
Mitochondrial Base Editing: Recent Advances towards Therapeutic Opportunities

 , , ,
, , , 
Abstract
1. Introduction
2. Mitochondrial Base Editing
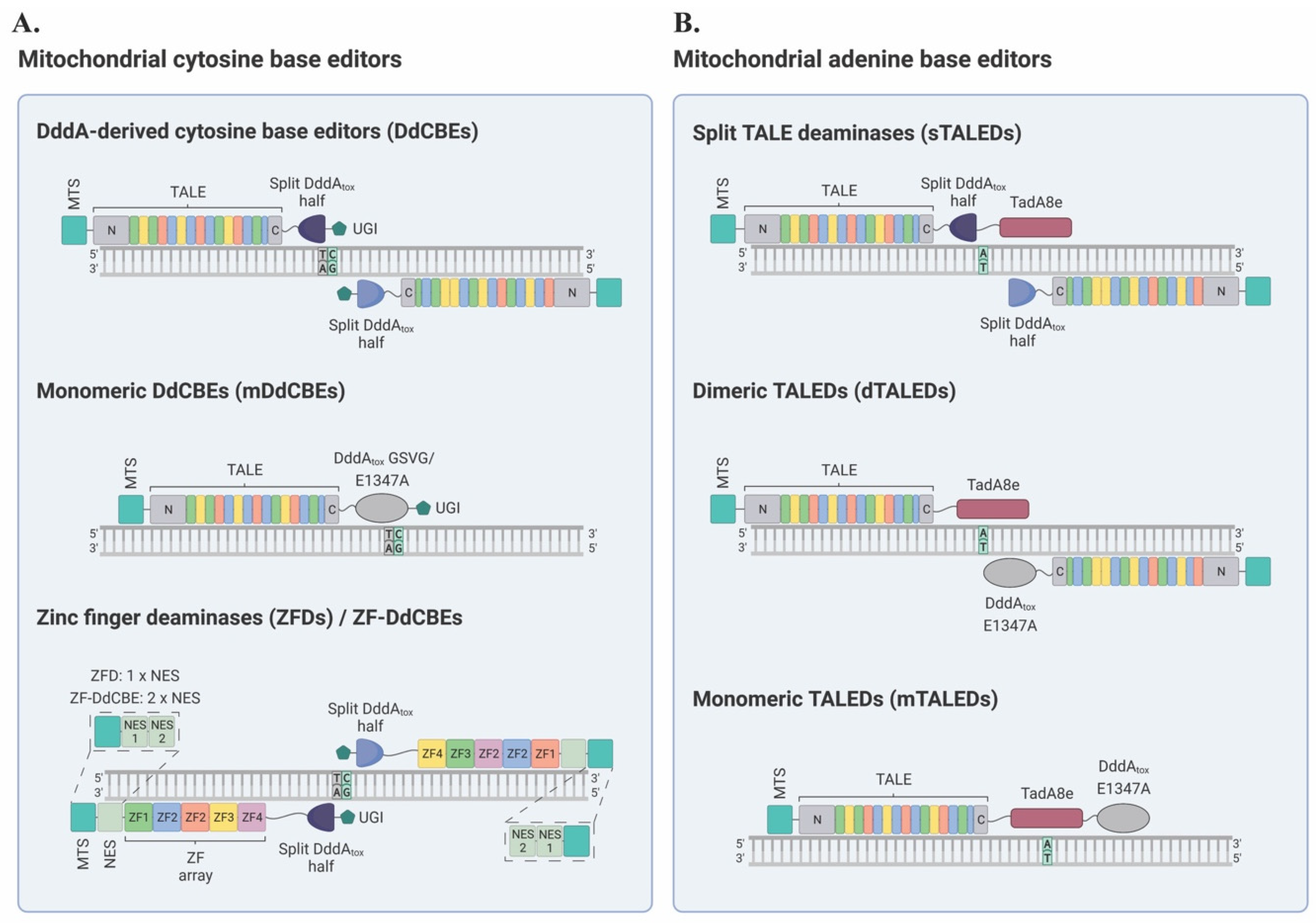
2.1. Mitochondrial Cytosine Base Editors
2.1.1. DddA-Derived Cytosine Base Editors (DdCBEs)
2.1.2. Zinc Finger Deaminases (ZFDs)
2.1.3. DddA6- and DddA11-Containing DdCBEs
2.1.4. Monomeric DdCBEs (mDdCBEs)
2.1.5. High-Fidelity DdCBEs (HiFi-DdCBEs)
2.1.6. Zinc Finger DdCBEs (ZF-DdCBEs)
2.1.7. Additional Strategies to Limit DdCBE-Induced Off-Target Mutagenesis
2.2. Mitochondrial Adenine Base Editors
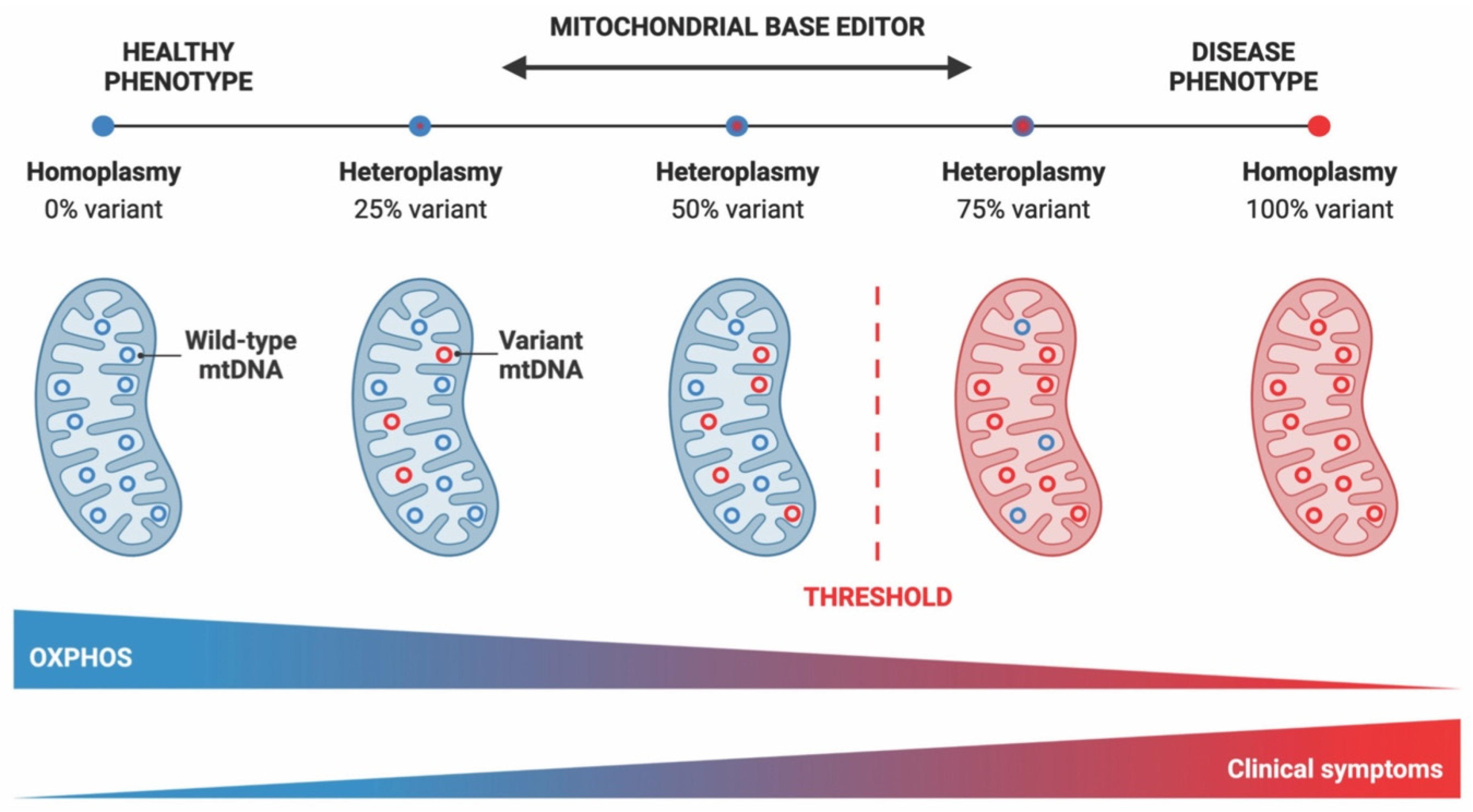
3. Mitochondrial DNA Base Editing for Therapy and Modeling of Mitochondrial Diseases
4. Animal Models for Mitochondrial Dysfunction Using Mitochondrial DNA Base Editors
5. Conclusions and Future Perspectives
Author Contributions
Funding
Conflicts of Interest
References
- Anderson, S.; Bankier, A.T.; Barrell, B.G.; de Bruijn, M.H.; Coulson, A.R.; Drouin, J.; Eperon, I.C.; Nierlich, D.P.; Roe, B.A.; Sanger, F.; et al. Sequence and organization of the human mitochondrial genome. Nature 1981, 290, 457–465. [Google Scholar] [CrossRef]
- Gorman, G.S.; Chinnery, P.F.; DiMauro, S.; Hirano, M.; Koga, Y.; McFarland, R.; Suomalainen, A.; Thorburn, D.R.; Zeviani, M.; Turnbull, D.M. Mitochondrial diseases. Nat. Rev. Dis. Prim. 2016, 2, 16080. [Google Scholar] [CrossRef] [PubMed]
- Pagliarini, D.J.; Calvo, S.E.; Chang, B.; Sheth, S.A.; Vafai, S.B.; Ong, S.-E.; Walford, G.A.; Sugiana, C.; Boneh, A.; Chen, W.K. A mitochondrial protein compendium elucidates complex I disease biology. Cell 2008, 134, 112–123. [Google Scholar] [CrossRef] [PubMed]
- Ott, M.; Amunts, A.; Brown, A. Organization and regulation of mitochondrial protein synthesis. Annu. Rev. Biochem. 2016, 85, 77–101. [Google Scholar] [CrossRef] [PubMed]
- Sabharwal, A.; Kar, B. Post-transcriptional regulation of genes and mitochondrial disorder. In Post-Transcriptional Gene Regulation in Human Disease; Elsevier: Amsterdam, The Netherlands, 2022; pp. 343–364. [Google Scholar]
- Rath, S.; Sharma, R.; Gupta, R.; Ast, T.; Chan, C.; Durham, T.J.; Goodman, R.P.; Grabarek, Z.; Haas, M.E.; Hung, W.H. MitoCarta3. 0: An updated mitochondrial proteome now with sub-organelle localization and pathway annotations. Nucleic Acids Res. 2021, 49, D1541–D1547. [Google Scholar] [CrossRef]
- Ryzhkova, A.I.; Sazonova, M.A.; Sinyov, V.V.; Galitsyna, E.V.; Chicheva, M.M.; Melnichenko, A.A.; Grechko, A.V.; Postnov, A.Y.; Orekhov, A.N.; Shkurat, T.P. Mitochondrial diseases caused by mtDNA mutations: A mini-review. Ther. Clin. Risk Manag. 2018, 14, 1933. [Google Scholar] [CrossRef]
- Rossmann, M.P.; Dubois, S.M.; Agarwal, S.; Zon, L.I. Mitochondrial function in development and disease. Dis. Model. Mech. 2021, 14, dmm048912. [Google Scholar] [CrossRef]
- Stewart, J.B. Current progress with mammalian models of mitochondrial DNA disease. J. Inherit. Metab. Dis. 2021, 44, 325–342. [Google Scholar] [CrossRef]
- Wallace, D.C. Mitochondrial DNA mutations in disease and aging. Environ. Mol. Mutagen. 2010, 51, 440–450. [Google Scholar] [CrossRef]
- Chinnery, P.F. Mitochondrial disorders overview. In GeneReviews®[Internet]; University of Washington, Seattle: Seattle, WA, USA, 2014. [Google Scholar]
- Yonova-Doing, E.; Calabrese, C.; Gomez-Duran, A.; Schon, K.; Wei, W.; Karthikeyan, S.; Chinnery, P.F.; Howson, J.M. An atlas of mitochondrial DNA genotype–phenotype associations in the UK Biobank. Nat. Genet. 2021, 53, 982–993. [Google Scholar] [CrossRef]
- Lott, M.T.; Leipzig, J.N.; Derbeneva, O.; Xie, H.M.; Chalkia, D.; Sarmady, M.; Procaccio, V.; Wallace, D.C. mtDNA variation and analysis using mitomap and mitomaster. Curr. Protoc. Bioinform. 2013, 44, 1.23.21–1.23.26. [Google Scholar] [CrossRef]
- Mi, L.; Shi, M.; Li, Y.-X.; Xie, G.; Rao, X.; Wu, D.; Cheng, A.; Niu, M.; Xu, F.; Yu, Y. DddA homolog search and engineering expand sequence compatibility of mitochondrial base editing. Nat. Commun. 2023, 14, 874. [Google Scholar] [CrossRef]
- Reeve, A.K.; Krishnan, K.J.; Turnbull, D. Mitochondrial DNA mutations in disease, aging, and neurodegeneration. Ann. N. Y. Acad. Sci. 2008, 1147, 21–29. [Google Scholar] [CrossRef]
- Gorelick, A.N.; Kim, M.; Chatila, W.K.; La, K.; Hakimi, A.A.; Berger, M.F.; Taylor, B.S.; Gammage, P.A.; Reznik, E. Respiratory complex and tissue lineage drive recurrent mutations in tumour mtDNA. Nat. Metab. 2021, 3, 558–570. [Google Scholar] [CrossRef]
- Cox, D.B.T.; Platt, R.J.; Zhang, F. Therapeutic genome editing: Prospects and challenges. Nat. Med. 2015, 21, 121–131. [Google Scholar] [CrossRef] [PubMed]
- Doudna, J.A. The promise and challenge of therapeutic genome editing. Nature 2020, 578, 229–236. [Google Scholar] [CrossRef]
- Kyriakouli, D.; Boesch, P.; Taylor, R.; Lightowlers, R. Progress and prospects: Gene therapy for mitochondrial DNA disease. Gene Ther. 2008, 15, 1017–1023. [Google Scholar] [CrossRef]
- Bacman, S.R.; Williams, S.L.; Pinto, M.; Peralta, S.; Moraes, C.T. Specific elimination of mutant mitochondrial genomes in patient-derived cells by mitoTALENs. Nat. Med. 2013, 19, 1111–1113. [Google Scholar] [CrossRef]
- Yahata, N.; Matsumoto, Y.; Omi, M.; Yamamoto, N.; Hata, R. TALEN-mediated shift of mitochondrial DNA heteroplasmy in MELAS-iPSCs with m. 13513G> A mutation. Sci. Rep. 2017, 7, 15557. [Google Scholar] [CrossRef]
- Srivastava, S.; Moraes, C.T. Manipulating mitochondrial DNA heteroplasmy by a mitochondrially targeted restriction endonuclease. Hum. Mol. Genet. 2001, 10, 3093–3099. [Google Scholar] [CrossRef]
- Bacman, S.R.; Kauppila, J.H.; Pereira, C.V.; Nissanka, N.; Miranda, M.; Pinto, M.; Williams, S.L.; Larsson, N.-G.; Stewart, J.B.; Moraes, C.T. MitoTALEN reduces mutant mtDNA load and restores tRNAAla levels in a mouse model of heteroplasmic mtDNA mutation. Nat. Med. 2018, 24, 1696–1700. [Google Scholar] [CrossRef] [PubMed]
- Bayona-Bafaluy, M.P.; Blits, B.; Battersby, B.J.; Shoubridge, E.A.; Moraes, C.T. Rapid directional shift of mitochondrial DNA heteroplasmy in animal tissues by a mitochondrially targeted restriction endonuclease. Proc. Natl. Acad. Sci. USA 2005, 102, 14392–14397. [Google Scholar] [CrossRef]
- Reddy, P.; Ocampo, A.; Suzuki, K.; Luo, J.; Bacman, S.R.; Williams, S.L.; Sugawara, A.; Okamura, D.; Tsunekawa, Y.; Wu, J. Selective elimination of mitochondrial mutations in the germline by genome editing. Cell 2015, 161, 459–469. [Google Scholar] [CrossRef]
- Gammage, P.A.; Viscomi, C.; Simard, M.-L.; Costa, A.S.; Gaude, E.; Powell, C.A.; Van Haute, L.; McCann, B.J.; Rebelo-Guiomar, P.; Cerutti, R. Genome editing in mitochondria corrects a pathogenic mtDNA mutation in vivo. Nat. Med. 2018, 24, 1691–1695. [Google Scholar] [CrossRef]
- Gammage, P.A.; Moraes, C.T.; Minczuk, M. Mitochondrial genome engineering: The revolution may not be CRISPR-Ized. Trends Genet. 2018, 34, 101–110. [Google Scholar] [CrossRef]
- Barrera-Paez, J.D.; Moraes, C.T. Mitochondrial genome engineering coming-of-age. Trends Genet. 2022, 38, 869–880. [Google Scholar] [CrossRef]
- Silva-Pinheiro, P.; Minczuk, M. The potential of mitochondrial genome engineering. Nat. Rev. Genet. 2022, 23, 199–214. [Google Scholar] [CrossRef]
- Yang, X.; Jiang, J.; Li, Z.; Liang, J.; Xiang, Y. Strategies for mitochondrial gene editing. Comput. Struct. Biotechnol. J. 2021, 19, 3319–3329. [Google Scholar] [CrossRef]
- Bian, W.-P.; Chen, Y.-L.; Luo, J.-J.; Wang, C.; Xie, S.-L.; Pei, D.-S. Knock-in strategy for editing human and zebrafish mitochondrial DNA using mito-CRISPR/Cas9 system. ACS Synth. Biol. 2019, 8, 621–632. [Google Scholar] [CrossRef]
- Hussain, S.-R.A.; Yalvac, M.E.; Khoo, B.; Eckardt, S.; McLaughlin, K.J. Adapting CRISPR/Cas9 system for targeting mitochondrial genome. Front. Genet. 2021, 12, 627050. [Google Scholar] [CrossRef]
- Jo, A.; Ham, S.; Lee, G.H.; Lee, Y.-I.; Kim, S.; Lee, Y.-S.; Shin, J.-H.; Lee, Y. Efficient mitochondrial genome editing by CRISPR/Cas9. BioMed Res. Int. 2015, 2015, 305716. [Google Scholar] [CrossRef] [PubMed]
- Amai, T.; Tsuji, T.; Ueda, M.; Kuroda, K. Development of a mito-CRISPR system for generating mitochondrial DNA-deleted strain in Saccharomyces cerevisiae. Biosci. Biotechnol. Biochem. 2021, 85, 895–901. [Google Scholar] [CrossRef] [PubMed]
- Jeandard, D.; Smirnova, A.; Tarassov, I.; Barrey, E.; Smirnov, A.; Entelis, N. Import of non-coding RNAs into human mitochondria: A critical review and emerging approaches. Cells 2019, 8, 286. [Google Scholar] [CrossRef] [PubMed]
- Schmiderer, L.; Yudovich, D.; Oburoglu, L.; Hjort, M.; Larsson, J. Site-specific CRISPR-based mitochondrial DNA manipulation is limited by gRNA import. Sci. Rep. 2022, 12, 18687. [Google Scholar] [CrossRef]
- Rees, H.A.; Liu, D.R. Base editing: Precision chemistry on the genome and transcriptome of living cells. Nat. Rev. Genet. 2018, 19, 770–788. [Google Scholar] [CrossRef]
- Porto, E.M.; Komor, A.C.; Slaymaker, I.M.; Yeo, G.W. Base editing: Advances and therapeutic opportunities. Nat. Rev. Drug Discov. 2020, 19, 839–859. [Google Scholar] [CrossRef]
- Mok, B.Y.; de Moraes, M.H.; Zeng, J.; Bosch, D.E.; Kotrys, A.V.; Raguram, A.; Hsu, F.; Radey, M.C.; Peterson, S.B.; Mootha, V.K.; et al. A bacterial cytidine deaminase toxin enables CRISPR-free mitochondrial base editing. Nature 2020, 583, 631–637. [Google Scholar] [CrossRef]
- Cho, S.-I.; Lee, S.; Mok, Y.G.; Lim, K.; Lee, J.; Lee, J.M.; Chung, E.; Kim, J.-S. Targeted A-to-G base editing in human mitochondrial DNA with programmable deaminases. Cell 2022, 185, 1764–1776.e12. [Google Scholar] [CrossRef]
- Ran, F.; Hsu, P.D.; Wright, J.; Agarwala, V.; Scott, D.A.; Zhang, F. Genome engineering using the CRISPR-Cas9 system. Nat. Protoc. 2013, 8, 2281–2308. [Google Scholar] [CrossRef]
- Cong, L.; Ran, F.A.; Cox, D.; Lin, S.; Barretto, R.; Habib, N.; Hsu, P.D.; Wu, X.; Jiang, W.; Marraffini, L.A. Multiplex genome engineering using CRISPR/Cas systems. Science 2013, 339, 819–823. [Google Scholar] [CrossRef]
- Harris, R.S.; Petersen-Mahrt, S.K.; Neuberger, M.S. RNA editing enzyme APOBEC1 and some of its homologs can act as DNA mutators. Mol. Cell 2002, 10, 1247–1253. [Google Scholar] [CrossRef]
- Mak, A.N.-S.; Bradley, P.; Cernadas, R.A.; Bogdanove, A.J.; Stoddard, B.L. The crystal structure of TAL effector PthXo1 bound to its DNA target. Science 2012, 335, 716–719. [Google Scholar] [CrossRef] [PubMed]
- Boch, J.; Scholze, H.; Schornack, S.; Landgraf, A.; Hahn, S.; Kay, S.; Lahaye, T.; Nickstadt, A.; Bonas, U. Breaking the code of DNA binding specificity of TAL-type III effectors. Science 2009, 326, 1509–1512. [Google Scholar] [CrossRef]
- Lee, H.; Lee, S.; Baek, G.; Kim, A.; Kang, B.-C.; Seo, H.; Kim, J.-S. Mitochondrial DNA editing in mice with DddA-TALE fusion deaminases. Nat. Commun. 2021, 12, 1190. [Google Scholar] [CrossRef]
- Qi, X.; Chen, X.; Guo, J.; Zhang, X.; Sun, H.; Wang, J.; Qian, X.; Li, B.; Tan, L.; Yu, L. Precision modeling of mitochondrial disease in rats via DdCBE-mediated mtDNA editing. Cell Discov. 2021, 7, 95. [Google Scholar] [CrossRef] [PubMed]
- Guo, J.; Zhang, X.; Chen, X.; Sun, H.; Dai, Y.; Wang, J.; Qian, X.; Tan, L.; Lou, X.; Shen, B. Precision modeling of mitochondrial diseases in zebrafish via DdCBE-mediated mtDNA base editing. Cell Discov. 2021, 7, 78. [Google Scholar] [CrossRef] [PubMed]
- Sabharwal, A.; Kar, B.; Restrepo-Castillo, S.; Holmberg, S.R.; Mathew, N.D.; Kendall, B.L.; Cotter, R.P.; WareJoncas, Z.; Seiler, C.; Nakamaru-Ogiso, E. The FusX TALE Base Editor (FusXTBE) for rapid mitochondrial DNA programming of human cells in vitro and zebrafish disease models in vivo. CRISPR J. 2021, 4, 799–821. [Google Scholar]
- Kang, B.-C.; Bae, S.-J.; Lee, S.; Lee, J.S.; Kim, A.; Lee, H.; Baek, G.; Seo, H.; Kim, J.; Kim, J.-S. Chloroplast and mitochondrial DNA editing in plants. Nat. Plants 2021, 7, 899–905. [Google Scholar] [CrossRef]
- Chen, X.; Liang, D.; Guo, J.; Zhang, J.; Sun, H.; Zhang, X.; Jin, J.; Dai, Y.; Bao, Q.; Qian, X. DdCBE-mediated mitochondrial base editing in human 3PN embryos. Cell Discov. 2022, 8, 8. [Google Scholar] [CrossRef]
- Wei, Y.; Xu, C.; Feng, H.; Xu, K.; Li, Z.; Hu, J.; Zhou, L.; Wei, Y.; Zuo, Z.; Zuo, E. Human cleaving embryos enable efficient mitochondrial base-editing with DdCBE. Cell Discov. 2022, 8, 7. [Google Scholar] [CrossRef]
- Moscou, M.J.; Bogdanove, A.J. A simple cipher governs DNA recognition by TAL effectors. Science 2009, 326, 1501. [Google Scholar] [CrossRef] [PubMed]
- Cermak, T.; Doyle, E.L.; Christian, M.; Wang, L.; Zhang, Y.; Schmidt, C.; Baller, J.A.; Somia, N.V.; Bogdanove, A.J.; Voytas, D.F. Efficient design and assembly of custom TALEN and other TAL effector-based constructs for DNA targeting. Nucleic Acids Res. 2011, 39, e82. [Google Scholar] [CrossRef] [PubMed]
- Magnusson, J.; Orth, M.; Lestienne, P.; Taanman, J.-W. Replication of mitochondrial DNA occurs throughout the mitochondria of cultured human cells. Exp. Cell Res. 2003, 289, 133–142. [Google Scholar] [CrossRef]
- Lin, Y.; Fine, E.J.; Zheng, Z.; Antico, C.J.; Voit, R.A.; Porteus, M.H.; Cradick, T.J.; Bao, G. SAPTA: A new design tool for improving TALE nuclease activity. Nucleic Acids Res. 2014, 42, e47. [Google Scholar] [CrossRef]
- Lei, Z.; Meng, H.; Liu, L.; Zhao, H.; Rao, X.; Yan, Y.; Wu, H.; Liu, M.; He, A.; Yi, C. Mitochondrial base editor induces substantial nuclear off-target mutations. Nature 2022, 606, 804–811. [Google Scholar] [CrossRef] [PubMed]
- Lim, K.; Cho, S.-I.; Kim, J.-S. Nuclear and mitochondrial DNA editing in human cells with zinc finger deaminases. Nat. Commun. 2022, 13, 366. [Google Scholar] [CrossRef]
- Gonzalez, B.; Schwimmer, L.J.; Fuller, R.P.; Ye, Y.; Asawapornmongkol, L.; Barbas, C.F. Modular system for the construction of zinc-finger libraries and proteins. Nat. Protoc. 2010, 5, 791–810. [Google Scholar] [CrossRef]
- Kim, S.; Lee, M.J.; Kim, H.; Kang, M.; Kim, J.-S. Preassembled zinc-finger arrays for rapid construction of ZFNs. Nat. Methods 2011, 8, 7. [Google Scholar] [CrossRef]
- Paschon, D.E.; Lussier, S.; Wangzor, T.; Xia, D.F.; Li, P.W.; Hinkley, S.J.; Scarlott, N.A.; Lam, S.C.; Waite, A.J.; Truong, L.N. Diversifying the structure of zinc finger nucleases for high-precision genome editing. Nat. Commun. 2019, 10, 1133. [Google Scholar] [CrossRef]
- Miller, J.C.; Patil, D.P.; Xia, D.F.; Paine, C.B.; Fauser, F.; Richards, H.W.; Shivak, D.A.; Bendaña, Y.R.; Hinkley, S.J.; Scarlott, N.A. Enhancing gene editing specificity by attenuating DNA cleavage kinetics. Nat. Biotechnol. 2019, 37, 945–952. [Google Scholar] [CrossRef]
- Mok, B.Y.; Kotrys, A.V.; Raguram, A.; Huang, T.P.; Mootha, V.K.; Liu, D.R. CRISPR-free base editors with enhanced activity and expanded targeting scope in mitochondrial and nuclear DNA. Nat. Biotechnol. 2022, 40, 1378–1387. [Google Scholar] [CrossRef] [PubMed]
- Thuronyi, B.W.; Koblan, L.W.; Levy, J.M.; Yeh, W.-H.; Zheng, C.; Newby, G.A.; Wilson, C.; Bhaumik, M.; Shubina-Oleinik, O.; Holt, J.R. Continuous evolution of base editors with expanded target compatibility and improved activity. Nat. Biotechnol. 2019, 37, 1070–1079. [Google Scholar] [CrossRef]
- Roth, T.B.; Woolston, B.M.; Stephanopoulos, G.; Liu, D.R. Phage-assisted evolution of Bacillus methanolicus methanol dehydrogenase 2. ACS Synth. Biol. 2019, 8, 796–806. [Google Scholar] [CrossRef]
- Miller, S.M.; Wang, T.; Liu, D.R. Phage-assisted continuous and non-continuous evolution. Nat. Protoc. 2020, 15, 4101–4127. [Google Scholar] [CrossRef] [PubMed]
- Kar, B.; Sabharwal, A.; Restrepo-Castillo, S.; Simone, B.W.; Clark, K.J.; Ekker, S.C. An optimized FusX assembly-based technique to introduce mitochondrial TC-to-TT variations in human cell lines. STAR Protoc. 2022, 3, 101288. [Google Scholar] [CrossRef] [PubMed]
- Fine, E.J.; Cradick, T.J.; Zhao, C.L.; Lin, Y.; Bao, G. An online bioinformatics tool predicts zinc finger and TALE nuclease off-target cleavage. Nucleic Acids Res. 2014, 42, e42. [Google Scholar] [CrossRef]
- Liao, Z.; Zhao, J.; Zhu, Y.; Yang, L.; Yang, A.; Sun, D.; Zhao, Z.; Wang, X.; Tao, Z.; Tang, X. The ND4 G11696A mutation may influence the phenotypic manifestation of the deafness-associated 12S rRNA A1555G mutation in a four-generation Chinese family. Biochem. Biophys. Res. Commun. 2007, 362, 670–676. [Google Scholar] [CrossRef]
- Gopal, R.K.; Calvo, S.E.; Shih, A.R.; Chaves, F.L.; McGuone, D.; Mick, E.; Pierce, K.A.; Li, Y.; Garofalo, A.; Van Allen, E.M. Early loss of mitochondrial complex I and rewiring of glutathione metabolism in renal oncocytoma. Proc. Natl. Acad. Sci. USA 2018, 115, E6283–E6290. [Google Scholar] [CrossRef]
- Mok, Y.G.; Lee, J.M.; Chung, E.; Lee, J.; Lim, K.; Cho, S.-I.; Kim, J.-S. Base editing in human cells with monomeric DddA-TALE fusion deaminases. Nat. Commun. 2022, 13, 4038. [Google Scholar] [CrossRef]
- Lee, S.; Lee, H.; Baek, G.; Kim, J.-S. Precision mitochondrial DNA editing with high-fidelity DddA-derived base editors. Nat. Biotechnol. 2022, 41, 378–386. [Google Scholar] [CrossRef]
- Willis, J.C.; Silva-Pinheiro, P.; Widdup, L.; Minczuk, M.; Liu, D.R. Compact zinc finger base editors that edit mitochondrial or nuclear DNA in vitro and in vivo. Nat. Commun. 2022, 13, 7204. [Google Scholar] [CrossRef]
- Lee, S.; Lee, H.; Baek, G.; Namgung, E.; Park, J.M.; Kim, S.; Hong, S.; Kim, J.-S. Enhanced mitochondrial DNA editing in mice using nuclear-exported TALE-linked deaminases and nucleases. Genome Biol. 2022, 23, 211. [Google Scholar] [CrossRef] [PubMed]
- Silva-Pinheiro, P.; Mutti, C.D.; Van Haute, L.; Powell, C.A.; Nash, P.A.; Turner, K.; Minczuk, M. A library of base editors for the precise ablation of all protein-coding genes in the mouse mitochondrial genome. Nat. Biomed. Eng. 2022, 1–12. [Google Scholar] [CrossRef] [PubMed]
- Beilstein, K.; Wittmann, A.; Grez, M.; Suess, B. Conditional control of mammalian gene expression by tetracycline-dependent hammerhead ribozymes. ACS Synth. Biol. 2015, 4, 526–534. [Google Scholar] [CrossRef] [PubMed]
- Liu, Z.; Chen, O.; Wall, J.; Zheng, M.; Zhou, Y.; Wang, L.; Ruth Vaseghi, H.; Qian, L.; Liu, J. Systematic comparison of 2A peptides for cloning multi-genes in a polycistronic vector. Sci. Rep. 2017, 7, 2193. [Google Scholar] [CrossRef]
- Gaudelli, N.M.; Lam, D.K.; Rees, H.A.; Solá-Esteves, N.M.; Barrera, L.A.; Born, D.A.; Edwards, A.; Gehrke, J.M.; Lee, S.-J.; Liquori, A.J. Directed evolution of adenine base editors with increased activity and therapeutic application. Nat. Biotechnol. 2020, 38, 892–900. [Google Scholar] [CrossRef]
- Mok, Y.G.; Hong, S.; Bae, S.-J.; Cho, S.-I.; Kim, J.-S. Targeted A-to-G base editing of chloroplast DNA in plants. Nat. Plants 2022, 8, 1378–1384. [Google Scholar] [CrossRef] [PubMed]
- Aihara, H.; Yin, L.; Shi, K. Structural basis of sequence-specific cytosine deamination by double-stranded DNA deaminase toxin DddA. Res. Sq. 2022. [Google Scholar] [CrossRef]
- Newby, G.A.; Yen, J.S.; Woodard, K.J.; Mayuranathan, T.; Lazzarotto, C.R.; Li, Y.; Sheppard-Tillman, H.; Porter, S.N.; Yao, Y.; Mayberry, K. Base editing of haematopoietic stem cells rescues sickle cell disease in mice. Nature 2021, 595, 295–302. [Google Scholar] [CrossRef]
- Newby, G.A.; Liu, D.R. In vivo somatic cell base editing and prime editing. Mol. Ther. 2021, 29, 3107–3124. [Google Scholar] [CrossRef]
- Schapira, A.H. Mitochondrial disease. Lancet 2006, 368, 70–82. [Google Scholar] [CrossRef]
- Greaves, L.C.; Reeve, A.K.; Taylor, R.W.; Turnbull, D.M. Mitochondrial DNA and disease. J. Pathol. 2012, 226, 274–286. [Google Scholar] [CrossRef]
- Jang, Y.-h.; Ahn, S.R.; Shim, J.-y.; Lim, K.-i. Engineering genetic systems for treating mitochondrial diseases. Pharmaceutics 2021, 13, 810. [Google Scholar] [CrossRef]
- Di Donfrancesco, A.; Massaro, G.; Di Meo, I.; Tiranti, V.; Bottani, E.; Brunetti, D. Gene therapy for mitochondrial diseases: Current status and future perspective. Pharmaceutics 2022, 14, 1287. [Google Scholar] [CrossRef]
- Wallace, D.C. Animal models for mitochondrial disease. In Mitochondrial DNA, Methods in Molecular Biology; Humana Press: Totowa, NJ, USA, 2002; pp. 3–54. [Google Scholar] [CrossRef]
- Wallace, D.C. Mitochondrial diseases in man and mouse. Science 1999, 283, 1482–1488. [Google Scholar] [CrossRef]
- Kauppila, J.H.; Baines, H.L.; Bratic, A.; Simard, M.-L.; Freyer, C.; Mourier, A.; Stamp, C.; Filograna, R.; Larsson, N.-G.; Greaves, L.C. A phenotype-driven approach to generate mouse models with pathogenic mtDNA mutations causing mitochondrial disease. Cell Rep. 2016, 16, 2980–2990. [Google Scholar] [CrossRef] [PubMed]
- Kasahara, A.; Ishikawa, K.; Yamaoka, M.; Ito, M.; Watanabe, N.; Akimoto, M.; Sato, A.; Nakada, K.; Endo, H.; Suda, Y. Generation of trans-mitochondrial mice carrying homoplasmic mtDNAs with a missense mutation in a structural gene using ES cells. Hum. Mol. Genet. 2006, 15, 871–881. [Google Scholar] [CrossRef] [PubMed]
- Inoue, K.; Nakada, K.; Ogura, A.; Isobe, K.; Goto, Y.-i.; Nonaka, I.; Hayashi, J.-I. Generation of mice with mitochondrial dysfunction by introducing mouse mtDNA carrying a deletion into zygotes. Nat. Genet. 2000, 26, 176–181. [Google Scholar] [CrossRef] [PubMed]
- Dunn, D.A.; Cannon, M.V.; Irwin, M.H.; Pinkert, C.A. Animal models of human mitochondrial DNA mutations. Biochim. Biophys. Acta (BBA) Gen. Subj. 2012, 1820, 601–607. [Google Scholar] [CrossRef] [PubMed]
- Khotina, V.A.; Vinokurov, A.Y.; Bagheri Ekta, M.; Sukhorukov, V.N.; Orekhov, A.N. Creation of Mitochondrial Disease Models Using Mitochondrial DNA Editing. Biomedicines 2023, 11, 532. [Google Scholar] [CrossRef]
- Chol, M.; Lebon, S.; Benit, P.; Chretien, D.; De Lonlay, P.; Goldenberg, A.; Odent, S.; Hertz-Pannier, L.; Vincent-Delorme, C.; Cormier-Daire, V. The mitochondrial DNA G13513A MELAS mutation in the NADH dehydrogenase 5 gene is a frequent cause of Leigh-like syndrome with isolated complex I deficiency. J. Med. Genet. 2003, 40, 188–191. [Google Scholar] [CrossRef] [PubMed]
- Shanske, S.; Coku, J.; Lu, J.; Ganesh, J.; Krishna, S.; Tanji, K.; Bonilla, E.; Naini, A.B.; Hirano, M.; DiMauro, S. The G13513A mutation in the ND5 gene of mitochondrial DNA as a common cause of MELAS or Leigh syndrome: Evidence from 12 cases. Arch. Neurol. 2008, 65, 368–372. [Google Scholar] [CrossRef] [PubMed]
- Guo, J.; Chen, X.; Liu, Z.; Sun, H.; Zhou, Y.; Dai, Y.; He, L.; Qian, X.; Wang, J.; Zhang, J. DdCBE mediates efficient and inheritable modifications in mouse mitochondrial genome. Mol. Ther. Nucleic Acids 2022, 27, 73–80. [Google Scholar] [CrossRef] [PubMed]
- Virgilio, R.; Ronchi, D.; Bordoni, A.; Fassone, E.; Bonato, S.; Donadoni, C.; Torgano, G.; Moggio, M.; Corti, S.; Bresolin, N. Mitochondrial DNA G8363A mutation in the tRNALys gene: Clinical, biochemical and pathological study. J. Neurol. Sci. 2009, 281, 85–92. [Google Scholar] [CrossRef] [PubMed]
- Anitori, R.; Manning, K.; Quan, F.; Weleber, R.G.; Buist, N.R.; Shoubridge, E.A.; Kennaway, N.G. Contrasting phenotypes in three patients with novel mutations in mitochondrial tRNA genes. Mol. Genet. Metab. 2005, 84, 176–188. [Google Scholar] [CrossRef] [PubMed]
- Bugiardini, E.; Bottani, E.; Marchet, S.; Poole, O.V.; Beninca, C.; Horga, A.; Woodward, C.; Lam, A.; Hargreaves, I.; Chalasani, A. Expanding the molecular and phenotypic spectrum of truncating MT-ATP6 mutations. Neurol. Genet. 2020, 6, e381. [Google Scholar] [CrossRef]
- Saha, K.; Sontheimer, E.J.; Brooks, P.; Dwinell, M.R.; Gersbach, C.A.; Liu, D.R.; Murray, S.A.; Tsai, S.Q.; Wilson, R.C.; Anderson, D.G. The NIH somatic cell genome editing program. Nature 2021, 592, 195–204. [Google Scholar] [CrossRef]




{kind=link}
{kind=link}
{kind=link}
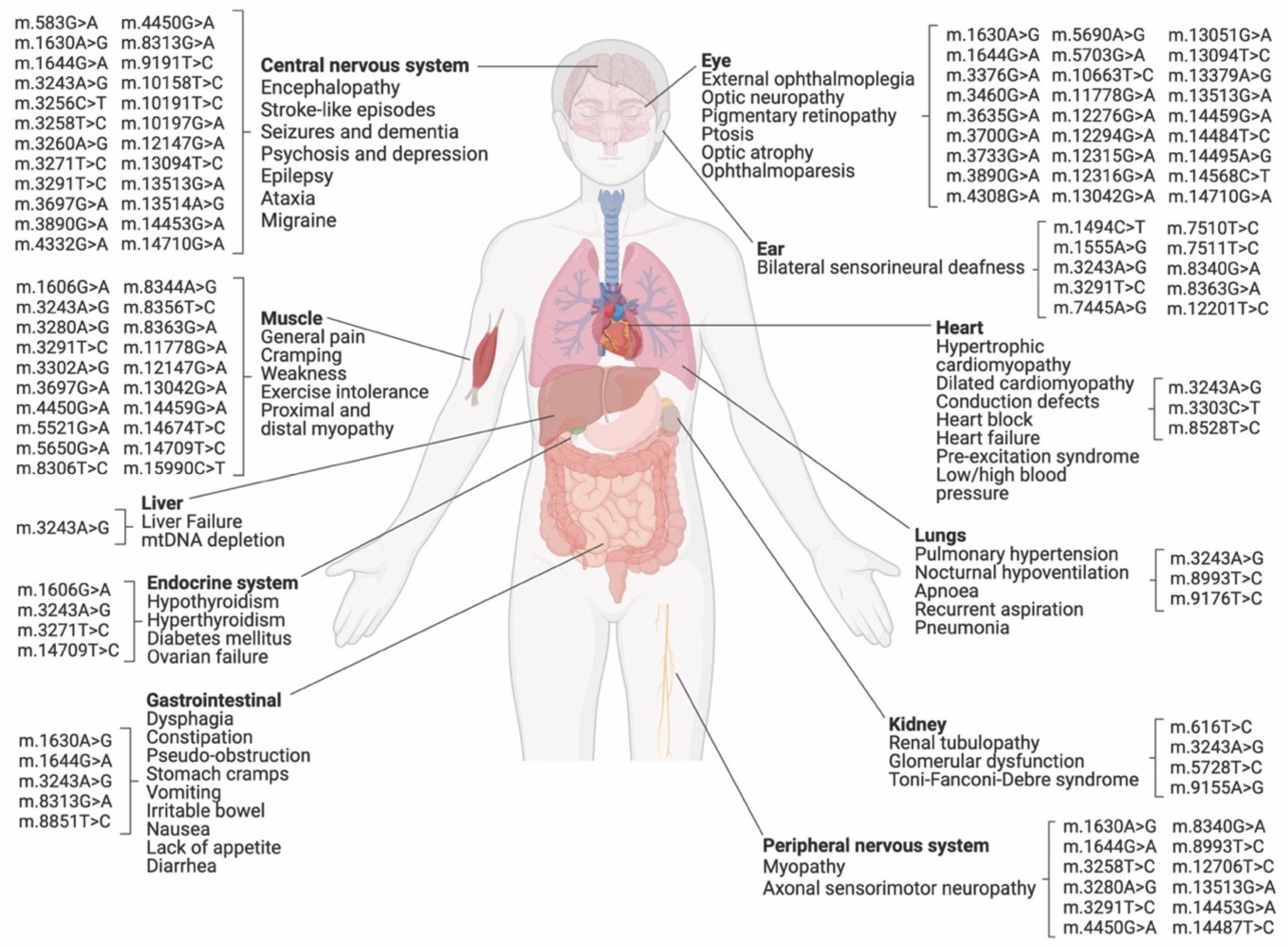
| Locus | Allele SNP | Associated Disease | Treatment via C-to-T Editing | Modeling via C-to-T Editing | Treatment via A-to-G Editing | Modeling via A-to-G Editing |
|---|---|---|---|---|---|---|
| MT-TF | m.583G>A | MELAS/MM & EXIT | ✓ | ✓ | ||
| m.616T>C | Maternally inherited epilepsy/mito tubulointerstitial kidney disease (MITKD)/Gitelman-like syndrome | ✓ | ✓ | |||
| MT-RNR1 | m.1494C>T | DEAF | ✓ | ✓ | ||
| m.1555A>G | DEAF; autism spectrum intellectual disability; possibly antiatherosclerotic | ✓ | ✓ | |||
| MT-TV | m.1606G>A | AMDF | ✓ | ✓ | ||
| m.1630A>G | MNGIE-like disease/MELAS | ✓ | ✓ | |||
| m.1644G>A | Leigh Syndrome/HCM/MELAS | ✓ | ✓ | |||
| MT-TL1 | m.3243A>G | MELAS/Leigh Syndrome/DMDF/MIDD/SNHL/CPEO/MM/FSGS/ASD/Cardiac + multi-organ dysfunction | ✓ | ✓ | ||
| m.3243A>T | MM/MELAS/SNHL/CPEO | |||||
| m.3256C>T | MELAS; possible atherosclerosis risk | ✓ | ✓ | |||
| m.3258T>C | MELAS/Myopathy | ✓ | ✓ | |||
| m.3260A>G | MMC/MELAS | ✓ | ✓ | |||
| m.3271T>C | MELAS/DM | ✓ | ✓ | |||
| m.3271delT | PEM/retinal dystrophy in MELAS | |||||
| m.3280A>G | Myopathy | ✓ | ✓ | |||
| m.3291T>C | MELAS/Myopathy/Deafness + Cognitive Impairment | ✓ | ✓ | |||
| m.3302A>G | MM | ✓ | ✓ | |||
| m.3303C>T | MMC | ✓ | ✓ | |||
| MT-ND1 | m.3376G>A | LHON MELAS overlap | ✓ | ✓ | ||
| m.3460G>A | LHON | ✓ | ✓ | |||
| m.3635G>A | LHON | ✓ | ✓ | |||
| m.3697G>A | MELAS/Leigh Syndrome/LDYT/BSN | ✓ | ✓ | |||
| m.3700G>A | LHON | ✓ | ✓ | |||
| m.3733G>A | LHON | ✓ | ✓ | |||
| m.3890G>A | Progressive Encephalomyopathy/Leigh Syndrome/Optic Atrophy | ✓ | ✓ | |||
| m.3902_3908 ACCTTGCinv | EXIT + myalgia/severe LA + cardiac/3-MGA aciduria/nephropathy + deafness + diabetes | |||||
| m.4171C>A | LHON/Leigh-like phenotype | |||||
| MT-TI | m.4298G>A | CPEO/MS | ✓ | ✓ | ||
| m.4300A>G | MICM | ✓ | ✓ | |||
| m.4308G>A | CPEO | ✓ | ✓ | |||
| MT-TQ | m.4332G>A | Encephalopathy/MELAS | ✓ | ✓ | ||
| MT-TM | m.4450G>A | Myopathy/MELAS/Leigh Syndrome | ✓ | ✓ | ||
| MT-TW | m.5521G>A | Mitochondrial myopathy | ✓ | ✓ | ||
| m.5537_5537insT | Leigh Syndrome | |||||
| MT-TA | m.5650G>A | Myopathy | ✓ | ✓ | ||
| MT-TN | m.5690A>G | CPEO + ptosis + proximal myopathy | ✓ | ✓ | ||
| m.5703G>A | CPEO/MM | ✓ | ✓ | |||
| m.5728T>C | Multiorgan failure/ myopathy | ✓ | ✓ | |||
| MT-CO1 | m.7445A>G | SNHL | ✓ | ✓ | ||
| MT-TS1 precursor | m.7445A>G | SNHL | ✓ | ✓ | ||
| MT-TS1 | m.7471_7472insC | PEM/AMDF/Motor neuron disease-like | ||||
| m.7497G>A | MM/EXIT | ✓ | ✓ | |||
| m.7510T>C | SNHL | ✓ | ✓ | |||
| m.7511T>C | SNHL/Deafness | ✓ | ✓ | |||
| MT-TK | m.8306T>C | Severe adult-onset multisymptom myopathy/ Myoclonic epilepsy | ✓ | ✓ | ||
| m.8313G>A | MNGIE/Progressive mito cytopathy | ✓ | ✓ | |||
| m.8340G>A | Myopathy/Exercise Intolerance/Eye disease + SNHL | ✓ | ✓ | |||
| m.8344A>G | MERRF.; Other-LD/Depressive mood disorder/leukoencephalopathy/HiCM | ✓ | ✓ | |||
| m.8356T>C | MERRF | ✓ | ✓ | |||
| m.8363G>A | MICM+DEAF/MERRF/Autism/Leigh Syndrome/Ataxia + Lipomas | ✓ | ✓ | |||
| MT-ATP8/6 | m.8528T>C | Infantile cardiomyopathy/hyperammonemia | ✓ | ✓ | ||
| MT-ATP6 | m.8851T>C | BSN/Leigh syndrome | ✓ | ✓ | ||
| m.8969G>A | Mitochondrial myopathy, lactic acidosis and sideroblastic anemia (MLASA)/IgG nephropathy | ✓ | ✓ | |||
| m.8993T>C | NARP/Leigh Disease/MILS/other | ✓ | ✓ | |||
| m.8993T>G | NARP/Leigh Disease/MILS/other | |||||
| m.9035T>C | Ataxia syndromes | ✓ | ✓ | |||
| m.9155A>G | MIDD, renal insufficiency | ✓ | ✓ | |||
| m.9176T>C | FBSN/Leigh Disease/Spinocerebellar Ataxia | ✓ | ✓ | |||
| m.9176T>G | Leigh Disease/Spastic Paraplegia/Spinocerebellar Ataxia | |||||
| m.9185T>C | Leigh Disease/Ataxia syndromes/NARP-like disease/Episodic | ✓ | ✓ | |||
| weakness and Charcot-Marie-Tooth | ||||||
| m.9191T>C | Leigh Disease | ✓ | ✓ | |||
| m.9205_9206delTA | Encephalopathy/Seizures/Lacticacidemia | |||||
| MT-TG | m.10010T>C | PEM | ✓ | ✓ | ||
| MT-ND3 | m.10158T>C | Leigh Disease/MELAS | ✓ | ✓ | ||
| m.10191T>C | Leigh Disease/Leigh-like Disease/ESOC | ✓ | ✓ | |||
| m.10197G>A | Leigh Disease/Dystonia/Stroke/LDYT | ✓ | ✓ | |||
| MT-ND4L | m.10663T>C | LHON | ✓ | ✓ | ||
| MT-ND4 | m.11777C>A | Leigh Disease | ||||
| m.11778G>A | LHON/Progressive Dystonia | ✓ | ✓ | |||
| MT-TH | m.12147G>A | MERRF-MELAS/Encephalopathy | ✓ | ✓ | ||
| m.12201T>C | Maternally inherited non-syndromic deafness | ✓ | ✓ | |||
| MT-TS2 | m.12258C>A | DMDF/RP + SNHL | ||||
| MT-TL2 | m.12276G>A | CPEO | ✓ | ✓ | ||
| m.12294G>A | CPEO/EXIT + Ophthalmoplegia | ✓ | ✓ | |||
| m.12315G>A | PEO/KSS/possible carotid atherosclerosis risk, trend toward myocardial infarction risk | ✓ | ✓ | |||
| m.12316G>A | CPEO | ✓ | ✓ | |||
| MT-ND5 | m.12706T>C | Leigh Disease | ✓ | ✓ | ||
| m.13042G>A | Optic neuropathy/retinopathy/LD | ✓ | ✓ | |||
| m.13051G>A | LHON | ✓ | ✓ | |||
| m.13094T>C | Ataxia + PEO/MELAS, LD, LHON, myoclonus, fatigue | ✓ | ✓ | |||
| m.13379A>G | LHON | ✓ | ✓ | |||
| m.13513G>A | Leigh Disease/MELAS/LHON-MELAS Overlap Syndrome/negative association w Carotid Atherosclerosis | ✓ | ✓ | |||
| m.13514A>G | Leigh Disease/MELAS/Ca2+ downregulation | ✓ | ✓ | |||
| MT-ND6 | m.14453G>A | MELAS/Leigh Disease | ✓ | ✓ | ||
| m.14459G>A | LDYT/Leigh Disease/dystonia/carotid atherosclerosis risk | ✓ | ✓ | |||
| m.14482C>A | LHON | |||||
| m.14482C>G | LHON | |||||
| m.14484T>C | LHON | ✓ | ✓ | |||
| m.14487T>C | Dystonia/Leigh Disease/ataxia/ptosis/epilepsy | ✓ | ✓ | |||
| m.14495A>G | LHON | ✓ | ✓ | |||
| m.14568C>T | LHON | ✓ | ✓ | |||
| MT-TE | m.14674T>C | Reversible COX deficiency myopathy | ✓ | ✓ | ||
| m.14709T>C | MM + DMDF/Encephalomyopathy/Dementia + diabetes + ophthalmoplegia | ✓ | ✓ | |||
| m.14710G>A | Encephalomyopathy + Retinopathy | ✓ | ✓ | |||
| MT-CYB | m.14849T>C | EXIT/Septo-Optic Dysplasia | ✓ | ✓ | ||
| m.15579A>G | Multisystem Disorder, EXIT | ✓ | ✓ | |||
| MT-TP | m.15990C>T | MM/PEO | ✓ | ✓ |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kar, B.; Castillo, S.R.; Sabharwal, A.; Clark, K.J.; Ekker, S.C. Mitochondrial Base Editing: Recent Advances towards Therapeutic Opportunities. Int. J. Mol. Sci. 2023, 24, 5798. https://doi.org/10.3390/ijms24065798
Kar B, Castillo SR, Sabharwal A, Clark KJ, Ekker SC. Mitochondrial Base Editing: Recent Advances towards Therapeutic Opportunities. International Journal of Molecular Sciences. 2023; 24(6):5798. https://doi.org/10.3390/ijms24065798
Chicago/Turabian StyleKar, Bibekananda, Santiago R. Castillo, Ankit Sabharwal, Karl J. Clark, and Stephen C. Ekker. 2023. "Mitochondrial Base Editing: Recent Advances towards Therapeutic Opportunities" International Journal of Molecular Sciences 24, no. 6: 5798. https://doi.org/10.3390/ijms24065798
APA StyleKar, B., Castillo, S. R., Sabharwal, A., Clark, K. J., & Ekker, S. C. (2023). Mitochondrial Base Editing: Recent Advances towards Therapeutic Opportunities. International Journal of Molecular Sciences, 24(6), 5798. https://doi.org/10.3390/ijms24065798