
Dopamine Transmission Imbalance in Neuroinflammation: Perspectives on Long-Term COVID-19

 , , ,
, , , 
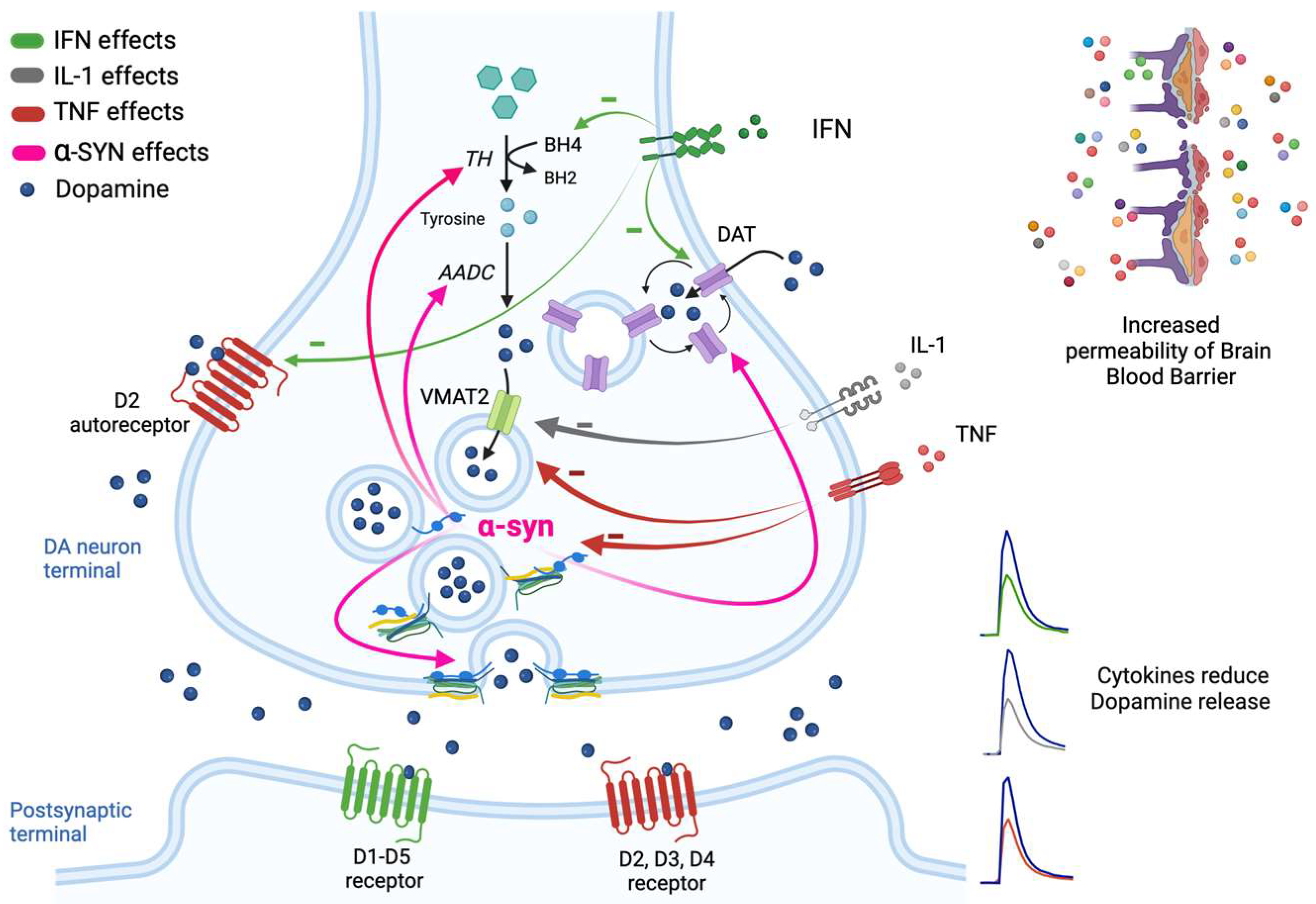{kind=link}
{kind=link}
Abstract
1. Introduction
2. Mechanisms of DA Release and Regulation in the Nigrostriatal Pathway
3. SARS-CoV-2 Viral Infection and Dysregulation of DA Neurotransmission
3.1. Mechanisms of SARS-CoV-2 Invasion and Associated Immune Responses
3.2. SARS-CoV-2-Induced Cytokine Storm
3.3. Cytokine-Mediated Changes in Neurotransmission and DA Release
3.4. Mechanisms Underlying the Cytokine-Induced Alterations in DA Release
3.5. Glia Activation and Modulation of DA Release
3.6. Upregulation of α-syn and Impairment of DA Release
4. Impact of Inflammation-Driven DA Dysregulation on PD Patients following SARS-CoV-2 Infection
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Greenhalgh, A.D.; David, S.; Bennett, F.C. Immune cell regulation of glia during CNS injury and disease. Nat. Rev. Neurosci. 2020, 21, 139–152. [Google Scholar] [CrossRef] [PubMed]
- Fani Maleki, A.; Rivest, S. Innate immune cells: Monocytes, monocyte-derived macrophages and microglia as therapeutic target for Alzheimer’s disease and multiple sclerosis. Front. Cell Neurosci. 2019, 13, 355. [Google Scholar] [CrossRef] [PubMed]
- Vainchtein, I.D.; Molofsky, A.V. Astrocytes and microglia: In sickness and in health. Trends Neurosci. 2020, 43, 144–154. [Google Scholar] [CrossRef] [PubMed]
- Xanthos, D.N.; Sandkühler, J. Neurogenic inflammation: Inflammatory CNS reactions in response to neuronal activity. Nat. Rev. Neurosci. 2014, 15, 43–53. [Google Scholar] [CrossRef] [PubMed]
- Habbas, S.; Santello, M.; Becker, D.; Stubbe, H.; Zappia, G.; Liaudet, N.; Klaus, F.R.; Kollias, G.; Fontana, A.; Pryce, C.R.; et al. Neuroinflammatory TNFα impairs memory via astrocyte signaling. Cell 2015, 163, 1730–1741. [Google Scholar] [CrossRef] [PubMed]
- Sheridan, G.K.; Murphy, K.J. Neuron-glia crosstalk in health and disease: Fractalike and CX3CR1 take centre stage. Open Biol. 2013, 3, 130181. [Google Scholar] [CrossRef]
- Dunn, A.J. Effects of cytokines and infections on brain neurochemistry. Clin. Neurosci. Res. 2006, 6, 52–68. [Google Scholar] [CrossRef]
- Emmi, A.; Rizzo, S.; Barzon, L.; Sandre, M.; Carturan, E.; Sinigaglia, A.; Riccetti, S.; Della Barbera, M.; Boscolo-Berto, R.; Cocco, P.; et al. Detection of SARS-CoV-2 viral proteins and genomic sequences in human brainstem nuclei. NPJ Parkinsons Dis. 2023, 9, 25. [Google Scholar] [CrossRef]
- Boura, I.; Chaudhuri, K.R. Coronavirus disease 2019 and related parkinsonism: The clinical evidence thus far. Mov. Disord. Clin. Pract. 2022, 9, 584–593. [Google Scholar] [CrossRef]
- Calculli, A.; Bocci, T.; Porcino, M.; Avenali, M.; Casellato, C.; Priori, A.; Pisani, A. Parkinson’s disease following COVID-19: Six cases report. Eur. J. Neurol. 2023. ahead of print. [Google Scholar] [CrossRef]
- Matsuda, W.; Furuta, T.; Nakamura, K.C.; Hioki, H.; Fujiyama, F.; Arai, R.; Kaneko, T. Single nigrostriatal dopaminergic neurons form widely spread and highly dense axonal arborizations in the neostriatum. J. Neurosci. 2009, 29, 444–453. [Google Scholar] [CrossRef] [PubMed]
- Rice, M.E.; Cragg, S.J.; Greenfield, S.A. Characteristics of electrically evoked somatodendritic dopamine release in substantia nigra and ventral tegmental area in vitro. J. Neurophysiol. 1997, 77, 853–862. [Google Scholar] [CrossRef] [PubMed]
- Cragg, S.J. Variable dopamine release probability and short-term plasticity between functional domains of the primate striatum. J. Neurosci. 2003, 23, 4378–4385. [Google Scholar] [CrossRef] [PubMed]
- Sulzer, D.; Cragg, S.J.; Rice, M.E. Striatal dopamine neurotransmission: Regulation of release and uptake. Basal. Ganglia 2016, 6, 123–148. [Google Scholar] [CrossRef] [PubMed]
- Chen, B.T.; Rice, M.E. Novel Ca2+ dependence and time course of somatodendritic dopamine release: Substantia nigra versus striatum. J. Neurosci. 2001, 21, 7841–7847. [Google Scholar] [CrossRef]
- Brimblecombe, K.R.; Gracie, C.J.; Platt, N.J.; Cragg, S.J. Gating of dopamine transmission by calcium and axonal N-, Q-, T-, and L-type voltage-gated calcium channels differs between striatal domains. J. Physiol. 2015, 593, 929–946. [Google Scholar] [CrossRef]
- Chen, B.T.; Moran, K.A.; Avshalumov, M.V.; Rice, M.E. Limited regulation of somatodendritic dopamine release by voltage-sensitive Ca channels contrasted with strong regulation of axonal dopamine release. J. Neurochem. 2006, 96, 645–655. [Google Scholar] [CrossRef]
- Liu, C.; Kaeser, P.S. Mechanisms and regulation of dopamine release. Curr. Opin. Neurobiol. 2019, 57, 46–53. [Google Scholar] [CrossRef]
- Liu, C.; Goel, P.; Kaeser, P.S. Spatial and temporal scales of dopamine transmission. Nat. Rev. Neurosci. 2021, 22, 345–358. [Google Scholar] [CrossRef]
- Pereira, D.B.; Sulzer, D. Mechanisms of dopamine quantal size regulation. Front. Biosci. 2012, 17, 2740–2767. [Google Scholar] [CrossRef]
- Pothos, E.N.; Larsen, K.E.; Krantz, D.E.; Liu, Y.; Haycock, J.W.; Setlik, W.; Gershon, M.D.; Edwards, R.H.; Sulzer, D. Synaptic vesicle transporter expression regulates vesicle phenotype and quantal size. J. Neurosci. 2000, 20, 7297–7306. [Google Scholar] [CrossRef] [PubMed]
- Omiatek, D.M.; Bressler, A.J.; Cans, A.S.; Andrews, A.M.; Heien, M.L.; Ewing, A.G. The real catecholamine content of secretory vesicles in the CNS revealed by electrochemical cytometry. Sci. Rep. 2013, 3, 1447. [Google Scholar] [CrossRef]
- Cartier, E.A.; Parra, L.A.; Baust, T.B.; Quiroz, M.; Salazar, G.; Faundez, V.; Egaña, L.; Torres, G.E. A biochemical and functional protein complex involving dopamine synthesis and transport into synaptic vesicles. J. Biol. Chem. 2010, 285, 1957–1966. [Google Scholar] [CrossRef] [PubMed]
- Brown, J.M.; Hanson, G.R.; Fleckenstein, A.E. Regulation of the vesicular monoamine transporter-2: A novel mechanism for cocaine and other psychostimulants. J. Pharmacol. Exp. Ther. 2001, 296, 762–767. [Google Scholar] [PubMed]
- Sandoval, V.; Riddle, E.L.; Hanson, G.R.; Fleckenstein, A.E. Methylphenidate redistributes vesicular monoamine transporter-2: Role of dopamine receptors. J. Neurosci. 2002, 22, 8705–8710. [Google Scholar] [CrossRef]
- Sandoval, V.; Riddle, E.L.; Hanson, G.R.; Fleckenstein, A.E. Methylphenidate alters vesicular monoamine transport and prevents methamphetamine-induced dopaminergic deficits. J. Pharmacol. Exp. Ther. 2003, 304, 1181–1187. [Google Scholar] [CrossRef]
- Nirenberg, M.J.; Chan, J.; Liu, Y.; Edwards, R.H.; Pickel, V.M. Ultrastructural localization of the vesicular monoamine transporter-2 in midbrain dopaminergic neurons: Potential sites for somatodendritic storage and release of dopamine. J. Neurosci. 1996, 16, 4135–4145. [Google Scholar] [CrossRef]
- Kaeser, P.S.; Regehr, W.G. Molecular mechanisms for synchronous, asynchronous, and spontaneous neurotransmitter release. Annu. Rev. Physiol. 2014, 76, 333–363. [Google Scholar] [CrossRef]
- Pang, Z.P.; Südhof, T.C. Cell biology of Ca2+-triggerd exocytosis. Curr. Opin. Cell Biol. 2010, 22, 496–505. [Google Scholar] [CrossRef]
- Bergquist, F.; Niazi, H.S.; Nissbrandt, H. Evidence for different exocytosis pathways in dendritic and terminal dopamine release in vivo. Brain Res. 2002, 950, 245–453. [Google Scholar] [CrossRef]
- Witkovsky, P.; Patel, J.C.; Lee, C.R.; Rice, M.E. Immunocytochemical identification of proteins involved in dopamine release from somatodendritic compartment of nigral dopaminergic neurons. Neuroscience 2009, 164, 488–496. [Google Scholar] [CrossRef] [PubMed]
- Mendez, J.A.; Bourque, M.J.; Fasano, C.; Kortleven, C.; Trudeau, L.E. Somatodendritic dopamine release requires synaptotagmin 4 and 7 and the participation of voltage-gated calcium channels. J. Biol. Chem. 2011, 286, 23928–23937. [Google Scholar] [CrossRef] [PubMed]
- Burré, J.; Sharma, M.; Tsetsenis, T.; Buchman, V.; Etherton, M.R.; Südhof, T.C. Alpha-synuclein promotes SNARE-complex assembly in vivo and in vitro. Science 2010, 329, 1663–1667. [Google Scholar] [CrossRef]
- Tritsch, N.X.; Sabatini, B.L. Dopaminergic modulation of synaptic transmission in cortex and striatum. Neuron 2012, 76, 33–50. [Google Scholar] [CrossRef] [PubMed]
- Palij, P.; Bull, D.R.; Sheehan, M.J.; Millar, J.; Stamford, J.; Kruk, Z.L.; Humphrey, P.P. Presynaptic regulation of dopamine release in corpus striatum monitored in vitro and in real time by fast cyclic voltammetry. Brain Res. 1990, 509, 172–174. [Google Scholar] [CrossRef]
- Bull, D.R.; Sheehan, M.J. Presynaptic regulation of electrically evoked dopamine overflow in nucleus accumbens: A pharmacological study using fast scan cyclic voltammetry in vitro. Naunyn Schmiedebergs Arch. Pharmacol. 1991, 343, 260–265. [Google Scholar] [CrossRef]
- Beckstead, M.J.; Grandy, D.K.; Wickman, K.; Williams, J.T. Vesicular dopamine release elicits an inhibitory postsynaptic current in midbrain dopamine neurons. Neuron 2004, 42, 939–946. [Google Scholar] [CrossRef]
- Nirenberg, M.J.; Vaughan, R.A.; Uhl, G.R.; Kuhar, M.J.; Pickel, V.M. The dopamine transporter is localized to dendritic and axonal plasma membranes of nigrostriatal dopaminergic neurons. J. Neurosci. 1996, 16, 436–447. [Google Scholar] [CrossRef]
- German, C.L.; Baladi, M.G.; McFadden, L.M.; Hanson, G.R.; Fleckenstein, A.E. Regulation of the dopamine and vesicular monoamine transporters: Pharmacological targets and implications for disease. Pharmacol. Rev. 2015, 67, 1005–1024. [Google Scholar] [CrossRef]
- Schmitz, Y.; Benoit-Marand, M.; Gonon, F.; Sulzer, D. Presynaptic regulation of dopaminergic neurotransmission. J. Neurochem. 2003, 87, 273–289. [Google Scholar] [CrossRef]
- Gulley, J.M.; Zahnizer, N.R. Rapid regulation of dopamine transporter function by substrates, blockers, and presynaptic receptor ligands. Eur. J. Pharmacol. 2003, 479, 139–152. [Google Scholar] [CrossRef] [PubMed]
- Schmitt, K.C.; Reith, M.E. Regulation of the dopamine transporter: Aspects relevant to psychostimulant drugs of abuse. Ann. N. Y. Acad. Sci. 2010, 1187, 316–340. [Google Scholar] [CrossRef] [PubMed]
- Yang, H.; Rao, Z. Structural biology of SARS-CoV-2 and implications for therapeutic development. Nat. Rev. Microbiol. 2021, 19, 685–700. [Google Scholar] [CrossRef]
- Hoffmann, M.; Kleine-Weber, H.; Schroeder, S.; Krüger, N.; Herrler, T.; Erichsen, S.; Schiergens, T.S.; Herrler, G.; Wu, N.H.; Nitsche, A.; et al. SARS-CoV-2 cell entry depends on ACE2 and TMRPSS2 and is blocked by a clinically proven protease inhibitor. Cell 2020, 181, 271–280. [Google Scholar] [CrossRef] [PubMed]
- Khan, S.; Shafiei, M.S.; Longoria, C.; Schoggins, J.W.; Savani, R.C.; Zaki, H. SARS-CoV-2 spike protein induces inflammation via TLR2-dependent activation of the NF-κB pathway. eLife 2021, 10, e68563. [Google Scholar] [CrossRef]
- Taylor, M.P.; Enquist, L.W. Axonal spread of neuroinvasive viral infections. Trends Microbiol. 2015, 23, 283–288. [Google Scholar] [CrossRef]
- Harschnitz, O.; Studer, L. Human stem cell models to study host-virus interactions in the central nervous system. Nat. Rev. Immunol. 2021, 21, 441–453. [Google Scholar] [CrossRef] [PubMed]
- Iroeglu, J.D.; Ifenatuoha, C.W.; Ijomone, O.M. Potential neurological impact of coronaviruses: Implications for the novel SARS-CoV-2. Neurol. Sci. 2020, 41, 1329–1337. [Google Scholar] [CrossRef] [PubMed]
- Pellegrini, L.; Albecka, A.; Mallery, D.L.; Kellner, M.J.; Paul, D.; Carter, A.P.; James, L.C.; Lancaster, M.A. SARS-CoV-2 infects the brain choroid plexus and disrupts the blood-CSF barrier in human brain organoids. Cell Stem Cell 2020, 27, 951–961. [Google Scholar] [CrossRef]
- Stein, S.R.; Ramelli, S.C.; Grazioli, A.; Chung, J.Y.; Singh, M.; Yinda, C.K.; Winkler, C.W.; Sun, J.; Dickey, J.M.; Ylaya, K.; et al. SARS-CoV-2 infection and persistence in the human body and brain at autopsy. Nature 2022, 612, 758–763. [Google Scholar] [CrossRef]
- Yang, L.; Han, Y.; Nilsson-Payant, B.E.; Gupta, V.; Wang, P.; Duan, X.; Tang, X.; Zhu, J.; Zhao, Z.; Jaffré, F.; et al. A human pluripotent stem cell-based platform to study SARS-CoV-2 tropism and model virus infection in human cells and organoids. Cell Stem Cell 2020, 27, 125–136.e7. [Google Scholar] [CrossRef] [PubMed]
- Jacob, F.; Pather, S.R.; Huang, W.K.; Zhang, F.; Wong, S.Z.H.; Zhou, H.; Cubitt, B.; Fan, W.; Chen, C.Z.; Xu, M.; et al. Human pluripotent stem cell-derived neural cells and brain organoids reveal SARS-CoV-2 neurotropism predominates in choroid plexus epithelium. Cell Stem Cell 2020, 27, 937–950.e9. [Google Scholar] [CrossRef] [PubMed]
- Song, E.; Zhang, C.; Israelow, B.; Lu-Calligan, A.; Vietes Prado, A.; Skriabine, S.; Lu, P.; Weizman, O.E.; Liu, F.; Dai, Y.; et al. Neuroinvasion of SARS-CoV-2 in human and mouse brain. J. Exp. Med. 2021, 218, e20202135. [Google Scholar] [CrossRef] [PubMed]
- Chen, R.; Wang, K.; Yu, J.; Howard, D.; French, L.; Chen, Z.; Wen, C.; Xu, Z. The spatial and cell-type distribution of SARS-CoV-2 receptor ACE2 in the human and mouse brains. Front. Neurol. 2021, 11, 573095. [Google Scholar] [CrossRef] [PubMed]
- Cantuti-Castelvetri, L.; Ojha, R.; Pedro, L.D.; Djannatian, M.; Franz, J.; Kuivanen, S.; van der Meer, F.; Kallio, K.; Kaya, T.; Anastasina, M.; et al. Neuropilin-1 facilitates SARS-CoV-2 cell entry and infectivity. Science 2020, 370, 856–860. [Google Scholar] [CrossRef]
- Simmons, G.; Gosalia, D.N.; Rennekamp, A.J.; Reeves, A.D.; Diamond, S.L.; Bates, P. Inhibitors of cathepsin L prevent severe acute respiratory syndrome coronavirus entry. Proc. Natl. Acad. Sci. USA 2005, 102, 11876–11881. [Google Scholar] [CrossRef]
- Coutard, B.; Valle, C.; de Lamballerie, X.; Canard, B.; Seidah, N.G.; Decroly, E. The spike glycoprotein of the new coronavirus 2019-nCoV contains a furin-like cleavage site absent in CoV of the same clade. Antivir. Res. 2020, 176, 104742. [Google Scholar] [CrossRef]
- Wang, K.; Chen, W.; Zhang, Z.; Deng, Y.; Lian, J.Q.; Du, P.; Wei, D.; Zhang, Y.; Sun, X.X.; Gong, L.; et al. CD147-spike protein is a novel route for SARS-CoV-2 infection to host cells. Signal Transduct. Target. Ther. 2020, 5, 283. [Google Scholar] [CrossRef]
- Méndez-Guerrero, A.; Laespada-García, M.I.; Gómez-Grande, A.; Ruiz-Ortiz, M.; Blanco-Palmero, V.A.; Azcarate-Diaz, F.J.; Rábano-Suárez, P.; Álvarez-Torres, E.; de Fuenmayor-Fernández de la Hoz, C.P.; Vega Pérez, D.; et al. Acute hypokinetic-rigid syndrome following SARS-CoV-2 infection. Neurology 2020, 95, e2109–e2118. [Google Scholar] [CrossRef]
- Cohen, M.E.; Eichel, R.; Steiner-Birmanns, B.; Janah, A.; Ioshpa, M.; Bar-Shalom, R.; Paul, J.J.; Gaber, H.; Skrahina, V.; Bornstein, N.M.; et al. A case of probable Parkinson’s disease after SARS-CoV-2 infection. Lancet Neurol. 2020, 19, 804–805. [Google Scholar] [CrossRef]
- Merello, M.; Bhatia, K.P.; Obeso, J.A. SARS-CoV-2 and the risk of Parkinson’s disease: Facts and fantasy. Lancet Neurol. 2021, 20, 94–95. [Google Scholar] [CrossRef] [PubMed]
- Freidin, M.; Bennett, M.V.; Kessler, J.A. Cultured sympathetic neurons synthesize and release the cytokine interleukin 1 beta. Proc. Natl. Acad. Sci. USA 1992, 89, 10440–10443. [Google Scholar] [CrossRef] [PubMed]
- Bartfai, T.; Schultzberg, M. Cytokines in neuronal cell types. Neurochem. Int. 1993, 22, 435–444. [Google Scholar] [CrossRef] [PubMed]
- Yamamoto, T.; Yamashita, A.; Yamada, K.; Hata, R.I. Immunohistochemical localization of chemokine CXCL14 in rat hypothalamic neurons. Neurosci. Lett. 2011, 487, 335–340. [Google Scholar] [CrossRef]
- Garay, P.A.; McAllister, A.K. Novel roles for immune molecules in neural development: Implications for neurodevelopmental disorders. Front. Synaptic Neurosci. 2010, 2, 136. [Google Scholar] [CrossRef]
- Yirmiya, R.; Goshen, I. Immune modulation of learning, memory, neural plasticity and neurogenesis. Brain Behav. Immun. 2011, 25, 181–213. [Google Scholar] [CrossRef]
- Fajgenbaum, D.C.; June, C.H. Cytokine storm. N. Engl. J. Med. 2020, 383, 2255–2273. [Google Scholar] [CrossRef]
- Normandin, E.; Holroyd, K.B.; Collens, S.I.; Shaw, B.M.; Siddle, K.J.; Adams, G.; Rudy, M.; Solomon, I.H.; Anahtar, M.N.; Lemieux, J.E.; et al. Intrathecal inflammatory responses in the absence of SARS-CoV-2 nucleic acid in the CNS of COVID-19 hospitalized patients. J. Neurol. Sci. 2021, 430, 120023. [Google Scholar] [CrossRef]
- Pilotto, A.; Masciocchi, S.; Volonghi, I.; De Giuli, V.; Caprioli, F.; Mariotto, S.; Ferrari, S.; Bozzetti, S.; Imarisio, A.; Risi, B.; et al. Severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2) encephalitis is a cytokine release syndrome: Evidences from cerebrospinal fluid analysis. Clin. Infest. Dis. 2021, 73, e3019–e3026. [Google Scholar] [CrossRef]
- Guasp, M.; Muñoz-Sánchez, G.; Martínez-Hernández, E.; Santana, D.; Carbayo, A.; Naranjo, L.; Bolos, U.; Framil, M.; Saiz, A.; Balasa, M.; et al. CSF biomarkers in COVID-19 associated encephalopathy and encephalitis predict long-term outcome. Front. Immunol. 2022, 13, 1600. [Google Scholar] [CrossRef]
- Steinman, L. Inflammatory cytokines at the summits of pathological signal cascades in brain diseases. Sci. Signal 2013, 6, pe3. [Google Scholar] [CrossRef] [PubMed]
- Kinouchi, K.; Brown, G.; Pasternak, G.; Donner, D.B. Identification and characterization of receptors for tumor necrosis factor-alpha in the brain. Biochem. Biopharm. Res. Commun. 1991, 181, 1532–1538. [Google Scholar] [CrossRef] [PubMed]
- Szelenyi, J. Cytokines and the central nervous system. Brain Res. Bull. 2001, 54, 329–338. [Google Scholar] [CrossRef] [PubMed]
- Ban, E.; Milon, G.; Prudhomme, N.; Fillion, G.; Haour, F. Receptors for interleukin-1 (alpha and beta) in mouse brain: Mapping and neuronal localization in hippocampus. Neuroscience 1991, 43, 21–30. [Google Scholar] [CrossRef]
- Takao, T.; Tracey, D.E.; Mitchell, W.M.; De Souza, E.B. Interleukin-1 receptors in mouse brain: Characterization and neuronal localization. Endocrinology 1990, 127, 3070–3078. [Google Scholar] [CrossRef]
- Schneider, H.; Pitossi, F.; Balschun, D.; Wagner, A.; del Rey, A.; Besedovsky, H.O. A neuromodulatory role of interleukin-1beta in the hippocampus. Proc. Natl. Acad. Sci. USA 1998, 95, 7778–7783. [Google Scholar] [CrossRef]
- Avital, A.; Goshen, I.; Kamsler, A.; Segal, M.; Iverfeldt, K.; Richter-Levin, G.; Yirmiya, R. Impaired interleukin-1 signaling is associated with deficits in hippocampal memory processes and neural plasticity. Hippocampus 2003, 13, 826–834. [Google Scholar] [CrossRef]
- Schmid, A.W.; Lynch, M.A.; Herron, C.E. The effects of IL-1 receptor antagonist on beta amyloid mediated depression of LTP in the rat CA1 in vivo. Hippocampus 2009, 19, 670–676. [Google Scholar] [CrossRef]
- Braida, D.; Sacerdote, P.; Panerai, A.E.; Bianchi, M.; Aloisi, A.M.; Iosuè, S.; Sala, M. Cognitive function in young and adult IL (interleukin)-6 deficient mice. Behav. Brain Res. 2004, 153, 423–429. [Google Scholar] [CrossRef]
- Curran, B.; O’Connor, J. The pro-inflammatory cytokine interleukin-18 impairs long-term potentiation and NMDA receptor-mediated transmission in the rat hippocampus in vitro. Neuroscience 2001, 108, 83–90. [Google Scholar] [CrossRef]
- Cumiskey, D.; Pickering, M.; O’Connor, J.J. Interleukin-18 mediated inhibition of LTP in the rat dentate gyrus is attenuated in the presence of nGluR antagonists. Neurosci. Lett. 2007, 412, 206–210. [Google Scholar] [CrossRef] [PubMed]
- Filiano, A.J.; Xu, Y.; Tustison, N.J.; Marsh, R.L.; Baker, W.; Smirnov, I.; Overall, C.C.; Gadani, S.P.; Turner, S.D.; Weng, Z.; et al. Unexpected role of interferon-γ in regulating neuronal connectivity and social behavior. Nature 2016, 535, 425–429. [Google Scholar] [CrossRef] [PubMed]
- Belarbi, K.; Jopson, T.; Tweedie, D.; Arellano, C.; Luo, W.; Greig, N.H.; Rosi, S. TNF-α protein synthesis inhibitor restores neuronal function and reverses cognitive deficits induced by chronic neuroinflammation. J. Neuroinflamm. 2012, 9, 23. [Google Scholar] [CrossRef] [PubMed]
- Albensi, B.C.; Mattson, M.P. Evidence for the involvement of TNF and NF-kappaB in hippocampal synaptic plasticity. Synapse 2000, 35, 151–159. [Google Scholar] [CrossRef]
- Chen, C.; Itakura, E.; Nelson, G.M.; Sheng, M.; Laurent, P.; Fenk, L.A.; Butcher, R.A.; Hedge, R.S.; de Bono, M. IL-17 is a neuromodulator of Caernorhabditis elegans sensory responses. Nature 2017, 542, 43–48. [Google Scholar] [CrossRef] [PubMed]
- Di Filippo, M.; Mancini, A.; Bellingacci, L.; Gaetani, L.; Mazzocchetti, P.; Zelante, T.; La Barbera, L.; De Luca, A.; Tantucci, M.; Tozzi, A.; et al. Interleukin-17 affects synaptic plasticity and cognition in an experimental model of multiple sclerosis. Cell Rep. 2021, 37, 110094. [Google Scholar] [CrossRef] [PubMed]
- Centonze, D.; Muzio, L.; Rossi, S.; Cavasinni, F.; De Chiara, V.; Bergami, A.; Musella, A.; D’Amelio, M.; Cavallucci, V.; Martorana, A.; et al. Inflammation triggers synaptic alteration and degeneration in experimental autoimmune encephalomyelitis. J. Neurosci. 2009, 29, 3442–3452. [Google Scholar] [CrossRef]
- Mandolesi, G.; Grasselli, G.; Musella, A.; Gentile, A.; Musumeci, G.; Sepman, H.; Haji, N.; Fresegna, D.; Bernardi, G.; Centonze, D. GABAergic signaling and connectivity on Purkinje cells are impaired in experimental autoimmune encephalomyelitis. Neurobiol. Dis. 2012, 46, 414–424. [Google Scholar] [CrossRef]
- Haji, N.; Mandolesi, G.; Gentile, A.; Sacchetti, L.; Fresegna, D.; Rossi, S.; Musella, A.; Sepman, H.; Motta, C.; Studer, V.; et al. TNF-α-mediated anxiety in a mouse model of multiple sclerosis. Exp. Neurol. 2012, 237, 296–303. [Google Scholar] [CrossRef]
- Mandolesi, G.; Musella, A.; Gentile, A.; Grasselli, G.; Haji, N.; Sepman, H.; Fresegna, D.; Bullitta, S.; De Vito, F.; Musumeci, G.; et al. Interleukin-1β alters glutamate transmission at purkinje cell synapses in a mouse model of multiple sclerosis. J. Neurosci. 2013, 33, 12105–12121. [Google Scholar] [CrossRef]
- Di Filippo, M.; Tozzi, A.; Arcangeli, S.; de Iure, A.; Durante, V.; Di Gregorio, M.; Gardoni, F.; Calabresi, P. Interferon-β1a modulates glutamate neurotransmission in the CNS through CaMKII and GluN2A-containing NMDA receptors. Neuropharmacology 2016, 100, 98–105. [Google Scholar] [CrossRef] [PubMed]
- Rossi, S.; Furlan, R.; De Chiara, V.; Motta, C.; Studer, V.; Mori, F.; Musella, A.; Bergami, A.; Muzio, L.; Bernardi, G.; et al. Interleukin-1β causes synaptic hyperexcitability in multiple sclerosis. Ann. Neurol. 2012, 71, 76–83. [Google Scholar] [CrossRef] [PubMed]
- Rossi, S.; Studer, V.; Motta, C.; De Chiara, V.; Barbieri, F.; Bernardi, G.; Centonze, D. Inflammation inhibits GABA transmission in multiple sclerosis. Mult. Scler. 2012, 18, 1633–1635. [Google Scholar] [CrossRef]
- Zalcman, S.; Green-Johnson, J.M.; Murray, L.; Nance, D.M.; Dyck, D.; Anisman, H.; Greenberg, A.H. Cytokine-specific central monoamine alterations induced by interleukin-1, -2 and -6. Brain Res. 1994, 643, 40–49. [Google Scholar] [CrossRef]
- Mohankumar, P.S.; Thyagarajan, S.; Quadri, S.K. Interleukin-1 stimulates the release of dopamine and dihydroxylphenylacetic acid from the hypothalamus in vivo. Life Sci. 1991, 48, 925–930. [Google Scholar] [CrossRef] [PubMed]
- Shintani, F.; Kanba, S.; Nakaki, T.; Nibuya, M.; Kinoshita, N.; Suzuki, E.; Yagi, G.; Kato, R.; Asai, M. Interleukin-1 beta augments release of norepinephrine, dopamine, and serotonin in the rat anterior hypothalamus. J. Neurosci. 1993, 13, 3574–3581. [Google Scholar] [CrossRef]
- Merali, Z.; Lacosta, S.; Anisman, H. Effects of interleukin-1beta and mild stress on alterations of norepinephrine, dopamine and serotonin neurotransmission: A regional microdialysis study. Brain Res. 1997, 761, 225–235. [Google Scholar] [CrossRef]
- Gentile, A.; Fresegna, D.; Federici, M.; Musella, A.; Rizzo, F.R.; Sepman, H.; Bullitta, S.; De Vito, F.; Haji, N.; Rossi, S.; et al. Dopaminergic dysfunction is associated with IL-1b-dependent mood alterations in experimental autoimmune encephalomyelitis. Neurobiol. Dis. 2015, 74, 347–358. [Google Scholar] [CrossRef]
- Alonso, R.; Chaudieu, I.; Diorio, J.; Krishnamurthy, A.; Quirion, R.; Boksa, P. Interleukin-2 modulates evoked release of [3H] dopamine in rat cultured mesencephalic cells. J. Neurochem. 1993, 61, 1284–1290. [Google Scholar] [CrossRef]
- Lapchak, P.A. A role for interleukin-2 in the regulation of striatal dopaminergic function. Neuroreport 1992, 3, 165–168. [Google Scholar] [CrossRef]
- Anisman, H.; Kokkinidis, L.; Merali, Z. Interleukin-2 decreases accumbal dopamine efflux and responding for rewarding lateral hypothalamic stimulation. Brain Res. 1996, 731, 1–11. [Google Scholar] [CrossRef] [PubMed]
- Petitto, J.M.; McCarthy, D.B.; Rinker, C.M.; Huang, Z.; Getty, T. Modulation of behavioral and neurochemical measures of forebrain dopamine function in mice by species-specific interleukin-2. J. Neuroimmunol. 1997, 73, 183–190. [Google Scholar] [CrossRef] [PubMed]
- Shuto, H.; Kataoka, Y.; Horikawa, T.; Fujihara, N.; Oishi, R. Repeated interferon-alpha administration inhibits dopaminergic neural activity in the mouse brain. Brain Res. 1997, 747, 348–351. [Google Scholar] [CrossRef] [PubMed]
- Felger, J.C.; Alagbe, O.; Hu, F.; Mook, D.; Freeman, A.A.; Sanchez, M.M.; Kalin, N.H.; Ratti, E.; Nemeroff, C.B.; Miller, A.H. Effects of interferon-alpha on rhesus monkeys: A nonhuman primate model of cytokine-induced depression. Biol. Psychiatry 2007, 62, 1324–1333. [Google Scholar] [CrossRef]
- Felger, J.C.; Miller, A.H. Cytokine effects on the basal ganglia and dopamine function: The subcortical source of inflammatory malaise. Front. Neuroendocrinol. 2012, 33, 315–327. [Google Scholar] [CrossRef]
- Felger, J.C.; Mun, J.; Kimmel, H.L.; Nye, J.A.; Drake, D.F.; Hernandez, C.R.; Freeman, A.A.; Rye, D.B.; Goodman, M.M.; Howell, L.L.; et al. Chronic interferon-a decreases dopamine 2 receptor binding and striatal dopamine release in association with anhedonia-like behavior in nonhuman primates. Neuropsychopharmacology 2013, 38, 2179–2187. [Google Scholar] [CrossRef] [PubMed]
- Felger, J.C.; Hernandez, C.R.; Miller, A.H. Levodopa reverses cytokine-induced reductions in striatal dopamine release. Int. J. Neuropsychopharmacol. 2015, 18, pyu084. [Google Scholar] [CrossRef] [PubMed]
- Kumai, T.; Tateischi, T.; Tanaka, M.; Watanable, M.; Shimizu, H.; Kobayashi, S. Effect of interferon-alpha on tyrosine hydroxylase and catecholamine levels in the brain of rats. Life Sci. 2000, 67, 663–669. [Google Scholar] [CrossRef]
- Skrzydelski, D.; Guyon, A.; Daugé, V.; Rovère, C.; Apartis, E.; Kitabgi, P.; Nahon, J.L.; Rostène, W.; Mélik Parsadaniantz, S. The chemokine stromal cell-derived factor-1/CXCL12 activates the nigrostriatal dopamine system. J. Neurochem. 2007, 102, 1175–1183. [Google Scholar] [CrossRef]
- Guyon, A.; Skrzydelsi, D.; Rovère, C.; Rostène, W.; Mélik Parsadaniantz, S.; Nahon, J.L. Stromal cell-derived factor-1alpha modulation of the excitability of rat substantia nigra dopaminergic neurons: Presynaptic mechanisms. J. Neurochem. 2006, 96, 1540–1550. [Google Scholar] [CrossRef]
- Guyon, A.; Skrzydelski, D.; Rovère, C.; Apartis, E.; Rostène, W.; Kitabgi, P.; Mélik Parsadaniantz, S.; Nahon, J.L. Stromal-cell-derived factor 1alpha/CXCL12 modulates high-threshold calcium currents in rat substantia nigra. Eur. J. Neurosci. 2008, 28, 862–870. [Google Scholar] [CrossRef] [PubMed]
- Vumma, R.; Johansson, J.; Venizelos, N. Proinflammatory cytokines and oxidative stress decrease the transport of dopamine precursor tyrosine in human fibroblasts. Neuropsychobiology 2017, 75, 178–184. [Google Scholar] [CrossRef]
- Kitagami, T.; Yamada, K.; Miura, H.; Hashimoto, R.; Nabeshima, T.; Ohta, T. Mechanism of systemically injected interferon-alpha impeding monoamine biosynthesis in rats: Role of nitric oxide as a signal crossing the blood-brain barrier. Brain Res. 2003, 978, 104–114. [Google Scholar] [CrossRef] [PubMed]
- Felger, J.C.; Treadway, M.T. Inflammation effects on motivation and motor activity: Role of dopamine. Neuropsychopharmacology 2017, 42, 216–241. [Google Scholar] [CrossRef]
- Li, W.; Knowlton, D.; Woodward, W.R.; Habecker, B.A. Regulation of noradrenergic function by inflammatory cytokines and depolarization. J. Neurochem. 2003, 86, 774–783. [Google Scholar] [CrossRef] [PubMed]
- Felger, J.C.; Li, L.; Marvan, P.J.; Woolwine, B.J.; Harrison, D.G.; Raison, C.L.; Miller, A.H. Tyrosine metabolism during interferon-alpha administration: Association with fatigue and CSF dopamine concentrations. Brain Behav. Immun. 2013, 31, 153–160. [Google Scholar] [CrossRef]
- Zoller, H.; Schloegl, A.; Schroecksnadel, S.; Wolfgang, V.; Fuchs, D. Interferon-alpha therapy in patients with hepatitis C virus infection increases plasma phenylalanine and the phenylalanine to tyrosine ratio. J. Interferon Cytokine Res. 2012, 32, 216–220. [Google Scholar] [CrossRef]
- Kazumori, H.; Ishihara, S.; Rumi, M.A.; Ortega-Cava, C.F.; Kadowaki, Y.; Kinoshita, Y. Transforming growth factor-alpha directly augments histidine decarboxylase and vesicular monoamine transporter 2 production in rat enterochromaffin-like cells. Am. J. Physiol. Gastrointest. Liver Physiol. 2004, 286, G508–G514. [Google Scholar] [CrossRef]
- Morón, J.A.; Zakharova, I.; Ferrer, J.V.; Merrill, G.A.; Hope, B.; Lafer, E.M.; Lin, Z.C.; Wang, J.B.; Javitch, J.A.; Galli, A.; et al. Mitogen-activated protein kinase regulates dopamine transporter surface expression and dopamine transporter capacity. J. Neurosci. 2003, 23, 8480–8488. [Google Scholar] [CrossRef]
- Crunfli, F.; Carregari, V.C.; Veras, F.P.; Silva, L.S.; Nogueira, M.H.; Antunes, A.S.L.M.; Vendramini, P.H.; Valença, A.G.F.; Brandão-Teles, C.; da Silva Zuccoli, G.; et al. Morphological, cellular, and molecular basis of brain infection in COVID-19 patients. Proc. Natl. Acad. Sci. USA 2022, 119, e2200960119. [Google Scholar] [CrossRef]
- Asanuma, M.; Miyazaki, I.; Murakami, S.; Diaz-Corrales, F.J.; Ogawa, N. Striatal astrocytes act as a reservoir for L-DOPA. PLoS ONE 2014, 9, e106362. [Google Scholar] [CrossRef] [PubMed]
- Adermark, L.; Lagström, O.; Loftén, A.; Licheri, V.; Havenäng, A.; Loi, E.A.; Stomberg, R.; Söderpalm, B.; Domi, A.; Ericson, M. Astrocytes modulate extracellular neurotransmitter levels and excitatory neurotransmission in dorsolateral striatum via dopamine D2 receptor signaling. Neuropsychopharmacology 2022, 47, 1493–1502. [Google Scholar] [CrossRef]
- Schwarcz, R.; Stone, T.W. The kynurenine pathway and the brain: Challenges, controversies and promises. Neuropharmacology 2017, 112, 237–247. [Google Scholar] [CrossRef] [PubMed]
- Rassoulpour, A.; Wu, H.Q.; Ferre, S.; Schwarcz, R. Nanomolar concentrations of kynurenic acid reduce extracellular dopamine levels in the striatum. J. Neurochem. 2005, 93, 762–765. [Google Scholar] [CrossRef] [PubMed]
- Amori, L.; Wu, H.Q.; Marinozzi, M.; Pellicciari, R.; Guidetti, P.; Schwarcz, R. Specific inhibition of kynurenate synthesis enhances extracellular dopamine levels in the rodent striatum. Neuroscience 2009, 159, 196–203. [Google Scholar] [CrossRef]
- Carpenedo, R.; Pittaluga, A.; Cozzi, A.; Attucci, S.; Galli, A.; Raiteri, M.; Moroni, F. Presynaptic kynurenate-sensitive receptors inhibit glutamate release. Eur. J. Neurosci. 2001, 13, 2141–2147. [Google Scholar] [CrossRef] [PubMed]
- Rice, M.E.; Patel, J.C.; Cragg, S.J. Dopamine release in the basal ganglia. Neuroscience 2011, 198, 112–137. [Google Scholar] [CrossRef]
- Avshalumov, M.V.; Chen, B.T.; Marshall, S.P.; Peña, D.M.; Rice, M.E. Glutamate-dependent inhibition of dopamine release in striatum is mediated by a new diffusible messenger, H2O2. J. Neurosci. 2003, 23, 2744–2750. [Google Scholar] [CrossRef]
- Bernard, V.; Bolam, J.P. Subcellular and subsynaptic distribution of the NR1 subunit of the NMDA receptor in the neostriatum and globus pallidus of the rat: Co-localization at synapses with the GluR2/3 subunit of the AMPA receptor. Eur. J. Neurosci. 1998, 10, 3721–3736. [Google Scholar] [CrossRef]
- Avshalumov, M.V.; Rice, M.E. Activation of ATP-sensitive K+ (K(ATP)) channels by H2O2 underlies glutamate-dependent inhibition of striatal dopamine release. Proc. Natl. Acad. Sci. USA 2003, 100, 11729–11734. [Google Scholar] [CrossRef]
- Avshalumov, M.V.; Patel, J.C.; Rice, M.E. AMPA receptor-dependent H2O2 generation in striatal medium spiny neurons but not dopamine axons: One source of a retrograde signal that can inhibit dopamine release. J. Neurophysiol. 2008, 100, 1590–1601. [Google Scholar] [CrossRef] [PubMed]
- Wu, K.E.; Fazal, F.M.; Parker, K.R.; Zou, J.; Chang, H.Y. RNA-GPS predicts SARS-CoV-2 RNA residency to host mitochondria and nucleolus. Cell Syst. 2020, 11, 102–108.e3. [Google Scholar] [CrossRef] [PubMed]
- Mander, P.K.; Jekabsone, A.; Brown, G.C. Microglia proliferation is regulated by hydrogen peroxide from NADPH oxidase. J. Immunol. 2006, 176, 1046–1052. [Google Scholar] [CrossRef] [PubMed]
- Saha, R.N.; Pahan, K. Regulation of inducible nitric oxide synthase gene in glial cells. Antioxid. Redox Signal 2006, 8, 929–947. [Google Scholar] [CrossRef]
- Hartung, H.; Threlfell, S.; Cragg, S.J. Nitric oxide donors enhance the frequency dependence of dopamine release in nucleus accumbens. Neuropsychopharmacology 2011, 36, 1811–1822. [Google Scholar] [CrossRef]
- Kiss, J.P.; Hennings, E.C.; Zsilla, G.; Vizi, E.S. A possible role of nitric oxide in the regulation of dopamine transporter function in the striatum. Neurochem. Int. 1999, 34, 345–450. [Google Scholar] [CrossRef]
- Ara, J.; Przedborski, S.; Naini, A.B.; Jackson-Lewis, V.; Trilifetti, R.R.; Horwitz, J.; Ischiropoulos, H. Inactivation of tyrosine hydroxylase by nitration following exposure to peroxynitrite and 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine (MPTP). Proc. Natl. Acad. Sci. USA 1998, 95, 7659–7663. [Google Scholar] [CrossRef]
- Lin, Y.F.; Raab-Graham, K.; Jan, Y.N.; Jan, L.Y. NO stimulation of ATP-sensitive potassium channels: Involvement of Ras/mitogen-activated protein kinase pathway and contribution to neuroprotection. Proc. Natl. Acad. Sci. USA 2004, 101, 7799–7804. [Google Scholar] [CrossRef]
- Aviner, R.; Frydman, J. Proteostasis in viral infection: Unfolding the complex virus-chaperone interplay. Cold Spring Harb. Perspect. Biol. 2020, 12, a034090. [Google Scholar] [CrossRef]
- Balchin, D.; Hayer-Hartl, M.; Hartl, F.U. In vivo aspects of protein folding and quality control. Science 2016, 353, aac4354. [Google Scholar] [CrossRef]
- Bojkova, D.; Klann, K.; Kock, B.; Widera, M.; Krause, D.; Ciesek, S.; Cinatl, J.; Münch, C. Proteomics of SARS-CoV-2-infected host cells reveals therapy targets. Nature 2020, 583, 469–472. [Google Scholar] [CrossRef]
- Marateaux, L.; Campanelli, J.T.; Scheller, R.H. Synuclein: A neuron-specific protein localized to the nucleus and presynaptic nerve terminal. J. Neurosci. 1988, 8, 2804–2815. [Google Scholar] [CrossRef] [PubMed]
- George, J.M. The synuccleins. Genome Biol. 2002, 3, reviews3002.1. [Google Scholar] [CrossRef] [PubMed]
- Perez, R.G.; Waymire, J.C.; Lin, E.; Liu, J.J.; Guo, F.; Zigmond, M.J. A role for alpha-synuclein in the regulation of dopamine biosynthesis. J. Neurosci. 2002, 22, 3090–3099. [Google Scholar] [CrossRef] [PubMed]
- Liu, D.; Jin, L.; Wang, H.; Zhao, H.; Zhao, C.; Duan, C.; Lu, L.; Wu, B.; Yu, S.; Chan, P.; et al. Silencing alpha-synuclein gene expression enhances tyrosine hydroxylase activity in MN9D cells. Neurochem. Res. 2008, 33, 1401–1409. [Google Scholar] [CrossRef]
- Tehranian, R.; Montoya, S.E.; Van Laar, A.D.; Hastings, T.G.; Perez, R.G. Alpha-synuclein inhibits aromatic amino acid decarboxylase activity in dopaminergic cells. J. Neurochem. 2006, 99, 1188–1196. [Google Scholar] [CrossRef]
- Somayaji, M.; Cataldi, S.; Choi, S.J.; Edwards, R.H.; Mosharov, E.V.; Sulzer, D. A dual role for α-synuclein in facilitation and depression of dopamine release from substantia nigra neurons in vivo. Proc. Natl. Acad. Sci. USA 2020, 117, 32701–32710. [Google Scholar] [CrossRef]
- Yavich, L.; Tanila, H.; Vepsäläinen, S.; Jäkälä, P. Role of alpha-synuclein in presynaptic dopamine recruitment. J. Neurosci. 2004, 24, 11165–11170. [Google Scholar] [CrossRef]
- Fountaine, T.M.; Wade-Martins, R. RNA interference-mediated knockdown of alpha-synuclein protects human dopaminergic neuroblastoma cells from MPP(+) toxicity and reduces dopamine transport. J. Neurosci. Res. 2007, 85, 351–363. [Google Scholar] [CrossRef]
- Wersinger, C.; Sidhu, A. Attenuation of dopamine transporter activity by alpha-synuclein. Neurosci. Lett. 2003, 340, 189–192. [Google Scholar] [CrossRef]
- Venda, L.L.; Cragg, S.J.; Buchman, V.L.; Wade-Martins, R. α-Synuclein and dopamine at the crossroads of Parkinson’s disease. Trends Neurosci. 2010, 33, 559–568. [Google Scholar] [CrossRef] [PubMed]
- Gao, N.; Li, Y.H.; Li, X.; Yu, S.; Fu, G.L.; Chen, B. Effect of alpha-synuclein on the promoter activity of tyrosine hydroxylase gene. Neurosci. Bull. 2007, 23, 53–57. [Google Scholar] [CrossRef] [PubMed]
- Peng, X.; Tehranian, R.; Dietrich, P.; Stefanis, L.; Perez, R.G. Alpha-synuclein activation of protein phosphatase 2A reduces tyrosine hydroxylase phosphorylation in dopaminergic cells. J. Cell Sci. 2005, 118, 3523–3530. [Google Scholar] [CrossRef] [PubMed]
- Masliah, E.; Rockenstein, E.; Veinbergs, I.; Mallory, M.; Hashimoto, M.; Takeda, A.; Sagara, Y.; Sisk, A.; Mucke, L. Dopaminergic loss and inclusion body formation in alpha-synuclein mice: Implications for neurodegenerative disorders. Science 2000, 287, 1265–1269. [Google Scholar] [CrossRef]
- Kirik, D.; Rosenblad, C.; Burger, C.; Lundberg, C.; Johansen, T.E.; Muzyczka, N.; Mandel, R.J.; Björklund, A. Parkinson-Like Neurodegeneration Induced by Targeted Overexpression of α-Synuclein in the Nigrostriatal System. J. Neurosci. 2002, 22, 2780–2791. [Google Scholar] [CrossRef]
- Yavich, L.; Oksman, M.; Tanila, H.; Kerokoski, P.; Hiltunen, M.; van Groen, T.; Puoliväli, J.; Männistö, P.T.; García-Horsman, A.; MacDonald, E.; et al. Locomotor activity and evoked dopamine release are reduced in mice overexpressing A30P-mutated human alpha-synuclein. Neurobiol. Dis. 2005, 20, 303–313. [Google Scholar] [CrossRef]
- Larsen, K.E.; Schmitz, Y.; Troyen, M.D.; Mosharov, E.; Dietrich, P.; Quazi, A.Z.; Savalle, M.; Nemani, V.; Chaudhry, F.A.; Edwards, R.H.; et al. Alpha-synuclein overexpression in PC12 and chromaffin cells impairs catecholamine release by interfering with a late step in exocytosis. J. Neurosci. 2006, 26, 11915–11922. [Google Scholar] [CrossRef]
- Nemani, V.M.; Lu, W.; Berge, V.; Nakamura, K.; Onoa, B.; Lee, M.K.; Chaudhry, F.A.; Nicoll, R.A.; Edwards, R.H. Increased expression of alpha-synuclein reduces neurotransmitter release by inhibiting synaptic vesicle reclustering after endocytosis. Neuron 2010, 65, 66–79. [Google Scholar] [CrossRef]
- Gitler, A.D.; Bevis, B.J.; Shorter, J.; Strathearn, K.E.; Hamamichi, S.; Su, L.J.; Caldwell, K.A.; Caldwell, G.A.; Rochet, J.C.; McCaffery, J.M.; et al. The Parkinson’s disease protein alpha-synuclein disrupts cellular Rab homeostasis. Proc. Natl. Acad. Sci. USA 2008, 105, 145–150. [Google Scholar] [CrossRef]
- Barbut, D.; Stolzenberg, E.; Zasloff, M. Gastrointestinal immunity and alpha-synuclein. J. Parkinson Dis. 2019, 9, S313–S322. [Google Scholar] [CrossRef]
- Greten-Harrison, B.; Polydoro, M.; Morimoto-Tomita, M.; Diao, L.; Williams, A.M.; Nie, E.H.; Makani, S.; Tian, N.; Castillo, P.E.; Buchman, V.L.; et al. αβγ-synuclein triple knockout mice reveal age-dependent neuronal dysfunction. Proc. Natl. Acad. Sci. USA 2010, 107, 19573–19578. [Google Scholar] [CrossRef] [PubMed]
- Beatman, E.L.; Massey, A.; Shives, K.D.; Burrack, K.S.; Chamanian, M.; Morrison, T.E.; Beckham, J.D. Alpha-synuclein expression restricts RNA viral infections in the brain. J. Virol. 2015, 90, 2767–2782. [Google Scholar] [CrossRef] [PubMed]
- Tomlinson, J.J.; Shutinoski, B.; Dong, L.; Meng, F.; Elleithy, D.; Lengacher, N.A.; Nguyen, A.P.; Cron, G.O.; Jiang, Q.; Roberson, E.D.; et al. Holocranohistochemistry enables the visualization of α-synuclein expression in the murine olfactory system and discovery of its systemic anti-microbial effects. J. Neural Transm. 2017, 124, 721–738. [Google Scholar] [CrossRef] [PubMed]
- Stolzenberg, E.; Berry, D.; Yang, D.; Lee, E.Y.; Kroemer, A.; Kaufman, S.; Wong, G.C.L.; Oppenheim, J.J.; Sen, S.; Fishbein, T.; et al. A role for neuronal alpha-synuclein in gastrointestinal immunity. J. Innate Immun. 2017, 9, 456–463. [Google Scholar] [CrossRef]
- Alam, M.M.; Yang, D.; Li, X.Q.; Liu, J.; Back, T.C.; Trivett, A.; Karim, B.; Barbut, D.; Zasloff, M.; Oppenheim, J.J. Alpha-synuclein, the culprit in Parkinson’s disease, is required for normal immune function. Cell Rep. 2022, 38, 110090. [Google Scholar] [CrossRef]
- Tanji, K.; Mori, F.; Imaizumi, T.; Yoshida, H.; Matsumiya, T.; Tamo, W.; Yoshimoto, M.; Odagiri, H.; Aasaki, M.; Takahashi, H.; et al. Upregulation of alpha-synuclein by lipopolysaccharide and interleukin-1 in human macrophages. Pathol. Int. 2002, 52, 572–577. [Google Scholar] [CrossRef]
- Bae, E.J.; Choi, M.; Kim, J.T.; Kim, D.K.; Jung, M.K.; Kim, C.; Kim, T.K.; Lee, J.S.; Jung, B.C.; Shin, S.J.; et al. TNF-α promotes α-synuclein propagation through stimulation of senescence-associated lysosomal exocytosis. Exp. Mol. Med. 2022, 54, 788–800. [Google Scholar] [CrossRef]
- Labrie, V.; Brundin, P. Alpha-synuclein to the rescue: Immune cell recruitment by alpha-synuclein during gastrointestinal infection. J. Innate Immun. 2017, 9, 437–440. [Google Scholar] [CrossRef]
- Shameli, A.; Xiao, W.; Zheng, Y.; Shyu, S.; Sumodi, J.; Meyerson, H.; Harding, C.V.; Maitta, R.W. A critical role for alpha-synuclein in development and function of T lymphocytes. Immunobiology 2016, 221, 333–340. [Google Scholar] [CrossRef]
- Kasen, A.; Houck, C.; Burmeister, A.R.; Sha, Q.; Brundin, L.; Brundin, P. Upregulation of α-synuclein following immune activation: Possible trigger of Parkinson’s disease. Neurobiol. Dis. 2022, 166, 105654. [Google Scholar] [CrossRef]
- Scheiblich, H.; Bousset, L.; Schwartz, S.; Griep, A.; Latz, E.; Melki, R.; Heneka, M.T. Microglial NLRP3 inflammasome activation upon TLR2 and TLR5 ligation by distinct α-synuclein assemblies. J. Immunol. 2021, 207, 2143–2154. [Google Scholar] [CrossRef] [PubMed]
- Trudler, D.; Nazor, K.L.; Eisele, Y.S.; Grabauskas, T.; Dolatabadi, N.; Parker, J.; Sultan, A.; Zhong, Z.; Goodwin, M.S.; Levites, Y.; et al. Soluble α-synuclein-antibody complexes activate the NLRP3 inflammasome in hiPSC-derived microglia. Proc. Natl. Acad. Sci. USA 2021, 118, e2025847118. [Google Scholar] [CrossRef] [PubMed]
- Lee, H.J.; Suk, J.E.; Bae, E.J.; Lee, S.J. Clearance and deposition of extracellular alpha-synuclein aggregates in microglia. Biochem. Biophys. Res. Commun. 2008, 372, 423–428. [Google Scholar] [CrossRef] [PubMed]
- Sarkar, S.; Dammer, E.B.; Malovic, E.; Olsen, A.L.; Raza, S.A.; Gao, T.; Xiao, H.; Oliver, D.L.; Duong, D.; Joers, V.; et al. Molecular Signatures of Neuroinflammation Induced by αSynuclein Aggregates in Microglial Cells. Front. Immunol. 2020, 11, 33. [Google Scholar] [CrossRef] [PubMed]
- Semerdzhiev, S.A.; Fakhree, M.A.A.; Segers-Nolten, I.; Blum, C.; Claessens, M.M.A.E. Interactions between SARS-CoV-2 N-protein and α-synclein accelerate amyloid formation. ACS Chem. Neurosci. 2022, 13, 143–150. [Google Scholar] [CrossRef]
- Wu, Z.; Zhang, X.; Huang, Z.; Ma, K. SARS-CoV-2 proteins interact with alpha synuclein and induce Lewy body-like pathology in vitro. Int. J. Mol. Sci. 2022, 23, 3394. [Google Scholar] [CrossRef]
- Albornoz, E.A.; Amarilla, A.A.; Modhiran, N.; Parker, S.; Li, X.X.; Wijesundara, D.K.; Aguado, J.; Zamora, A.P.; McMillan, C.L.D.; Liang, B.; et al. SARS-CoV-2 drives NLRP3 inflammasome activation in human microglia through spike protein. Mol. Psychiatry 2022. ahead of print. [Google Scholar] [CrossRef]
- Fellner, L.; Irschick, R.; Schanda, K.; Reindl, M.; Klimaschewski, L.; Poewe, W.; Wenning, G.K.; Stefanova, N. Toll-like receptor 4 is required for a-synuclein dependent activation of microglia and astroglia. Glia 2013, 61, 349–360. [Google Scholar] [CrossRef]
- Zhao, Y.; Kuang, M.; Li, J.; Zhu, L.; Jia, Z.; Guo, X.; Hu, Y.; Kong, J.; Yin, H.; Wang, X.; et al. SARS-CoV-2 spike protein interacts with and activates TLR41. Cell Res. 2021, 31, 818–820. [Google Scholar] [CrossRef]
- Spillantini, M.G.; Schmidt, M.L.; Lee, V.M.; Trojanowski, J.Q.; Jakes, R.; Goedert, M. Alpha-synuclein in Lewy bodies. Nature 1997, 388, 839–840. [Google Scholar] [CrossRef]
- Dickson, D.W.; Fujishiro, H.; Orr, C.; DelleDonne, A.; Josephs, K.A.; Frigerio, R.; Burnett, M.; Parisi, J.E.; Klos, K.J.; Ahlskog, J.E. Neuropathology of non-motor features of Parkinson disease. Parkinsonism Relat. Disord. 2009, 15 (Suppl. 3), S1–S5. [Google Scholar] [CrossRef] [PubMed]
- Wolters, E.C. Non-motor extranigral signs and symptoms in Parkinson’s disease. Parkinsonism Relat. Disord. 2009, 15 (Suppl. 3), S6–S12. [Google Scholar] [CrossRef] [PubMed]
- Sulzer, D. Multiple hit hypotheses for dopamine neuron loss in Parkinson’s disease. Trends Neurosci. 2007, 30, 244–250. [Google Scholar] [CrossRef]
- Picconi, B.; Pisani, A.; Barone, I.; Bonsi, P.; Centonze, D.; Bernardi, G.; Calabresi, P. Pathological synaptic plasticity in the striatum: Implications for Parkinson’s disease. Neurotoxicology 2005, 26, 779–783. [Google Scholar] [CrossRef] [PubMed]
- Imbriani, P.; Martella, G.; Bonsi, P.; Pisani, A. Oxidative stress and synaptic dysfunction in rodent models of Parkinson’s disease. Neurobiol. Dis. 2022, 173, 105851. [Google Scholar] [CrossRef]
- Blauwendraat, C.; Nalls, M.A.; Singleton, A.B. The genetic architecture of Parkinson’s disease. Lancet Neurol. 2020, 19, 170–178. [Google Scholar] [CrossRef]
- Kline, E.M.; Houser, M.C.; Herrick, M.K.; Seibler, P.; Klein, C.; West, A.; Tansey, M.G. Genetic and environmental factors in Parkinson’s disease converge on immune function and inflammation. Mov. Disord. 2021, 36, 25–36. [Google Scholar] [CrossRef]
- Harms, A.S.; Ferreira, S.A.; Romero-Ramos, M. Periphery and brain, innate and adaptive immunity in Parkinson’s disease. Acta Neuropathol. 2021, 141, 527–545. [Google Scholar] [CrossRef]
- Hirsch, E.C.; Standaert, D.G. Ten unsolved questions about neuroinflammation in Parkinson’s disease. Mov. Disord. 2020, 36, 16–24. [Google Scholar] [CrossRef]
- Smeyne, R.J.; Noyce, A.J.; Byrne, M.; Savica, R.; Marras, C. Infection and risk of Parkinson’s disease. J. Parkinsons Dis. 2021, 11, 31–43. [Google Scholar] [CrossRef]
- Casals, J.; Elizan, T.S.; Yahr, M.D. Postencephalitic parkinsonism—A review. J. Neural. Transm. 1998, 105, 645–676. [Google Scholar] [CrossRef] [PubMed]
- Krusz, J.C.; Koller, W.C.; Ziegler, D.K. Historical review: Abnormal movements associated with epidemic encephalitis lethargica. Mov. Disord. 1987, 2, 137–141. [Google Scholar] [CrossRef] [PubMed]
- Hoffman, L.A.; Vilensky, J.A. Encephalitis lethargica: 100 years after the epidemic. Brain 2017, 140, 2246–2251. [Google Scholar] [CrossRef] [PubMed]
- Cadar, D.; Jellinger, K.A.; Riederer, P.; Strobel, S.; Monoranu, C.M.; Tappe, D. No metagenomics evidence of causative viral pathogens in postencephalitic parkinsonism following encephalitis lethargica. Microrganisms 2021, 9, 1716. [Google Scholar] [CrossRef]
- Leta, V.; Urso, D.; Batzu, L.; Lau, Y.H.; Mathew, D.; Boura, I.; Raeder, V.; Falup-Pecurariu, C.; van Wamelen, D.; Ray Chaudhuri, K. Viruses, parkinsonism and Parkinson’s disease: The past, present and future. J. Neural. Transm. 2022, 129, 1119–1132. [Google Scholar] [CrossRef]
- Jang, H.; Boltz, D.A.; Webster, R.G.; Smeyne, R.J. Viral Parkinsonism. Biochim. Biophys. Acta 2009, 1792, 714–721. [Google Scholar] [CrossRef]
- Bopeththa, B.V.K.M.; Ralapanawa, U. Post encephalic parkinsonism following dengue viral infection. BMC Res. Notes 2017, 10, 655. [Google Scholar] [CrossRef]
- Guan, J.; Lu, Z.; Zhou, Q. Reversible parkinsonism due to involvement of substantia nigra in Epstein-Barr virus encephalitis. Mov. Disord. 2012, 27, 156–157. [Google Scholar] [CrossRef]
- Pradhan, S.; Pandey, N.; Shashank, S.; Gupta, R.K.; Mathur, A. Parkinsonism due to predominant involvement of substantia nigra in Japanese encephalitis. Neurology 1999, 53, 1782–1786. [Google Scholar] [CrossRef]
- Pasha, S.A.; Pasha, S.A.; Suhasini, T.; Rao, D.A. Hepatitis E virus-associated acute encephalitic Parkinsonism. J. Assoc. Physicians India 2018, 66, 92–93. [Google Scholar]
- Leta, V.; Boura, I.; van Wamelan, D.J.; Rodriguez-Violante, M.; Antonini, A.; Chaudhuri, K.R. COVID-19 and Parkinson’s disease: Acute clinical implications, long-COVID and post-COVID-19 parkinsonism. Int. Rev. Neurobiol. 2022, 165, 63–89. [Google Scholar] [CrossRef] [PubMed]
- Ellul, M.A.; Benjamin, L.; Singh, B.; Lant, S.; Michael, B.D.; Easton, A.; Kneen, R.; Defres, S.; Sejvar, J.; Solomon, T. Neurological associations of COVID-19. Lancet Neurol. 2020, 19, 767–783. [Google Scholar] [CrossRef] [PubMed]
- Morassi, M.; Palmerini, F.; Nici, S.; Magni, E.; Savelli, G.; Guerra, U.P.; Chieregato, M.; Morbelli, S.; Vogrig, A. SARS-CoV-2-related encephalitis with prominent parkinsonism: Clinical and FDG-PET correlates in two patients. J. Neurol. 2021, 268, 3980–3987. [Google Scholar] [CrossRef] [PubMed]
- Jellinger, K.A. Absence of alpha-synuclein pathology in postencephalitic parkinsonism. Acta Neuropathol. 2009, 118, 371–379. [Google Scholar] [CrossRef]
- Brydon, L.; Harrison, N.A.; Walker, C.; Steptoe, A.; Critchley, H.D. Peripheral inflammation is associated with altered substantia nigra activity and psychomotor slowing in humans. Biol. Psychiatry 2008, 63, 1022–1029. [Google Scholar] [CrossRef]
- Capuron, L.; Pagnoni, G.; Demetrashvili, M.F.; Lawson, D.H.; Fornwalt, F.B.; Woolwine, B.; Berns, G.S.; Nemeroff, C.B.; Miller, A.H. Basal ganglia hypermetabolism and symptoms of fatigue during interferon-alpha therapy. Neuropsychopharmacology 2007, 32, 2384–2392. [Google Scholar] [CrossRef]
- Capuron, L.; Pagnoni, G.; Drake, D.F.; Woolwine, B.J.; Spivey, J.R.; Crowe, R.J.; Votaw, J.R.; Goodman, M.M.; Miller, A.H. Dopaminergic mechanisms of reduced basal ganglia responses to hedonic reward during interferon alfa administration. Arch. Gen. Psychiatry 2012, 69, 1044–1053. [Google Scholar] [CrossRef]
- Majer, M.; Welberg, L.A.M.; Capuron, L.; Pagnoni, G.; Raison, C.L.; Miller, A.H. IFN-alpha-induced motor slowing is associated with increased depression and fatigue in patients with chronic hepatitis C. Brain Behav. Immun. 2008, 22, 870–880. [Google Scholar] [CrossRef]
- Capuron, L.; Dantzer, R. Cytokines and depression: The need for a new paradigm. Brain Behav. Immun. 2003, 17 (Suppl. 1), S119–S124. [Google Scholar] [CrossRef]
- Raison, C.L.; Borison, A.V.; Majer, M.; Drake, D.F.; Pagnoni, G.; Woolwine, B.J.; Vogt, G.J.; Massung, B.; Miller, A.H. Activation of central nervous system inflammatory pathways by interferon-alpha: Relationship to monoamines and depression. Biol. Psychiatry 2009, 65, 296–303. [Google Scholar] [CrossRef]
- Felger, J.C.; Li, Z.; Haroon, E.; Woolwine, B.J.; Jung, M.Y.; Hu, X.; Miller, A.H. Inflammation is associated with decreased functional connectivity within corticostriatal reward circuitry in depression. Mol. Psychiatry 2016, 21, 1358–1365. [Google Scholar] [CrossRef] [PubMed]
- Goldsmith, D.R.; Haroon, E.; Woolwine, B.J.; Jung, M.Y.; Wommack, E.C.; Harvey, P.D.; Treadway, M.T.; Felger, J.C.; Miller, A.H. Inflammatory markers are associated with decreased psychomotor speed in patients with major depressive disorder. Brain Behav. Immun. 2016, 56, 281–288. [Google Scholar] [CrossRef]
- Juengling, F.D.; Ebert, D.; Gut, O.; Engelbrecht, M.A.; Rasenack, J.; Nitzsche, E.U.; Bauer, J.; Lieb, K. Prefrontal cortical hypometabolism during low-dose interferon alpha treatment. Psychopharmacology 2000, 152, 383–389. [Google Scholar] [CrossRef] [PubMed]
- Eidelberg, D.; Moeller, J.R.; Dhawan, V.; Spetsieris, P.; Takikawa, S.; Ishikawa, T.; Chaly, T.; Robeson, W.; Margouleff, D.; Przedborski, S. The metabolic topography of parkinsonism. J. Cereb. Blood Flow Metab. 1994, 14, 783–801. [Google Scholar] [CrossRef] [PubMed]
- Mentis, M.J.; McIntosh, A.R.; Perrine, K.; Dhawan, V.; Berlin, B.; Feigin, A.; Edwards, C.; Mattis, P.; Eidelberg, D. Relationships among the metabolic patterns that correlate with mnemonic, visuospatial, and mood symptoms in Parkinson’s disease. Am. J Psychiatry 2002, 159, 746–754. [Google Scholar] [CrossRef] [PubMed]
- Spetsieris, P.G.; Moeller, J.R.; Dhawan, V.; Ishikawa, T.; Eidelberg, D. Visualizing the evolution of abnormal metabolic networks in the brain using PET. Comput. Med. Imaging Graph. 1995, 19, 295–306. [Google Scholar] [CrossRef] [PubMed]
- Wichmann, T.; DeLong, M.R. Oscillations in the basal ganglia. Nature 1999, 400, 621–622. [Google Scholar] [CrossRef]
- Reite, M.; Laudenslager, M.; Jones, J.; Crnic, L.; Kaemingk, K. Interferon decreases REM latency. Biol. Psychiatry 1987, 22, 104–107. [Google Scholar] [CrossRef]
- Kostić, V.S.; Susić, V.; Covicković-Sternić, N.; Marinković, Z.; Janković, S. Reduced rapid eye movement sleep latency in patients with Parkinson’s disease. J. Neurol. 1989, 236, 421–423. [Google Scholar] [CrossRef]
- Kumakura, Y.; Cumming, P. PET studies of cerebral levodopa metabolism: A review of clinical findings and modeling approaches. Neuroscientist 2009, 15, 635–650. [Google Scholar] [CrossRef]
- Kumakura, Y.; Gjedde, A.; Danielsen, E.H.; Christensen, S.; Cumming, P. Dopamine storage capacity in caudate and putamen of patients with early Parkinson’s disease: Correlation with asymmetry of motor symptoms. J. Cereb. Blood Flow Metab. 2006, 26, 358–370. [Google Scholar] [CrossRef] [PubMed]
- Bersano, A.; Aghemo, A.; Rumi, M.G.; Ballabio, E.; Candelise, L.; Colombo, M. Recovery after L-DOPA treatment in peginterferon and ribavirin induced parkinsonism. Eur. J. Intern. Med. 2008, 19, 370–371. [Google Scholar] [CrossRef] [PubMed]
- Sarasombath, P.; Sumida, K.; Kaku, D.A. Parkinsonism associated with interferon alpha therapy for chronic myelogenous leukemia. Hawaii Med. J. 2002, 61, 57. [Google Scholar] [PubMed]


Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Mancini, M.; Natoli, S.; Gardoni, F.; Di Luca, M.; Pisani, A. Dopamine Transmission Imbalance in Neuroinflammation: Perspectives on Long-Term COVID-19. Int. J. Mol. Sci. 2023, 24, 5618. https://doi.org/10.3390/ijms24065618
Mancini M, Natoli S, Gardoni F, Di Luca M, Pisani A. Dopamine Transmission Imbalance in Neuroinflammation: Perspectives on Long-Term COVID-19. International Journal of Molecular Sciences. 2023; 24(6):5618. https://doi.org/10.3390/ijms24065618
Chicago/Turabian StyleMancini, Maria, Silvia Natoli, Fabrizio Gardoni, Monica Di Luca, and Antonio Pisani. 2023. "Dopamine Transmission Imbalance in Neuroinflammation: Perspectives on Long-Term COVID-19" International Journal of Molecular Sciences 24, no. 6: 5618. https://doi.org/10.3390/ijms24065618
APA StyleMancini, M., Natoli, S., Gardoni, F., Di Luca, M., & Pisani, A. (2023). Dopamine Transmission Imbalance in Neuroinflammation: Perspectives on Long-Term COVID-19. International Journal of Molecular Sciences, 24(6), 5618. https://doi.org/10.3390/ijms24065618