
Pharmacological Chaperones and Protein Conformational Diseases: Approaches of Computational Structural Biology




Abstract
1. Protein Conformational Diseases
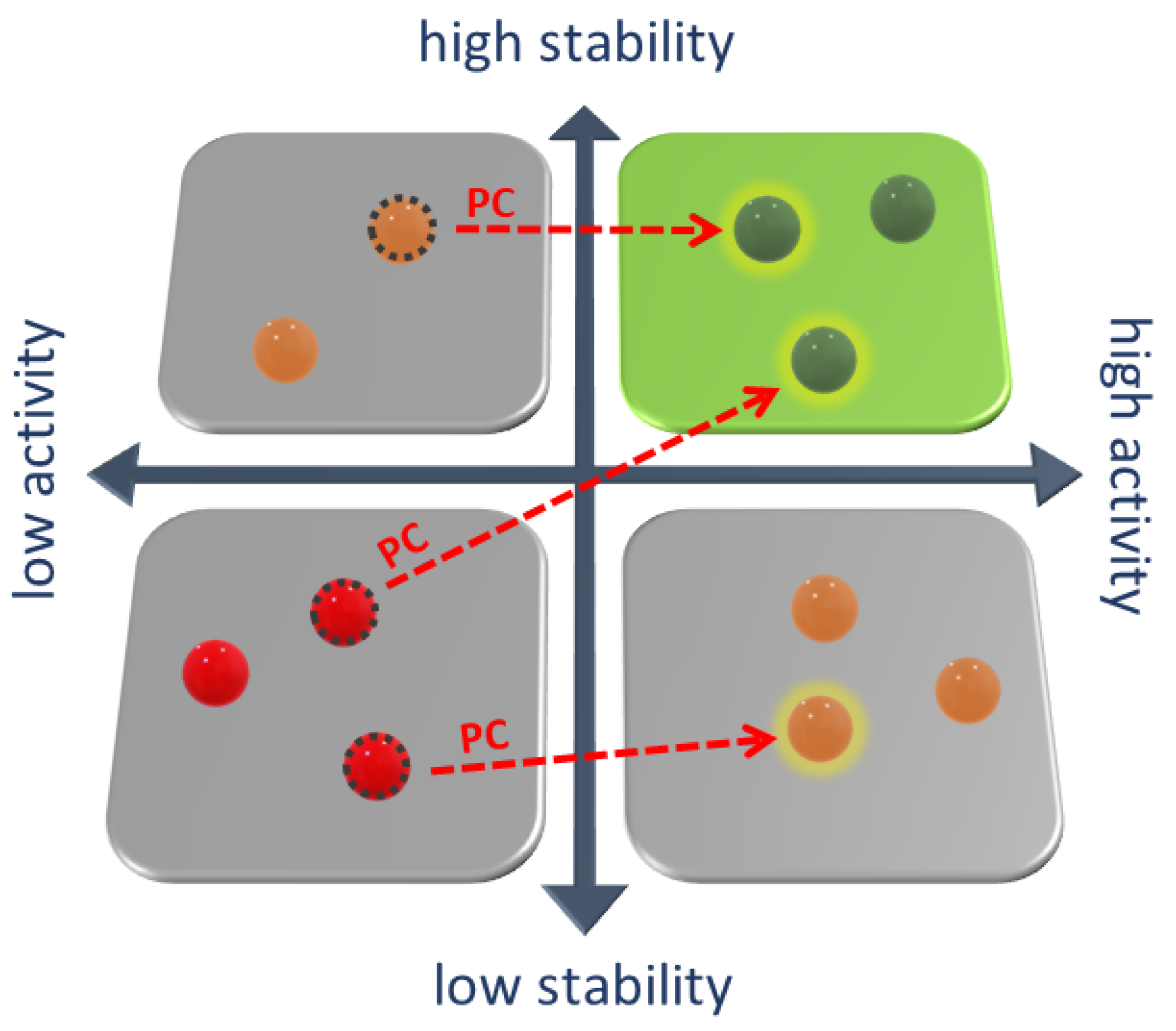
2. Pharmacological Chaperones
2.1. Competitive Inhibitors
2.2. Enzyme Cofactors
2.3. Allosteric Ligands

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Name of Disease | Pharmacological Chaperones | Clinical Status |
|---|---|---|
| Transthyretin-related hereditary amyloidosis | Vyndamax (Tafamidis) | Market approved |
| Phenylketonuria | Kuvan (tetrahydrobiopterin or BH4) | Market approved |
| Fabry disease | Migalistat (1-deoxygalactonojirimycin or Galafold) | Market approved |
| Gaucher disease, Type 1 | Afegostat tartrate (Isofagomine or AT2101) | Phase 2 NCT00446550 |
| Gaucher disease, Type 1 | Ambroxol | Phase 2 NCT03950050 |
| Gaucher disease, Type 1 | NCGC607 | Preclinical cell-based study [43] |
| Pompe disease | Duvoglustat | Phase 2 NCT00688597 |
| Pompe disease (late-onset) | Miglustat (AT2221) (with alglucosidase alfa, ATB200) | Phase 3 NCT03729362 |
| Gangliosidoses, GM1 | N-octyl 4-epi-β-valienamine | Preclinical in vivo study [44] |
| Gangliosidoses, GM1 | 1,5-dideoxy-1,5-iminoribitol C-glycoside | Preclinical cell-based study [45] |
| Gangliosidoses, GM2 Sandhoff disease Tay-Sachs disease | Pyrimethamine | Phase ½ NCT01102686 |
| Mucopolysaccharidosis IIIC | Glucosamine | Preclinical in vivo study [46] |
| Batten disease | CS38 | Preclinical cell-based study [47] |
2.4. Alternative Binders
3. Drug Repositioning
4. Predicting the Pathogenicity of Variants and the Effect on Protein Stability
5. Protein Instability Prediction by MD Simulation
6. Pocket Prediction Tools
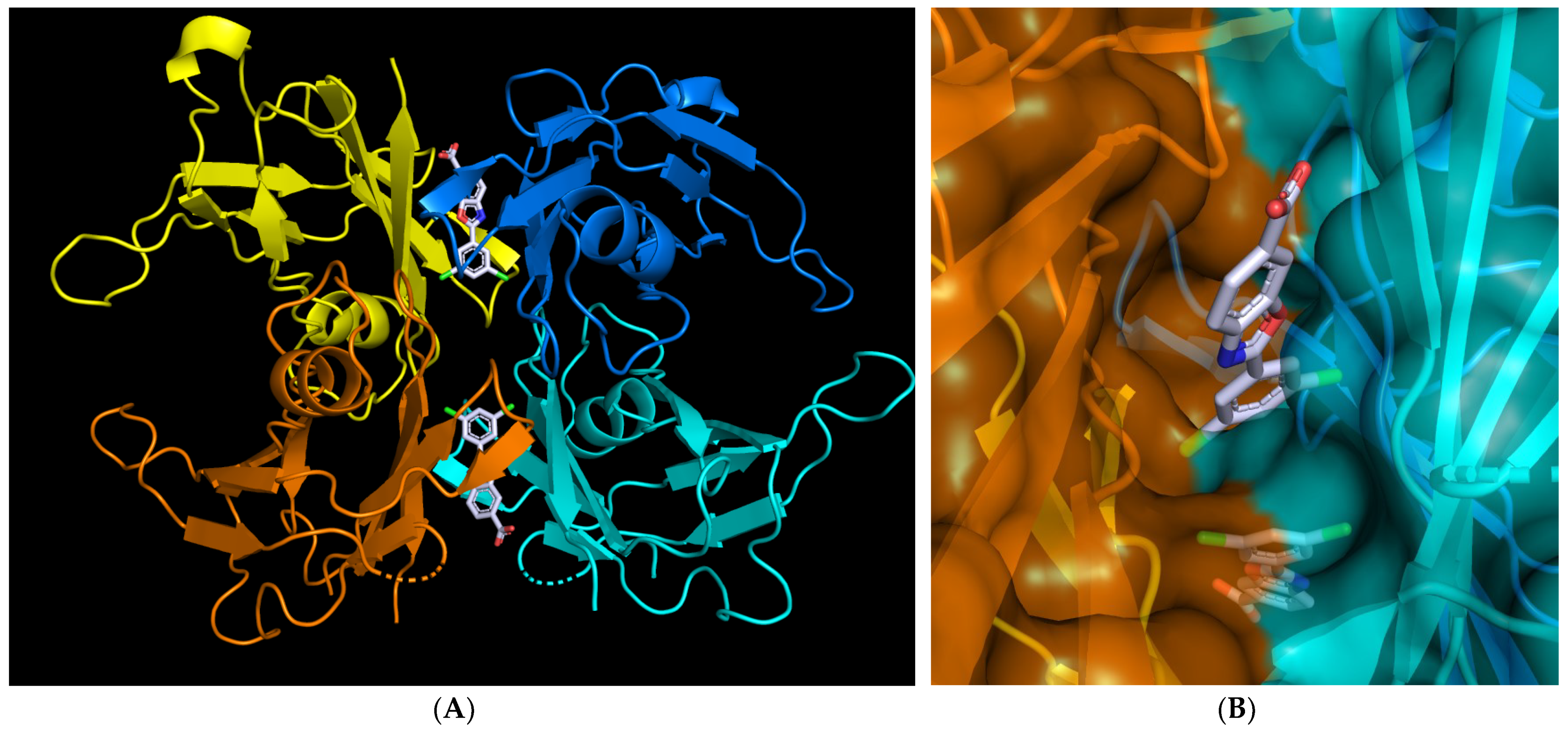
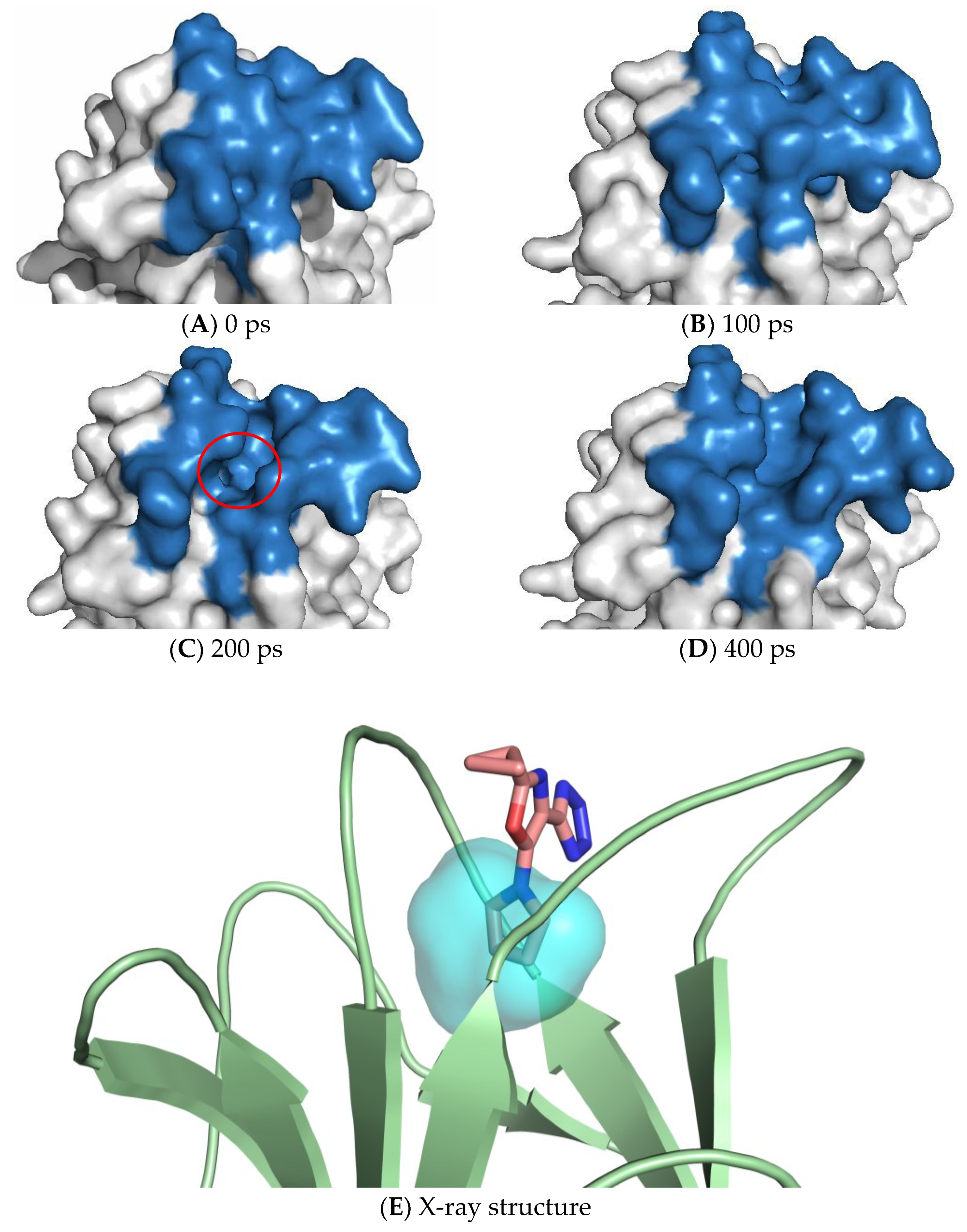
7. Exploiting Transient Pockets for PC Binding
8. Druggability of Pockets
9. Virtual Screening of Compound Libraries in Search of PCs
10. The Impact of Artificial Intelligence
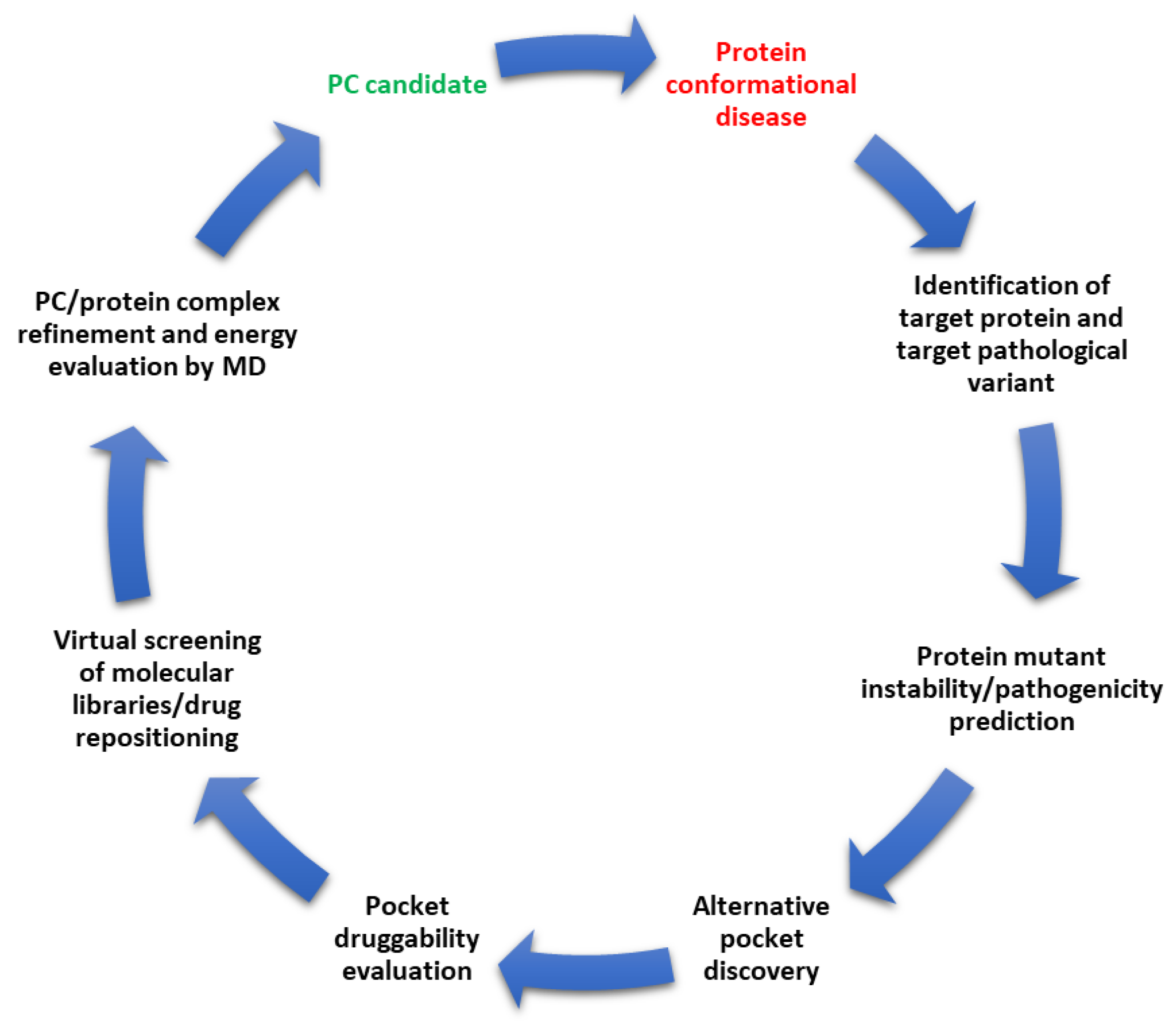
11. Conclusions and Future Directions
Funding
Institutional Review Board Statement
Informed Consent Statement
Acknowledgments
Conflicts of Interest
References
- Reynaud, E. Protein Misfolding and Degenerative Diseases. Nat. Educ. 2010, 3, 28. [Google Scholar]
- Soto, C. Protein Misfolding and Disease; Protein Refolding and Therapy. FEBS Lett. 2001, 498, 204–207. [Google Scholar] [CrossRef] [PubMed]
- Prabantu, V.M.; Naveenkumar, N.; Srinivasan, N. Influence of Disease-Causing Mutations on Protein Structural Networks. Front. Mol. Biosci. 2021, 7, 492. [Google Scholar] [CrossRef] [PubMed]
- Valastyan, J.S.; Lindquist, S. Mechanisms of Protein-Folding Diseases at a Glance. Dis. Model. Mech. 2014, 7, 9–14. [Google Scholar] [CrossRef]
- Bross, P.; Andresen, B.S.; Corydon, T.J.; Gregersen, N. Protein Misfolding and Degradation in Genetic Disease. Hum. Mutat. 1999, 14, 186. [Google Scholar] [CrossRef]
- Miosge, L.A.; Field, M.A.; Sontani, Y.; Cho, V.; Johnson, S.; Palkova, A.; Balakishnan, B.; Liang, R.; Zhang, Y.; Lyon, S.; et al. Comparison of Predicted and Actual Consequences of Missense Mutations. Proc. Natl. Acad. Sci. USA 2015, 112, E5189–E5198. [Google Scholar] [CrossRef]
- Andrews, T.D.; Sjollema, G.; Goodnow, C.C. Understanding the Immunological Impact of the Human Mutation Explosion. Trends Immunol. 2013, 34, 99–106. [Google Scholar] [CrossRef]
- Peng, Y.; Alexov, E. Protein Conformational Disease: Visit the Facts at a Glance. eLS 2017. [Google Scholar] [CrossRef]
- Conde-Giménez, M.; Galano-Frutos, J.J.; Galiana-Cameo, M.; Mahía, A.; Victor, B.L.; Salillas, S.; Velázquez-Campoy, A.; Brito, R.M.M.; Gálvez, J.A.; Díaz-de-Villegas, M.D.; et al. Alchemical Design of Pharmacological Chaperones with Higher Affinity for Phenylalanine Hydroxylase. Int. J. Mol. Sci. 2022, 23, 4502. [Google Scholar] [CrossRef]
- Chen, A.; Pan, Y.; Chen, J. Clinical, Genetic, and Experimental Research of Hyperphenylalaninemia. Front. Genet. 2022, 13, 1051153. [Google Scholar] [CrossRef] [PubMed]
- Grasso, D.; Geminiani, M.; Galderisi, S.; Iacomelli, G.; Peruzzi, L.; Marzocchi, B.; Santucci, A.; Bernini, A. Untargeted NMR Metabolomics Reveals Alternative Biomarkers and Pathways in Alkaptonuria. Int. J. Mol. Sci. 2022, 23, 5805. [Google Scholar] [CrossRef] [PubMed]
- Zatkova, A.; Sedlackova, T.; Radvansky, J.; Polakova, H.; Nemethova, M.; Aquaron, R.; Dursun, I.; Usher, J.L.; Kadasi, L. Identification of 11 Novel Homogentisate 1,2 Dioxygenase Variants in Alkaptonuria Patients and Establishment of a Novel LOVD-Based HGD Mutation Database. JIMD Rep. 2012, 4, 55–65. [Google Scholar] [CrossRef]
- Echaniz-Laguna, A.; Cauquil, C.; Labeyrie, C.; Adams, D. Treating Hereditary Transthyretin Amyloidosis: Present & Future Challenges. Rev. Neurol. 2023, 179, 30–34. [Google Scholar] [CrossRef] [PubMed]
- Shaimardanova, A.A.; Solovyeva, V.V.; Issa, S.S.; Rizvanov, A.A. Gene Therapy of Sphingolipid Metabolic Disorders. Int. J. Mol. Sci. 2023, 24, 3627. [Google Scholar] [CrossRef]
- Chang, Y.F.; Imam, J.S.; Wilkinson, M.F. The Nonsense-Mediated Decay RNA Surveillance Pathway. Annu. Rev. Biochem. 2007, 76, 51–74. [Google Scholar] [CrossRef] [PubMed]
- Bauer, N.C.; Doetsch, P.W.; Corbett, A.H. Mechanisms Regulating Protein Localization. Traffic 2015, 16, 1039–1061. [Google Scholar] [CrossRef] [PubMed]
- Aymami, J.; Barril, X.; Rodríguez-Pascau, L.; Martinell, M. Pharmacological Chaperones for Enzyme Enhancement Therapy in Genetic Diseases. Pharm. Pat. Anal. 2013, 2, 109–124. [Google Scholar] [CrossRef] [PubMed]
- Muntau, A.C.; Leandro, J.; Staudigl, M.; Mayer, F.; Gersting, S.W. Innovative Strategies to Treat Protein Misfolding in Inborn Errors of Metabolism: Pharmacological Chaperones and Proteostasis Regulators. J. Inherit. Metab. Dis. 2014, 37, 505–523. [Google Scholar] [CrossRef]
- Ringe, D.; Petsko, G.A. What Are Pharmacological Chaperones and Why Are They Interesting? J. Biol. 2009, 8, 80. [Google Scholar] [CrossRef]
- Parenti, G.; Moracci, M.; Fecarotta, S.; Andria, G. Pharmacological chaperone therapy for lysosomal storage diseases. Future Med. Chem. 2014, 6, 1031. [Google Scholar] [CrossRef]
- Leeson, P.D.; Springthorpe, B. The Influence of Drug-like Concepts on Decision-Making in Medicinal Chemistry. Nat. Rev. Drug. Discov. 2007, 6, 881–890. [Google Scholar] [CrossRef] [PubMed]
- Parenti, G.; Andria, G.; Valenzano, K.J. Pharmacological Chaperone Therapy: Preclinical Development, Clinical Translation, and Prospects for the Treatment of Lysosomal Storage Disorders. Mol. Ther. 2015, 23, 1138–1148. [Google Scholar] [CrossRef] [PubMed]
- Asano, N.; Ishii, S.; Kizu, H.; Ikeda, K.; Yasuda, K.; Kato, A.; Martin, O.R.; Fan, J.Q. In Vitro Inhibition and Intracellular Enhancement of Lysosomal Alpha-Galactosidase A Activity in Fabry Lymphoblasts by 1-Deoxygalactonojirimycin and Its Derivatives. Eur. J. Biochem. 2000, 267, 4179–4186. [Google Scholar] [CrossRef]
- Yam, G.H.-F.; Zuber, C.; Roth, J. A Synthetic Chaperone Corrects the Trafficking Defect and Disease Phenotype in a Protein Misfolding Disorder. FASEB J. 2005, 19, 12–18. [Google Scholar] [CrossRef]
- Fan, J.Q.; Ishii, S.; Asano, N.; Suzuki, Y. Accelerated Transport and Maturation of Lysosomal Alpha-Galactosidase A in Fabry Lymphoblasts by an Enzyme Inhibitor. Nat. Med. 1999, 5, 112–115. [Google Scholar] [CrossRef] [PubMed]
- Yam, G.H.F.; Bosshard, N.; Zuber, C.; Steinmann, B.; Roth, J. Pharmacological Chaperone Corrects Lysosomal Storage in Fabry Disease Caused by Trafficking-Incompetent Variants. Am. J. Physiol. Cell Physiol. 2006, 290, C1076–C1082. [Google Scholar] [CrossRef]
- Lieberman, R.L.; Wustman, B.A.; Huertas, P.; Powe, A.C.; Pine, C.W.; Khanna, R.; Schlossmacher, M.G.; Ringe, D.; Petsko, G.A. Structure of Acid Beta-Glucosidase with Pharmacological Chaperone Provides Insight into Gaucher Disease. Nat. Chem. Biol. 2007, 3, 101–107. [Google Scholar] [CrossRef]
- Khanna, R.; Benjamin, E.R.; Pellegrino, L.; Schilling, A.; Rigat, B.A.; Soska, R.; Nafar, H.; Ranes, B.E.; Feng, J.; Lun, Y.; et al. The Pharmacological Chaperone Isofagomine Increases Activity of the Gaucher Disease L444P Mutant Form of β-Glucosidase. FEBS J. 2010, 277, 1618. [Google Scholar] [CrossRef] [PubMed]
- Staudigl, M.; Gersting, S.W.; Danecka, M.K.; Messing, D.D.; Woidy, M.; Pinkas, D.; Kemter, K.F.; Blau, N.; Muntau, A.C. The Interplay between Genotype, Metabolic State and Cofactor Treatment Governs Phenylalanine Hydroxylase Function and Drug Response. Hum. Mol. Genet. 2011, 20, 2628–2641. [Google Scholar] [CrossRef]
- Michals-Matalon, K. Sapropterin Dihydrochloride, 6-R-L-Erythro-5,6,7,8-Tetrahydrobiopterin, in the Treatment of Phenylketonuria. Expert Opin. Investig. Drugs 2008, 17, 245–251. [Google Scholar] [CrossRef]
- Pey, A.L.; Pérez, B.; Desviat, L.R.; Martínez, M.A.; Aguado, C.; Erlandsen, H.; Gámez, A.; Stevens, R.C.; Thórólfsson, M.; Ugarte, M.; et al. Mechanisms Underlying Responsiveness to Tetrahydrobiopterin in Mild Phenylketonuria Mutations. Hum. Mutat. 2004, 24, 388–399. [Google Scholar] [CrossRef]
- Plumadore, E.; Lombardo, L.; Cabral, K.P. Pharmacotherapy Review: Emerging Treatment Modalities in Transthyretin Cardiac Amyloidosis. Am. J. Health-Syst. Pharm. 2022, 79, 52–62. [Google Scholar] [CrossRef]
- Lamb, Y.N.; Deeks, E.D. Tafamidis: A Review in Transthyretin Amyloidosis with Polyneuropathy. Drugs 2019, 79, 863–874. [Google Scholar] [CrossRef] [PubMed]
- Lamb, Y.N. Tafamidis: A Review in Transthyretin Amyloid Cardiomyopathy. Am. J. Cardiovasc. Drugs 2021, 21, 113–121. [Google Scholar] [CrossRef]
- Bulawa, C.E.; Connelly, S.; DeVit, M.; Wang, L.; Weigel, C.; Fleming, J.A.; Packman, J.; Powers, E.T.; Wiseman, R.L.; Foss, T.R.; et al. Tafamidis, a Potent and Selective Transthyretin Kinetic Stabilizer That Inhibits the Amyloid Cascade. Proc. Natl. Acad. Sci. USA 2012, 109, 9629–9634. [Google Scholar] [CrossRef]
- Garcia-Pavia, P.; Domínguez, F.; Gonzalez-Lopez, E. Transthyretin Amyloid Cardiomyopathy. Med. Clin. 2021, 156, 126–134. [Google Scholar] [CrossRef]
- Coelho, T.; Maia, L.F.; Da Silva, A.M.; Cruz, M.W.; Planté-Bordeneuve, V.; Lozeron, P.; Suhr, O.B.; Campistol, J.M.; Conceição, I.M.; Schmidt, H.H.J.; et al. Tafamidis for Transthyretin Familial Amyloid Polyneuropathy: A Randomized, Controlled Trial. Neurology 2012, 79, 785–792. [Google Scholar] [CrossRef] [PubMed]
- Merlini, G.; Planté-Bordeneuve, V.; Judge, D.P.; Schmidt, H.; Obici, L.; Perlini, S.; Packman, J.; Tripp, T.; Grogan, D.R. Effects of Tafamidis on Transthyretin Stabilization and Clinical Outcomes in Patients with Non-Val30Met Transthyretin Amyloidosis. J. Cardiovasc. Transl. Res. 2013, 6, 1011–1020. [Google Scholar] [CrossRef]
- Maurer, M.S.; Grogan, D.R.; Judge, D.P.; Mundayat, R.; Packman, J.; Lombardo, I.; Quyyumi, A.A.; Aarts, J.; Falk, R.H. Tafamidis in Transthyretin Amyloid Cardiomyopathy: Effects on Transthyretin Stabilization and Clinical Outcomes. Circ. Heart Fail. 2015, 8, 519–526. [Google Scholar] [CrossRef] [PubMed]
- Kon, T.; Misumi, Y.; Nishijima, H.; Honda, M.; Suzuki, C.; Baba, M.; Inomata, Y.; Obayashi, K.; Ando, Y.; Tomiyama, M. Effects of Liver Transplantation and Tafamidis in Hereditary Transthyretin Amyloidosis Caused by Transthyretin Leu55Pro Mutation: A Case Report. Amyloid 2015, 22, 203–204. [Google Scholar] [CrossRef]
- Scott, L.J. Tafamidis: A Review of Its Use in Familial Amyloid Polyneuropathy. Drugs 2014, 74, 1371–1378. [Google Scholar] [CrossRef]
- Suhr, O.B.; Conceição, I.M.; Karayal, O.N.; Mandel, F.S.; Huertas, P.E.; Ericzon, B.G. Post Hoc Analysis of Nutritional Status in Patients with Transthyretin Familial Amyloid Polyneuropathy: Impact of Tafamidis. Neurol. Ther. 2014, 3, 101–112. [Google Scholar] [CrossRef] [PubMed]
- Aflaki, E.; Borger, D.K.; Moaven, N.; Stubblefield, B.K.; Rogers, S.A.; Patnaik, S.; Schoenen, F.J.; Westbroek, W.; Zheng, W.; Sullivan, P.; et al. A New Glucocerebrosidase Chaperone Reduces α-Synuclein and Glycolipid Levels in IPSC-Derived Dopaminergic Neurons from Patients with Gaucher Disease and Parkinsonism. J. Neurosci. 2016, 36, 7441–7452. [Google Scholar] [CrossRef]
- Suzuki, Y.; Ichinomiya, S.; Kurosawa, M.; Matsuda, J.; Ogawa, S.; Iida, M.; Kubo, T.; Tabe, M.; Itoh, M.; Higaki, K.; et al. Therapeutic Chaperone Effect of N-Octyl 4-Epi-β-Valienamine on Murine G(M1)-Gangliosidosis. Mol. Genet. Metab. 2012, 106, 92–98. [Google Scholar] [CrossRef]
- Siriwardena, A.; Sonawane, D.P.; Bande, O.P.; Markad, P.R.; Yonekawa, S.; Tropak, M.B.; Ghosh, S.; Chopade, B.A.; Mahuran, D.J.; Dhavale, D.D. Synthesis of 1,5-Dideoxy-1,5-Iminoribitol C-Glycosides through a Nitrone-Olefin Cycloaddition Domino Strategy: Identification of Pharmacological Chaperones of Mutant Human Lysosomal β-Galactosidase. J. Org. Chem. 2014, 79, 4398–4404. [Google Scholar] [CrossRef] [PubMed]
- Pan, X.; Taherzadeh, M.; Bose, P.; Heon-Roberts, R.; Nguyen, A.L.A.; Xu, T.; Pará, C.; Yamanaka, Y.; Priestman, D.A.; Platt, F.M.; et al. Glucosamine Amends CNS Pathology in Mucopolysaccharidosis IIIC Mouse Expressing Misfolded HGSNAT. J. Exp. Med. 2022, 219, e20211860. [Google Scholar] [CrossRef]
- Dawson, G.; Schroeder, C.; Dawson, P.E. Palmitoyl:Protein Thioesterase (PPT1) Inhibitors Can Act as Pharmacological Chaperones in Infantile Batten Disease. Biochem. Biophys. Res. Commun. 2010, 395, 66–69. [Google Scholar] [CrossRef]
- Bier, D.; Bartel, M.; Sies, K.; Halbach, S.; Higuchi, Y.; Haranosono, Y.; Brummer, T.; Kato, N.; Ottmann, C. Small-Molecule Stabilization of the 14-3-3/Gab2 Protein-Protein Interaction (PPI) Interface. ChemMedChem 2016, 11, 911–918. [Google Scholar] [CrossRef]
- Inglese, J.; Shamu, C.E.; Guy, R.K. Reporting Data from High-Throughput Screening of Small-Molecule Libraries. Nat. Chem. Biol. 2007, 3, 438–441. [Google Scholar] [CrossRef] [PubMed]
- Langedijk, J.; Mantel-Teeuwisse, A.K.; Slijkerman, D.S.; Schutjens, M.H.D.B. Drug Repositioning and Repurposing: Terminology and Definitions in Literature. Drug Discov. Today 2015, 20, 1027–1034. [Google Scholar] [CrossRef] [PubMed]
- Croset, S. Drug Repositioning and Indication Discovery Using Description Logics. Ph.D. Thesis, University of Cambridge, Cambridge, UK, 2014. [Google Scholar] [CrossRef]
- Khanapure, A.; Chuki, P.; De Sousa, A. Drug Repositioning: Old Drugs For New Indications. Indian J. Appl. Res. 2011, 4, 462–466. [Google Scholar] [CrossRef]
- Paul, S.M.; Mytelka, D.S.; Dunwiddie, C.T.; Persinger, C.C.; Munos, B.H.; Lindborg, S.R.; Schacht, A.L. How to Improve RD Productivity: The Pharmaceutical Industry’s Grand Challenge. Nat. Rev. Drug Discov. 2010, 9, 203–214. [Google Scholar] [CrossRef]
- Ashburn, T.T.; Thor, K.B. Drug Repositioning: Identifying and Developing New Uses for Existing Drugs. Nat. Rev. Drug Discov. 2004, 3, 673–683. [Google Scholar] [CrossRef] [PubMed]
- Chong, C.R.; Sullivan, D.J. New Uses for Old Drugs. Nature 2007, 448, 645–646. [Google Scholar] [CrossRef]
- Hernandez, J.J.; Pryszlak, M.; Smith, L.; Yanchus, C.; Kurji, N.; Shahani, V.M.; Molinski, S.V. Giving Drugs a Second Chance: Overcoming Regulatory and Financial Hurdles in Repurposing Approved Drugs As Cancer Therapeutics. Front. Oncol. 2017, 7, 273. [Google Scholar] [CrossRef] [PubMed]
- Ma, D.L.; Chan, D.S.H.; Leung, C.H. Drug Repositioning by Structure-Based Virtual Screening. Chem. Soc. Rev. 2013, 42, 2130–2141. [Google Scholar] [CrossRef]
- Maegawa, G.H.B.; Tropak, M.B.; Buttner, J.D.; Rigat, B.A.; Fuller, M.; Pandit, D.; Tang, L.; Kornhaber, G.J.; Hamuro, Y.; Clarke, J.T.R.; et al. Identification and Characterization of Ambroxol as an Enzyme Enhancement Agent for Gaucher Disease. J. Biol. Chem. 2009, 284, 23502–23516. [Google Scholar] [CrossRef]
- Bendikov-Bar, I.; Maor, G.; Filocamo, M.; Horowitz, M. Ambroxol as a Pharmacological Chaperone for Mutant Glucocerebrosidase. Blood. Cells Mol. Dis. 2013, 50, 141–145. [Google Scholar] [CrossRef]
- Narita, A.; Shirai, K.; Itamura, S.; Matsuda, A.; Ishihara, A.; Matsushita, K.; Fukuda, C.; Kubota, N.; Takayama, R.; Shigematsu, H.; et al. Ambroxol Chaperone Therapy for Neuronopathic Gaucher Disease: A Pilot Study. Ann. Clin. Transl. Neurol. 2016, 3, 200–215. [Google Scholar] [CrossRef]
- Istaiti, M.; Revel-Vilk, S.; Becker-Cohen, M.; Dinur, T.; Ramaswami, U.; Castillo-Garcia, D.; Ceron-Rodriguez, M.; Chan, A.; Rodic, P.; Tincheva, R.S.; et al. Upgrading the Evidence for the Use of Ambroxol in Gaucher Disease and GBA Related Parkinson: Investigator Initiated Registry Based on Real Life Data. Am. J. Hematol. 2021, 96, 545–551. [Google Scholar] [CrossRef]
- Berger, A.C.; Olson, S.; Johnson, S.G.; Beachy, S.H. Drug Repurposing and Repositioning: Workshop Summary; Drug Repurposing and Repositioning; National Academies Press: Washington, DC, USA, 2014. [Google Scholar] [CrossRef]
- Ng, P.C.; Henikoff, S. Predicting Deleterious Amino Acid Substitutions. Genome Res. 2001, 11, 863–874. [Google Scholar] [CrossRef] [PubMed]
- Thomas, P.D.; Ebert, D.; Muruganujan, A.; Mushayahama, T.; Albou, L.P.; Mi, H. PANTHER: Making Genome-Scale Phylogenetics Accessible to All. Protein Sci. 2022, 31, 8–22. [Google Scholar] [CrossRef] [PubMed]
- Snap2—Rost Lab Open. Available online: https://rostlab.org/owiki/index.php/Snap2 (accessed on 3 January 2023).
- Calabrese, R.; Capriotti, E.; Fariselli, P.; Martelli, P.L.; Casadio, R. Functional Annotations Improve the Predictive Score of Human Disease-Related Mutations in Proteins. Hum. Mutat. 2009, 30, 1237–1244. [Google Scholar] [CrossRef] [PubMed]
- Adzhubei, I.A.; Schmidt, S.; Peshkin, L.; Ramensky, V.E.; Gerasimova, A.; Bork, P.; Kondrashov, A.S.; Sunyaev, S.R. A Method and Server for Predicting Damaging Missense Mutations. Nat. Methods 2010, 7, 248–249. [Google Scholar] [CrossRef]
- Shihab, H.A.; Gough, J.; Cooper, D.N.; Stenson, P.D.; Barker, G.L.A.; Edwards, K.J.; Day, I.N.M.; Gaunt, T.R. Predicting the Functional, Molecular, and Phenotypic Consequences of Amino Acid Substitutions Using Hidden Markov Models. Hum. Mutat. 2013, 34, 57–65. [Google Scholar] [CrossRef] [PubMed]
- Pappalardo, M.; Wass, M.N. VarMod: Modelling the Functional Effects of Non-Synonymous Variants. Nucleic Acids. Res. 2014, 42, W331–W336. [Google Scholar] [CrossRef]
- Pejaver, V.; Urresti, J.; Lugo-Martinez, J.; Pagel, K.A.; Lin, G.N.; Nam, H.-J.; Mort, M.; Cooper, D.N.; Sebat, J.; Iakoucheva, L.M.; et al. MutPred2: Inferring the Molecular and Phenotypic Impact of Amino Acid Variants. Nat. Commun. 2020, 11, 5918. [Google Scholar] [CrossRef] [PubMed]
- Wong, K.C.; Zhang, Z. SNPdryad: Predicting Deleterious Non-Synonymous Human SNPs Using Only Orthologous Protein Sequences. Bioinformatics 2014, 30, 1112–1119. [Google Scholar] [CrossRef]
- Zhou, H.; Gao, M.; Skolnick, J. ENTPRISE: An Algorithm for Predicting Human Disease-Associated Amino Acid Substitutions from Sequence Entropy and Predicted Protein Structures. PLoS ONE 2016, 11, e0150965. [Google Scholar] [CrossRef]
- Reva, B.; Antipin, Y.; Sander, C. Determinants of Protein Function Revealed by Combinatorial Entropy Optimization. Genome Biol. 2007, 8, R232. [Google Scholar] [CrossRef]
- Cheng, J.; Randall, A.; Baldi, P. Prediction of Protein Stability Changes for Single-Site Mutations Using Support Vector Machines. Proteins 2006, 62, 1125–1132. [Google Scholar] [CrossRef]
- Parthiban, V.; Gromiha, M.M.; Schomburg, D. CUPSAT: Prediction of Protein Stability upon Point Mutations. Nucleic Acids Res. 2006, 34, W239–W242. [Google Scholar] [CrossRef] [PubMed]
- Fariselli, P.; Martelli, P.L.; Savojardo, C.; Casadio, R. INPS: Predicting the Impact of Non-Synonymous Variations on Protein Stability from Sequence. Bioinformatics 2015, 31, 2816–2821. [Google Scholar] [CrossRef] [PubMed]
- Savojardo, C.; Fariselli, P.; Martelli, P.L.; Casadio, R. INPS-MD: A Web Server to Predict Stability of Protein Variants from Sequence and Structure. Bioinformatics 2016, 32, 2542–2544. [Google Scholar] [CrossRef]
- Yates, C.M.; Filippis, I.; Kelley, L.A.; Sternberg, M.J.E. SuSPect: Enhanced Prediction of Single Amino Acid Variant (SAV) Phenotype Using Network Features. J. Mol. Biol. 2014, 426, 2692–2701. [Google Scholar] [CrossRef]
- Pandurangan, A.P.; Ochoa-Montaño, B.; Ascher, D.B.; Blundell, T.L. SDM: A Server for Predicting Effects of Mutations on Protein Stability. Nucleic Acids Res. 2017, 45, W229–W235. [Google Scholar] [CrossRef]
- Rodrigues, C.H.M.; Myung, Y.; Pires, D.E.V.; Ascher, D.B. MCSM-PPI2: Predicting the Effects of Mutations on Protein–Protein Interactions. Nucleic Acids Res. 2019, 47, W338–W344. [Google Scholar] [CrossRef] [PubMed]
- Pires, D.E.V.; Ascher, D.B.; Blundell, T.L. DUET: A Server for Predicting Effects of Mutations on Protein Stability Using an Integrated Computational Approach. Nucleic Acids Res. 2014, 42, W314–W319. [Google Scholar] [CrossRef]
- Pires, D.E.V.; Rodrigues, C.H.M.; Ascher, D.B. MCSM-Membrane: Predicting the Effects of Mutations on Transmembrane Proteins. Nucleic Acids Res. 2020, 48, W147–W153. [Google Scholar] [CrossRef]
- Pires, D.E.V.; Ascher, D.B. MCSM-AB: A Web Server for Predicting Antibody–Antigen Affinity Changes upon Mutation with Graph-Based Signatures. Nucleic Acids Res. 2016, 44, W469–W473. [Google Scholar] [CrossRef]
- Rodrigues, C.H.; Pires, D.E.; Ascher, D.B.; David Ascher, C.B. DynaMut2: Assessing Changes in Stability and Flexibility upon Single and Multiple Point Missense Mutations. Protein Sci. 2021, 30, 60–69. [Google Scholar] [CrossRef] [PubMed]
- Andreotti, G.; Guarracino, M.R.; Cammisa, M.; Correra, A.; Cubellis, M.V. Prediction of the Responsiveness to Pharmacological Chaperones: Lysosomal Human Alpha-Galactosidase, a Case of Study. Orphanet J. Rare Dis. 2010, 5, 36. [Google Scholar] [CrossRef] [PubMed]
- Eng, C.M.; Niehaus, D.J.; Enriquez, A.L.; Burgert, T.S.; Ludman, M.D.; Desnick, R.J. Fabry Disease: Twenty-Three Mutations Including Sense and Antisense CPG Alterations and Identification of a Deletional Hot-Spot in the α-Galactosidase A Gene. Hum. Mol. Genet. 1994, 3, 1795–1799. [Google Scholar] [CrossRef]
- du Moulin, M.; Koehn, A.F.; Golsari, A.; Dulz, S.; Atiskova, Y.; Patten, M.; Münch, J.; Avanesov, M.; Ullrich, K.; Muschol, N. The Mutation p.D313Y Is Associated with Organ Manifestation in Fabry Disease. Clin. Genet. 2017, 92, 528–533. [Google Scholar] [CrossRef]
- Zompola, C.; Palaiodimou, L.; Kokotis, P.; Papadopoulou, M.; Theodorou, A.; Sabanis, N.; Theodoroula, E.; Papathanasiou, M.; Tsivgoulis, G.; Chroni, E. The Mutation D313Y May Be Associated with Nervous System Manifestations in Fabry Disease. J. Neurol. Sci. 2020, 412, 116757. [Google Scholar] [CrossRef]
- Lukas, J.; Giese, A.K.; Markoff, A.; Grittner, U.; Kolodny, E.; Mascher, H.; Lackner, K.J.; Meyer, W.; Wree, P.; Saviouk, V.; et al. Functional Characterisation of Alpha-Galactosidase a Mutations as a Basis for a New Classification System in Fabry Disease. PLoS Genet. 2013, 9, e1003632. [Google Scholar] [CrossRef]
- Koulousios, K.; Stylianou, K.; Pateinakis, P.; Zamanakou, M.; Loules, G.; Manou, E.; Kyriklidou, P.; Katsinas, C.; Ouzouni, A.; Kyriazis, J.; et al. Fabry Disease Due to D313Y and Novel GLA Mutations. BMJ Open 2017, 7, e017098. [Google Scholar] [CrossRef]
- Van Der Spoel, D.; Lindahl, E.; Hess, B.; Groenhof, G.; Mark, A.E.; Berendsen, H.J.C. GROMACS: Fast, Flexible, and Free. J. Comput. Chem. 2005, 26, 1701–1718. [Google Scholar] [CrossRef]
- Pronk, S.; Páll, S.; Schulz, R.; Larsson, P.; Bjelkmar, P.; Apostolov, R.; Shirts, M.R.; Smith, J.C.; Kasson, P.M.; Van Der Spoel, D.; et al. GROMACS 4.5: A High-Throughput and Highly Parallel Open Source Molecular Simulation Toolkit. Bioinformatics 2013, 29, 845–854. [Google Scholar] [CrossRef] [PubMed]
- Case, D.A.; Cheatham, T.E., III; Darden, T.; Gohlke, H.; Luo, R.; Merz, K.M., Jr.; Onufriev, A.; Simmerling, C.B.; Wang, B.; Woods, R. The Amber biomolecular simulation programs. J. Computat. Chem. 2005, 26, 1668. [Google Scholar] [CrossRef]
- Phillips, J.C.; Hardy, D.J.; Maia, J.D.C.; Stone, J.E.; Ribeiro, J.V.; Bernardi, R.C.; Buch, R.; Fiorin, G.; Hénin, J.; Jiang, W.; et al. Scalable Molecular Dynamics on CPU and GPU Architectures with NAMD. J. Chem. Phys. 2020, 153, 044130. [Google Scholar] [CrossRef] [PubMed]
- Bowers, K.; Chow, E.; Xu, H.; Dror, R.; Eastwood, M.; Gregersen, B.; Klepeis, J.; Kolossváry, I.; Moraes, M.; Sacerdoti, F.; et al. Molecular Dynamics—Scalable Algorithms for Molecular Dynamics Simulations on Commodity Clusters. In Proceedings of the SC’06, 2006 ACM/IEEE Conference on Supercomputing, Tampa, FL, USA, 11–17 November 2006; p. 84. [Google Scholar]
- Rackers, J.A.; Wang, Z.; Lu, C.; Laury, M.L.; Lagardère, L.; Schnieders, M.J.; Piquemal, J.P.; Ren, P.; Ponder, J.W. Tinker 8: Software Tools for Molecular Design. J. Chem. Theory. Comput. 2018, 14, 5273–5289. [Google Scholar] [CrossRef] [PubMed]
- Berman, H.M.; Westbrook, J.; Feng, Z.; Gilliland, G.; Bhat, T.N.; Weissig, H.; Shindyalov, I.N.; Bourne, P.E. The Protein Data Bank. Nucleic Acids Res. 2000, 28, 235–242. [Google Scholar] [CrossRef]
- Schrödinger LLC. The PyMOL Molecular Graphics System, Version~1.8; 2015. [Google Scholar]
- Boyce, S.E.; Mobley, D.L.; Rocklin, G.J.; Graves, A.P.; Dill, K.A.; Shoichet, B.K. Predicting Ligand Binding Affinity with Alchemical Free Energy Methods in a Polar Model Binding Site. J. Mol. Biol. 2009, 394, 747–763. [Google Scholar] [CrossRef]
- Lundborg, M.; Lidmar, J.; Hess, B. The Accelerated Weight Histogram Method for Alchemical Free Energy Calculations. J. Chem. Phys. 2021, 154, 204103. [Google Scholar] [CrossRef] [PubMed]
- Bennett, C.H. Efficient Estimation of Free Energy Differences from Monte Carlo Data. J. Comput. Phys. 1976, 22, 245–268. [Google Scholar] [CrossRef]
- Matsunaga, Y.; Kamiya, M.; Oshima, H.; Jung, J.; Ito, S.; Sugita, Y. Use of Multistate Bennett Acceptance Ratio Method for Free-Energy Calculations from Enhanced Sampling and Free-Energy Perturbation. Biophys. Rev. 2022, 14, 1503–1512. [Google Scholar] [CrossRef]
- Lindahl, V.; Lidmar, J.; Hess, B. Accelerated Weight Histogram Method for Exploring Free Energy Landscapes. J. Chem. Phys. 2014, 141, 044110. [Google Scholar] [CrossRef]
- Daggett, V. Molecular Dynamics Simulations of the Protein Unfolding/Folding Reaction. Acc. Chem. Res. 2002, 35, 422–429. [Google Scholar] [CrossRef]
- Childers, M.C.; Daggett, V. Validating Molecular Dynamics Simulations against Experimental Observables in Light of Underlying Conformational Ensembles. J. Phys. Chem. B 2018, 122, 6673–6689. [Google Scholar] [CrossRef]
- Mayor, U.; Johnson, C.M.; Daggett, V.; Fersht, A.R.; Kazmirski, S.L.; Wong, K.B.; Freund, S.M.V.; Tan, Y.-J. Protein Folding and Unfolding in Microseconds to Nanoseconds by Experiment and Simulation. Proc. Natl. Acad. Sci. USA 2000, 97, 13518–13522. [Google Scholar] [CrossRef] [PubMed]
- Zhang, D.; Lazim, R. Application of Conventional Molecular Dynamics Simulation in Evaluating the Stability of Apomyoglobin in Urea Solution. Sci. Rep. 2017, 7, 44651. [Google Scholar] [CrossRef]
- Dechene, M.; Wink, G.; Smith, M.; Swartz, P.; Mattos, C. Multiple Solvent Crystal Structures of Ribonuclease A: An Assessment of the Method. Proteins Struct. Funct. Bioinform. 2009, 76, 861–881. [Google Scholar] [CrossRef] [PubMed]
- Agarwal, S.; Smith, M.; De La Rosa, I.; Verba, K.A.; Swartz, P.; Segura-Totten, M.; Mattos, C. Development of a Structure-Analysis Pipeline Using Multiple-Solvent Crystal Structures of Barrier-to-Autointegration Factor. Acta Cryst. D Struct. Biol. 2020, 76, 1001–1014. [Google Scholar] [CrossRef]
- Mattos, C.; Bellamacina, C.R.; Peisach, E.; Pereira, A.; Vitkup, D.; Petsko, G.A.; Ringe, D. Multiple Solvent Crystal Structures: Probing Binding Sites, Plasticity and Hydration. J. Mol. Biol. 2006, 357, 1471–1482. [Google Scholar] [CrossRef]
- Nguyen, T.T.; Venditti, V. An Allosteric Pocket for Inhibition of Bacterial Enzyme I Identified by NMR-Based Fragment Screening. J. Struct. Biol. X 2020, 4, 100034. [Google Scholar] [CrossRef]
- Bernini, A.; Venditti, V.; Spiga, O.; Niccolai, N. Probing Protein Surface Accessibility with Solvent and Paramagnetic Molecules. Prog. Nucl. Magn. Reson. Spectrosc. 2009, 54, 278–289. [Google Scholar] [CrossRef]
- Consortium, T.U.; Bateman, A.; Martin, M.-J.; Orchard, S.; Magrane, M.; Ahmad, S.; Alpi, E.; Bowler-Barnett, E.H.; Britto, R.; Bye-A-Jee, H.; et al. UniProt: The Universal Protein Knowledgebase in 2023. Nucleic Acids Res. 2013, 1, 13–14. [Google Scholar] [CrossRef]
- Jumper, J.; Evans, R.; Pritzel, A.; Green, T.; Figurnov, M.; Ronneberger, O.; Tunyasuvunakool, K.; Bates, R.; Žídek, A.; Potapenko, A.; et al. Highly Accurate Protein Structure Prediction with AlphaFold. Nature 2021, 596, 583–589. [Google Scholar] [CrossRef]
- Tian, W.; Chen, C.; Lei, X.; Zhao, J.; Liang, J. CASTp 3.0: Computed Atlas of Surface Topography of Proteins. Nucleic Acids Res. 2018, 46, W363–W367. [Google Scholar] [CrossRef] [PubMed]
- McGreig, J.E.; Uri, H.; Antczak, M.; Sternberg, M.J.E.; Michaelis, M.; Wass, M.N. 3DLigandSite: Structure-Based Prediction of Protein–Ligand Binding Sites. Nucleic Acids Res. 2022, 50, W13–W20. [Google Scholar] [CrossRef]
- Kelley, L.A.; Mezulis, S.; Yates, C.M.; Wass, M.N.; Sternberg, M.J.E. The Phyre2 Web Portal for Protein Modeling, Prediction and Analysis. Nat. Protoc. 2015, 10, 845–858. [Google Scholar] [CrossRef] [PubMed]
- Mcguffin, L.J.; Adiyaman, R.; Maghrabi, A.H.A.; Shuid, A.N.; Brackenridge, D.A.; Nealon, J.O.; Philomina, L.S. IntFOLD: An Integrated Web Resource for High Performance Protein Structure and Function Prediction. Nucleic Acids Res. 2019, 47, W408–W413. [Google Scholar] [CrossRef] [PubMed]
- Jiménez, J.; Doerr, S.; Martínez-Rosell, G.; Rose, A.S.; De Fabritiis, G. DeepSite: Protein-Binding Site Predictor Using 3D-Convolutional Neural Networks. Bioinformatics 2017, 33, 3036–3042. [Google Scholar] [CrossRef] [PubMed]
- Wu, Q.; Peng, Z.; Zhang, Y.; Yang, J. COACH-D: Improved Protein-Ligand Binding Sites Prediction with Refined Ligand-Binding Poses through Molecular Docking. Nucleic Acids Res. 2018, 46, W438–W442. [Google Scholar] [CrossRef] [PubMed]
- Jendele, L.; Krivak, R.; Skoda, P.; Novotny, M.; Hoksza, D. PrankWeb: A Web Server for Ligand Binding Site Prediction and Visualization. Nucleic Acids Res. 2019, 47, W345–W349. [Google Scholar] [CrossRef] [PubMed]
- Citro, V.; Peña-García, J.; Den-Haan, H.; Pérez-Sánchez, H.; Del Prete, R.; Liguori, L.; Cimmaruta, C.; Lukas, J.; Cubellis, M.V.; Andreotti, G. Identification of an Allosteric Binding Site on Human Lysosomal Alpha-Galactosidase Opens the Way to New Pharmacological Chaperones for Fabry Disease. PLoS ONE 2016, 11, e0165463. [Google Scholar] [CrossRef] [PubMed]
- Benz, J.; Rufer, A.C.; Huber, S.; Ehler, A.; Hug, M.; Topp, A.; Guba, W.; Hofmann, E.C.; Jagasia, R.; Rodríguez Sarmiento, R.M. Novel β-Glucocerebrosidase Activators That Bind to a New Pocket at a Dimer Interface and Induce Dimerization. Angew. Chem. Int. Ed. 2021, 60, 5436–5442. [Google Scholar] [CrossRef]
- Oleinikovas, V.; Saladino, G.; Cossins, B.P.; Gervasio, F.L. Understanding Cryptic Pocket Formation in Protein Targets by Enhanced Sampling Simulations. J. Am. Chem. Soc. 2016, 138, 14257–14263. [Google Scholar] [CrossRef] [PubMed]
- Wells, J.A.; McClendon, C.L. Reaching for High-Hanging Fruit in Drug Discovery at Protein–Protein Interfaces. Nature 2007, 450, 1001–1009. [Google Scholar] [CrossRef] [PubMed]
- Eyrisch, S.; Helms, V. Transient Pockets on Protein Surfaces Involved in Protein-Protein Interaction. J. Med. Chem. 2007, 50, 3457–3464. [Google Scholar] [CrossRef]
- Seddon, G.M.; Bywater, R.P. Accelerated Simulation of Unfolding and Refolding of a Large Single Chain Globular Protein. Open Biol. 2012, 2, 120087. [Google Scholar] [CrossRef]
- Eyrisch, S.; Helms, V. What Induces Pocket Openings on Protein Surface Patches Involved in Protein-Protein Interactions? J. Comput. Aided Mol. Des. 2009, 23, 73–86. [Google Scholar] [CrossRef]
- Joerger, A.C.; Bauer, M.R.; Wilcken, R.; Baud, M.G.J.; Harbrecht, H.; Exner, T.E.; Boeckler, F.M.; Spencer, J.; Fersht, A.R. Exploiting Transient Protein States for the Design of Small-Molecule Stabilizers of Mutant P53. Structure 2015, 23, 2246–2255. [Google Scholar] [CrossRef]
- Wassman, C.D.; Baronio, R.; Demir, Ö.; Wallentine, B.D.; Chen, C.K.; Hall, L.V.; Salehi, F.; Lin, D.W.; Chung, B.P.; Wesley Hatfield, G.; et al. Computational Identification of a Transiently Open L1/S3 Pocket for Reactivation of Mutant P53. Nat. Commun. 2013, 4, 1407. [Google Scholar] [CrossRef] [PubMed]
- Bernini, A.; Henrici De Angelis, L.; Morandi, E.; Spiga, O.; Santucci, A.; Assfalg, M.; Molinari, H.; Pillozzi, S.; Arcangeli, A.; Niccolai, N. Searching for Protein Binding Sites from Molecular Dynamics Simulations and Paramagnetic Fragment-Based NMR Studies. Biochim. Biophys. Acta 2014, 1844, 561–566. [Google Scholar] [CrossRef] [PubMed]
- Pietrucci, F.; Vargiu, A.V.; Kranjc, A. HIV-1 Protease Dimerization Dynamics Reveals a Transient Druggable Binding Pocket at the Interface. Sci. Rep. 2015, 5, 18555. [Google Scholar] [CrossRef]
- Bernini, A.; Galderisi, S.; Spiga, O.; Bernardini, G.; Niccolai, N.; Manetti, F.; Santucci, A. Toward a Generalized Computational Workflow for Exploiting Transient Pockets as New Targets for Small Molecule Stabilizers: Application to the Homogentisate 1,2-Dioxygenase Mutants at the Base of Rare Disease Alkaptonuria. Comput. Biol. Chem. 2017, 70, 133–141. [Google Scholar] [CrossRef] [PubMed]
- Schmidtke, P.; Bidon-chanal, A.; Luque, F.J.; Barril, X. MDpocket: Open-Source Cavity Detection and Characterization on Molecular Dynamics Trajectories. Bioinformatics 2011, 27, 3276–3285. [Google Scholar] [CrossRef]
- Brady, G.P.; Stouten, P.F.W. Fast Prediction and Visualization of Protein Binding Pockets with PASS. J. Comput. Aided Mol. Des. 2000, 14, 383–401. [Google Scholar] [CrossRef] [PubMed]
- Schmidtke, P.; Le Guilloux, V.; Maupetit, J.; Tufféry, P. Fpocket: Online Tools for Protein Ensemble Pocket Detection and Tracking. Nucleic Acids Res. 2010, 38, W582. [Google Scholar] [CrossRef] [PubMed]
- Lipinski, C.A.; Lombardo, F.; Dominy, B.W.; Feeney, P.J. Experimental and Computational Approaches to Estimate Solubility and Permeability in Drug Discovery and Development Settings. Adv. Drug Deliv. Rev. 2001, 46, 3–26. [Google Scholar] [CrossRef] [PubMed]
- Shultz, M.D. Two Decades under the Influence of the Rule of Five and the Changing Properties of Approved Oral Drugs. J. Med. Chem. 2019, 62, 1701–1714. [Google Scholar] [CrossRef]
- Hussein, H.A.; Borrel, A.; Geneix, C.; Petitjean, M.; Regad, L.; Camproux, A.C. PockDrug-Server: A New Web Server for Predicting Pocket Druggability on Holo and Apo Proteins. Nucleic Acids Res. 2015, 43, W436–W442. [Google Scholar] [CrossRef] [PubMed]
- Krasowski, A.; Muthas, D.; Sarkar, A.; Schmitt, S.; Brenk, R. DrugPred: A Structure-Based Approach to Predict Protein Druggability Developed Using an Extensive Nonredundant Data Set. J. Chem. Inf. Model. 2011, 51, 2829–2842. [Google Scholar] [CrossRef] [PubMed]
- Volkamer, A.; Kuhn, D.; Rippmann, F.; Rarey, M. DoGSiteScorer: A Web Server for Automatic Binding Site Prediction, Analysis and Druggability Assessment. Bioinformatics 2012, 28, 2074–2075. [Google Scholar] [CrossRef] [PubMed]
- Anand, P.; Nagarajan, D.; Mukherjee, S.; Chandra, N. PLIC: Protein–Ligand Interaction Clusters. Database 2014, 2014, bau029. [Google Scholar] [CrossRef]
- Van Dongen, S.; Abreu-Goodger, C. Using MCL to Extract Clusters from Networks. Methods Mol. Biol. 2012, 804, 281–295. [Google Scholar]
- Sobolev, V.; Eyal, E.; Gerzon, S.; Potapov, V.; Babor, M.; Prilusky, J.; Edelman, M. SPACE: A Suite of Tools for Protein Structure Prediction and Analysis Based on Complementarity and Environment. Nucleic Acids Res. 2005, 33, W39–W43. [Google Scholar] [CrossRef]
- Morris, G.M.; Goodsell, D.S.; Halliday, R.S.; Huey, R.; Hart, W.E.; Belew, R.K.; Olson, A.J. Automated Docking Using a Lamarckian Genetic Algorithm and and Empirical Binding Free Energy Function. J. Comp. Chem. 1998, 19, 1639–1662. [Google Scholar] [CrossRef]
- Xu, Y.; Wang, S.; Hu, Q.; Gao, S.; Ma, X.; Zhang, W.; Shen, Y.; Chen, F.; Lai, L.; Pei, J. CavityPlus: A Web Server for Protein Cavity Detection with Pharmacophore Modelling, Allosteric Site Identification and Covalent Ligand Bind ing Ability Prediction. Nucleic Acids Res. 2018, 46, W374–W379. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Shen, Y.; Wang, S.; Li, S.; Zhang, W.; Liu, X.; Lai, L.; Pei, J.; Li, H. PharmMapper 2017 Update: A Web Server for Potential Drug Target Identification with a Comprehensive Target Pharmacophore Database. Nucleic Acids Res. 2017, 45, W356–W360. [Google Scholar] [CrossRef] [PubMed]
- Liu, X.; Ouyang, S.; Yu, B.; Liu, Y.; Huang, K.; Gong, J.; Zheng, S.; Li, Z.; Li, H.; Jiang, H. PharmMapper Server: A Web Server for Potential Drug Target Identification Using Pharmacophore Mapping Approach. Nucleic Acids Res. 2010, 38, W609. [Google Scholar] [CrossRef]
- Bender, B.J.; Gahbauer, S.; Luttens, A.; Lyu, J.; Webb, C.M.; Stein, R.M.; Fink, E.A.; Balius, T.E.; Carlsson, J.; Irwin, J.J.; et al. A Practical Guide to Large-Scale Docking. Nat. Protoc. 2021, 16, 4799–4832. [Google Scholar] [CrossRef] [PubMed]
- Zhu, T.; Cao, S.; Su, P.C.; Patel, R.; Shah, D.; Chokshi, H.B.; Szukala, R.; Johnson, M.E.; Hevener, K.E. Hit Identification and Optimization in Virtual Screening: Practical Recommendations Based on a Critical Literature Analysis. J. Med. Chem. 2013, 56, 6560–6572. [Google Scholar] [CrossRef]
- Goodwin, S.; Shahtahmassebi, G.; Hanley, Q.S. Statistical Models for Identifying Frequent Hitters in High Throughput Screening. Sci. Rep. 2020, 10, 17200. [Google Scholar] [CrossRef] [PubMed]
- Irwin, J.J.; Tang, K.G.; Young, J.; Dandarchuluun, C.; Wong, B.R.; Khurelbaatar, M.; Moroz, Y.S.; Mayfield, J.; Sayle, R.A. ZINC20—A Free Ultralarge-Scale Chemical Database for Ligand Discovery. J. Chem. Inf. Model. 2020, 60, 6065–6073. [Google Scholar] [CrossRef]
- Kim, S.; Chen, J.; Cheng, T.; Gindulyte, A.; He, J.; He, S.; Li, Q.; Shoemaker, B.A.; Thiessen, P.A.; Yu, B.; et al. PubChem in 2021: New Data Content and Improved Web Interfaces. Nucleic Acids Res. 2021, 49, D1388–D1395. [Google Scholar] [CrossRef]
- Wishart, D.S.; Feunang, Y.D.; Guo, A.C.; Lo, E.J.; Marcu, A.; Grant, J.R.; Sajed, T.; Johnson, D.; Li, C.; Sayeeda, Z.; et al. DrugBank 5.0: A Major Update to the DrugBank Database for 2018. Nucleic Acids Res. 2018, 46, D1074–D1082. [Google Scholar] [CrossRef]
- Gaulton, A.; Hersey, A.; Nowotka, M.L.; Patricia Bento, A.; Chambers, J.; Mendez, D.; Mutowo, P.; Atkinson, F.; Bellis, L.J.; Cibrian-Uhalte, E.; et al. The ChEMBL Database in 2017. Nucleic Acids Res. 2017, 45, D945–D954. [Google Scholar] [CrossRef]
- Pihan, E.; Colliandre, L.; Guichou, J.F.; Douguet, D. E-Drug3D: 3D Structure Collections Dedicated to Drug Repurposing and Fragment-Based Drug Design. Bioinformatics 2012, 28, 1540–1541. [Google Scholar] [CrossRef] [PubMed]
- Siramshetty, V.B.; Eckert, O.A.; Gohlke, B.O.; Goede, A.; Chen, Q.; Devarakonda, P.; Preissner, S.; Preissner, R. SuperDRUG2: A One Stop Resource for Approved/Marketed Drugs. Nucleic Acids Res. 2018, 46, D1137–D1143. [Google Scholar] [CrossRef]
- Chen, X.; Liu, M.; Gilson, M. BindingDB: A Web-Accessible Molecular Recognition Database. Comb. Chem. High Throughput Screen. 2001, 4, 719–725. [Google Scholar] [CrossRef]
- Wishart, D.S.; Guo, A.C.; Oler, E.; Wang, F.; Anjum, A.; Peters, H.; Dizon, R.; Sayeeda, Z.; Tian, S.; Lee, B.L.; et al. HMDB 5.0: The Human Metabolome Database for 2022. Nucleic Acids Res. 2022, 50, D622–D631. [Google Scholar] [CrossRef]
- Goto, S.; Okuno, Y.; Hattori, M.; Nishioka, T.; Kanehisa, M. LIGAND: Database of Chemical Compounds and Reactions in Biological Pathways. Nucleic Acids Res. 2002, 30, 402–404. [Google Scholar] [CrossRef] [PubMed]
- Grygorenko, O.O.; Radchenko, D.S.; Dziuba, I.; Chuprina, A.; Gubina, K.E.; Moroz, Y.S. Generating Multibillion Chemical Space of Readily Accessible Screening Compounds. iScience 2020, 23, 101681. [Google Scholar] [CrossRef]
- Ruddigkeit, L.; Van Deursen, R.; Blum, L.C.; Reymond, J.L. Enumeration of 166 Billion Organic Small Molecules in the Chemical Universe Database GDB-17. J. Chem. Inf. Model. 2012, 52, 2864–2875. [Google Scholar] [CrossRef] [PubMed]
- Weininger, D. SMILES, a Chemical Language and Information System: 1: Introduction to Methodology and Encoding Rules. J. Chem. Inf. Comput. Sci. 1988, 28, 31–36. [Google Scholar] [CrossRef]
- Santos, L.H.S.; Ferreira, R.S.; Caffarena, E.R. Integrating Molecular Docking and Molecular Dynamics Simulations. Methods Mol. Biol. 2019, 2053, 13–34. [Google Scholar]
- Cerón-Carrasco, J.P.; Mozzarelli, A.; Moro, S.; Salmaso, V. Bridging Molecular Docking to Molecular Dynamics in Exploring Ligand-Protein Recognition Process: An Overview. Front. Pharmacol. 1980, 1, 923. [Google Scholar] [CrossRef]
- Morris, G.M.; Ruth, H.; Lindstrom, W.; Sanner, M.F.; Belew, R.K.; Goodsell, D.S.; Olson, A.J. AutoDock4 and AutoDockTools4: Automated Docking with Selective Receptor Flexibility. J. Comput. Chem. 2009, 30, 2785. [Google Scholar] [CrossRef] [PubMed]
- Huey, R.; Morris, G.M.; Olson, A.J.; Goodsell, D.S. A Semiempirical Free Energy Force Field with Charge-Based Desolvation. J. Comput. Chem. 2007, 28, 1145–1152. [Google Scholar] [CrossRef]
- Eberhardt, J.; Santos-Martins, D.; Tillack, A.F.; Forli, S. AutoDock Vina 1.2.0: New Docking Methods, Expanded Force Field, and Python Bindings. J. Chem. Inf. Model. 2021, 61, 3891–3898. [Google Scholar] [CrossRef] [PubMed]
- Trott, O.; Olson, A.J. AutoDock Vina: Improving the Speed and Accuracy of Docking with a New Scoring Function, Efficient Optimization and Multithreading. J. Comput. Chem. 2010, 31, 455–461. [Google Scholar] [CrossRef]
- Ravindranath, P.A.; Forli, S.; Goodsell, D.S.; Olson, A.J.; Sanner, M.F. AutoDockFR: Advances in Protein-Ligand Docking with Explicitly Specified Binding Site Flexibility. PLoS Comput. Biol. 2015, 11, e1004586. [Google Scholar] [CrossRef] [PubMed]
- McGann, M. FRED Pose Prediction and Virtual Screening Accuracy. J. Chem. Inf. Model. 2011, 51, 578–596. [Google Scholar] [CrossRef]
- Das, S.; Shimshi, M.; Raz, K.; Nitoker Eliaz, N.; Mhashal, A.R.; Ansbacher, T.; Major, D.T. EnzyDock: Protein-Ligand Docking of Multiple Reactive States along a Reaction Coordinate in Enzymes. J. Chem. Theory Comput. 2019, 15, 5116–5134. [Google Scholar] [CrossRef] [PubMed]
- Flexible Protein–Ligand Docking by Global Energy Optimization in Internal Coordinates—Totrov-1997-Proteins: Structure, Function, and Bioinformatics—Wiley Online Library. Available online: https://onlinelibrary.wiley.com/doi/10.1002/(SICI)1097-0134(1997)1+%3C215::AID-PROT29%3E3.0.CO;2-Q (accessed on 31 January 2023).
- Kramer, B.; Rarey, M.; Lengauer, T. Evaluation of the FLEXX Incremental Construction Algorithm for Protein-Ligand Docking. Proteins Struct. Funct. Bioinform. 1999, 37, 228–241. [Google Scholar] [CrossRef]
- Friesner, R.A.; Banks, J.L.; Murphy, R.B.; Halgren, T.A.; Klicic, J.J.; Mainz, D.T.; Repasky, M.P.; Knoll, E.H.; Shelley, M.; Perry, J.K.; et al. Glide: A New Approach for Rapid, Accurate Docking and Scoring. 1. Method and Assessment of Docking Accuracy. J. Med. Chem. 2004, 47, 1739–1749. [Google Scholar] [CrossRef]
- Verdonk, M.L.; Cole, J.C.; Hartshorn, M.J.; Murray, C.W.; Taylor, R.D. Improved Protein–Ligand Docking Using GOLD. Proteins Struct. Funct. Bioinform. 2003, 52, 609–623. [Google Scholar] [CrossRef] [PubMed]
- Huang, S.Y.; Zou, X. Ensemble Docking of Multiple Protein Structures: Considering Protein Structural Variations in Molecular Docking. Proteins Struct. Funct. Bioinform. 2007, 66, 399–421. [Google Scholar] [CrossRef]
- Vilar, S.; Cozza, G.; Moro, S. Medicinal Chemistry and the Molecular Operating Environment (MOE): Application of QSAR and Molecular Docking to Drug Discovery. Curr. Top. Med. Chem. 2008, 8, 1555–1572. [Google Scholar] [CrossRef]
- Murugan, N.A.; Podobas, A.; Gadioli, D.; Vitali, E.; Palermo, G.; Markidis, S. A Review on Parallel Virtual Screening Softwares for High-Performance Computers. Pharmaceuticals 2022, 15, 63. [Google Scholar] [CrossRef]
- Pagadala, N.S.; Syed, K.; Tuszynski, J. Software for Molecular Docking: A Review. Biophys. Rev. 2017, 9, 91. [Google Scholar] [CrossRef]
- Wang, Z.; Sun, H.; Yao, X.; Li, D.; Xu, L.; Li, Y.; Tian, S.; Hou, T. Comprehensive Evaluation of Ten Docking Programs on a Diverse Set of Protein–Ligand Complexes: The Prediction Accuracy of Sampling Power and Scoring Power. Phys. Chem. Chem. Phys. 2016, 18, 12964–12975. [Google Scholar] [CrossRef] [PubMed]
- Cross, J.B.; Thompson, D.C.; Rai, B.K.; Baber, J.C.; Fan, K.Y.; Hu, Y.; Humblet, C. Comparison of Several Molecular Docking Programs: Pose Prediction and Virtual Screening Accuracy. J. Chem. Inf. Model. 2009, 49, 1455–1474. [Google Scholar] [CrossRef]
- Çlnaroǧlu, S.S.; Timuçin, E. Comparative Assessment of Seven Docking Programs on a Nonredundant Metalloprotein Subset of the PDBbind Refined. J. Chem. Inf. Model. 2019, 59, 3846–3859. [Google Scholar] [CrossRef] [PubMed]
- Forli, S.; Huey, R.; Pique, M.E.; Sanner, M.F.; Goodsell, D.S.; Olson, A.J. Computational Protein–Ligand Docking and Virtual Drug Screening with the AutoDock Suite. Nat. Protoc. 2016, 11, 905–919. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.C. Beware of Docking! Trends. Pharm. Sci. 2015, 36, 78–95. [Google Scholar] [CrossRef]
- Wang, E.; Sun, H.; Wang, J.; Wang, Z.; Liu, H.; Zhang, J.Z.H.; Hou, T. End-Point Binding Free Energy Calculation with MM/PBSA and MM/GBSA: Strategies and Applications in Drug Design. Chem. Rev. 2019, 119, 9478–9508. [Google Scholar] [CrossRef]
- Thirumal Kumar, D.; Iyer, S.; Christy, J.P.; Siva, R.; Tayubi, I.A.; George Priya Doss, C.; Zayed, H. A Comparative Computational Approach toward Pharmacological Chaperones (NN-DNJ and Ambroxol) on N370S and L444P Mutations Causing Gaucher’s Disease. Adv. Protein Chem. Struct. Biol. 2019, 114, 315–339. [Google Scholar] [CrossRef] [PubMed]
- Nakagome, I.; Kato, A.; Yamaotsu, N.; Yoshida, T.; Ozawa, S.I.; Adachi, I.; Hirono, S.S. Design of a New α-1-C-Alkyl-DAB Derivative Acting as a Pharmacological Chaperone for β-Glucocerebrosidase Using Ligand Docking and Molecular Dynamics Simulation. Molecules 2018, 23, 2683. [Google Scholar] [CrossRef] [PubMed]
- Yilmazer, B.; Yagci, Z.B.; Bakar, E.; Ozden, B.; Ulgen, K.; Ozkirimli, E. Investigation of Novel Pharmacological Chaperones for Gaucher Disease. J. Mol. Graph. Model. 2017, 76, 364–378. [Google Scholar] [CrossRef]
- Kato, A.; Nakagome, I.; Sato, K.; Yamamoto, A.; Adachi, I.; Nash, R.J.; Fleet, G.W.J.; Natori, Y.; Watanabe, Y.; Imahori, T.; et al. Docking Study and Biological Evaluation of Pyrrolidine-Based Iminosugars as Pharmacological Chaperones for Gaucher Disease. Org. Biomol. Chem. 2016, 14, 1039–1048. [Google Scholar] [CrossRef]
- Kato, A.; Nakagome, I.; Nakagawa, S.; Koike, Y.; Nash, R.J.; Adachi, I.; Hirono, S. Docking and SAR Studies of Calystegines: Binding Orientation and Influence on Pharmacological Chaperone Effects for Gaucher’s Disease. Bioorg. Med. Chem. 2014, 22, 2435–2441. [Google Scholar] [CrossRef]
- Kato, A.; Nakagome, I.; Nakagawa, S.; Kinami, K.; Adachi, I.; Jenkinson, S.F.; Désiré, J.; Blériot, Y.; Nash, R.J.; Fleet, G.W.J.; et al. In Silico Analyses of Essential Interactions of Iminosugars with the Hex A Active Site and Evaluation of Their Pharmacological Chaperone Effects for Tay-Sachs Disease. Org. Biomol. Chem. 2017, 15, 9297–9304. [Google Scholar] [CrossRef] [PubMed]
- Olarte-Avellaneda, S.; Cepeda Del Castillo, J.; Rojas-Rodriguez, A.F.; Sánchez, O.; Rodríguez-López, A.; Suárez García, D.A.; Pulido, L.M.S.; Alméciga-Díaz, C.J. Bromocriptine as a Novel Pharmacological Chaperone for Mucopolysaccharidosis IV A. ACS Med. Chem. Lett. 2020, 11, 1377–1385. [Google Scholar] [CrossRef] [PubMed]
- Ballester, P.J.; Mitchell, J.B.O. A Machine Learning Approach to Predicting Protein-Ligand Binding Affinity with Applications to Molecular Docking. Bioinformatics 2010, 26, 1169. [Google Scholar] [CrossRef] [PubMed]
- Yang, C.; Chen, E.A.; Zhang, Y. Protein–Ligand Docking in the Machine-Learning Era. Molecules 2022, 27, 4568. [Google Scholar] [CrossRef]
- Crampon, K.; Giorkallos, A.; Deldossi, M.; Baud, S.; Steffenel, L.A. Machine-Learning Methods for Ligand–Protein Molecular Docking. Drug Discov. Today 2022, 27, 151–164. [Google Scholar] [CrossRef]
- Torres, P.H.M.; Sodero, A.C.R.; Jofily, P.; Silva-Jr, F.P. Key Topics in Molecular Docking for Drug Design. Int. J. Mol. Sci. 2019, 20, 4574. [Google Scholar] [CrossRef]
- Amendola, G.; Cosconati, S. PyRMD: A New Fully Automated AI-Powered Ligand-Based Virtual Screening Tool. J. Chem. Inf. Model. 2021, 61, 3835–3845. [Google Scholar] [CrossRef]
- Kumar, N.; Acharya, V. Machine Intelligence-Driven Framework for Optimized Hit Selection in Virtual Screening. J. Cheminform. 2022, 14, 48. [Google Scholar] [CrossRef]
- Tripathi, M.K.; Nath, A.; Singh, T.P.; Ethayathulla, A.S.; Kaur, P. Evolving Scenario of Big Data and Artificial Intelligence (AI) in Drug Discovery. Mol. Divers. 2021, 25, 1439–1460. [Google Scholar] [CrossRef] [PubMed]
- Arul Murugan, N.; Ruba Priya, G.; Narahari Sastry, G.; Markidis, S. Artificial Intelligence in Virtual Screening: Models versus Experiments. Drug Discov. Today 2022, 27, 1913–1923. [Google Scholar] [CrossRef]
- Böhm, H.J.; Flohr, A.; Stahl, M. Scaffold Hopping. Drug Discov. Today Technol. 2004, 1, 217–224. [Google Scholar] [CrossRef]
- Bajorath, J. Computational Scaffold Hopping: Cornerstone for the Future of Drug Design? Future Med. Chem. 2017, 9, 629–631. [Google Scholar] [CrossRef] [PubMed]
- Gentile, F.; Agrawal, V.; Hsing, M.; Ton, A.T.; Ban, F.; Norinder, U.; Gleave, M.E.; Cherkasov, A. Deep Docking: A Deep Learning Platform for Augmentation of Structure Based Drug Discovery. ACS. Cent. Sci. 2020, 6, 939–949. [Google Scholar] [CrossRef]
- Yaacoub, J.C.; Gleave, J.; Gentile, F.; Stern, A.; Cherkasov, A. DD-GUI: A Graphical User Interface for Deep Learning-Accelerated Virtual Screening of Large Chemical Libraries (Deep Docking). Bioinformatics 2022, 38, 1146–1148. [Google Scholar] [CrossRef]
- Graff, D.E.; Shakhnovich, E.I.; Coley, C.W. Accelerating High-Throughput Virtual Screening through Molecular Pool-Based Active Learning. Chem. Sci. 2021, 12, 7866–7881. [Google Scholar] [CrossRef] [PubMed]
- Gorgulla, C.; Boeszoermenyi, A.; Wang, Z.F.; Fischer, P.D.; Coote, P.W.; Padmanabha Das, K.M.; Malets, Y.S.; Radchenko, D.S.; Moroz, Y.S.; Scott, D.A.; et al. An Open-Source Drug Discovery Platform Enables Ultra-Large Virtual Screens. Nature 2020, 580, 663–668. [Google Scholar] [CrossRef]
- Gorgulla, C.; Çınaroğlu, S.S.; Fischer, P.D.; Fackeldey, K.; Wagner, G.; Arthanari, H. VirtualFlow Ants—Ultra-Large Virtual Screenings with Artificial Intelligence Driven Docking Algorithm Based on Ant Colony Optimization. Int. J. Mol. Sci. 2021, 22, 5807. [Google Scholar] [CrossRef] [PubMed]
- Sadybekov, A.A.; Sadybekov, A.V.; Liu, Y.; Iliopoulos-Tsoutsouvas, C.; Huang, X.P.; Pickett, J.; Houser, B.; Patel, N.; Tran, N.K.; Tong, F.; et al. Synthon-Based Ligand Discovery in Virtual Libraries of over 11 Billion Compounds. Nature 2022, 601, 452–459. [Google Scholar] [CrossRef]
- Schneider, G.; Clark, D.E. Automated De Novo Drug Design: Are We Nearly There Yet? Angew. Chem. Int. Ed. 2019, 58, 10792–10803. [Google Scholar] [CrossRef] [PubMed]
- Merk, D.; Friedrich, L.; Grisoni, F.; Schneider, G. De Novo Design of Bioactive Small Molecules by Artificial Intelligence. Mol. Inform. 2018, 37, 1700153. [Google Scholar] [CrossRef]
- Lee, Y.J.; Kahng, H.; Kim, S.B. Generative Adversarial Networks for De Novo Molecular Design. Mol. Inform. 2021, 40, 2100045. [Google Scholar] [CrossRef] [PubMed]
- Popova, M.; Isayev, O.; Tropsha, A. Deep Reinforcement Learning for de Novo Drug Design. Sci. Adv. 2018, 4, eaap7885. [Google Scholar] [CrossRef]
- Palazzesi, F.; Pozzan, A. Deep Learning Applied to Ligand-Based De Novo Drug Design. Methods Mol. Biol. 2022, 2390, 273–299. [Google Scholar] [CrossRef]





| Name | URL | Method |
|---|---|---|
| SIFT | https://sift.bii.a-star.edu.sg/ (accessed on 10 January 2023) | Sequence conservation and amino acids properties |
| Panther | http://pantherdb.org/tools/csnpScoreForm.jsp (accessed on 10 January 2023) | Evolutionary preservation |
| SNAP2 | https://rostlab.org/services/snap/ (accessed on 10 January 2023) | Neural networks |
| SNPs&GO | https://snps-and-go.biocomp.unibo.it/snps-and-go/ (accessed on 10 January 2023) | Support vector machines |
| PolyPhen-2 | http://genetics.bwh.harvard.edu/pph2/ (accessed on 10 January 2023) | Naïve Bayes classifier |
| FatHMM | http://fathmm.biocompute.org.uk/ (accessed on 10 January 2023) | Hidden Markov models |
| VarMod | http://www.wasslab.org/varmod/ (accessed on 10 January 2023) | Support vector machines |
| MutPred2 | http://mutpred.mutdb.org/ (accessed on 10 January 2023) | Neural networks |
| SNPdryad | https://maayanlab.cloud/datasets2tools/landing/tool/SNPdryad (accessed on 10 January 2023) | Sequence alignment using protein orthologs |
| ENTPRISE | http://cssb2.biology.gatech.edu/ENTPRISE/ (accessed on 10 January 2023) | Sequence entropy and predicted protein structures |
| MutationAssessor | http://mutationassessor.org/r3/ (accessed on 10 January 2023) | Evolutionary preservation |
| MUpro | https://mupro.proteomics.ics.uci.edu/ (accessed on 10 January 2023) | Support vector machines |
| CUPSAT | http://cupsat.tu-bs.de/ (accessed on 10 January 2023) | Amino acid–atom potentials and torsion angle distribution |
| INPS | https://inpsmd.biocomp.unibo.it/inpsSuite (accessed on 10 January 2023) | Support vector machines |
| SuSPect | http://www.sbg.bio.ic.ac.uk/~suspect/ (accessed on 10 January 2023) | Support vector machines |
| SDM | http://marid.bioc.cam.ac.uk/sdm2/prediction (accessed on 10 January 2023) | Graph-based signatures |
| mCSM-ppi2 | https://biosig.lab.uq.edu.au/mcsm_ppi2/ (accessed on 10 January 2023) | Graph-based signatures |
| DUET | http://biosig.unimelb.edu.au/duet/ (accessed on 10 January 2023) | Support vector machines |
| mCSM-Membrane | https://biosig.lab.uq.edu.au/mcsm_membrane/ (accessed on 10 January 2023) | Graph-based signatures |
| mCSM-AB | https://biosig.lab.uq.edu.au/mcsm_ab/ (accessed on 10 January 2023) | Graph-based signatures |
| DynaMut2 | https://biosig.lab.uq.edu.au/dynamut2/ (accessed on 10 January 2023) | Graph-based signatures and normal mode dynamics |
| Name | URL | Method |
|---|---|---|
| CASTp | http://sts.bioe.uic.edu/castp (accessed on 10 January 2023) | grid-based geometry |
| 3DLigandSite | https://www.wass-michaelislab.org/3dligandsite (accessed on 10 January 2023) | template-based |
| IntFOLD | https://www.reading.ac.uk/bioinf/IntFOLD (accessed on 10 January 2023) | template-based |
| DeepSite | https://playmolecule.com/deepsite (accessed on 10 January 2023) | template-based, neural networks |
| COACH-D | https://yanglab.nankai.edu.cn/COACH-D (accessed on 10 January 2023) | consensus, SVM |
| PrankWeb | http://prankweb.cz (accessed on 10 January 2023) | template-free, random forest |
| Name | URL |
|---|---|
| PockDrug | http://pockdrug.rpbs.univ-paris-diderot.fr/ (accessed on 10 January 2023) |
| Fpocket | https://fpocket.sourceforge.net/ (accessed on 10 January 2023) |
| DoGSiteScorer | https://proteins.plus/ (accessed on 10 January 2023) |
| CavityPlus | http://www.pkumdl.cn:8000/cavityplus/ (accessed on 10 January 2023) |
| PharmMapper | http://www.lilab-ecust.cn/pharmmapper/ (accessed on 10 January 2023) |
| PLIC | http://proline.biochem.iisc.ernet.in/PLIC/ (accessed on 10 January 2023) |
| Name | URL |
|---|---|
| ZINC20 | https://zinc20.docking.org/ (accessed on 10 January 2023) |
| PubChem | https://pubchem.ncbi.nlm.nih.gov/ (accessed on 10 January 2023) |
| DrugBank | https://go.drugbank.com/ (accessed on 10 January 2023) |
| ChEMBL | https://www.ebi.ac.uk/chembl/ (accessed on 10 January 2023) |
| e-Drug3D | https://chemoinfo.ipmc.cnrs.fr/MOLDB/index.php (accessed on 10 January 2023) |
| SuperDRUG2 | http://bioinf.charite.de/superdrug (accessed on 10 January 2023) |
| BindingDB | https://www.bindingdb.org/bind/index.jsp (accessed on 10 January 2023) |
| HMDB | https://hmdb.ca/ (accessed on 10 January 2023) |
| Ligand | https://www.genome.jp/kegg/compound/ (accessed on 10 January 2023) |
| REAL | https://enamine.net/compound-collections/real-compounds (accessed on 10 January 2023) |
| GDB17 | https://gdb.unibe.ch/downloads/ (accessed on 10 January 2023) |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Grasso, D.; Galderisi, S.; Santucci, A.; Bernini, A. Pharmacological Chaperones and Protein Conformational Diseases: Approaches of Computational Structural Biology. Int. J. Mol. Sci. 2023, 24, 5819. https://doi.org/10.3390/ijms24065819
Grasso D, Galderisi S, Santucci A, Bernini A. Pharmacological Chaperones and Protein Conformational Diseases: Approaches of Computational Structural Biology. International Journal of Molecular Sciences. 2023; 24(6):5819. https://doi.org/10.3390/ijms24065819
Chicago/Turabian StyleGrasso, Daniela, Silvia Galderisi, Annalisa Santucci, and Andrea Bernini. 2023. "Pharmacological Chaperones and Protein Conformational Diseases: Approaches of Computational Structural Biology" International Journal of Molecular Sciences 24, no. 6: 5819. https://doi.org/10.3390/ijms24065819
APA StyleGrasso, D., Galderisi, S., Santucci, A., & Bernini, A. (2023). Pharmacological Chaperones and Protein Conformational Diseases: Approaches of Computational Structural Biology. International Journal of Molecular Sciences, 24(6), 5819. https://doi.org/10.3390/ijms24065819