
Retinal Organoids from an AIPL1 CRISPR/Cas9 Knockout Cell Line Successfully Recapitulate the Molecular Features of LCA4 Disease
 , ,
, , {kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Results
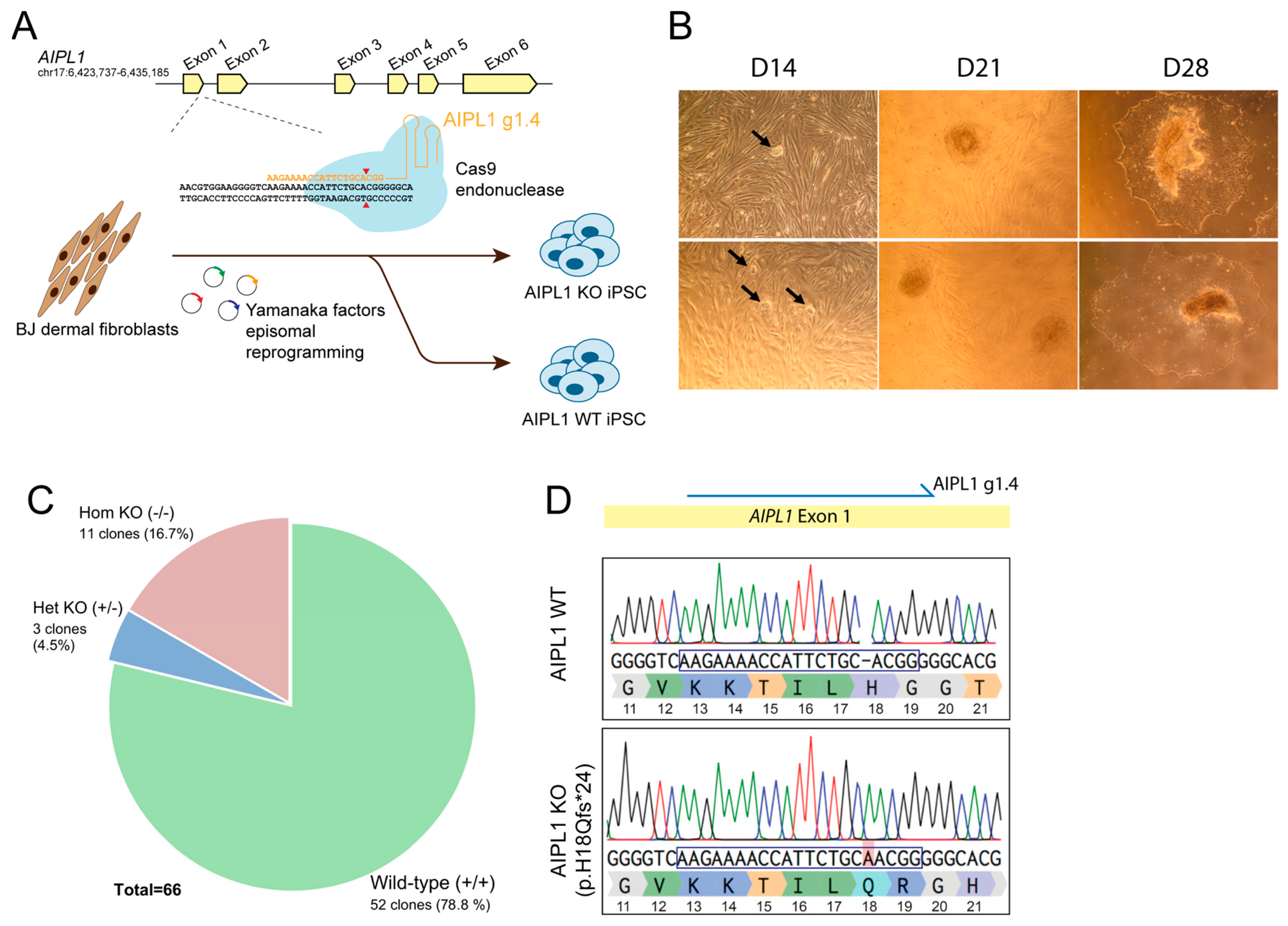
2.1. Generation of Isogenic AIPL1 Knockout iPSC by Simultaneous Reprogramming and CRISPR/Cas9 Gene Editing
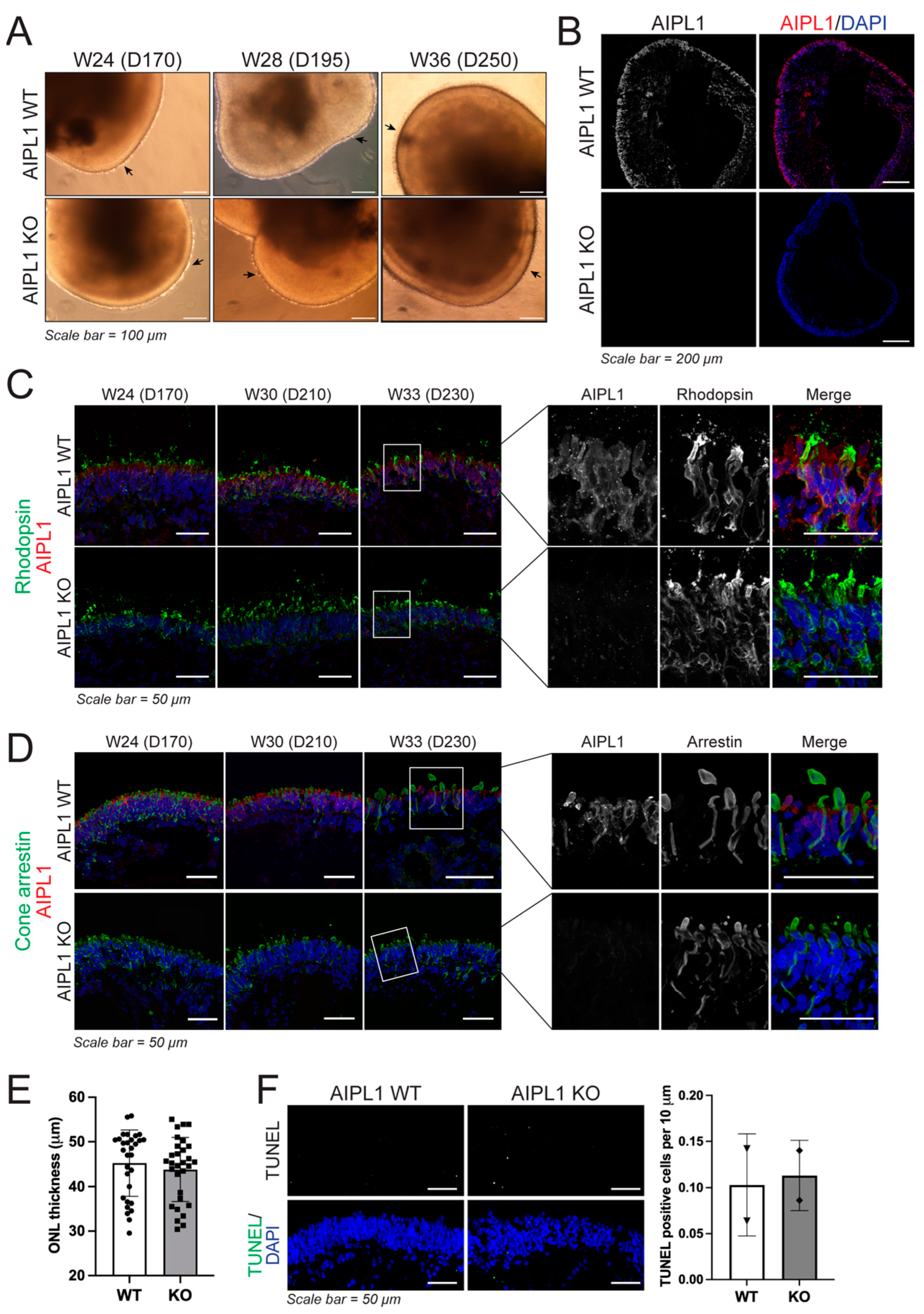
2.2. Characterisation of Retinal Organoid Structure from AIPL1 KO iPSC
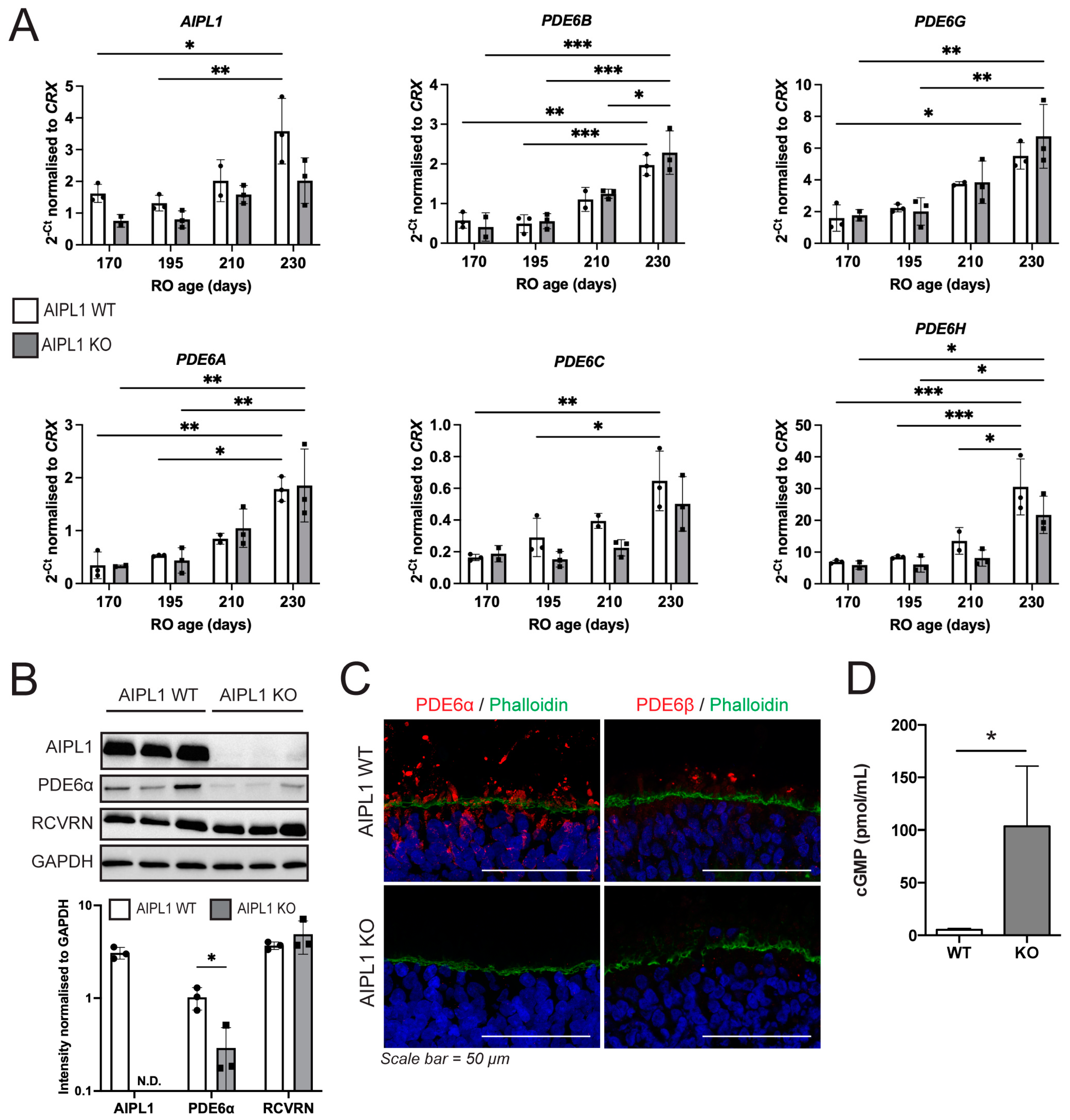
2.3. AIPL1 Knockout Results in RO with an LCA4 Phenotype
3. Discussion
4. Materials and Methods
4.1. Cell Culture
4.2. Design of CRISPR/Cas9 Sequences for AIPL1 Knockout
4.3. Generation of AIPL1 Knockout Isogenic iPSC
4.4. Generation of Retinal Organoids (RO)
4.5. Immunofluorescence and Imaging
4.6. Image Quantification
4.7. RNA Extraction and Quantitative PCR (qPCR)
4.8. Western Blotting
4.9. Quantification of cGMP
Supplementary Materials
Author Contributions
Funding
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- den Hollander, A.I.; Roepman, R.; Koenekoop, R.K.; Cremers, F.P. Leber congenital amaurosis: Genes, proteins and disease mechanisms. Prog. Retin. Eye Res. 2008, 27, 391–419. [Google Scholar] [CrossRef]
- Kumaran, N.; Moore, A.T.; Weleber, R.G.; Michaelides, M. Leber congenital amaurosis/early-onset severe retinal dystrophy: Clinical features, molecular genetics and therapeutic interventions. Br. J. Ophthalmol. 2017, 101, 1147–1154. [Google Scholar] [CrossRef] [PubMed]
- Sohocki, M.M.; Bowne, S.J.; Sullivan, L.S.; Blackshaw, S.; Cepko, C.L.; Payne, A.M.; Bhattacharya, S.S.; Khaliq, S.; Qasim Mehdi, S.; Birch, D.G.; et al. Mutations in a new photoreceptor-pineal gene on 17p cause Leber congenital amaurosis. Nat. Genet. 2000, 24, 79–83. [Google Scholar] [CrossRef] [PubMed]
- van der Spuy, J.; Chapple, J.P.; Clark, B.J.; Luthert, P.J.; Sethi, C.S.; Cheetham, M.E. The Leber congenital amaurosis gene product AIPL1 is localized exclusively in rod photoreceptors of the adult human retina. Hum. Mol. Genet. 2002, 11, 823–831. [Google Scholar] [CrossRef]
- Liu, X.; Bulgakov, O.V.; Wen, X.H.; Woodruff, M.L.; Pawlyk, B.; Yang, J.; Fain, G.L.; Sandberg, M.A.; Makino, C.L.; Li, T. AIPL1, the protein that is defective in Leber congenital amaurosis, is essential for the biosynthesis of retinal rod cGMP phosphodiesterase. Proc. Natl. Acad. Sci. USA 2004, 101, 13903–13908. [Google Scholar] [CrossRef] [PubMed]
- Hidalgo-de-Quintana, J.; Evans, R.J.; Cheetham, M.E.; van der Spuy, J. The Leber congenital amaurosis protein AIPL1 functions as part of a chaperone heterocomplex. Investig. Ophthalmol. Vis. Sci. 2008, 49, 2878–2887. [Google Scholar] [CrossRef]
- Kolandaivelu, S.; Huang, J.; Hurley, J.B.; Ramamurthy, V. AIPL1, a protein associated with childhood blindness, interacts with alpha-subunit of rod phosphodiesterase (PDE6) and is essential for its proper assembly. J. Biol. Chem. 2009, 284, 30853–30861. [Google Scholar] [CrossRef]
- Sacristan-Reviriego, A.; Bellingham, J.; Prodromou, C.; Boehm, A.N.; Aichem, A.; Kumaran, N.; Bainbridge, J.; Michaelides, M.; van der Spuy, J. The integrity and organization of the human AIPL1 functional domains is critical for its role as a HSP90-dependent co-chaperone for rod PDE6. Hum. Mol. Genet. 2017, 26, 4465–4480. [Google Scholar] [CrossRef]
- Cote, R.H. Photoreceptor phosphodiesterase (PDE6): Activation and inactivation mechanisms during visual transduction in rods and cones. Pflügers Arch.–Eur. J. Physiol. 2021, 473, 1377–1391. [Google Scholar] [CrossRef]
- Ramamurthy, V.; Niemi, G.A.; Reh, T.A.; Hurley, J.B. Leber congenital amaurosis linked to AIPL1: A mouse model reveals destabilization of cGMP phosphodiesterase. Proc. Natl. Acad. Sci. USA 2004, 101, 13897–13902. [Google Scholar] [CrossRef]
- Kirschman, L.T.; Kolandaivelu, S.; Frederick, J.M.; Dang, L.; Goldberg, A.F.; Baehr, W.; Ramamurthy, V. The Leber congenital amaurosis protein, AIPL1, is needed for the viability and functioning of cone photoreceptor cells. Hum. Mol. Genet. 2010, 19, 1076–1087. [Google Scholar] [CrossRef] [PubMed]
- Kolandaivelu, S.; Singh, R.K.; Ramamurthy, V. AIPL1, A protein linked to blindness, is essential for the stability of enzymes mediating cGMP metabolism in cone photoreceptor cells. Hum. Mol. Genet. 2014, 23, 1002–1012. [Google Scholar] [CrossRef] [PubMed]
- Majumder, A.; Gopalakrishna, K.N.; Cheguru, P.; Gakhar, L.; Artemyev, N.O. Interaction of aryl hydrocarbon receptor-interacting protein-like 1 with the farnesyl moiety. J. Biol. Chem. 2013, 288, 21320–21328. [Google Scholar] [CrossRef] [PubMed]
- Yadav, R.P.; Majumder, A.; Gakhar, L.; Artemyev, N.O. Extended conformation of the proline-rich domain of human aryl hydrocarbon receptor-interacting protein-like 1: Implications for retina disease. J. Neurochem. 2015, 135, 165–175. [Google Scholar] [CrossRef] [PubMed]
- Yadav, R.P.; Gakhar, L.; Yu, L.; Artemyev, N.O. Unique structural features of the AIPL1-FKBP domain that support prenyl lipid binding and underlie protein malfunction in blindness. Proc. Natl. Acad. Sci. USA 2017, 114, E6536–E6545. [Google Scholar] [CrossRef] [PubMed]
- Yadav, R.P.; Boyd, K.; Yu, L.; Artemyev, N.O. Interaction of the tetratricopeptide repeat domain of aryl hydrocarbon receptor-interacting protein-like 1 with the regulatory Pgamma subunit of phosphodiesterase 6. J. Biol. Chem. 2019, 294, 15795–15807. [Google Scholar] [CrossRef] [PubMed]
- Sacristan-Reviriego, A.; Le, H.M.; Georgiou, M.; Meunier, I.; Bocquet, B.; Roux, A.F.; Prodromou, C.; Bainbridge, J.; Michaelides, M.; van der Spuy, J. Clinical and functional analyses of AIPL1 variants reveal mechanisms of pathogenicity linked to different forms of retinal degeneration. Sci. Rep. 2020, 10, 17520. [Google Scholar] [CrossRef]
- Zhong, X.; Gutierrez, C.; Xue, T.; Hampton, C.; Vergara, M.N.; Cao, L.H.; Peters, A.; Park, T.S.; Zambidis, E.T.; Meyer, J.S.; et al. Generation of three-dimensional retinal tissue with functional photoreceptors from human iPSCs. Nat. Commun. 2014, 5, 4047. [Google Scholar] [CrossRef]
- Gonzalez-Cordero, A.; Kruczek, K.; Naeem, A.; Fernando, M.; Kloc, M.; Ribeiro, J.; Goh, D.; Duran, Y.; Blackford, S.J.I.; Abelleira-Hervas, L.; et al. Recapitulation of Human Retinal Development from Human Pluripotent Stem Cells Generates Transplantable Populations of Cone Photoreceptors. Stem Cell Rep. 2017, 9, 820–837. [Google Scholar] [CrossRef]
- Lukovic, D.; Artero Castro, A.; Kaya, K.D.; Munezero, D.; Gieser, L.; Davo-Martinez, C.; Corton, M.; Cuenca, N.; Swaroop, A.; Ramamurthy, V.; et al. Retinal Organoids derived from hiPSCs of an AIPL1-LCA Patient Maintain Cytoarchitecture despite Reduced levels of Mutant AIPL1. Sci. Rep. 2020, 10, 5426. [Google Scholar] [CrossRef]
- Leung, A.; Sacristan-Reviriego, A.; Perdigao, P.R.L.; Sai, H.; Georgiou, M.; Kalitzeos, A.; Carr, A.F.; Coffey, P.J.; Michaelides, M.; Bainbridge, J.; et al. Investigation of PTC124-mediated translational readthrough in a retinal organoid model of AIPL1-associated Leber congenital amaurosis. Stem Cell Rep. 2022, 17, 2187–2202. [Google Scholar] [CrossRef]
- Lukovic, D.; Artero Castro, A.; Leon, M.; Del Buey Furio, V.; Corton, M.; Ayuso, C.; Erceg, S. Generation of a human iPSC line from a patient with Leber congenital amaurosis caused by mutation in AIPL1. Stem Cell Res. 2018, 33, 151–155. [Google Scholar] [CrossRef]
- Perdigao, P.R.L.; van der Spuy, J. Gene and Cell Therapy for AIPL1-Associated Leber Congenital Amaurosis: Challenges and Prospects. Adv. Exp. Med. Biol. 2019, 1185, 97–101. [Google Scholar]
- Howden, S.E.; Maufort, J.P.; Duffin, B.M.; Elefanty, A.G.; Stanley, E.G.; Thomson, J.A. Simultaneous Reprogramming and Gene Correction of Patient Fibroblasts. Stem Cell Rep. 2015, 5, 1109–1118. [Google Scholar] [CrossRef]
- Popp, M.W.; Maquat, L.E. Leveraging Rules of Nonsense-Mediated mRNA Decay for Genome Engineering and Personalized Medicine. Cell 2016, 165, 1319–1322. [Google Scholar] [CrossRef]
- Singh, R.K.; Kolandaivelu, S.; Ramamurthy, V. Early alteration of retinal neurons in Aipl1-/- animals. Investig. Ophthalmol. Vis. Sci. 2014, 55, 3081–3092. [Google Scholar] [CrossRef]
- Vandenberghe, L.H.; Auricchio, A. Novel adeno-associated viral vectors for retinal gene therapy. Gene Ther. 2012, 19, 162–168. [Google Scholar] [CrossRef]
- Bordet, T.; Behar-Cohen, F. Ocular gene therapies in clinical practice: Viral vectors and nonviral alternatives. Drug Discov. Today 2019, 24, 1685–1693. [Google Scholar] [CrossRef]
- Pierce, E.A.; Bennett, J. The Status of RPE65 Gene Therapy Trials: Safety and Efficacy. Cold Spring Harb. Perspect. Med. 2015, 5, a017285. [Google Scholar] [CrossRef]
- Gao, J.; Hussain, R.M.; Weng, C.Y. Voretigene Neparvovec in Retinal Diseases: A Review of the Current Clinical Evidence. Clin. Ophthalmol. 2020, 14, 3855–3869. [Google Scholar] [CrossRef]
- Tan, M.H.; Smith, A.J.; Pawlyk, B.; Xu, X.; Liu, X.; Bainbridge, J.B.; Basche, M.; McIntosh, J.; Tran, H.V.; Nathwani, A.; et al. Gene therapy for retinitis pigmentosa and Leber congenital amaurosis caused by defects in AIPL1: Effective rescue of mouse models of partial and complete Aipl1 deficiency using AAV2/2 and AAV2/8 vectors. Hum. Mol. Genet. 2009, 18, 2099–2114. [Google Scholar] [CrossRef]
- Testa, F.; Surace, E.M.; Rossi, S.; Marrocco, E.; Gargiulo, A.; Di Iorio, V.; Ziviello, C.; Nesti, A.; Fecarotta, S.; Bacci, M.L.; et al. Evaluation of Italian Patients with Leber Congenital Amaurosis due to AIPL1 Mutations Highlights the Potential Applicability of Gene Therapy. Investig. Opthalmol. Vis. Sci. 2011, 52, 5618–5624. [Google Scholar] [CrossRef] [PubMed]
- Sun, X.; Pawlyk, B.; Xu, X.; Liu, X.; Bulgakov, O.V.; Adamian, M.; Sandberg, M.A.; Khani, S.C.; Tan, M.H.; Smith, A.J.; et al. Gene therapy with a promoter targeting both rods and cones rescues retinal degeneration caused by AIPL1 mutations. Gene Ther. 2010, 17, 117–131. [Google Scholar] [CrossRef] [PubMed]
- Garita-Hernandez, M.; Routet, F.; Guibbal, L.; Khabou, H.; Toualbi, L.; Riancho, L.; Reichman, S.; Duebel, J.; Sahel, J.A.; Goureau, O.; et al. AAV-Mediated Gene Delivery to 3D Retinal Organoids Derived from Human Induced Pluripotent Stem Cells. Int. J. Mol. Sci. 2020, 21, 994. [Google Scholar] [CrossRef] [PubMed]
- Gonzalez-Cordero, A.; Goh, D.; Kruczek, K.; Naeem, A.; Fernando, M.; Kleine Holthaus, S.M.; Takaaki, M.; Blackford, S.J.I.; Kloc, M.; Agundez, L.; et al. Assessment of AAV Vector Tropisms for Mouse and Human Pluripotent Stem Cell-Derived RPE and Photoreceptor Cells. Hum. Gene Ther. 2018, 29, 1124–1139. [Google Scholar] [CrossRef] [PubMed]
- Lane, A.; Jovanovic, K.; Shortall, C.; Ottaviani, D.; Panes, A.B.; Schwarz, N.; Guarascio, R.; Hayes, M.J.; Palfi, A.; Chadderton, N.; et al. Modeling and Rescue of RP2 Retinitis Pigmentosa Using iPSC-Derived Retinal Organoids. Stem Cell Rep. 2020, 15, 67–79. [Google Scholar] [CrossRef]
- Tolone, A.; Belhadj, S.; Rentsch, A.; Schwede, F.; Paquet-Durand, F. The cGMP Pathway and Inherited Photoreceptor Degeneration: Targets, Compounds, and Biomarkers. Genes 2019, 10, 453. [Google Scholar] [CrossRef]
- Paquet-Durand, F.; Hauck, S.M.; van Veen, T.; Ueffing, M.; Ekstrom, P. PKG activity causes photoreceptor cell death in two retinitis pigmentosa models. J. Neurochem. 2009, 108, 796–810. [Google Scholar] [CrossRef]
- Vighi, E.; Trifunovic, D.; Veiga-Crespo, P.; Rentsch, A.; Hoffmann, D.; Sahaboglu, A.; Strasser, T.; Kulkarni, M.; Bertolotti, E.; van den Heuvel, A.; et al. Combination of cGMP analogue and drug delivery system provides functional protection in hereditary retinal degeneration. Proc. Natl. Acad. Sci. USA 2018, 115, E2997–E3006. [Google Scholar] [CrossRef]
- Wucherpfennig, S.; Haq, W.; Popp, V.; Kesh, S.; Das, S.; Melle, C.; Rentsch, A.; Schwede, F.; Paquet-Durand, F.; Nache, V. cGMP Analogues with Opposing Actions on CNG Channels Selectively Modulate Rod or Cone Photoreceptor Function. Pharmaceutics 2022, 14, 2102. [Google Scholar] [CrossRef]
- Kutluer, M.; Huang, L.; Marigo, V. Targeting molecular pathways for the treatment of inherited retinal degeneration. Neural Regen. Res. 2020, 15, 1784–1791. [Google Scholar] [PubMed]
- Roy, A.; Tolone, A.; Hilhorst, R.; Groten, J.; Tomar, T.; Paquet-Durand, F. Kinase activity profiling identifies putative downstream targets of cGMP/PKG signaling in inherited retinal neurodegeneration. Cell Death Discov. 2022, 8, 93. [Google Scholar] [CrossRef] [PubMed]
- Sahaboglu, A.; Sharif, A.; Feng, L.; Secer, E.; Zrenner, E.; Paquet-Durand, F. Temporal progression of PARP activity in the Prph2 mutant rd2 mouse: Neuroprotective effects of the PARP inhibitor PJ34. PLoS ONE 2017, 12, e0181374. [Google Scholar] [CrossRef] [PubMed]
- Yan, J.; Gunter, A.; Das, S.; Muhlfriedel, R.; Michalakis, S.; Jiao, K.; Seeliger, M.W.; Paquet-Durand, F. Inherited Retinal Degeneration: PARP-Dependent Activation of Calpain Requires CNG Channel Activity. Biomolecules 2022, 12, 455. [Google Scholar] [CrossRef] [PubMed]
- Ran, F.A.; Hsu, P.D.; Wright, J.; Agarwala, V.; Scott, D.A.; Zhang, F. Genome engineering using the CRISPR-Cas9 system. Nat. Protoc. 2013, 8, 2281–2308. [Google Scholar] [CrossRef] [PubMed]



Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Perdigão, P.R.L.; Ollington, B.; Sai, H.; Leung, A.; Sacristan-Reviriego, A.; van der Spuy, J. Retinal Organoids from an AIPL1 CRISPR/Cas9 Knockout Cell Line Successfully Recapitulate the Molecular Features of LCA4 Disease. Int. J. Mol. Sci. 2023, 24, 5912. https://doi.org/10.3390/ijms24065912
Perdigão PRL, Ollington B, Sai H, Leung A, Sacristan-Reviriego A, van der Spuy J. Retinal Organoids from an AIPL1 CRISPR/Cas9 Knockout Cell Line Successfully Recapitulate the Molecular Features of LCA4 Disease. International Journal of Molecular Sciences. 2023; 24(6):5912. https://doi.org/10.3390/ijms24065912
Chicago/Turabian StylePerdigão, Pedro R. L., Bethany Ollington, Hali Sai, Amy Leung, Almudena Sacristan-Reviriego, and Jacqueline van der Spuy. 2023. "Retinal Organoids from an AIPL1 CRISPR/Cas9 Knockout Cell Line Successfully Recapitulate the Molecular Features of LCA4 Disease" International Journal of Molecular Sciences 24, no. 6: 5912. https://doi.org/10.3390/ijms24065912
APA StylePerdigão, P. R. L., Ollington, B., Sai, H., Leung, A., Sacristan-Reviriego, A., & van der Spuy, J. (2023). Retinal Organoids from an AIPL1 CRISPR/Cas9 Knockout Cell Line Successfully Recapitulate the Molecular Features of LCA4 Disease. International Journal of Molecular Sciences, 24(6), 5912. https://doi.org/10.3390/ijms24065912