
Drug Repositioning as a Therapeutic Strategy against Streptococcus pneumoniae: Cell Membrane as Potential Target
 ,
,  and
and
Abstract
1. Introduction
2. Results
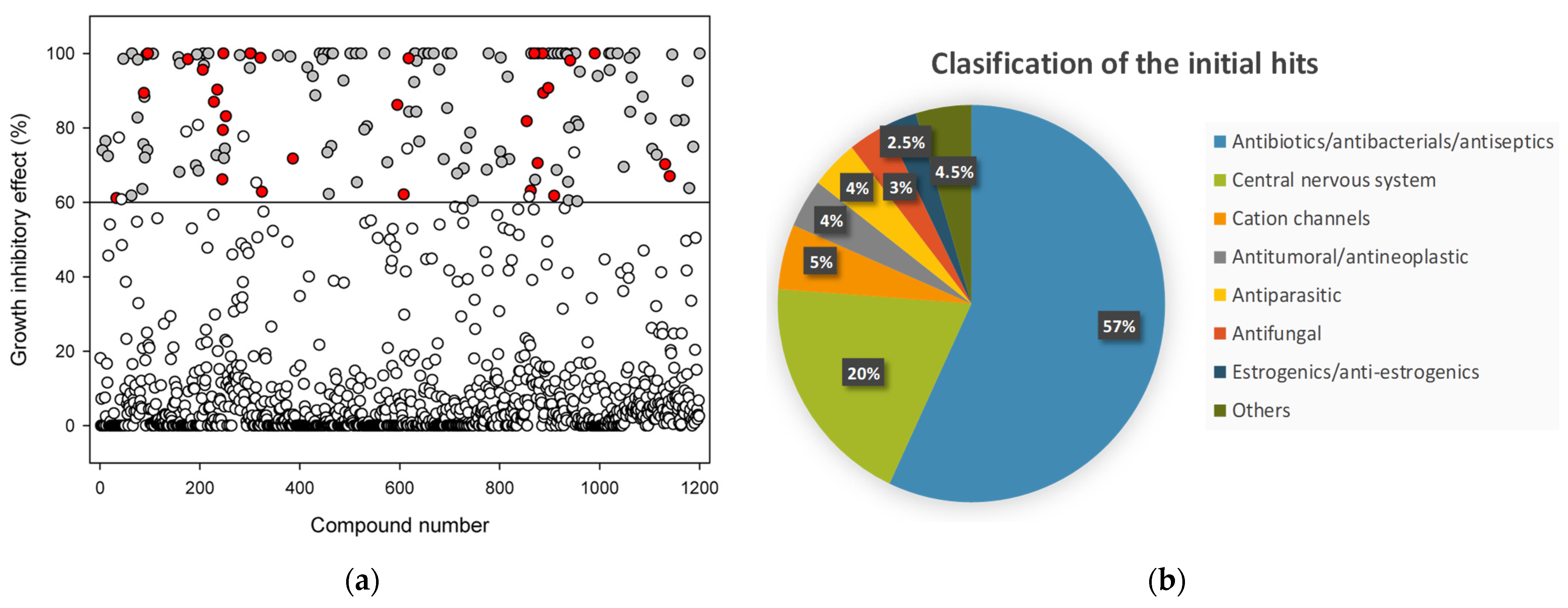
2.1. Library Screening
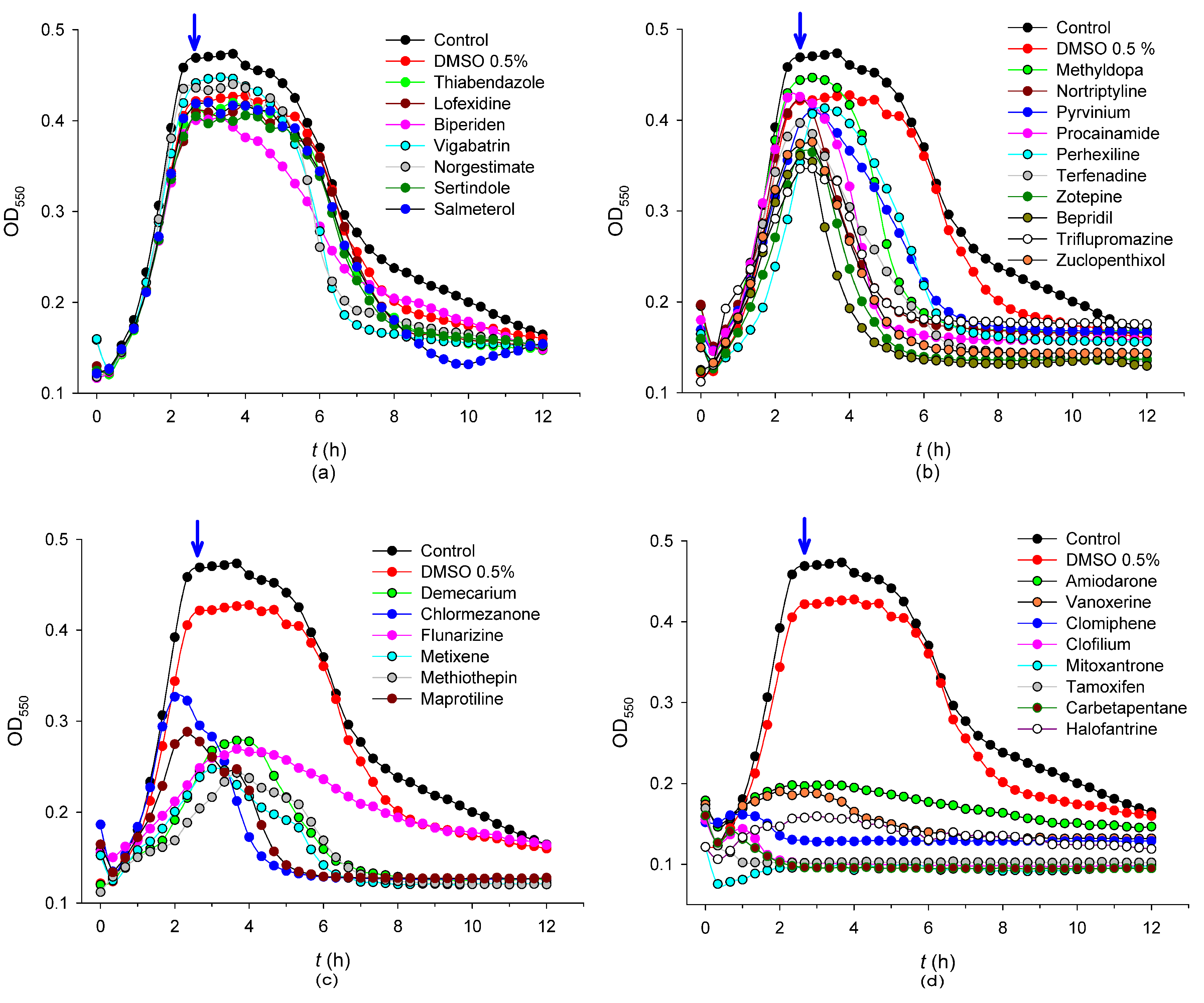
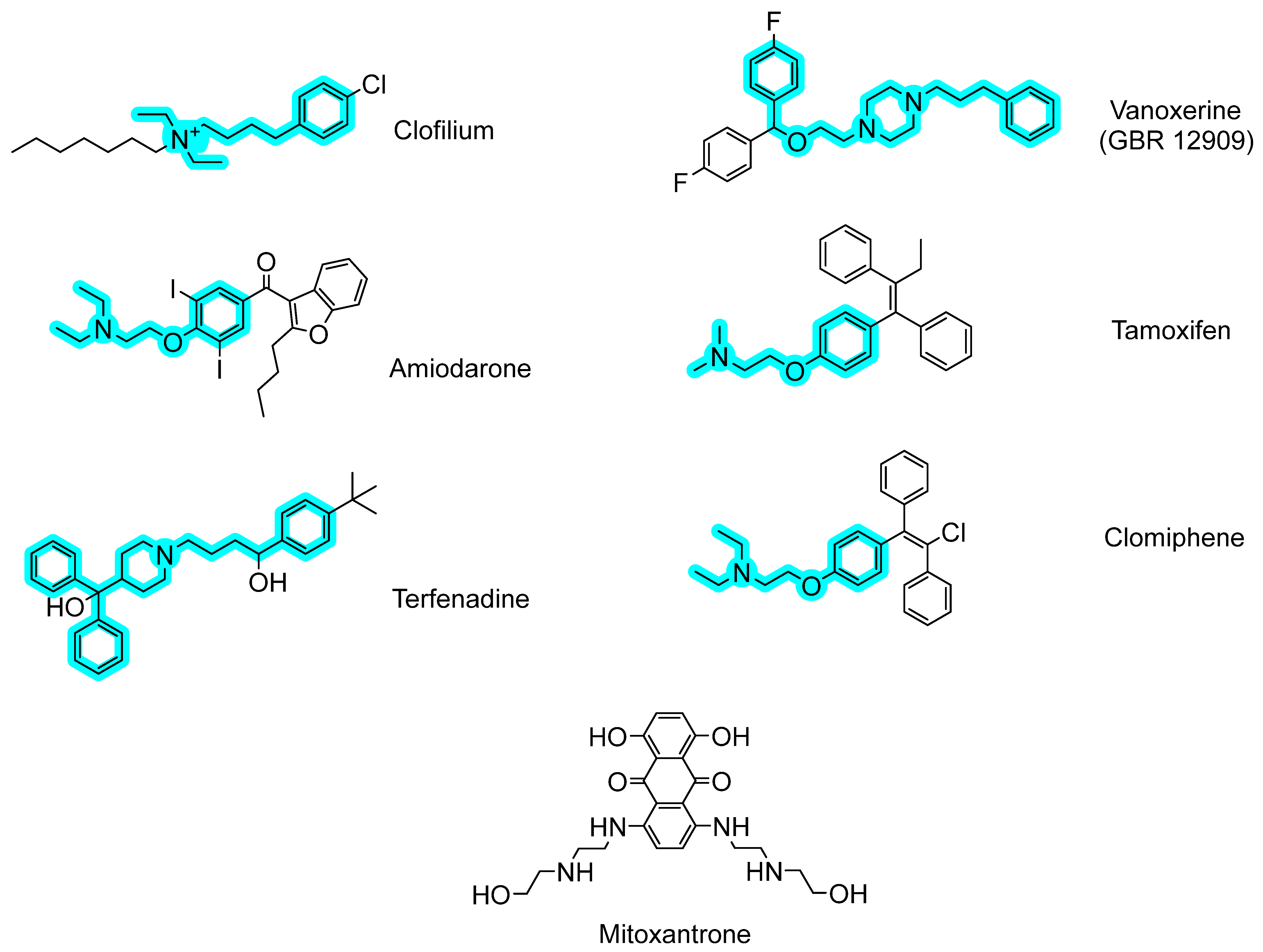
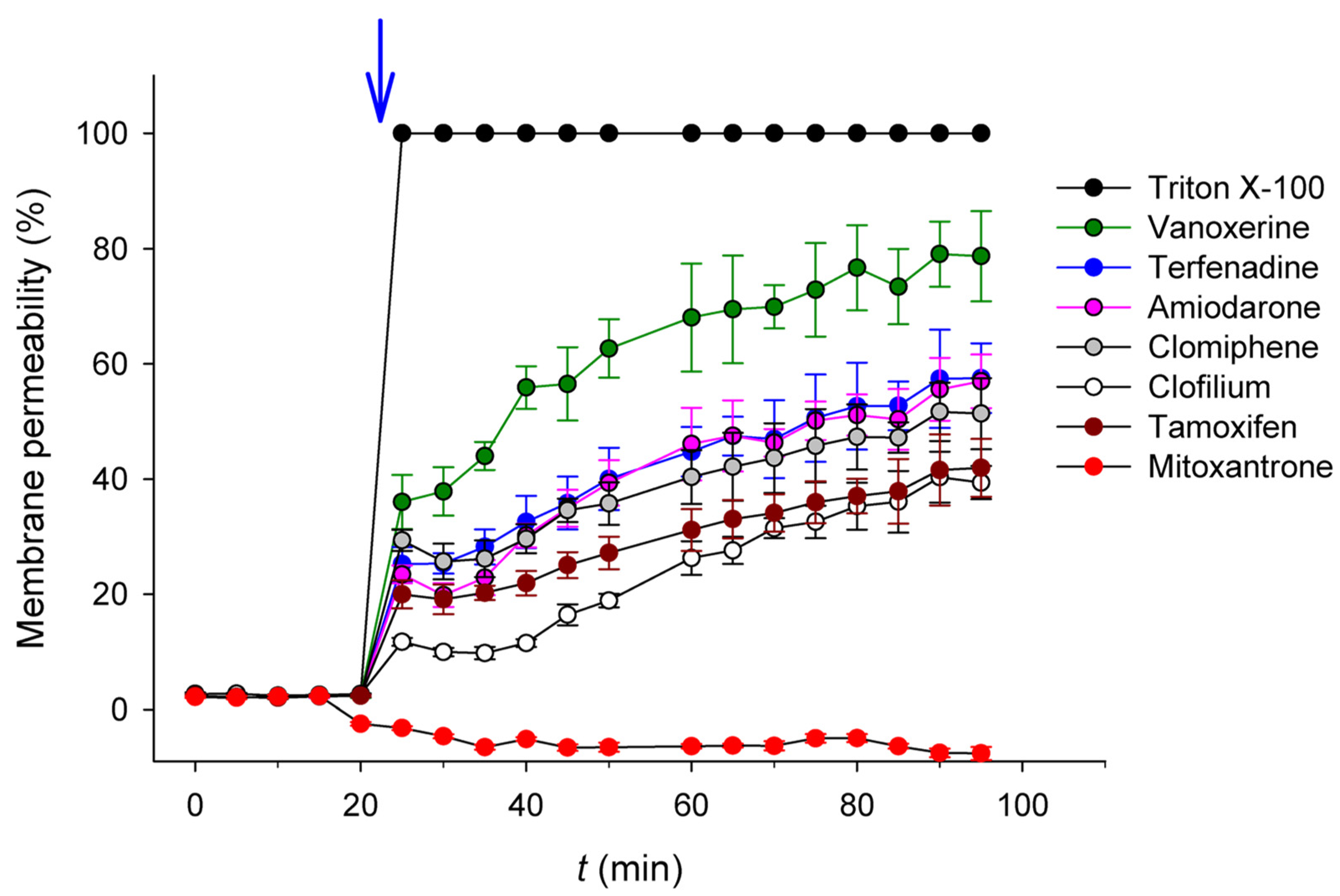
2.2. Antibacterial Mechanisms of the Selected Hits: Membrane Destabilization Studies
3. Discussion
4. Materials and Methods
4.1. Bacterial Strains and Growth Conditions
4.2. Library Screening
4.3. Susceptibility Tests
4.4. Theoretical Calculations
4.5. Cell Permeability Assays
5. Patents
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- World Health Organization Antimicrobial Resistance. Available online: https://www.who.int/news-room/fact-sheets/detail/antimicrobial-resistance (accessed on 17 March 2003).
- Centers for Disease Control and Prevention. Antibiotic Resistance Threats in the United States; U.S. Department of Health and Human Services: Atlanta, GA, USA, 2019.
- GBD 2019 Antimicrobial Resistance Collaborators. Global Burden of Bacterial Antimicrobial Resistance in 2019: A Systematic Analysis. Lancet 2022, 399, 629–655. [Google Scholar] [CrossRef] [PubMed]
- Loughran, A.J.; Orihuela, C.J.; Tuomanen, E.I. Streptococcus pneumoniae: Invasion and Inflammation. Microbiol. Spectr. 2019, 7. [Google Scholar] [CrossRef] [PubMed]
- Ikuta, K.S.; Swetschinski, L.R.; Robles Aguilar, G.; Sharara, F.; Mestrovic, T.; Gray, A.P.; Davis Weaver, N.; Wool, E.E.; Han, C.; Gershberg Hayoon, A.; et al. Global Mortality Associated with 33 Bacterial Pathogens in 2019: A Systematic Analysis for the Global Burden of Disease Study 2019. Lancet 2022, 400, 2221–2248. [Google Scholar] [CrossRef]
- Zhu, X.; Ge, Y.; Wu, T.; Zhao, K.; Chen, Y.; Wu, B.; Zhu, F.; Zhu, B.; Cui, L. Co-Infection with Respiratory Pathogens among COVID-2019 Cases. Virus Res. 2020, 285, 198005. [Google Scholar] [CrossRef] [PubMed]
- Nieto-Moro, M.; Ecclesia, F.G.; Tomé-Masa, I.; De Lama Caro-Patón, G.; Leoz-Gordillo, I.; Cabrero-Hernández, M.; García-Salido, A. SARS-CoV-2 and Streptococcuspneumoniae Coinfection as a Cause of Severe Pneumonia in an Infant. Pediatr. Pulmonol. 2020, 55, 2198–2200. [Google Scholar] [CrossRef]
- World Health Organization. Prioritization of Pathogens to Guide Discovery, Research and Development of New Antibiotics for Drug-Resistant Bacterial Infections, Including Tuberculosis. Available online: https://apps.who.int/iris/handle/10665/311820 (accessed on 19 December 2022).
- Roig-Molina, E.; Domenech, M.; Retamosa, M.D.G.; Nácher-Vázquez, M.; Rivas, L.; Maestro, B.; García, P.; García, E.; Sanz, J.M. Widening the Antimicrobial Spectrum of Esters of Bicyclic Amines: In Vitro Effect on Gram-Positive Streptococcus pneumoniae and Gram-Negative Non-Typeable Haemophilus influenzae Biofilms. Biochim. Biophys. Acta—Gen. Subj. 2019, 1863, 96–104. [Google Scholar] [CrossRef]
- Oprea, T.I.; Mestres, J. Drug Repurposing: Far beyond New Targets for Old Drugs. AAPS J. 2012, 14, 759–763. [Google Scholar] [CrossRef]
- Rangel-Vega, A.; Bernstein, L.R.; Mandujano-Tinoco, E.A.; García-Contreras, S.J.; García-Contreras, R. Drug Repurposing as an Alternative for the Treatment of Recalcitrant Bacterial Infections. Front. Microbiol. 2015, 6, 282. [Google Scholar] [CrossRef]
- Torres, N.S.; Abercrombie, J.J.; Srinivasan, A.; Lopez-Ribot, J.L.; Ramasubramanian, A.K.; Leung, K.P. Screening a Commercial Library of Pharmacologically Active Small Molecules against Staphylococcus aureus Biofilms. Antimicrob. Agents Chemother. 2016, 60, 5663–5672. [Google Scholar] [CrossRef]
- Hind, C.K.; Dowson, C.G.; Sutton, J.M.; Jackson, T.; Clifford, M.; Garner, R.C.; Czaplewski, L. Evaluation of a Library of FDA-Approved Drugs for Their Ability to Potentiate Antibiotics against Multidrug-Resistant Gram-Negative Pathogens. Antimicrob. Agents Chemother. 2019, 63, e00769-19. [Google Scholar] [CrossRef]
- Yousfi, H.; Cassagne, C.; Ranque, S.; Rolain, J.M.; Bittar, F. Repurposing of Ribavirin as an Adjunct Therapy against Invasive Candida Strains in an in Vitro Study. Antimicrob. Agents Chemother. 2019, 63, e00263-19. [Google Scholar] [CrossRef]
- Touret, F.; Gilles, M.; Barral, K.; Nougairède, A.; van Helden, J.; Decroly, E.; de Lamballerie, X.; Coutard, B. In Vitro Screening of a FDA Approved Chemical Library Reveals Potential Inhibitors of SARS-CoV-2 Replication. Sci. Rep. 2020, 10, 13093. [Google Scholar] [CrossRef]
- Roig-Molina, E.; Sánchez-Angulo, M.; Seele, J.; García-Asencio, F.; Nau, R.; Sanz, J.M.; Maestro, B. Searching for Antipneumococcal Targets: Choline-Binding Modules as Phagocytosis Enhancers. ACS Infect. Dis. 2020, 6, 954–974. [Google Scholar] [CrossRef]
- Alkhalifa, S.; Jennings, M.C.; Granata, D.; Klein, M.; Wuest, W.M.; Minbiole, K.P.C.; Carnevale, V. Analysis of the Destabilization of Bacterial Membranes by Quaternary Ammonium Compounds: A Combined Experimental and Computational Study. ChemBioChem 2020, 21, 1510–1516. [Google Scholar] [CrossRef]
- Huang, E.; Yousef, A.E. The Lipopeptide Antibiotic Paenibacterin Binds to the Bacterial Outer Membrane and Exerts Bactericidal Activity through Cytoplasmic Membrane Damage. Appl. Environ. Microbiol. 2014, 80, 2700–2704. [Google Scholar] [CrossRef] [PubMed]
- Cheng, M.; Huang, J.X.; Ramu, S.; Butler, M.S.; Cooper, M.A. Ramoplanin at Bactericidal Concentrations Induces Bacterial Membrane Depolarization in Staphylococcus aureus. Antimicrob. Agents Chemother. 2014, 58, 6819–6827. [Google Scholar] [CrossRef] [PubMed]
- Hansch, C.; Dunn, W.J. Linear Relationships between Lipophilic Character and Biological Activity of Drugs. J. Pharm. Sci. 1972, 61, 1–19. [Google Scholar] [CrossRef]
- Roth, B.L.; Poot, M.; Yue, S.T.; Millard, P.J. Bacterial Viability and Antibiotic Susceptibility Testing with SYTOX Green Nucleic Acid Stain. Appl. Environ. Microbiol. 1997, 63, 2421–2431. [Google Scholar] [CrossRef]
- Hackel, M.; Lascols, C.; Bouchillon, S.; Hilton, B.; Morgenstern, D.; Purdy, J. Serotype Prevalence and Antibiotic Resistance in Streptococcus pneumoniae Clinical Isolates among Global Populations. Vaccine 2013, 31, 4881–4887. [Google Scholar] [CrossRef] [PubMed]
- Cornick, J.E.; Bentley, S.D. Streptococcus pneumoniae: The Evolution of Antimicrobial Resistance to Beta-Lactams, Fluoroquinolones and Macrolides. Microbes Infect. 2012, 14, 573–583. [Google Scholar] [CrossRef]
- Moujaber, G.E.; Osman, M.; Rafei, R.; Dabboussi, F.; Hamze, M. Molecular Mechanisms and Epidemiology of Resistance in Streptococcus pneumoniae in the Middle East Region. J. Med. Microbiol. 2017, 66, 847–858. [Google Scholar] [CrossRef] [PubMed]
- Vandevelde, N.M.; Tulkens, P.M.; Diaz Iglesias, Y.; Verhaegen, J.; Rodriguez-Villalobos, H.; Philippart, I.; Cadrobbi, J.; Coppens, N.; Boel, A.; Van Vaerenbergh, K.; et al. Characterisation of a Collection of Streptococcus pneumoniae Isolates from Patients Suffering from Acute Exacerbations of Chronic Bronchitis: In Vitro Susceptibility to Antibiotics and Biofilm Formation in Relation to Antibiotic Efflux and Serotypes/Serog. Int. J. Antimicrob. Agents 2014, 44, 209–217. [Google Scholar] [CrossRef]
- Wang, H.; Huebner, R.; Chen, M.; Klugman, K. Antibiotic Susceptibility Patterns of Streptococcus pneumoniae in China and Comparison of MICs by Agar Dilution and E-Test Methods. Antimicrob. Agents Chemother. 1998, 42, 2633–2636. [Google Scholar] [CrossRef]
- Zhang, Z.; Chen, M.; Yu, Y.; Pan, S.; Liu, Y. Antimicrobial Susceptibility among Streptococcus pneumoniae and Haemophilus influenzae Collected Globally between 2015 and 2017 as Part of the Tigecycline Evaluation and Surveillance Trial (TEST). Infect. Drug Resist. 2019, 12, 1209–1220. [Google Scholar] [CrossRef] [PubMed]
- Ittzes, B.; Szentkiralyi, E.; Szabo, Z.; Batai, I.Z.; Gyorffy, O.; Kovacs, T.; Batai, I.; Kerenyi, M. Amiodarone That Has Antibacterial Effect against Human Pathogens May Represent a Novel Catheter Lock. Acta Microbiol. Immunol. Hung. 2020, 67, 133–137. [Google Scholar] [CrossRef] [PubMed]
- Rosa, S.M.L.J.; Antunes-Madeira, M.C.; Jurado, A.S.; Madeira, V.V.M.C. Amiodarone Interactions with Membrane Lipids and with Growth of Bacillus stearothermophilus Used as a Model. Appl. Biochem. Biotechnol. 2000, 87, 165–175. [Google Scholar] [CrossRef] [PubMed]
- Luxo, C.; Jurado, A.S.; Madeira, V.M.C.; Silva, M.T. Tamoxifen Induces Ultrastructural Alterations in Membranes of Bacillus stearothermophilus. In Proceedings of the Toxicology In Vitro, Pergamon, Turkey, 1 October 2003; Volume 17, pp. 623–628. [Google Scholar]
- Knauf, G.A.; Cunningham, A.L.; Kazi, M.I.; Riddington, I.M.; Crofts, A.A.; Cattoir, V.; Trent, M.S.; Davies, B.W. Exploring the Antimicrobial Action of Quaternary Amines against Acinetobacter baumannii. mBio 2018, 9, e02394-17. [Google Scholar] [CrossRef]
- Gilbert, P.; Moore, L.E. Cationic Antiseptics: Diversity of Action under a Common Epithet. J. Appl. Microbiol. 2005, 99, 703–715. [Google Scholar] [CrossRef]
- Feng, X.; Zhu, W.; Schurig-Briccio, L.A.; Lindert, S.; Shoen, C.; Hitchings, R.; Li, J.; Wang, Y.; Baig, N.; Zhou, T.; et al. Antiinfectives Targeting Enzymes and the Proton Motive Force. Proc. Natl. Acad. Sci. USA 2015, 112, E7073–E7082. [Google Scholar] [CrossRef]
- Montoya, M.C.; Krysan, D.J. Repurposing Estrogen Receptor Antagonists for the Treatment of Infectious Disease. mBio 2018, 9, e02272-18. [Google Scholar] [CrossRef]
- Hurdle, J.G.; O’Neill, A.J.; Chopra, I.; Lee, R.E. Targeting Bacterial Membrane Function: An Underexploited Mechanism for Treating Persistent Infections. Nat. Rev. Microbiol. 2011, 9, 62–75. [Google Scholar] [CrossRef] [PubMed]
- De Gracia Retamosa, M.; Díez-Martínez, R.; Maestro, B.; García-Fernández, E.; De Waal, B.; Meijer, E.W.; García, P.; Sanz, J.M. Aromatic Esters of Bicyclic Amines as Antimicrobials against Streptococcus pneumoniae. Angew. Chemie. Int. Ed. 2015, 54, 13673–13677. [Google Scholar] [CrossRef] [PubMed]
- Perlmutter, J.I.; Forbes, L.T.; Krysan, D.J.; Ebsworth-Mojica, K.; Colquhoun, J.M.; Wang, J.L.; Dunman, P.M.; Flaherty, D.P. Repurposing the Antihistamine Terfenadine for Antimicrobial Activity against Staphylococcus aureus. J. Med. Chem. 2014, 57, 8540–8562. [Google Scholar] [CrossRef] [PubMed]
- Kanvatirth, P.; Jeeves, R.E.; Bacon, J.; Besra, G.S.; Alderwick, L.J. Utilisation of the Prestwick Chemical Library to Identify Drugs That Inhibit the Growth of Mycobacteria. PLoS ONE 2019, 14, e0213713. [Google Scholar] [CrossRef]
- Machado, D.; Pires, D.; Perdigão, J.; Couto, I.; Portugal, I.; Martins, M.; Amaral, L.; Anes, E.; Viveiros, M. Ion Channel Blockers as Antimicrobial Agents, Efflux Inhibitors, and Enhancers of Macrophage Killing Activity against Drug Resistant Mycobacterium tuberculosis. PLoS ONE 2016, 11, e0149326. [Google Scholar] [CrossRef] [PubMed]
- Saputo, S.; Faustoferri, R.C.; Quivey, R.G. A Drug Repositioning Approach Reveals That Streptococcus mutans Is Susceptible to a Diverse Range of Established Antimicrobials and Nonantibiotics. Antimicrob. Agents Chemother. 2018, 62, e01674-17. [Google Scholar] [CrossRef]
- Prakash, O.; Hussain, K.; Kumar, R.; Wadhwa, D.; Sharma, C.; Aneja, K.R. Synthesis and Antimicrobial Evaluation of New 1,4-Dihydro-4-Pyrazolylpyridines and 4-Pyrazolylpyridines. Org. Med. Chem. Lett. 2011, 1, 5. [Google Scholar] [CrossRef]
- National Institute of Diabetes and Digestive and Kidney Diseases (NIDDK). LiverTox; National Institute of Diabetes and Digestive and Kidney Diseases: Bethesda, MD, USA, 2012.
- Soo, V.; Kwan, B.; Quezada, H.; Castillo-Juárez, I.; Pérez-Eretza, B.; García-Contreras, S.; Martínez-Vázquez, M.; Wood, T.; García-Contreras, R. Repurposing of Anticancer Drugs for the Treatment of Bacterial Infections. Curr. Top. Med. Chem. 2016, 17, 1157–1176. [Google Scholar] [CrossRef]
- Jeswani, G.; Paul, S.D. Recent Advances in the Delivery of Chemotherapeutic Agents. In Nano- and Microscale Drug Delivery Systems: Design and Fabrication; Elsevier: Amsterdam, The Netherlands, 2017; pp. 281–298. ISBN 9780323527279. [Google Scholar]
- Zhang, H.; Liu, S.; Yue, J.; Sun, S.; Lv, Q.; Jian, S.; Xie, Y.; Han, L.; Zhang, F.; Dai, Y.; et al. In Vitro Antimicrobial Activity of Diacerein on 76 Isolates of Gram-Positive Cocci from Bacterial Keratitis Patients and in Vivo Study of Diacerein Eye Drops on Staphylococcus aureus Keratitis in Mice. Antimicrob. Agents Chemother. 2019, 63, e01874-18. [Google Scholar] [CrossRef]
- Zhang, J.; Redman, N.; Litke, A.P.; Zeng, J.; Zhan, J.; Chan, K.Y.; Chang, C.W.T. Synthesis and Antibacterial Activity Study of a Novel Class of Cationic Anthraquinone Analogs. Bioorg. Med. Chem. 2011, 19, 498–503. [Google Scholar] [CrossRef]
- Henriksson, R.; Holm, S.; Littbrand, B. Interactions between Antibiotics and Antineoplastic Drugs on Antibacterial Activity in Vitro. Acta Oncol. 1990, 29, 43–46. [Google Scholar] [CrossRef] [PubMed]
- Murdock, K.C.; Child, R.G.; Fabio, P.F.; Angier, R.B.; Wallace, R.E.; Durr, F.E.; Citarella, R.V. Antitumor Agents. 1. 1,4-Bis[(Aminoalkyl)Amino]-9,10-Anthracenediones. J. Med. Chem. 1979, 22, 1024–1030. [Google Scholar] [CrossRef]
- Awasthi, P.; Vatsal, M.; Sharma, A. Structural and Biological Study of Synthesized Anthraquinone Series of Compounds with Sulfonamide Feature. J. Biomol. Struct. Dyn. 2019, 37, 4465–4480. [Google Scholar] [CrossRef] [PubMed]
- Huff, A.C.; Kreuzer, K.N. Evidence for a Common Mechanism of Action for Antitumor and Antibacterial Agents That Inhibit Type II DNA Topoisomerases. J. Biol. Chem. 1990, 265, 20496–20505. [Google Scholar] [CrossRef] [PubMed]
- Valenzuela, M.V.; Domenech, M.; Mateos-Martínez, P.; González-Camacho, F.; de la Campa, A.G.; García, M.T. Antibacterial Activity of a DNA Topoisomerase I Inhibitor versus Fluoroquinolones in Streptococcus pneumoniae. PLoS ONE 2020, 15, e0241780. [Google Scholar] [CrossRef]
- Choi, B.Y.; Kim, H.Y.; Lee, K.H.; Cho, Y.H.; Kong, G. Clofilium, a Potassium Channel Blocker, Induces Apoptosis of Human Promyelocytic Leukemia (HL-60) Cells via Bcl-2-Insensitive Activation of Caspase-3. Cancer Lett. 1999, 147, 85–93. [Google Scholar] [CrossRef]
- Marraffa, J.M. Amiodarone. Encycl. Toxicol. Third Ed. 2022, 197–199. [Google Scholar] [CrossRef]
- Tsai-Turton, M. Tamoxifen. Encycl. Toxicol. Third Ed. 2022, 471–473. [Google Scholar] [CrossRef]
- Yasuda, S.U.; Yasuda, R.P. Affinities of Brompheniramine, Chlorpheniramine, and Terfenadine at the Five Human Muscarinic Cholinergic Receptor Subtypes. Pharmacotherapy 1999, 19, 447–451. [Google Scholar] [CrossRef]
- Pepperell, R.; Burger, H. Clomiphene. Med. J. Aust. 1988, 149, 50. [Google Scholar] [CrossRef]
- ClinicalTrials.gov Study Record|Beta ClinicalTrials.Gov. Available online: https://beta.clinicaltrials.gov/study/NCT02454283 (accessed on 13 March 2023).
- Ingwersen, S.H.; Snel, S.; Mant, T.G.K.; Edwards, D. Nonlinear Multiple-dose Pharmacokinetics of the Dopamine Reuptake Inhibitor Vanoxerine. J. Pharm. Sci. 1993, 82, 1164–1166. [Google Scholar] [CrossRef] [PubMed]
- Hoskins, J.; Alborn, J.; Arnold, J.; Blaszczak, L.C.; Burgett, S.; Dehoff, B.S.; Estrem, S.T.; Fritz, L.; Fu, D.J.; Fuller, W.; et al. Genome of the Bacterium Streptococcus pneumoniae Strain R6. J. Bacteriol. 2001, 183, 5709–5717. [Google Scholar] [CrossRef] [PubMed]
- Lanie, J.A.; Ng, W.L.; Kazmierczak, K.M.; Andrzejewski, T.M.; Davidsen, T.M.; Wayne, K.J.; Tettelin, H.; Glass, J.I.; Winkler, M.E. Genome Sequence of Avery’s Virulent Serotype 2 Strain D39 of Streptococcus pneumoniae and Comparison with That of Unencapsulated Laboratory Strain R6. J. Bacteriol. 2007, 189, 38–51. [Google Scholar] [CrossRef] [PubMed]
- Lacks, S.; Hotchkiss, R.D. A Study of the Genetic Material Determining an Enzyme Activity in Pneumococcus. BBA—Biochim. Biophys. Acta 1960, 39, 508–518. [Google Scholar] [CrossRef] [PubMed]
- CLSI—Clinical & Laboratory Standards Institute. Performance Standards for Antimicrobial Susceptibility Testing: An Informational Supplement for Global Application Developed through the Clinical and Laboratory Standards Institute Consensus Process. Supplement M100, 27th ed.; Wayne, P., Ed.; CLSI—Clinical & Laboratory Standards Institute: Wayne, PA, USA, 2017; ISBN 1562388045. [Google Scholar]
- Mueller, J.H.; Hinton, J. A Protein-Free Medium for Primary Isolation of the Gonococcus and Meningococcus. Proc. Soc. Exp. Biol. Med. 1941, 48, 330–333. [Google Scholar] [CrossRef]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Compound | Decrease in OD550nm (%) 1 | UFC (mL−1 × 10−8) 2 | MIC (R6) (µg mL−1) | MIC (D39) (µg mL−1) | Permeability (%) at 90 min | cLogP |
|---|---|---|---|---|---|---|
| Control (no addition) | - | 6.6 ± 0.6 | - | - | - | - |
| DMSO 0.5% | - | 4.2 ± 0.3 | - | - | - | - |
| Nortriptyline hydrochloride 25 µM | 48 ± 5 | 6.9 ± 0.2 | ND 3 | ND | ND | 3.94 |
| Methyldopa (L, -) | 3 ± 1 | 6.8 ± 0.5 | ND | ND | ND | 1.23 |
| Pyrvinium pamoate | 19 ± 3 | 5.9 ± 0.3 | ND | ND | ND | 1.94 |
| Procainamide hydrochloride | 31 ± 2 | 5.7 ± 0.8 | ND | ND | ND | 0.99 |
| Maprotiline hydrochloride | 62 ± 4 | 5.7 ± 0.5 | ND | ND | ND | 4.01 |
| Triflupromazine hydrochloride | 41 ± 5 | 5.6 ± 0.8 | ND | ND | ND | 5.25 |
| Demecarium bromide | 46 ± 4 | 5.3 ± 0.1 | ND | ND | ND | −1.42 |
| Chlormezanone | 78 ± 9 | 4.1 ± 0.4 | ND | ND | ND | 1.44 |
| Flunarizine dihydrochloride | 50 ± 3 | 4.1 ± 0.4 | ND | ND | ND | 6.09 |
| Metixene hydrochloride | 64 ± 7 | 3.5 ± 0.5 | ND | ND | ND | 4.68 |
| Methiothepin maleate | 58 ± 7 | 3.3 ± 0.4 | ND | ND | ND | 4.14 |
| Perhexiline maleate | 10 ± 2 | 3.3 ± 0.3 | ND | ND | ND | 6.21 |
| Carbetapentane citrate | >99 | 2.8 ± 0.2 | ND | ND | ND | 3.83 |
| Bepridil hydrochloride | 72 ± 6 | 2.4 ± 0.2 | ND | ND | ND | 4.96 |
| Zotepine | 58 ± 7 | 2.3 ± 0.1 | ND | ND | ND | 5.52 |
| Zuclopenthixol hydrochloride | 49 ± 2 | 1.7 ± 0.6 | ND | ND | ND | 4.69 |
| Halofantrine hydrochloride | 83 ± 8 | 1.9 ± 0.4 | ND | ND | ND | 8.55 |
| Clofilium tosylate | >99 | 0.36 ± 0.02 (9%) | 51 | 38 | 52 | 3.34 |
| Vanoxerine | 80 ± 8 | 0.29 ± 0.01 (7%) | 26 | 26 | 98 | 6.04 |
| Mitoxantrone dihydrochloride | >99 | 0.17 ± 0.02 (4%) | >26 | >52 | <0.1 | 0.36 |
| Amiodarone hydrochloride | 71 ± 9 | 0.011 ± 0.004 (0.3%) | 34 | 51 | 71 | 8.31 |
| Tamoxifen citrate | 99 ± 1 | <0.001 (<0.02%) | >28 | >56 | 52 | 6.06 |
| Terfenadine | 38 ± 5 | <0.001 (<0.02%) | 12 | 47 | 71 | 6.17 |
| Clomiphene citrate (Z, E) | 91 ± 8 | <0.001 (<0.02%) | 30 | 45 | 64 | 6.53 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ortiz-Miravalles, L.; Sánchez-Angulo, M.; Sanz, J.M.; Maestro, B. Drug Repositioning as a Therapeutic Strategy against Streptococcus pneumoniae: Cell Membrane as Potential Target. Int. J. Mol. Sci. 2023, 24, 5831. https://doi.org/10.3390/ijms24065831
Ortiz-Miravalles L, Sánchez-Angulo M, Sanz JM, Maestro B. Drug Repositioning as a Therapeutic Strategy against Streptococcus pneumoniae: Cell Membrane as Potential Target. International Journal of Molecular Sciences. 2023; 24(6):5831. https://doi.org/10.3390/ijms24065831
Chicago/Turabian StyleOrtiz-Miravalles, Laura, Manuel Sánchez-Angulo, Jesús M. Sanz, and Beatriz Maestro. 2023. "Drug Repositioning as a Therapeutic Strategy against Streptococcus pneumoniae: Cell Membrane as Potential Target" International Journal of Molecular Sciences 24, no. 6: 5831. https://doi.org/10.3390/ijms24065831
APA StyleOrtiz-Miravalles, L., Sánchez-Angulo, M., Sanz, J. M., & Maestro, B. (2023). Drug Repositioning as a Therapeutic Strategy against Streptococcus pneumoniae: Cell Membrane as Potential Target. International Journal of Molecular Sciences, 24(6), 5831. https://doi.org/10.3390/ijms24065831