Recessive Isolated Fingernail Dysplasia in a Pakistani Family. Br. J. Derm. 2018, 178, 556–558. [Google Scholar] [CrossRef]
- Yu, Y.; Moretti, I.F.; Grzeschik, N.A.; Sibon, O.C.M.; Schepers, H. Coenzyme A Levels Influence Protein Acetylation, CoAlation and 4’-Phosphopantetheinylation: Expanding the Impact of a Metabolic Nexus Molecule. Biochim. Biophys. Acta Mol. Cell Res. 2021, 1868, 118965. [Google Scholar] [CrossRef] [PubMed]
- Shi, L.; Tu, B.P. Acetyl-CoA and the Regulation of Metabolism: Mechanisms and Consequences. Curr. Opin. Cell Biol. 2015, 33, 125–131. [Google Scholar] [CrossRef]
- Pietrocola, F.; Galluzzi, L.; Bravo-San Pedro, J.M.; Madeo, F.; Kroemer, G. Acetyl Coenzyme A: A Central Metabolite and Second Messenger. Cell Metab. 2015, 21, 805–821. [Google Scholar] [CrossRef] [PubMed]
- Barnes, C.E.; English, D.M.; Cowley, S.M. Acetylation & Co: An Expanding Repertoire of Histone Acylations Regulates Chromatin and Transcription. Essays Biochem. 2019, 63, 97–107. [Google Scholar] [CrossRef] [PubMed]
- Ali, I.; Conrad, R.J.; Verdin, E.; Ott, M. Lysine Acetylation Goes Global: From Epigenetics to Metabolism and Therapeutics. Chem. Rev. 2018, 118, 1216–1252. [Google Scholar] [CrossRef]
- Shang, S.; Liu, J.; Hua, F. Protein Acylation: Mechanisms, Biological Functions and Therapeutic Targets. Signal Transduct. Target. 2022, 7, 1–30. [Google Scholar] [CrossRef]
- Xu, Y.; Shi, Z.; Bao, L. An Expanding Repertoire of Protein Acylations. Mol. Cell. Proteom. 2022, 21, 100193. [Google Scholar] [CrossRef]
- Beld, J.; Sonnenschein, E.C.; Vickery, C.R.; Noel, J.P.; Burkart, M.D. The Phosphopantetheinyl Transferases: Catalysis of a Post-Translational Modification Crucial for Life. Nat. Prod. Rep. 2014, 31, 61–108. [Google Scholar] [CrossRef]
- Strickland, K.C.; Hoeferlin, L.A.; Oleinik, N.V.; Krupenko, N.I.; Krupenko, S.A. Acyl Carrier Protein-Specific 4’-Phosphopantetheinyl Transferase Activates 10-Formyltetrahydrofolate Dehydrogenase. J. Biol. Chem. 2010, 285, 1627–1633. [Google Scholar] [CrossRef]
- Masud, A.J.; Kastaniotis, A.J.; Rahman, M.T.; Autio, K.J.; Hiltunen, J.K. Mitochondrial Acyl Carrier Protein (ACP) at the Interface of Metabolic State Sensing and Mitochondrial Function. Biochim. Biophys. Acta Mol. Cell Res. 2019, 1866, 118540. [Google Scholar] [CrossRef]
- Lambrechts, R.A.; Schepers, H.; Yu, Y.; van der Zwaag, M.; Autio, K.J.; Vieira-Lara, M.A.; Bakker, B.M.; Tijssen, M.A.; Hayflick, S.J.; Grzeschik, N.A.; et al. CoA-dependent Activation of Mitochondrial Acyl Carrier Protein Links Four Neurodegenerative Diseases. EMBO Mol. Med. 2019, 11, e10488. [Google Scholar] [CrossRef] [PubMed]
- Tsuchiya, Y.; Peak-Chew, S.Y.; Newell, C.; Miller-Aidoo, S.; Mangal, S.; Zhyvoloup, A.; Bakovic´, J.; Malanchuk, O.; Pereira, G.C.; Kotiadis, V.; et al. Protein CoAlation: A Redox-Regulated Protein Modification by Coenzyme A in Mammalian Cells. Biochem. J. 2017, 474, 2489–2508. [Google Scholar] [CrossRef] [PubMed]
- Gout, I. Coenzyme A, Protein CoAlation and Redox Regulation in Mammalian Cells. Biochem. Soc. Trans. 2018, 46, 721–728. [Google Scholar] [CrossRef]
- Brezavar, D.; Bonnen, P.E. Incidence of PKAN Determined by Bioinformatic and Population-Based Analysis of ~140,000 Humans. Mol. Genet. Metab. 2019, 128, 463–469. [Google Scholar] [CrossRef] [PubMed]
- Hartig, M.B.; Hörtnagel, K.; Garavaglia, B.; Zorzi, G.; Kmiec, T.; Klopstock, T.; Rostasy, K.; Svetel, M.; Kostic, V.S.; Schuelke, M.; et al. Genotypic and Phenotypic Spectrum of PANK2 Mutations in Patients with Neurodegeneration with Brain Iron Accumulation. Ann. Neurol. 2006, 59, 248–256. [Google Scholar] [CrossRef]
- Choayb, S.; Adil, H.; Ali Mohamed, D.; Allali, N.; Chat, L.; El Haddad, S. Eye of the Tiger Sign in Pantothenate Kinase-Associated Neurodegeneration. Case Rep. Radiol. 2021, 2021, 6633217. [Google Scholar] [CrossRef]
- Delgado, R.F.; Sanchez, P.R.; Speckter, H.; Then, E.P.; Jimenez, R.; Oviedo, J.; Dellani, P.R.; Foerster, B.; Stoeter, P. Missense PANK2 Mutation without “Eye of the Tiger” Sign: MR Findings in a Large Group of Patients with Pantothenate Kinase-Associated Neurodegeneration (PKAN). J. Magn. Reson. Imaging 2012, 35, 788–794. [Google Scholar] [CrossRef]
- Dehghan Manshadi, M.; Rohani, M.; Rezaei, A.; Aryani, O. A Case of MPAN with “Eye of the Tiger Sign,” Mimicking PKAN. Mov. Disord. Clin. Pr. 2022, 9, 693–695. [Google Scholar] [CrossRef]
- Gregory, A.; Hayflick, S.J. Pantothenate Kinase-Associated Neurodegeneration. In GeneReviews®; Adam, M.P., Everman, D.B., Mirzaa, G.M., Pagon, R.A., Wallace, S.E., Bean, L.J., Gripp, K.W., Amemiya, A., Eds.; University of Washington, Seattle: Seattle, WA, USA, 1993. [Google Scholar]
- Kruer, M.C.; Hiken, M.; Gregory, A.; Malandrini, A.; Clark, D.; Hogarth, P.; Grafe, M.; Hayflick, S.J.; Woltjer, R.L. Novel Histopathologic Findings in Molecularly-Confirmed Pantothenate Kinase-Associated Neurodegeneration. Brain 2011, 134, 947–958. [Google Scholar] [CrossRef]
- Li, A.; Paudel, R.; Johnson, R.; Courtney, R.; Lees, A.J.; Holton, J.L.; Hardy, J.; Revesz, T.; Houlden, H. Pantothenate Kinase-Associated Neurodegeneration Is Not a Synucleinopathy. Neuropathol. Appl. Neurobiol. 2013, 39, 121–131. [Google Scholar] [CrossRef]
- Zhang, Y.-M.; Rock, C.O.; Jackowski, S. Biochemical Properties of Human Pantothenate Kinase 2 Isoforms and Mutations Linked to Pantothenate Kinase-Associated Neurodegeneration*. J. Biol. Chem. 2006, 281, 107–114. [Google Scholar] [CrossRef]
- Hörtnagel, K.; Prokisch, H.; Meitinger, T. An Isoform of HPANK2, Deficient in Pantothenate Kinase-Associated Neurodegeneration, Localizes to Mitochondria. Hum. Mol. Genet. 2003, 12, 321–327. [Google Scholar] [CrossRef] [PubMed]
- Kotzbauer, P.T.; Truax, A.C.; Trojanowski, J.Q.; Lee, V.M.-Y. Altered Neuronal Mitochondrial Coenzyme A Synthesis in Neurodegeneration with Brain Iron Accumulation Caused by Abnormal Processing, Stability, and Catalytic Activity of Mutant Pantothenate Kinase 2. J. Neurosci. 2005, 25, 689–698. [Google Scholar] [CrossRef] [PubMed]
- Kurian, M.A.; Hayflick, S.J. Pantothenate Kinase-Associated Neurodegeneration (PKAN) and PLA2G6-Associated Neurodegeneration (PLAN): Review of Two Major Neurodegeneration with Brain Iron Accumulation (NBIA) Phenotypes. Int. Rev. Neurobiol. 2013, 110, 49–71. [Google Scholar] [CrossRef]
- Aoun, M.; Corsetto, P.A.; Nugue, G.; Montorfano, G.; Ciusani, E.; Crouzier, D.; Hogarth, P.; Gregory, A.; Hayflick, S.; Zorzi, G.; et al. Changes in Red Blood Cell Membrane Lipid Composition: A New Perspective into the Pathogenesis of PKAN. Mol. Genet. Metab. 2017, 121, 180–189. [Google Scholar] [CrossRef] [PubMed]
- Leoni, V.; Strittmatter, L.; Zorzi, G.; Zibordi, F.; Dusi, S.; Garavaglia, B.; Venco, P.; Caccia, C.; Souza, A.L.; Deik, A.; et al. Metabolic Consequences of Mitochondrial Coenzyme A Deficiency in Patients with PANK2 Mutations. Mol. Genet. Metab. 2012, 105, 463–471. [Google Scholar] [CrossRef] [PubMed]
- Hayflick, S.J.; Jeong, S.Y.; Sibon, O.C.M. PKAN Pathogenesis and Treatment. Mol. Genet. Metab. 2022, 137, 283–291. [Google Scholar] [CrossRef]
- Campanella, A.; Privitera, D.; Guaraldo, M.; Rovelli, E.; Barzaghi, C.; Garavaglia, B.; Santambrogio, P.; Cozzi, A.; Levi, S. Skin Fibroblasts from Pantothenate Kinase-Associated Neurodegeneration Patients Show Altered Cellular Oxidative Status and Have Defective Iron-Handling Properties. Hum. Mol. Genet. 2012, 21, 4049–4059. [Google Scholar] [CrossRef]
- Santambrogio, P.; Dusi, S.; Guaraldo, M.; Rotundo, L.I.; Broccoli, V.; Garavaglia, B.; Tiranti, V.; Levi, S. Mitochondrial Iron and Energetic Dysfunction Distinguish Fibroblasts and Induced Neurons from Pantothenate Kinase-Associated Neurodegeneration Patients. Neurobiol. Dis. 2015, 81, 144–153. [Google Scholar] [CrossRef]
- Álvarez-Córdoba, M.; Fernández Khoury, A.; Villanueva-Paz, M.; Gómez-Navarro, C.; Villalón-García, I.; Suárez-Rivero, J.M.; Povea-Cabello, S.; de la Mata, M.; Cotán, D.; Talaverón-Rey, M.; et al. Pantothenate Rescues Iron Accumulation in Pantothenate Kinase-Associated Neurodegeneration Depending on the Type of Mutation. Mol. Neurobiol. 2019, 56, 3638–3656. [Google Scholar] [CrossRef]
- Poli, M.; Derosas, M.; Luscieti, S.; Cavadini, P.; Campanella, A.; Verardi, R.; Finazzi, D.; Arosio, P. Pantothenate Kinase-2 (Pank2) Silencing Causes Cell Growth Reduction, Cell-Specific Ferroportin Upregulation and Iron Deregulation. Neurobiol. Dis. 2010, 39, 204–210. [Google Scholar] [CrossRef] [PubMed]
- Orellana, D.I.; Santambrogio, P.; Rubio, A.; Yekhlef, L.; Cancellieri, C.; Dusi, S.; Giannelli, S.G.; Venco, P.; Mazzara, P.G.; Cozzi, A.; et al. Coenzyme A Corrects Pathological Defects in Human Neurons of PANK2-associated Neurodegeneration. EMBO Mol. Med. 2016, 8, 1197–1211. [Google Scholar] [CrossRef] [PubMed]
- Santambrogio, P.; Ripamonti, M.; Paolizzi, C.; Panteghini, C.; Carecchio, M.; Chiapparini, L.; Raimondi, M.; Rubio, A.; Di Meo, I.; Cozzi, A.; et al. Harmful Iron-Calcium Relationship in Pantothenate Kinase Associated Neurodegeneration. Int. J. Mol. Sci. 2020, 21, 3664. [Google Scholar] [CrossRef] [PubMed]
- Shi, X.; Zheng, F.; Ye, X.; Li, X.; Zhao, Q.; Lin, Z.; Hu, Y.; Wang, J. Basal Ganglia Calcification and Novel Compound Heterozygous Mutations in the PANK2 Gene in a Chinese Boy with Classic Pantothenate Kinase-Associated Neurodegeneration. Medicine 2018, 97, e0316. [Google Scholar] [CrossRef]
- Santambrogio, P.; Ripamonti, M.; Cozzi, A.; Raimondi, M.; Cavestro, C.; Di Meo, I.; Rubio, A.; Taverna, S.; Tiranti, V.; Levi, S. Massive Iron Accumulation in PKAN-Derived Neurons and Astrocytes: Light on the Human Pathological Phenotype. Cell Death Dis. 2022, 13, 185. [Google Scholar] [CrossRef]
- Ceccatelli Berti, C.; Gilea, A.I.; De Gregorio, M.A.; Goffrini, P. Exploring Yeast as a Study Model of Pantothenate Kinase-Associated Neurodegeneration and for the Identification of Therapeutic Compounds. Int. J. Mol. Sci. 2020, 22, 293. [Google Scholar] [CrossRef]
- Ceccatelli Berti, C.; Gihaz, S.; Figuccia, S.; Choi, J.-Y.; Pal, A.C.; Goffrini, P.; Ben Mamoun, C. Evidence for a Conserved Function of Eukaryotic Pantothenate Kinases in the Regulation of Mitochondrial Homeostasis and Oxidative Stress. Int. J. Mol. Sci. 2022, 24, 435. [Google Scholar] [CrossRef]
- Bosveld, F.; Rana, A.; van der Wouden, P.E.; Lemstra, W.; Ritsema, M.; Kampinga, H.H.; Sibon, O.C.M. De Novo CoA Biosynthesis Is Required to Maintain DNA Integrity during Development of the Drosophila Nervous System. Hum. Mol. Genet. 2008, 17, 2058–2069. [Google Scholar] [CrossRef]
- Siudeja, K.; Srinivasan, B.; Xu, L.; Rana, A.; de Jong, J.; Nollen, E.A.A.; Jackowski, S.; Sanford, L.; Hayflick, S.; Sibon, O.C.M. Impaired Coenzyme A Metabolism Affects Histone and Tubulin Acetylation in Drosophila and Human Cell Models of Pantothenate Kinase Associated Neurodegeneration. EMBO Mol. Med. 2011, 3, 755–766. [Google Scholar] [CrossRef]
- Wu, Z.; Li, C.; Lv, S.; Zhou, B. Pantothenate Kinase-Associated Neurodegeneration: Insights from a Drosophila Model. Hum. Mol. Genet. 2009, 18, 3659–3672. [Google Scholar] [CrossRef]
- Afshar, K.; Gönczy, P.; DiNardo, S.; Wasserman, S.A. Fumble Encodes a Pantothenate Kinase Homolog Required for Proper Mitosis and Meiosis in Drosophila Melanogaster. Genetics 2001, 157, 1267–1276. [Google Scholar] [CrossRef] [PubMed]
- Huang, Y.; Wan, Z.; Tang, Y.; Xu, J.; Laboret, B.; Nallamothu, S.; Yang, C.; Liu, B.; Lu, R.O.; Lu, B.; et al. Pantothenate Kinase 2 Interacts with PINK1 to Regulate Mitochondrial Quality Control via Acetyl-CoA Metabolism. Nat. Commun. 2022, 13, 2412. [Google Scholar] [CrossRef] [PubMed]
- Zizioli, D.; Tiso, N.; Guglielmi, A.; Saraceno, C.; Busolin, G.; Giuliani, R.; Khatri, D.; Monti, E.; Borsani, G.; Argenton, F.; et al. Knock-down of Pantothenate Kinase 2 Severely Affects the Development of the Nervous and Vascular System in Zebrafish, Providing New Insights into PKAN Disease. Neurobiol. Dis. 2016, 85, 35–48. [Google Scholar] [CrossRef] [PubMed]
- Pagani, F.; Trivedi, A.; Khatri, D.; Zizioli, D.; Garrafa, E.; Mitola, S.; Finazzi, D. Silencing of Pantothenate Kinase 2 Reduces Endothelial Cell Angiogenesis. Mol. Med. Rep. 2018, 18, 4739–4746. [Google Scholar] [CrossRef] [PubMed]
- Khatri, D.; Zizioli, D.; Trivedi, A.; Borsani, G.; Monti, E.; Finazzi, D. Overexpression of Human Mutant PANK2 Proteins Affects Development and Motor Behavior of Zebrafish Embryos. Neuromol. Med. 2019, 21, 120–131. [Google Scholar] [CrossRef]
- Mignani, L.; Zizioli, D.; Khatri, D.; Facchinello, N.; Schiavone, M.; De Palma, G.; Finazzi, D. Bi-Allelic Mutations in Zebrafish Pank2 Gene Lead to Testicular Atrophy and Perturbed Behavior without Signs of Neurodegeneration. Int. J. Mol. Sci. 2022, 23, 12914. [Google Scholar] [CrossRef]
- Kuo, Y.-M.; Duncan, J.L.; Westaway, S.K.; Yang, H.; Nune, G.; Xu, E.Y.; Hayflick, S.J.; Gitschier, J. Deficiency of Pantothenate Kinase 2 (Pank2) in Mice Leads to Retinal Degeneration and Azoospermia. Hum. Mol. Genet. 2005, 14, 49–57. [Google Scholar] [CrossRef]
- Brunetti, D.; Dusi, S.; Morbin, M.; Uggetti, A.; Moda, F.; D’Amato, I.; Giordano, C.; d’Amati, G.; Cozzi, A.; Levi, S.; et al. Pantothenate Kinase-Associated Neurodegeneration: Altered Mitochondria Membrane Potential and Defective Respiration in Pank2 Knock-out Mouse Model. Hum. Mol. Genet. 2012, 21, 5294–5305. [Google Scholar] [CrossRef]
- Kuo, Y.M.; Hayflick, S.J.; Gitschier, J. Deprivation of Pantothenic Acid Elicits a Movement Disorder and Azoospermia in a Mouse Model of Pantothenate Kinase-Associated Neurodegeneration. J. Inherit. Metab. Dis. 2007, 30, 310–317. [Google Scholar] [CrossRef]
- Brunetti, D.; Dusi, S.; Giordano, C.; Lamperti, C.; Morbin, M.; Fugnanesi, V.; Marchet, S.; Fagiolari, G.; Sibon, O.; Moggio, M.; et al. Pantethine Treatment Is Effective in Recovering the Disease Phenotype Induced by Ketogenic Diet in a Pantothenate Kinase-Associated Neurodegeneration Mouse Model. Brain 2014, 137, 57–68. [Google Scholar] [CrossRef]
- Jeong, S.Y.; Hogarth, P.; Placzek, A.; Gregory, A.M.; Fox, R.; Zhen, D.; Hamada, J.; van der Zwaag, M.; Lambrechts, R.; Jin, H.; et al. 4′-Phosphopantetheine Corrects CoA, Iron, and Dopamine Metabolic Defects in Mammalian Models of PKAN. EMBO Mol. Med. 2019, 11, e10489. [Google Scholar] [CrossRef] [PubMed]
- Leonardi, R.; Rehg, J.E.; Rock, C.O.; Jackowski, S. Pantothenate Kinase 1 Is Required to Support the Metabolic Transition from the Fed to the Fasted State. PLoS ONE 2010, 5, e11107. [Google Scholar] [CrossRef] [PubMed]
- Garcia, M.; Leonardi, R.; Zhang, Y.-M.; Rehg, J.E.; Jackowski, S. Germline Deletion of Pantothenate Kinases 1 and 2 Reveals the Key Roles for CoA in Postnatal Metabolism. PLoS ONE 2012, 7, e40871. [Google Scholar] [CrossRef]
- Sharma, L.K.; Subramanian, C.; Yun, M.-K.; Frank, M.W.; White, S.W.; Rock, C.O.; Lee, R.E.; Jackowski, S. A Therapeutic Approach to Pantothenate Kinase Associated Neurodegeneration. Nat. Commun. 2018, 9, 4399. [Google Scholar] [CrossRef] [PubMed]
- Subramanian, C.; Yao, J.; Frank, M.W.; Rock, C.O.; Jackowski, S. A Pantothenate Kinase-Deficient Mouse Model Reveals a Gene Expression Program Associated with Brain Coenzyme a Reduction. Biochim. Biophys. Acta (BBA)-Mol. Basis Dis. 2020, 1866, 165663. [Google Scholar] [CrossRef]
- Álvarez-Córdoba, M.; Talaverón-Rey, M.; Villalón-García, I.; Povea-Cabello, S.; Suárez-Rivero, J.M.; Suárez-Carrillo, A.; Munuera-Cabeza, M.; Salas, J.J.; Sánchez-Alcázar, J.A. Down Regulation of the Expression of Mitochondrial Phosphopantetheinyl-Proteins in Pantothenate Kinase-Associated Neurodegeneration: Pathophysiological Consequences and Therapeutic Perspectives. Orphanet J. Rare Dis. 2021, 16, 201. [Google Scholar] [CrossRef] [PubMed]
- Rowland, E.A.; Snowden, C.K.; Cristea, I.M. Protein Lipoylation: An Evolutionarily Conserved Metabolic Regulator of Health and Disease. Curr. Opin. Chem. Biol. 2018, 42, 76–85. [Google Scholar] [CrossRef]
- Ceccatelli Berti, C.; di Punzio, G.; Dallabona, C.; Baruffini, E.; Goffrini, P.; Lodi, T.; Donnini, C. The Power of Yeast in Modelling Human Nuclear Mutations Associated with Mitochondrial Diseases. Genes 2021, 12, 300. [Google Scholar] [CrossRef]
- Drecourt, A.; Babdor, J.; Dussiot, M.; Petit, F.; Goudin, N.; Garfa-Traoré, M.; Habarou, F.; Bole-Feysot, C.; Nitschké, P.; Ottolenghi, C.; et al. Impaired Transferrin Receptor Palmitoylation and Recycling in Neurodegeneration with Brain Iron Accumulation. Am. J. Hum. Genet. 2018, 102, 266–277. [Google Scholar] [CrossRef]
- Ripamonti, M.; Santambrogio, P.; Racchetti, G.; Cozzi, A.; Di Meo, I.; Tiranti, V.; Levi, S. PKAN HiPS-Derived Astrocytes Show Impairment of Endosomal Trafficking: A Potential Mechanism Underlying Iron Accumulation. Front. Cell. Neurosci. 2022, 16, 878103. [Google Scholar] [CrossRef]
- Zhyvoloup, A.; Nemazanyy, I.; Babich, A.; Panasyuk, G.; Pobigailo, N.; Vudmaska, M.; Naidenov, V.; Kukharenko, O.; Palchevskii, S.; Savinska, L.; et al. Molecular Cloning of CoA Synthase: THE MISSING LINK IN CoA BIOSYNTHESIS*210. J. Biol. Chem. 2002, 277, 22107–22110. [Google Scholar] [CrossRef] [PubMed]
- Nemazanyy, I.; Panasyuk, G.; Breus, O.; Zhyvoloup, A.; Filonenko, V.; Gout, I.T. Identification of a Novel CoA Synthase Isoform, Which Is Primarily Expressed in the Brain. Biochem. Biophys. Res. Commun. 2006, 341, 995–1000. [Google Scholar] [CrossRef] [PubMed]
- Lin, C.-C.; Kitagawa, M.; Tang, X.; Hou, M.-H.; Wu, J.; Qu, D.C.; Srinivas, V.; Liu, X.; Thompson, J.W.; Mathey-Prevot, B.; et al. CoA Synthase Regulates Mitotic Fidelity via CBP-Mediated Acetylation. Nat. Commun. 2018, 9, 1039. [Google Scholar] [CrossRef] [PubMed]
- Orphanet: COASY Protein Associated Neurodegeneration. Available online: https://www.orpha.net/consor/cgi-bin/OC_Exp.php?lng=EN&Expert=397725 (accessed on 8 March 2023).
- Annesi, G.; Gagliardi, M.; Iannello, G.; Quattrone, A.; Iannello, G.; Quattrone, A. Mutational Analysis of COASY in an Italian Patient with NBIA. Park. Relat. Disord. 2016, 28, 150–151. [Google Scholar] [CrossRef]
- Evers, C.; Seitz, A.; Assmann, B.; Opladen, T.; Karch, S.; Hinderhofer, K.; Granzow, M.; Paramasivam, N.; Eils, R.; Diessl, N.; et al. Diagnosis of CoPAN by Whole Exome Sequencing: Waking up a Sleeping Tiger’s Eye. Am. J. Med. Genet. Part A 2017, 173, 1878–1886. [Google Scholar] [CrossRef]
- van Dijk, T.; Ferdinandusse, S.; Ruiter, J.P.N.; Alders, M.; Mathijssen, I.B.; Parboosingh, J.S.; Innes, A.M.; Meijers-Heijboer, H.; Poll-The, B.T.; Bernier, F.P.; et al. Biallelic Loss of Function Variants in COASY Cause Prenatal Onset Pontocerebellar Hypoplasia, Microcephaly, and Arthrogryposis. Eur. J. Hum. Genet. 2018, 26, 1752–1758. [Google Scholar] [CrossRef]
- Mishra, R.; Kulshreshtha, S.; Mandal, K.; Khurana, A.; Diego-Álvarez, D.; Pradas, L.; Saxena, R.; Phadke, S.; Moirangthem, A.; Masih, S.; et al. COASY Related Pontocerebellar Hypoplasia Type 12: A Common Indian Mutation with Expansion of the Phenotypic Spectrum. Am. J. Med. Genet. A 2022, 188, 2339–2350. [Google Scholar] [CrossRef]
- Rosati, J.; Johnson, J.; Stander, Z.; White, A.; Tortorelli, S.; Bailey, D.; Fong, C.-T.; Lee, B.H. Progressive Brain Atrophy and Severe Neurodevelopmental Phenotype in Siblings with Biallelic COASY Variants. Am. J. Med. Genet. A 2022, 141, 842–845. [Google Scholar] [CrossRef]
- Lee, J.H.; Gurney, S.; Pang, D.; Temkin, A.; Park, N.; Janicki, S.C.; Zigman, W.B.; Silverman, W.; Tycko, B.; Schupf, N. Polymorphisms in HSD17B1: Early Onset and Increased Risk of Alzheimer’s Disease in Women with Down Syndrome. Curr. Gerontol. Geriatr. Res. 2012, 2012, e361218. [Google Scholar] [CrossRef]
- Kolarova, H.; Tan, J.; Strom, T.M.; Meitinger, T.; Wagner, M.; Klopstock, T. Lifetime Risk of Autosomal Recessive Neurodegeneration with Brain Iron Accumulation (NBIA) Disorders Calculated from Genetic Databases. EBioMedicine 2022, 77, 103869. [Google Scholar] [CrossRef]
- Olzhausen, J.; Moritz, T.; Neetz, T.; Schüller, H.-J. Molecular Characterization of the Heteromeric Coenzyme A-Synthesizing Protein Complex (CoA-SPC) in the Yeast Saccharomyces Cerevisiae. FEMS Yeast Res. 2013, 13, 565–573. [Google Scholar] [CrossRef] [PubMed]
- Ceccatelli Berti, C.; Lazzaretti, M.; Dusi, S.; Tosi, E.; Tiranti, V.; Goffrini, P. Modeling Human Coenzyme A Synthase Mutation in Yeast Reveals Altered Mitochondrial Function, Lipid Content and Iron Metabolism. Microb. Cell 2015, 2, 126–135. [Google Scholar] [CrossRef] [PubMed]
- Khatri, D.; Zizioli, D.; Tiso, N.; Facchinello, N.; Vezzoli, S.; Gianoncelli, A.; Memo, M.; Monti, E.; Borsani, G.; Finazzi, D. Down-Regulation of Coasy, the Gene Associated with NBIA-VI, Reduces Bmp Signaling, Perturbs Dorso-Ventral Patterning and Alters Neuronal Development in Zebrafish. Sci. Rep. 2016, 6, 37660. [Google Scholar] [CrossRef] [PubMed]
- Di Meo, I.; Cavestro, C.; Pedretti, S.; Fu, T.; Ligorio, S.; Manocchio, A.; Lavermicocca, L.; Santambrogio, P.; Ripamonti, M.; Levi, S.; et al. Neuronal Ablation of CoA Synthase Causes Motor Deficits, Iron Dyshomeostasis, and Mitochondrial Dysfunctions in a CoPAN Mouse Model. Int. J. Mol. Sci. 2020, 21, 9707. [Google Scholar] [CrossRef]
- Ferrandon, S.; DeVecchio, J.; Duraes, L.; Chouhan, H.; Karagkounis, G.; Davenport, J.; Orloff, M.; Liska, D.; Kalady, M.F. CoA Synthase (COASY) Mediates Radiation Resistance via PI3K Signaling in Rectal Cancer. Cancer Res. 2020, 80, 334–346. [Google Scholar] [CrossRef]
- Nemazanyy, I.; Panasyuk, G.; Zhyvoloup, A.; Panayotou, G.; Gout, I.T.; Filonenko, V. Specific Interaction between S6K1 and CoA Synthase: A Potential Link between the MTOR/S6K Pathway, CoA Biosynthesis and Energy Metabolism. FEBS Lett. 2004, 578, 357–362. [Google Scholar] [CrossRef]
- Kharabsheh, H.A.; Scott, J.E. CoAsy Knockdown in TNBC Cell Lines Resulted in No Overt Effect on Cell Proliferation in Vitro. Biochem. Biophys. Res. Commun. 2020, 530, 136–141. [Google Scholar] [CrossRef]
- Breus, O.; Panasyuk, G.; Gout, I.T.; Filonenko, V.; Nemazanyy, I. CoA Synthase Is in Complex with P85αPI3K and Affects PI3K Signaling Pathway. Biochem. Biophys. Res. Commun. 2009, 385, 581–585. [Google Scholar] [CrossRef]
- Breus, O.; Panasyuk, G.; Gout, I.T.; Filonenko, V.; Nemazanyy, I. CoA Synthase Is Phosphorylated on Tyrosines in Mammalian Cells, Interacts with and Is Dephosphorylated by Shp2PTP. Mol. Cell. Biochem. 2010, 335, 195–202. [Google Scholar] [CrossRef]
- Gudkova, D.; Panasyuk, G.; Nemazanyy, I.; Zhyvoloup, A.; Monteil, P.; Filonenko, V.; Gout, I. EDC4 Interacts with and Regulates the Dephospho-CoA Kinase Activity of CoA Synthase. FEBS Lett. 2012, 586, 3590–3595. [Google Scholar] [CrossRef]
- Kobayashi, N.; Shinagawa, S.; Nagata, T.; Shimada, K.; Shibata, N.; Ohnuma, T.; Kasanuki, K.; Arai, H.; Yamada, H.; Nakayama, K.; et al. Usefulness of DNA Methylation Levels in COASY and SPINT1 Gene Promoter Regions as Biomarkers in Diagnosis of Alzheimer’s Disease and Amnestic Mild Cognitive Impairment. PLoS ONE 2016, 11, e0168816. [Google Scholar] [CrossRef] [PubMed]
- Kobayashi, N.; Shinagawa, S.; Niimura, H.; Kida, H.; Nagata, T.; Tagai, K.; Shimada, K.; Oka, N.; Shikimoto, R.; Noda, Y.; et al. Increased Blood COASY DNA Methylation Levels a Potential Biomarker for Early Pathology of Alzheimer’s Disease. Sci. Rep. 2020, 10, 12217. [Google Scholar] [CrossRef] [PubMed]
- Iuso, A.; Zhang, F.; Rusha, E.; Campbell, B.; Dorn, T.; Zanuttigh, E.; Haas, D.; Anikster, Y.; Lederer, G.; Pertek, A.; et al. Generation of Two Human IPSC Lines, HMGUi003-A and MRIi028-A, Carrying Pathogenic Biallelic Variants in the PPCS Gene. Stem. Cell Res. 2022, 61, 102773. [Google Scholar] [CrossRef] [PubMed]
- Daugherty, M.; Polanuyer, B.; Farrell, M.; Scholle, M.; Lykidis, A.; de Crécy-Lagard, V.; Osterman, A. Complete Reconstitution of the Human Coenzyme A Biosynthetic Pathway via Comparative Genomics*. J. Biol. Chem. 2002, 277, 21431–21439. [Google Scholar] [CrossRef]
- Hogarth, P.; Kurian, M.A.; Gregory, A.; Csányi, B.; Zagustin, T.; Kmiec, T.; Wood, P.; Klucken, A.; Scalise, N.; Sofia, F.; et al. Consensus Clinical Management Guideline for Pantothenate Kinase-Associated Neurodegeneration (PKAN). Mol. Genet. Metab. 2017, 120, 278–287. [Google Scholar] [CrossRef]
- Munshi, M.I.; Yao, S.J.; Ben Mamoun, C. Redesigning Therapies for Pantothenate Kinase–Associated Neurodegeneration. J. Biol. Chem. 2022, 298, 101577. [Google Scholar] [CrossRef]
- Álvarez-Córdoba, M.; Reche-López, D.; Cilleros-Holgado, P.; Talaverón-Rey, M.; Villalón-García, I.; Povea-Cabello, S.; Suárez-Rivero, J.M.; Suárez-Carrillo, A.; Munuera-Cabeza, M.; Piñero-Pérez, R.; et al. Therapeutic Approach with Commercial Supplements for Pantothenate Kinase-Associated Neurodegeneration with Residual PANK2 Expression Levels. Orphanet J. Rare Dis. 2022, 17, 311. [Google Scholar] [CrossRef]
- Werning, M.; Müllner, E.W.; Mlynek, G.; Dobretzberger, V.; Djinovic-Carugo, K.; Baron, D.M.; Prokisch, H.; Büchner, B.; Klopstock, T.; Salzer, U. PKAN Neurodegeneration and Residual PANK2 Activities in Patient Erythrocytes. Ann. Clin. Transl. Neurol. 2020, 7, 1340–1351. [Google Scholar] [CrossRef]
- Tóth, F.; Cseh, E.K.; Vécsei, L. Natural Molecules and Neuroprotection: Kynurenic Acid, Pantethine and α-Lipoic Acid. Int. J. Mol. Sci. 2021, 22, 403. [Google Scholar] [CrossRef]
- Balibar, C.J.; Hollis-Symynkywicz, M.F.; Tao, J. Pantethine Rescues Phosphopantothenoylcysteine Synthetase and Phosphopantothenoylcysteine Decarboxylase Deficiency in Escherichia Coli but Not in Pseudomonas Aeruginosa. J. Bacteriol. 2011, 193, 3304–3312. [Google Scholar] [CrossRef]
- Chang, X.; Zhang, J.; Jiang, Y.; Yao, B.; Wang, J.; Wu, Y. Pilot Trial on the Efficacy and Safety of Pantethine in Children with Pantothenate Kinase-Associated Neurodegeneration: A Single-Arm, Open-Label Study. Orphanet J. Rare Dis. 2020, 15, 248. [Google Scholar] [CrossRef] [PubMed]
- Elbaum, D.; Beconi, M.G.; Monteagudo, E.; Marco, A.D.; Quinton, M.S.; Lyons, K.A.; Vaino, A.; Harper, S. Fosmetpantotenate (RE-024), a Phosphopantothenate Replacement Therapy for Pantothenate Kinase-Associated Neurodegeneration: Mechanism of Action and Efficacy in Nonclinical Models. PLoS ONE 2018, 13, e0192028. [Google Scholar] [CrossRef] [PubMed]
- Christou, Y.-P.; Tanteles, G.A.; Kkolou, E.; Ormiston, A.; Konstantopoulos, K.; Beconi, M.; Marshall, R.D.; Plotkin, H.; Kleopa, K.A. Open-Label Fosmetpantotenate, a Phosphopantothenate Replacement Therapy in a Single Patient with Atypical PKAN. Case Rep. Neurol. Med. 2017, 2017, 3247034. [Google Scholar] [CrossRef]
- Klopstock, T.; Videnovic, A.; Bischoff, A.T.; Bonnet, C.; Cif, L.; Comella, C.; Correa-Vela, M.; Escolar, M.L.; Fraser, J.L.; Gonzalez, V.; et al. Fosmetpantotenate Randomized Controlled Trial in Pantothenate Kinase–Associated Neurodegeneration. Mov. Disord. 2021, 36, 1342–1352. [Google Scholar] [CrossRef] [PubMed]
- Klopstock, T.; Escolar, M.L.; Marshall, R.D.; Perez-Dueñas, B.; Tuller, S.; Videnovic, A.; Greblikas, F. The FOsmetpantotenate Replacement Therapy (FORT) Randomized, Double-Blind, Placebo-Controlled Pivotal Trial: Study Design and Development Methodology of a Novel Primary Efficacy Outcome in Patients with Pantothenate Kinase-Associated Neurodegeneration. Clin. Trials 2019, 16, 410–418. [Google Scholar] [CrossRef] [PubMed]
- Auciello, G.; Di Marco, A.; Gonzalez Paz, O.; Malancona, S.; Harper, S.; Beconi, M.; Rossetti, I.; Ciammaichella, A.; Fezzardi, P.; Vecchi, A.; et al. Cyclic Phosphopantothenic Acid Prodrugs for Treatment of Pantothenate Kinase-Associated Neurodegeneration. J. Med. Chem. 2020, 63, 15785–15801. [Google Scholar] [CrossRef]
- Di Meo, I.; Colombelli, C.; Srinivasan, B.; de Villiers, M.; Hamada, J.; Jeong, S.Y.; Fox, R.; Woltjer, R.L.; Tepper, P.G.; Lahaye, L.L.; et al. Acetyl-4’-Phosphopantetheine Is Stable in Serum and Prevents Phenotypes Induced by Pantothenate Kinase Deficiency. Sci. Rep. 2017, 7, 11260. [Google Scholar] [CrossRef]
- Jackowski, S. Proposed Therapies for Pantothenate-Kinase-Associated Neurodegeneration. J. Exp. Neurosci. 2019, 13, 1179069519851118. [Google Scholar] [CrossRef]
- Sharma, L.K.; Leonardi, R.; Lin, W.; Boyd, V.A.; Goktug, A.; Shelat, A.A.; Chen, T.; Jackowski, S.; Rock, C.O. A High-Throughput Screen Reveals New Small-Molecule Activators and Inhibitors of Pantothenate Kinases. J. Med. Chem. 2015, 58, 1563–1568. [Google Scholar] [CrossRef]
- Subramanian, C.; Frank, M.W.; Tangallapally, R.; Yun, M.-K.; White, S.W.; Lee, R.E.; Rock, C.O.; Jackowski, S. Relief of CoA Sequestration and Restoration of Mitochondrial Function in a Mouse Model of Propionic Acidemia. J. Inherit. Metab. Dis. 2023, 46, 28–42. [Google Scholar] [CrossRef]
- Santambrogio, P.; Cozzi, A.; Di Meo, I.; Cavestro, C.; Vergara, C.; Rodríguez-Pascau, L.; Martinell, M.; Pizcueta, P.; Tiranti, V.; Levi, S. PPAR Gamma Agonist Leriglitazone Recovers Alterations Due to Pank2-Deficiency in HiPS-Derived Astrocytes. Pharmaceutics 2023, 15, 202. [Google Scholar] [CrossRef]
- Kersten, S.; Desvergne, B.; Wahli, W. Roles of PPARs in Health and Disease. Nature 2000, 405, 421–424. [Google Scholar] [CrossRef] [PubMed]
- Rodríguez-Pascau, L.; Vilalta, A.; Cerrada, M.; Traver, E.; Forss-Petter, S.; Weinhofer, I.; Bauer, J.; Kemp, S.; Pina, G.; Pascual, S.; et al. The Brain Penetrant PPARγ Agonist Leriglitazone Restores Multiple Altered Pathways in Models of X-Linked Adrenoleukodystrophy. Sci. Transl. Med. 2021, 13, eabc0555. [Google Scholar] [CrossRef] [PubMed]
- Rodríguez-Pascau, L.; Britti, E.; Calap-Quintana, P.; Dong, Y.N.; Vergara, C.; Delaspre, F.; Medina-Carbonero, M.; Tamarit, J.; Pallardó, F.V.; Gonzalez-Cabo, P.; et al. PPAR Gamma Agonist Leriglitazone Improves Frataxin-Loss Impairments in Cellular and Animal Models of Friedreich Ataxia. Neurobiol. Dis. 2021, 148, 105162. [Google Scholar] [CrossRef]
- Pandolfo, M.; Reetz, K.; Darling, A.; Rodriguez de Rivera, F.J.; Henry, P.-G.; Joers, J.; Lenglet, C.; Adanyeguh, I.; Deelchand, D.; Mochel, F.; et al. Efficacy and Safety of Leriglitazone in Patients With Friedreich Ataxia: A Phase 2 Double-Blind, Randomized Controlled Trial (FRAMES). Neurol. Genet. 2022, 8, e200034. [Google Scholar] [CrossRef] [PubMed]
- Köhler, W.; Engelen, M.; Eichler, F.; Lachmann, R.; Fatemi, A.; Sampson, J.; Salsano, E.; Gamez, J.; Molnar, M.J.; Pascual, S.; et al. Safety and Efficacy of Leriglitazone for Preventing Disease Progression in Men with Adrenomyeloneuropathy (ADVANCE): A Randomised, Double-Blind, Multi-Centre, Placebo-Controlled Phase 2–3 Trial. Lancet Neurol. 2023, 22, 127–136. [Google Scholar] [CrossRef]
- Yan, H.; Zou, T.; Tuo, Q.; Xu, S.; Li, H.; Belaidi, A.A.; Lei, P. Ferroptosis: Mechanisms and Links with Diseases. Signal Transduct. Target. 2021, 6, 49. [Google Scholar] [CrossRef]
- Entezari, S.; Haghi, S.M.; Norouzkhani, N.; Sahebnazar, B.; Vosoughian, F.; Akbarzadeh, D.; Islampanah, M.; Naghsh, N.; Abbasalizadeh, M.; Deravi, N. Iron Chelators in Treatment of Iron Overload. J. Toxicol 2022, 2022, 4911205. [Google Scholar] [CrossRef]
- Klopstock, T.; Tricta, F.; Neumayr, L.; Karin, I.; Zorzi, G.; Fradette, C.; Kmieć, T.; Büchner, B.; Steele, H.E.; Horvath, R.; et al. Safety and Efficacy of Deferiprone for Pantothenate Kinase-Associated Neurodegeneration: A Randomised, Double-Blind, Controlled Trial and an Open-Label Extension Study. Lancet Neurol. 2019, 18, 631–642. [Google Scholar] [CrossRef]
- Romano, N.; Baiardi, G.; Pinto, V.M.; Quintino, S.; Gianesin, B.; Sasso, R.; Diociasi, A.; Mattioli, F.; Marchese, R.; Abbruzzese, G.; et al. Long-Term Neuroradiological and Clinical Evaluation of NBIA Patients Treated with a Deferiprone Based Iron-Chelation Therapy. J. Clin. Med. 2022, 11, 4524. [Google Scholar] [CrossRef]
- Ayton, S.; Bush, A.I. Decreasing Iron Neurotoxicity in Pantothenate Kinase-Associated Neurodegeneration. Lancet Neurol. 2019, 18, 616–617. [Google Scholar] [CrossRef] [PubMed]




{kind=link}
{kind=link}
{kind=link}
| Gene (* OMIM) | Protein Function | Cellular Localization | Disease (# OMIM) | Inheritance | Clinical Features |
|---|---|---|---|---|---|
| PANK2 (* 606157) | Pantothenate kinase | Mitochondrial intermembrane space | Pantothenate kinase-associated neurodegeneration, PKAN (# 234200) | AR | Early (classical) or late (atypical) onset dystonia, spasticity, cognitive decline, pigmentary retinopathy. Iron overload in GP (eye of the tiger sign). |
| PPCS (* 609853) | Phosphopantothenoyl cysteine synthetase | Cytosol | Dilated cardiomyopathy 2C, CMD2C (# 618189) | AR | Early onset dilated cardiomyopathy, hypotonia, necrotizing myopathy, intermittent rhabdomyolysis. |
| PPCDC (* 609854) | Phosphopantothenoyl cysteine decarboxylase | Cytosol | Dilated cardiomyopathy | AR | Early onset dilated cardiomyopathy, lactic acidosis, neurological involvement with lethargy and limb hypertonia. |
| COASY (* 609855) | Phosphopantetheine adenylyl-transferase (PPAT) and dephospho-CoA kinase (DPCK) | Mitochondrial outer membrane, mitochondrial matrix, cytosol, nucleus | COASY protein- associated neurodegeneration, CoPAN (# 615643) | AR | Early onset spastic-dystonic paraparesis, dysarthria, obsessive-compulsive behavior. Iron overload in GP. |
| Pontocerebellar hypoplasia type 12, PCH12 (# 618266) | AR | Perinatal onset lethal microcephaly and arthrogryposis. |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Cavestro, C.; Diodato, D.; Tiranti, V.; Di Meo, I. Inherited Disorders of Coenzyme A Biosynthesis: Models, Mechanisms, and Treatments. Int. J. Mol. Sci. 2023, 24, 5951. https://doi.org/10.3390/ijms24065951
Cavestro C, Diodato D, Tiranti V, Di Meo I. Inherited Disorders of Coenzyme A Biosynthesis: Models, Mechanisms, and Treatments. International Journal of Molecular Sciences. 2023; 24(6):5951. https://doi.org/10.3390/ijms24065951
Chicago/Turabian StyleCavestro, Chiara, Daria Diodato, Valeria Tiranti, and Ivano Di Meo. 2023. "Inherited Disorders of Coenzyme A Biosynthesis: Models, Mechanisms, and Treatments" International Journal of Molecular Sciences 24, no. 6: 5951. https://doi.org/10.3390/ijms24065951
APA StyleCavestro, C., Diodato, D., Tiranti, V., & Di Meo, I. (2023). Inherited Disorders of Coenzyme A Biosynthesis: Models, Mechanisms, and Treatments. International Journal of Molecular Sciences, 24(6), 5951. https://doi.org/10.3390/ijms24065951