
Modulation of Endothelial Function by TMAO, a Gut Microbiota-Derived Metabolite

Abstract
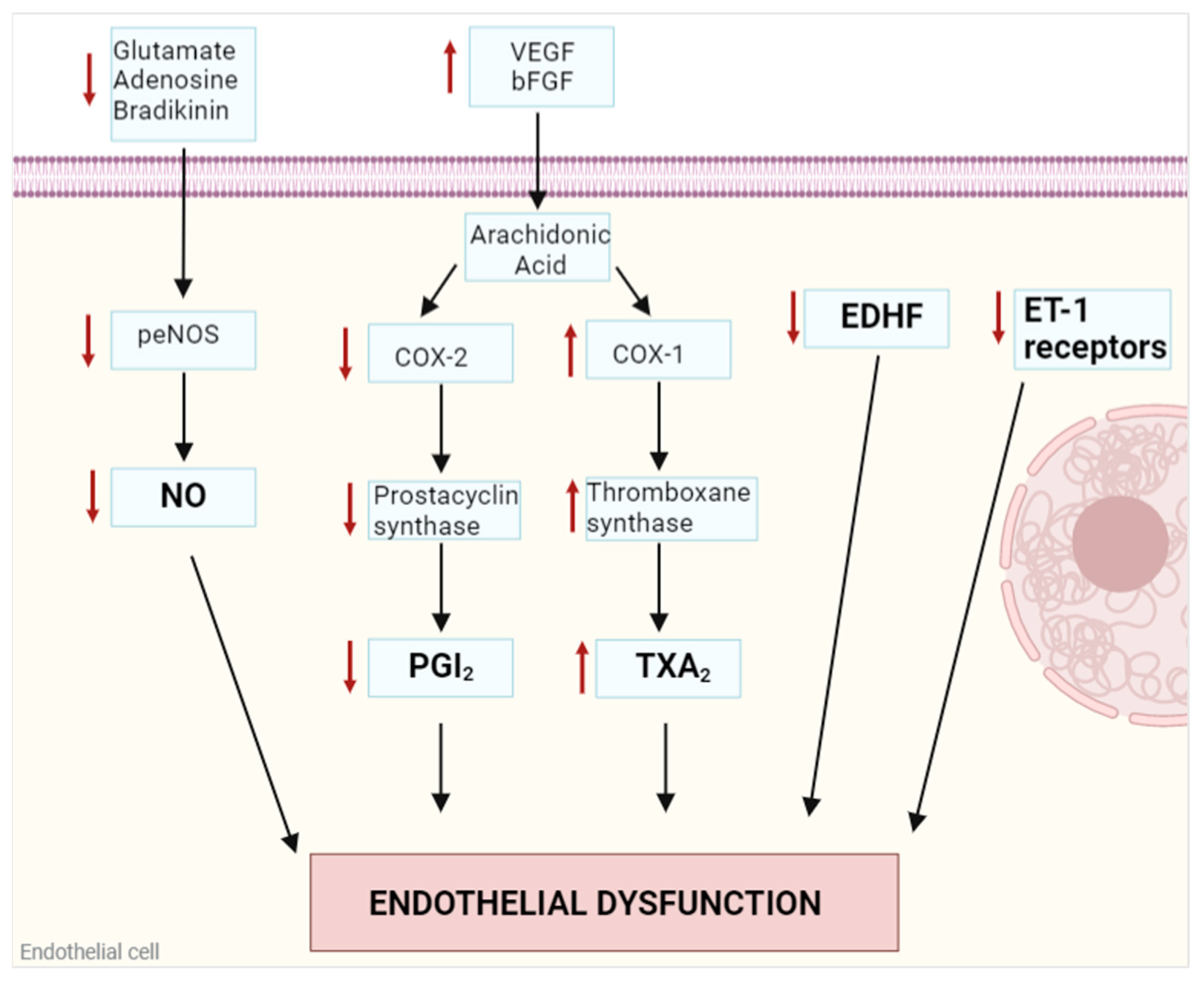
1. Endothelial Dysfunction
2. Trimethylamine N-Oxide (TMAO)
2.1. Sources
2.2. Biological Functions
2.3. TMAO and Atherosclerotic Risk
3. TMAO-Mediated Angiocrine Factors Modulation
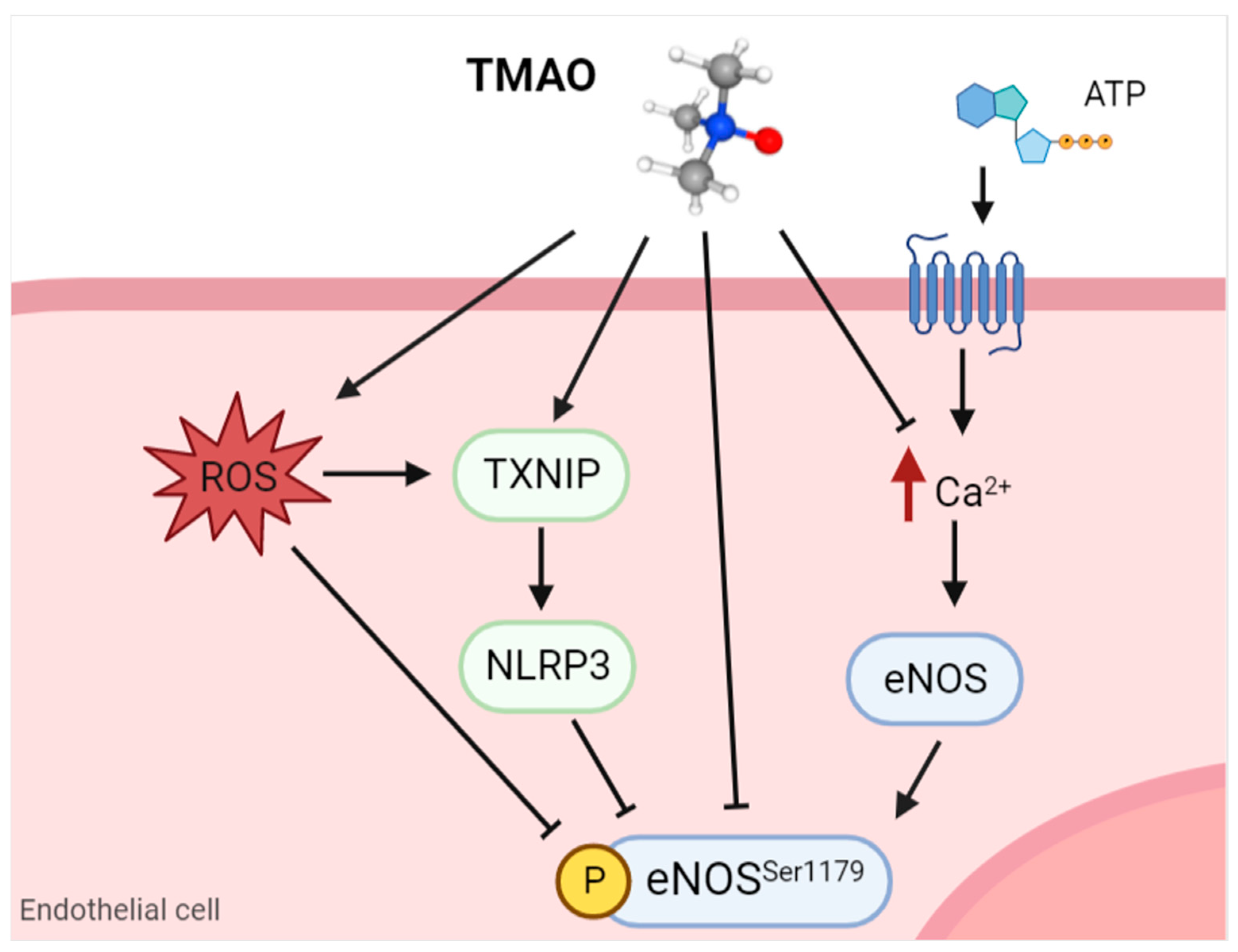
3.1. Nitric Oxide
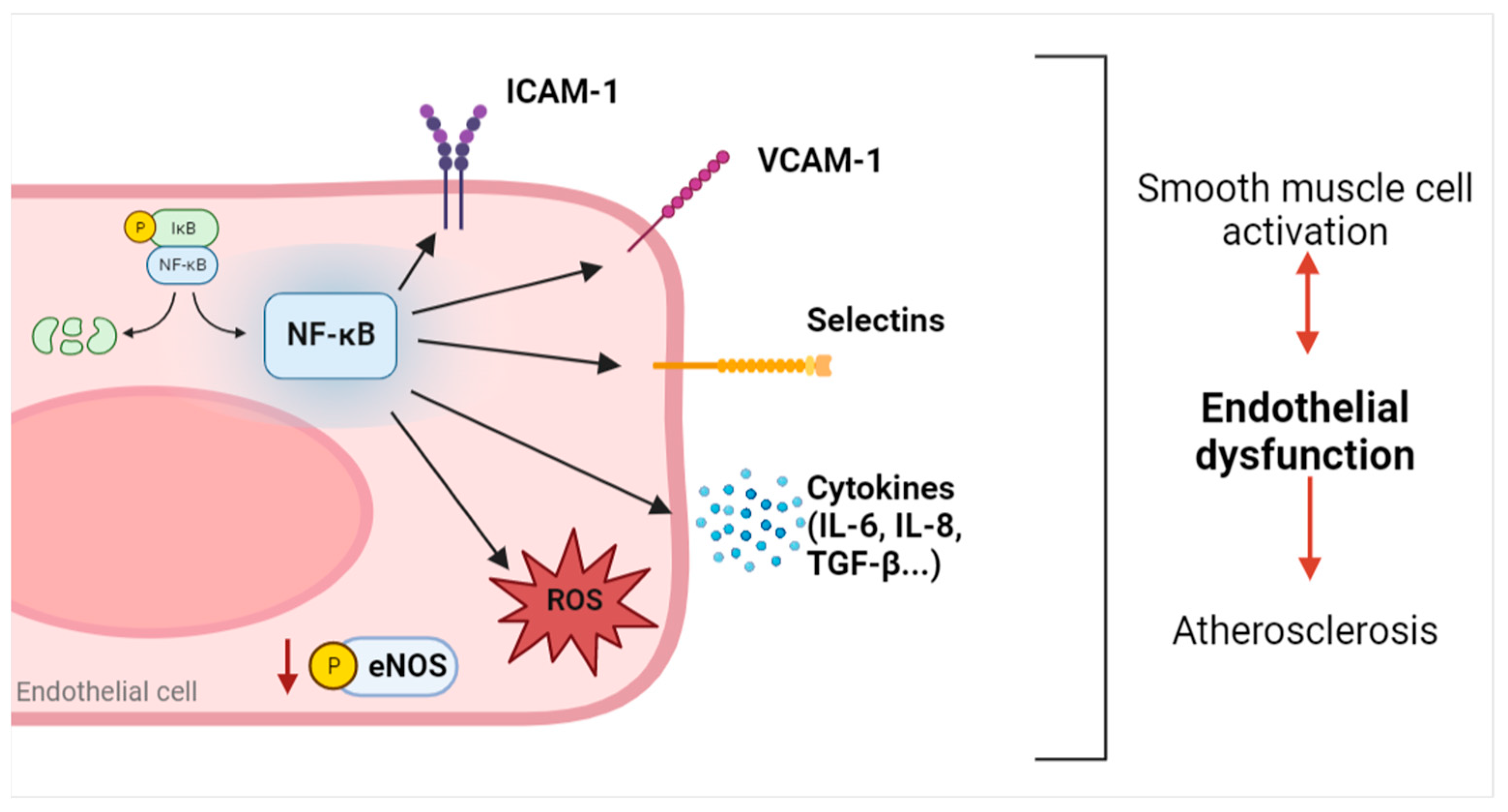
3.2. Adhesion Molecules
3.3. IL-6
4. TMAO Modulation of Angiocrine Factors Involved in Other Pathological Onsets
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Madonna, R. Angiocrine endothelium: From physiology to atherosclerosis and cardiac repair. Vasc. Pharmacol. 2022, 144, 106993. [Google Scholar] [CrossRef] [PubMed]
- Rafii, S.; Butler, J.M.; Ding, B.-S. Angiocrine functions of organ-specific endothelial cells. Nature 2016, 529, 316–325. [Google Scholar] [CrossRef] [PubMed]
- Pasquier, J.; Ghiabi, P.; Chouchane, L.; Razzouk, K.; Rafii, S.; Rafii, A. Angiocrine endothelium: From physiology to cancer. J. Transl. Med. 2020, 18, 52. [Google Scholar] [CrossRef] [PubMed]
- Deanfield, J.E.; Halcox, J.P.; Rabelink, T.J. Endothelial function and dysfunction: Testing and clinical relevance. Circulation 2007, 115, 1285–1295. [Google Scholar] [CrossRef] [PubMed]
- Godo, S.; Shimokawa, H. Endothelial Functions. Arter. Thromb. Vasc. Biol. 2017, 37, e108–e114. [Google Scholar] [CrossRef]
- Dudzinski, D.M.; Michel, T. Life history of eNOS: Partners and pathways. Cardiovasc. Res. 2007, 75, 247–260. [Google Scholar] [CrossRef]
- Kolluru, G.K.; Siamwala, J.H.; Chatterjee, S. eNOS phosphorylation in health and disease. Biochimie 2010, 92, 1186–1198. [Google Scholar] [CrossRef] [PubMed]
- Wilson, C.; Lee, M.D.; McCarron, J.G. Acetylcholine released by endothelial cells facilitates flow-mediated dilatation. J. Physiol. 2016, 594, 7267–7307. [Google Scholar] [CrossRef]
- Su, J.B. Role of Bradykinin in the Regulation of Endothelial Nitric Oxide Synthase Expression by Cardiovascular Drugs. Curr. Pharm. Des. 2018, 23, 6215–6222. [Google Scholar] [CrossRef]
- Fernández, N.; Sánchez, M.A.; Martínez, M.A.; García-Villalón, A.L.; Monge, L.; Gómez, B.; Diéguez, G. Role of nitric oxide in vascular tone and in reactivity to isoproterenol and adenosine in the goat coronary circulation. Eur. J. Pharmacol. 2000, 387, 93–99. [Google Scholar] [CrossRef]
- Negri, S.; Faris, P.; Pellavio, G.; Botta, L.; Orgiu, M.; Forcaia, G.; Sancini, G.; Laforenza, U.; Moccia, F. Group 1 metabotropic glutamate receptors trigger glutamate-induced intracellular Ca2+ signals and nitric oxide release in human brain microvascular endothelial cells. Cell. Mol. Life Sci. 2019, 77, 2235–2253. [Google Scholar] [CrossRef] [PubMed]
- Sandoo, A.; van Zanten, J.J.C.S.V.; Metsios, G.S.; Carroll, D.; Kitas, G.D. The Endothelium and Its Role in Regulating Vascular Tone. Open Cardiovasc. Med. J. 2010, 4, 302–312. [Google Scholar] [CrossRef] [PubMed]
- Mottola, A.; Antoniotti, S.; Lovisolo, D.; Munaron, L. Regulation of noncapacitative calcium entry by arachidonic acid and nitric oxide in endothelial cells. FASEB J. 2005, 19, 2075–2077. [Google Scholar] [CrossRef]
- Oyama, J.-I.; Node, K. Endothelium-derived hyperpolarizing factor and hypertension. Hypertens. Res. 2013, 36, 852–853. [Google Scholar] [CrossRef] [PubMed]
- Garland, C.J.; Dora, K.A. EDH: Endothelium-dependent hyperpolarization and microvascular signalling. Acta Physiol. 2016, 219, 152–161. [Google Scholar] [CrossRef] [PubMed]
- Houde, M.; Desbiens, L.; D’Orléans-Juste, P. Chapter Five—Endothelin-1: Biosynthesis, Signaling and Vasoreactivity. In Advances in Pharmacology; Khalil, R.A., Ed.; Endothelium; Academic Press: Cambridge, MA, USA, 2016; Volume 77, pp. 143–175. [Google Scholar]
- Theofilis, P.; Sagris, M.; Oikonomou, E.; Antonopoulos, A.; Siasos, G.; Tsioufis, C.; Tousoulis, D. Inflammatory Mechanisms Contributing to Endothelial Dysfunction. Biomedicines 2021, 9, 781. [Google Scholar] [CrossRef] [PubMed]
- Zhang, C. The role of inflammatory cytokines in endothelial dysfunction. Basic Res. Cardiol. 2008, 103, 398–406. [Google Scholar] [CrossRef]
- Tabruyn, S.P.; Mémet, S.; Avé, P.; Verhaeghe, C.; Mayo, K.H.; Struman, I.; Martial, J.A.; Griffioen, A.W. NF-κB activation in endothelial cells is critical for the activity of angiostatic agents. Mol. Cancer Ther. 2009, 8, 2645–2654. [Google Scholar] [CrossRef]
- Mussbacher, M.; Salzmann, M.; Brostjan, C.; Hoesel, B.; Schoergenhofer, C.; Datler, H.; Hohensinner, P.; Basílio, J.; Petzelbauer, P.; Assinger, A.; et al. Cell Type-Specific Roles of NF-ΚB Linking Inflammation and Thrombosis. Front. Immunol. 2019, 10, 85. [Google Scholar] [CrossRef]
- de Winther, M.P.; Kanters, E.; Kraal, G.; Hofker, M.H. Nuclear Factor κB Signaling in Atherogenesis. Arter. Thromb. Vasc. Biol. 2005, 25, 904–914. [Google Scholar] [CrossRef]
- Grumbach, I.; Chen, W.; Mertens, S.; Harrison, D. A negative feedback mechanism involving nitric oxide and nuclear factor kappa-B modulates endothelial nitric oxide synthase transcription. J. Mol. Cell. Cardiol. 2005, 39, 595–603. [Google Scholar] [CrossRef] [PubMed]
- Lee, K.-S.; Kim, J.; Kwak, S.-N.; Lee, K.-S.; Lee, D.-K.; Ha, K.-S.; Won, M.-H.; Jeoung, D.; Lee, H.; Kwon, Y.-G.; et al. Functional role of NF-κB in expression of human endothelial nitric oxide synthase. Biochem. Biophys. Res. Commun. 2014, 448, 101–107. [Google Scholar] [CrossRef] [PubMed]
- Mallavia, B.; Recio, C.; Oguiza, A.; Ortiz-Muñoz, G.; Lazaro, I.; Lopez-Parra, V.; Lopez-Franco, O.; Schindler, S.; Depping, R.; Egido, J.; et al. Peptide Inhibitor of NF-κB Translocation Ameliorates Experimental Atherosclerosis. Am. J. Pathol. 2013, 182, 1910–1921. [Google Scholar] [CrossRef]
- Cook-Mills, J.M.; Marchese, M.E.; Abdala-Valencia, H. Vascular Cell Adhesion Molecule-1 Expression and Signaling During Disease: Regulation by Reactive Oxygen Species and Antioxidants. Antioxid. Redox Signal. 2011, 15, 1607–1638. [Google Scholar] [CrossRef]
- Habas, K.; Shang, L. Alterations in intercellular adhesion molecule 1 (ICAM-1) and vascular cell adhesion molecule 1 (VCAM-1) in human endothelial cells. Tissue Cell 2018, 54, 139–143. [Google Scholar] [CrossRef]
- Lawson, C.; Wolf, S. ICAM-1 signaling in endothelial cells. Pharmacol. Rep. 2009, 61, 22–32. [Google Scholar] [CrossRef] [PubMed]
- Hadi, H.A.R.; Carr, C.S.; Al Suwaidi, J. Endothelial Dysfunction: Cardiovascular Risk Factors, Therapy, and Outcome. Vasc. Health Risk Manag. 2005, 1, 183–198. [Google Scholar] [PubMed]
- Ahmadmehrabi, S.; Tang, W.W. Gut microbiome and its role in cardiovascular diseases. Curr. Opin. Cardiol. 2017, 32, 761–766. [Google Scholar] [CrossRef] [PubMed]
- Jonsson, A.L.; Bäckhed, F. Role of gut microbiota in atherosclerosis. Nat. Rev. Cardiol. 2016, 14, 79–87. [Google Scholar] [CrossRef]
- Verhaar, B.; Prodan, A.; Nieuwdorp, M.; Muller, M. Gut Microbiota in Hypertension and Atherosclerosis: A Review. Nutrients 2020, 12, 2982. [Google Scholar] [CrossRef]
- Yubero-Serrano, E.M.; Fernandez-Gandara, C.; Garcia-Rios, A.; Rangel-Zuñiga, O.A.; Gutierrez-Mariscal, F.M.; Torres-Peña, J.D.; Marin, C.; Lopez-Moreno, J.; Castaño, J.P.; Delgado-Lista, J.; et al. Mediterranean diet and endothelial function in patients with coronary heart disease: An analysis of the CORDIOPREV randomized controlled trial. PLOS Med. 2020, 17, e1003282. [Google Scholar] [CrossRef] [PubMed]
- van Bussel, B.C.; Henry, R.M.; Ferreira, I.; van Greevenbroek, M.M.; van der Kallen, C.J.; Twisk, J.W.; Feskens, E.J.; Schalkwijk, C.G.; DA Stehouwer, C. A Healthy Diet Is Associated with Less Endothelial Dysfunction and Less Low-Grade Inflammation over a 7-Year Period in Adults at Risk of Cardiovascular Disease. J. Nutr. 2015, 145, 532–540. [Google Scholar] [CrossRef]
- Schwingshackl, L.; Hoffmann, G. Mediterranean dietary pattern, inflammation and endothelial function: A systematic review and meta-analysis of intervention trials. Nutr. Metab. Cardiovasc. Dis. 2014, 24, 929–939. [Google Scholar] [CrossRef] [PubMed]
- Spence, J.D.; Srichaikul, K.; Kristie; Jenkins, D.J.A. Cardiovascular Harm From Egg Yolk and Meat: More Than Just Cholesterol and Saturated Fat. J. Am. Hear. Assoc. 2021, 10, e017066. [Google Scholar] [CrossRef] [PubMed]
- Murray, K.O.; Mahoney, S.A.; Venkatasubramanian, R.; Seals, D.R.; Clayton, Z.S. Aging, aerobic exercise, and cardiovascular health: Barriers, alternative strategies and future directions. Exp. Gerontol. 2023, 173, 112105. [Google Scholar] [CrossRef] [PubMed]
- Li, C.; Liang, Y.; Qiao, Y. Messengers From the Gut: Gut Microbiota-Derived Metabolites on Host Regulation. Front. Microbiol. 2022, 13, 863407. [Google Scholar] [CrossRef]
- Agus, A.; Clément, K.; Sokol, H. Gut microbiota-derived metabolites as central regulators in metabolic disorders. Gut 2020, 70, 1174–1182. [Google Scholar] [CrossRef] [PubMed]
- Brunt, V.E.; Greenberg, N.T.; Sapinsley, Z.J.; Casso, A.G.; Richey, J.J.; VanDongen, N.S.; Gioscia-Ryan, R.A.; Ziemba, B.P.; Neilson, A.P.; Davy, K.P.; et al. Suppression of trimethylamine N-oxide with DMB mitigates vascular dysfunction, exercise intolerance, and frailty associated with a Western-style diet in mice. J. Appl. Physiol. 2022, 133, 798–813. [Google Scholar] [CrossRef]
- Argyridou, S.; Bernieh, D.; Henson, J.; Edwardson, C.L.; Davies, M.J.; Khunti, K.; Suzuki, T.; Yates, T. Associations between physical activity and trimethylamine N-oxide in those at risk of type 2 diabetes. BMJ Open Diabetes Res. Care 2020, 8, e001359. [Google Scholar] [CrossRef]
- Zhang, H.; Meng, J.; Yu, H. Trimethylamine N-oxide Supplementation Abolishes the Cardioprotective Effects of Voluntary Exercise in Mice Fed a Western Diet. Front. Physiol. 2017, 8, 944. [Google Scholar] [CrossRef]
- Janeiro, M.H.; Ramírez, M.J.; Milagro, F.I.; Martínez, J.A.; Solas, M. Implication of Trimethylamine N-Oxide (TMAO) in Disease: Potential Biomarker or New Therapeutic Target. Nutrients 2018, 10, 1398. [Google Scholar] [CrossRef] [PubMed]
- Simó, C.; García-Cañas, V. Dietary bioactive ingredients to modulate the gut microbiota-derived metabolite TMAO. New opportunities for functional food development. Food Funct. 2020, 11, 6745–6776. [Google Scholar] [CrossRef]
- Zuo, K.; Liu, X.; Wang, P.; Jiao, J.; Han, C.; Liu, Z.; Yin, X.; Li, J.; Yang, X. Metagenomic data-mining reveals enrichment of trimethylamine-N-oxide synthesis in gut microbiome in atrial fibrillation patients. BMC Genom. 2020, 21, 526. [Google Scholar] [CrossRef]
- Wang, Z.; Bergeron, N.; Levison, B.S.; Li, X.S.; Chiu, S.; Jia, X.; Koeth, R.A.; Li, L.; Wu, Y.; Tang, W.H.W.; et al. Impact of chronic dietary red meat, white meat, or non-meat protein on trimethylamine N-oxide metabolism and renal excretion in healthy men and women. Eur. Heart J. 2019, 40, 583–594. [Google Scholar] [CrossRef] [PubMed]
- Sanchez-Gimenez, R.; Ahmed-Khodja, W.; Molina, Y.; Peiró, O.M.; Bonet, G.; Carrasquer, A.; Fragkiadakis, G.A.; Bulló, M.; Bardaji, A.; Papandreou, C. Gut Microbiota-Derived Metabolites and Cardiovascular Disease Risk: A Systematic Review of Prospective Cohort Studies. Nutrients 2022, 14, 2654. [Google Scholar] [CrossRef] [PubMed]
- Catucci, G.; Querio, G.; Sadeghi, S.J.; Gilardi, G.; Levi, R. Enzymatically Produced Trimethylamine N-Oxide: Conserving It or Eliminating It. Catalysts 2019, 9, 1028. [Google Scholar] [CrossRef]
- Saaoud, F.; Liu, L.; Xu, K.; Cueto, R.; Shao, Y.; Lu, Y.; Sun, Y.; Snyder, N.W.; Wu, S.; Yang, L.; et al. Aorta- and liver-generated TMAO enhances trained immunity for increased inflammation via ER stress/mitochondrial ROS/glycolysis pathways. J. Clin. Investig. 2023, 8, e158183. [Google Scholar] [CrossRef]
- Bennett, B.J.; de Aguiar Vallim, T.Q.; Wang, Z.; Shih, D.M.; Meng, Y.; Gregory, J.; Allayee, H.; Lee, R.; Graham, M.; Crooke, R.; et al. Trimethylamine-N-Oxide, a Metabolite Associated with Atherosclerosis, Exhibits Complex Genetic and Dietary Regulation. Cell Metab. 2013, 17, 49–60. [Google Scholar] [CrossRef] [PubMed]
- Teft, W.A.; Morse, B.L.; Leake, B.F.; Wilson, A.; Mansell, S.E.; Hegele, R.A.; Ho, R.H.; Kim, R.B. Identification and Characterization of Trimethylamine-N-oxide Uptake and Efflux Transporters. Mol. Pharm. 2016, 14, 310–318. [Google Scholar] [CrossRef]
- Miyake, T.; Mizuno, T.; Mochizuki, T.; Kimura, M.; Matsuki, S.; Irie, S.; Ieiri, I.; Maeda, K.; Kusuhara, H. Involvement of Organic Cation Transporters in the Kinetics of Trimethylamine N-oxide. J. Pharm. Sci. 2017, 106, 2542–2550. [Google Scholar] [CrossRef]
- Samodelov, S.L.; Kullak-Ublick, G.A.; Gai, Z.; Visentin, M. Organic Cation Transporters in Human Physiology, Pharmacology, and Toxicology. Int. J. Mol. Sci. 2020, 21, 7890. [Google Scholar] [CrossRef] [PubMed]
- Ufnal, M.; Zadlo, A.; Ostaszewski, R. TMAO: A small molecule of great expectations. Nutrition 2015, 31, 1317–1323. [Google Scholar] [CrossRef]
- Ganguly, P.; Polak, J.; Van Der Vegt, N.F.A.; Heyda, J.; Shea, J.-E. Protein Stability in TMAO and Mixed Urea–TMAO Solutions. J. Phys. Chem. B 2020, 124, 6181–6197. [Google Scholar] [CrossRef]
- Hu, C.Y.; Lynch, G.C.; Kokubo, H.; Pettitt, B.M. TrimethylamineN-oxide influence on the backbone of proteins: An oligoglycine model. Proteins: Struct. Funct. Bioinform. 2009, 78, 695–704. [Google Scholar] [CrossRef]
- Cho, S.S.; Reddy, G.; Straub, J.E.; Thirumalai, D. Entropic Stabilization of Proteins by TMAO. J. Phys. Chem. B 2011, 115, 13401–13407. [Google Scholar] [CrossRef] [PubMed]
- Liao, Y.-T.; Manson, A.C.; DeLyser, M.R.; Noid, W.G.; Cremer, P.S. Trimethylamine N -oxide stabilizes proteins via a distinct mechanism compared with betaine and glycine. Proc. Natl. Acad. Sci. USA 2017, 114, 2479–2484. [Google Scholar] [CrossRef]
- Monhemi, H.; Hoang, H.N.; Standley, D.M.; Matsuda, T.; Housaindokht, M.R. The protein-stabilizing effects of TMAO in aqueous and non-aqueous conditions. Phys. Chem. Chem. Phys. 2022, 24, 21178–21187. [Google Scholar] [CrossRef]
- Schmidt, A.C.; Leroux, J.-C. Treatments of trimethylaminuria: Where we are and where we might be heading. Drug Discov. Today 2020, 25, 1710–1717. [Google Scholar] [CrossRef]
- Roddy, D.; McCarthy, P.; Nerney, D.; Mulligan-Rabbitt, J.; Smith, E.; Treacy, E.P. Impact of trimethylaminuria on daily psychosocial functioning. JIMD Rep. 2020, 57, 67–75. [Google Scholar] [CrossRef] [PubMed]
- Jaworska, K.; Hering, D.; Mosieniak, G.; Bielak-Zmijewska, A.; Pilz, M.; Konwerski, M.; Gasecka, A.; Kapłon-Cieślicka, A.; Filipiak, K.; Sikora, E.; et al. TMA, A Forgotten Uremic Toxin, but Not TMAO, Is Involved in Cardiovascular Pathology. Toxins 2019, 11, 490. [Google Scholar] [CrossRef] [PubMed]
- Maksymiuk, K.M.; Szudzik, M.; Gawryś-Kopczyńska, M.; Onyszkiewicz, M.; Samborowska, E.; Mogilnicka, I.; Ufnal, M. Trimethylamine, a gut bacteria metabolite and air pollutant, increases blood pressure and markers of kidney damage including proteinuria and KIM-1 in rats. J. Transl. Med. 2022, 20, 470. [Google Scholar] [CrossRef]
- Naghipour, S.; Cox, A.J.; Peart, J.N.; Du Toit, E.F.; Headrick, J.P. Trimethylamine N-oxide: Heart of the microbiota–CVD nexus? Nutr. Res. Rev. 2020, 34, 125–146. [Google Scholar] [CrossRef]
- Wang, B.; Qiu, J.; Lian, J.; Yang, X.; Zhou, J. Gut Metabolite Trimethylamine-N-Oxide in Atherosclerosis: From Mechanism to Therapy. Front. Cardiovasc. Med. 2021, 8, 723886. [Google Scholar] [CrossRef]
- Thomas, M.S.; Fernandez, M.L. Trimethylamine N-Oxide (TMAO), Diet and Cardiovascular Disease. Curr. Atheroscler. Rep. 2021, 23, 470. [Google Scholar] [CrossRef] [PubMed]
- Wang, Z.; Klipfell, E.; Bennett, B.J.; Koeth, R.; Levison, B.S.; DuGar, B.; Feldstein, A.E.; Britt, E.B.; Fu, X.; Chung, Y.-M.; et al. Gut Flora Metabolism of Phosphatidylcholine Promotes Cardiovascular Disease. Nature 2011, 472, 57–63. [Google Scholar] [CrossRef] [PubMed]
- Kong, W.; Ma, J.; Lin, Y.; Chen, W. Positive Association of Plasma Trimethylamine-N-Oxide and Atherosclerosis in Patient with Acute Coronary Syndrome. Cardiovasc. Ther. 2022, 2022, 2484018. [Google Scholar] [CrossRef]
- Haghikia, A.; Li, X.S.; Liman, T.G.; Bledau, N.; Schmidt, D.; Zimmermann, F.; Kränkel, N.; Widera, C.; Sonnenschein, K.; Haghikia, A.; et al. Gut Microbiota–Dependent Trimethylamine N -Oxide Predicts Risk of Cardiovascular Events in Patients With Stroke and Is Related to Proinflammatory Monocytes. Arter. Thromb. Vasc. Biol. 2018, 38, 2225–2235. [Google Scholar] [CrossRef]
- Brunt, V.E.; Gioscia-Ryan, R.A.; Casso, A.G.; VanDongen, N.S.; Ziemba, B.P.; Sapinsley, Z.J.; Richey, J.J.; Zigler, M.C.; Neilson, A.P.; Davy, K.P.; et al. Trimethylamine-N-Oxide Promotes Age-Related Vascular Oxidative Stress and Endothelial Dysfunction in Mice and Healthy Humans. Hypertension 2020, 76, 101–112. [Google Scholar] [CrossRef] [PubMed]
- Koay, Y.C.; Chen, Y.-C.; A Wali, J.; Luk, A.W.S.; Li, M.; Doma, H.; Reimark, R.; Zaldivia, M.T.K.; Habtom, H.T.; Franks, A.; et al. Plasma levels of trimethylamine-N-oxide can be increased with ‘healthy’ and ‘unhealthy’ diets and do not correlate with the extent of atherosclerosis but with plaque instability. Cardiovasc. Res. 2020, 117, 435–449. [Google Scholar] [CrossRef]
- Jomard, A.; Liberale, L.; Doytcheva, P.; Reiner, M.F.; Müller, D.; Visentin, M.; Bueter, M.; Lüscher, T.F.; Vettor, R.; Lutz, T.A.; et al. Effects of acute administration of trimethylamine N-oxide on endothelial function: A translational study. Sci. Rep. 2022, 12, 8664. [Google Scholar] [CrossRef]
- Papandreou, C.; Moré, M.; Bellamine, A. Trimethylamine N-Oxide in Relation to Cardiometabolic Health—Cause or Effect? Nutrients 2020, 12, 1330. [Google Scholar] [CrossRef] [PubMed]
- Querio, G.; Antoniotti, S.; Geddo, F.; Levi, R.; Gallo, M.P. Trimethylamine N-Oxide (TMAO) Impairs Purinergic Induced Intracellular Calcium Increase and Nitric Oxide Release in Endothelial Cells. Int. J. Mol. Sci. 2022, 23, 3982. [Google Scholar] [CrossRef]
- Sun, X.; Jiao, X.; Ma, Y.; Liu, Y.; Zhang, L.; He, Y.; Chen, Y. Trimethylamine N-oxide induces inflammation and endothelial dysfunction in human umbilical vein endothelial cells via activating ROS-TXNIP-NLRP3 inflammasome. Biochem. Biophys. Res. Commun. 2016, 481, 63–70. [Google Scholar] [CrossRef]
- Seldin, M.M.; Meng, Y.; Qi, H.; Zhu, W.; Wang, Z.; Hazen, S.L.; Lusis, A.J.; Shih, D.M. Trimethylamine N-Oxide Promotes Vascular Inflammation Through Signaling of Mitogen-Activated Protein Kinase and Nuclear Factor-κB. J. Am. Hear. Assoc. 2016, 5, e002767. [Google Scholar] [CrossRef]
- Ma, G.; Pan, B.; Chen, Y.; Guo, C.; Zhao, M.; Zheng, L.; Chen, B. Trimethylamine N-oxide in atherogenesis: Impairing endothelial self-repair capacity and enhancing monocyte adhesion. Biosci. Rep. 2017, 37, BSR20160244. [Google Scholar] [CrossRef] [PubMed]
- Geng, J.; Yang, C.; Wang, B.; Zhang, X.; Hu, T.; Gu, Y.; Li, J. Trimethylamine N-oxide promotes atherosclerosis via CD36-dependent MAPK/JNK pathway. Biomed. Pharmacother. 2018, 97, 941–947. [Google Scholar] [CrossRef] [PubMed]
- Zhou, S.; Xue, J.; Shan, J.; Hong, Y.; Zhu, W.; Nie, Z.; Zhang, Y.; Ji, N.; Luo, X.; Zhang, T.; et al. Gut-Flora-Dependent Metabolite Trimethylamine-N-Oxide Promotes Atherosclerosis-Associated Inflammation Responses by Indirect ROS Stimulation and Signaling Involving AMPK and SIRT. Nutrients 2022, 14, 3338. [Google Scholar] [CrossRef] [PubMed]
- De Caterina, R.; Libby, P.; Peng, H.B.; Thannickal, V.J.; Rajavashisth, T.B.; Gimbrone, M.A., Jr.; Shin, W.S.; Liao, J.K. Nitric oxide decreases cytokine-induced endothelial activation. Nitric oxide selectively reduces endothelial expression of adhesion molecules and proinflammatory cytokines. J. Clin. Investig. 1995, 96, 60–68. [Google Scholar] [CrossRef] [PubMed]
- Costabile, G.; Vetrani, C.; Bozzetto, L.; Giacco, R.; Bresciani, L.; Del Rio, D.; Vitale, M.; Della Pepa, G.; Brighenti, F.; Riccardi, G.; et al. Plasma TMAO increase after healthy diets: Results from 2 randomized controlled trials with dietary fish, polyphenols, and whole-grain cereals. Am. J. Clin. Nutr. 2021, 114, 1342–1350. [Google Scholar] [CrossRef]
- Nakashima, Y.; Raines, E.W.; Plump, A.S.; Breslow, J.L.; Ross, R. Upregulation of VCAM-1 and ICAM-1 at Atherosclerosis-Prone Sites on the Endothelium in the ApoE-Deficient Mouse. Arter. Thromb. Vasc. Biol. 1998, 18, 842–851. [Google Scholar] [CrossRef]
- Cybulsky, M.I.; Iiyama, K.; Li, H.; Zhu, S.; Chen, M.; Iiyama, M.; Davis, V.; Gutierrez-Ramos, J.-C.; Connelly, P.; Milstone, D.S. A major role for VCAM-1, but not ICAM-1, in early atherosclerosis. J. Clin. Investig. 2001, 107, 1255–1262. [Google Scholar] [CrossRef] [PubMed]
- de Freitas, I.A.; Lima, N.D.A.; Jr, G.B.D.S.; Jr, R.L.D.C.; Patel, P.; Lima, C.C.D.V.; Lino, D.O.D.C. Novel biomarkers in the prognosis of patients with atherosclerotic coronary artery disease. Rev. Port. de Cardiol. 2020, 39, 667–672. [Google Scholar] [CrossRef]
- Schieffer, B.; Selle, T.; Hilfiker, A.; Hilfiker-Kleiner, D.; Grote, K.; Tietge, U.J.F.; Trautwein, C.; Luchtefeld, M.; Schmittkamp, C.; Heeneman, S.; et al. Impact of Interleukin-6 on Plaque Development and Morphology in Experimental Atherosclerosis. Circulation 2004, 110, 3493–3500. [Google Scholar] [CrossRef]
- Ridker, P.M.; Rane, M. Interleukin-6 Signaling and Anti-Interleukin-6 Therapeutics in Cardiovascular Disease. Circ. Res. 2021, 128, 1728–1746. [Google Scholar] [CrossRef] [PubMed]
- Villar-Fincheira, P.; Sanhueza-Olivares, F.; Norambuena-Soto, I.; Cancino-Arenas, N.; Hernandez-Vargas, F.; Troncoso, R.; Gabrielli, L.; Chiong, M. Role of Interleukin-6 in Vascular Health and Disease. Front. Mol. Biosci. 2021, 8, 641734. [Google Scholar] [CrossRef] [PubMed]
- Camaré, C.; Pucelle, M.; Nègre-Salvayre, A.; Salvayre, R. Angiogenesis in the atherosclerotic plaque. Redox Biol. 2017, 12, 18–34. [Google Scholar] [CrossRef] [PubMed]
- Ozorowski, M.; Wiciński, M.; Wróbel, Ł.; Fajkiel-Madajczyk, A. Cholecalciferol supplementation lowers leptin and TMAO but increases NO and VEGF-A levels in obese vitamin D deficient patients: Is it one of the potential cardioprotective mechanisms of vitamin D? Nutr. Metab. 2022, 19, 31. [Google Scholar] [CrossRef] [PubMed]
- Yang, S.; Dai, H.; Lu, Y.; Li, R.; Gao, C.; Pan, S. Trimethylamine N-Oxide Promotes Cell Proliferation and Angiogenesis in Colorectal Cancer. J. Immunol. Res. 2022, 2022, 7043856. [Google Scholar] [CrossRef]
- Yang, W.; Zhang, S.; Zhu, J.; Jiang, H.; Jia, D.; Ou, T.; Qi, Z.; Zou, Y.; Qian, J.; Sun, A.; et al. Gut microbe-derived metabolite trimethylamine N-oxide accelerates fibroblast-myofibroblast differentiation and induces cardiac fibrosis. J. Mol. Cell. Cardiol. 2019, 134, 119–130. [Google Scholar] [CrossRef]
- El-Deeb, O.S.; Atef, M.M.; Hafez, Y.M. The interplay between microbiota-dependent metabolite trimethylamine N -oxide, Transforming growth factor β /SMAD signaling and inflammasome activation in chronic kidney disease patients: A new mechanistic perspective. J. Cell. Biochem. 2019, 120, 14476–14485. [Google Scholar] [CrossRef]
- Li, X.; Sun, Y.; Zhang, X.; Wang, J. Reductions in gut microbiota-derived metabolite trimethylamine N-oxide in the circulation may ameliorate myocardial infarction-induced heart failure in rats, possibly by inhibiting interleukin-8 secretion. Mol. Med. Rep. 2019, 20, 779–786. [Google Scholar] [CrossRef] [PubMed]
- Baginski, A.M.; Farmer, N.; Baumer, Y.; Wallen, G.R.; Powell-Wiley, T.M. Interleukin-8 (IL-8) as a Potential Mediator of an Association between Trimethylamine N-Oxide (TMAO) and Proprotein Convertase Subtilisin/Kexin Type 9 (PCSK9) among African Americans at Risk of Cardiovascular Disease. Metabolites 2022, 12, 1196. [Google Scholar] [CrossRef]
- MacPherson, M.E.; Hov, J.R.; Ueland, T.; Dahl, T.B.; Kummen, M.; Otterdal, K.; Holm, K.; Berge, R.K.; Mollnes, T.E.; Trøseid, M.; et al. Gut Microbiota-Dependent Trimethylamine N-Oxide Associates With Inflammation in Common Variable Immunodeficiency. Front. Immunol. 2020, 11, 574500. [Google Scholar] [CrossRef] [PubMed]
- Zhang, X.; Li, Y.; Yang, P.; Liu, X.; Lu, L.; Chen, Y.; Zhong, X.; Li, Z.; Liu, H.; Ou, C.; et al. Trimethylamine-N-Oxide Promotes Vascular Calcification Through Activation of NLRP3 (Nucleotide-Binding Domain, Leucine-Rich-Containing Family, Pyrin Domain-Containing-3) Inflammasome and NF-κB (Nuclear Factor κB) Signals. Arter. Thromb. Vasc. Biol. 2020, 40, 751–765. [Google Scholar] [CrossRef] [PubMed]




{kind=link}
{kind=link}
{kind=link}
| Angiocrine Factor | Modulation Effect by TMAO | References |
|---|---|---|
| NO | Reduction in eNOS phosphorylation and NO release in HUVECs and BAE-1 cells | [73,74] |
| Reduction in eNOS phosphorylation and NO release in the carotid artery of C57BL/6N mice | [69] | |
| No variation in eNOS phosphorylation and NO release in HAECs and rat aortas | [71] | |
| Adhesion molecules | Enhancement of ICAM-1 and E-selectin expression in HAECs | [48,75] |
| Induction of VCAM-1 expression in HUVECs and mice aortic endothelial cells | [76] | |
| IL-6 | Enhanced expression at the mRNA level in aortas from C57BL/6J mice and cultured HEACs and VSMCs | [75] |
| Increased expression at the mRNA level in the aortic arch of C57BL/6J mice | [77] | |
| Increased expression at mRNA and protein level in HUVECs and VSMCs | [78] |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Querio, G.; Antoniotti, S.; Geddo, F.; Levi, R.; Gallo, M.P. Modulation of Endothelial Function by TMAO, a Gut Microbiota-Derived Metabolite. Int. J. Mol. Sci. 2023, 24, 5806. https://doi.org/10.3390/ijms24065806
Querio G, Antoniotti S, Geddo F, Levi R, Gallo MP. Modulation of Endothelial Function by TMAO, a Gut Microbiota-Derived Metabolite. International Journal of Molecular Sciences. 2023; 24(6):5806. https://doi.org/10.3390/ijms24065806
Chicago/Turabian StyleQuerio, Giulia, Susanna Antoniotti, Federica Geddo, Renzo Levi, and Maria Pia Gallo. 2023. "Modulation of Endothelial Function by TMAO, a Gut Microbiota-Derived Metabolite" International Journal of Molecular Sciences 24, no. 6: 5806. https://doi.org/10.3390/ijms24065806
APA StyleQuerio, G., Antoniotti, S., Geddo, F., Levi, R., & Gallo, M. P. (2023). Modulation of Endothelial Function by TMAO, a Gut Microbiota-Derived Metabolite. International Journal of Molecular Sciences, 24(6), 5806. https://doi.org/10.3390/ijms24065806