
NAADP-Evoked Ca2+ Signaling Leads to Mutant Huntingtin Aggregation and Autophagy Impairment in Murine Astrocytes

 , ,
, ,  , , , and
, , , and
Abstract
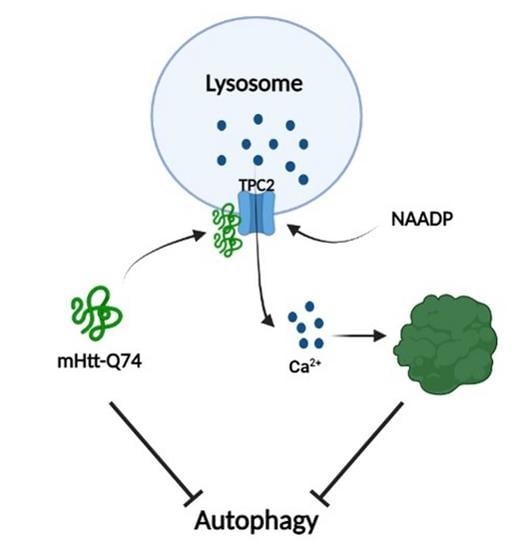
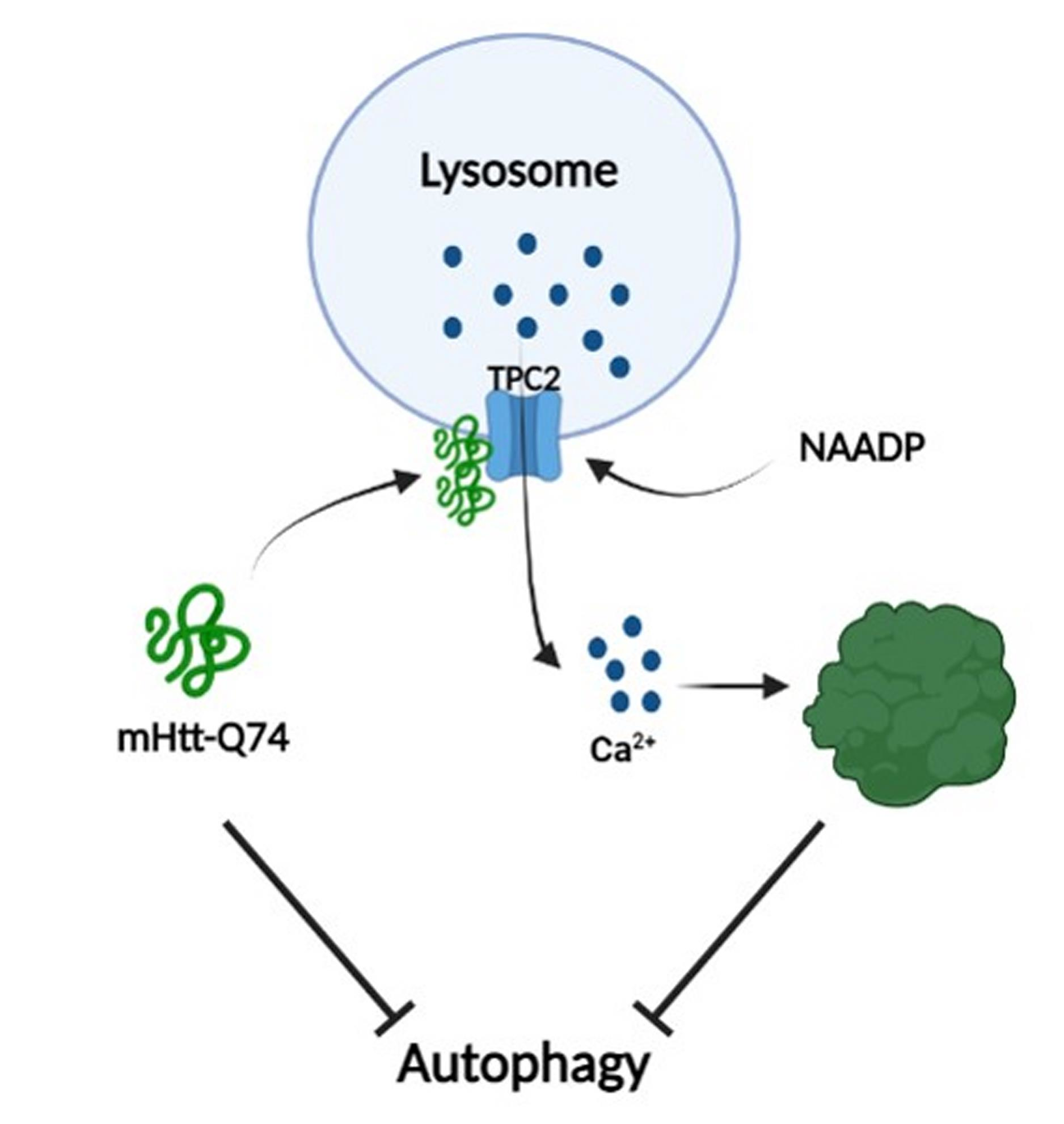{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Results
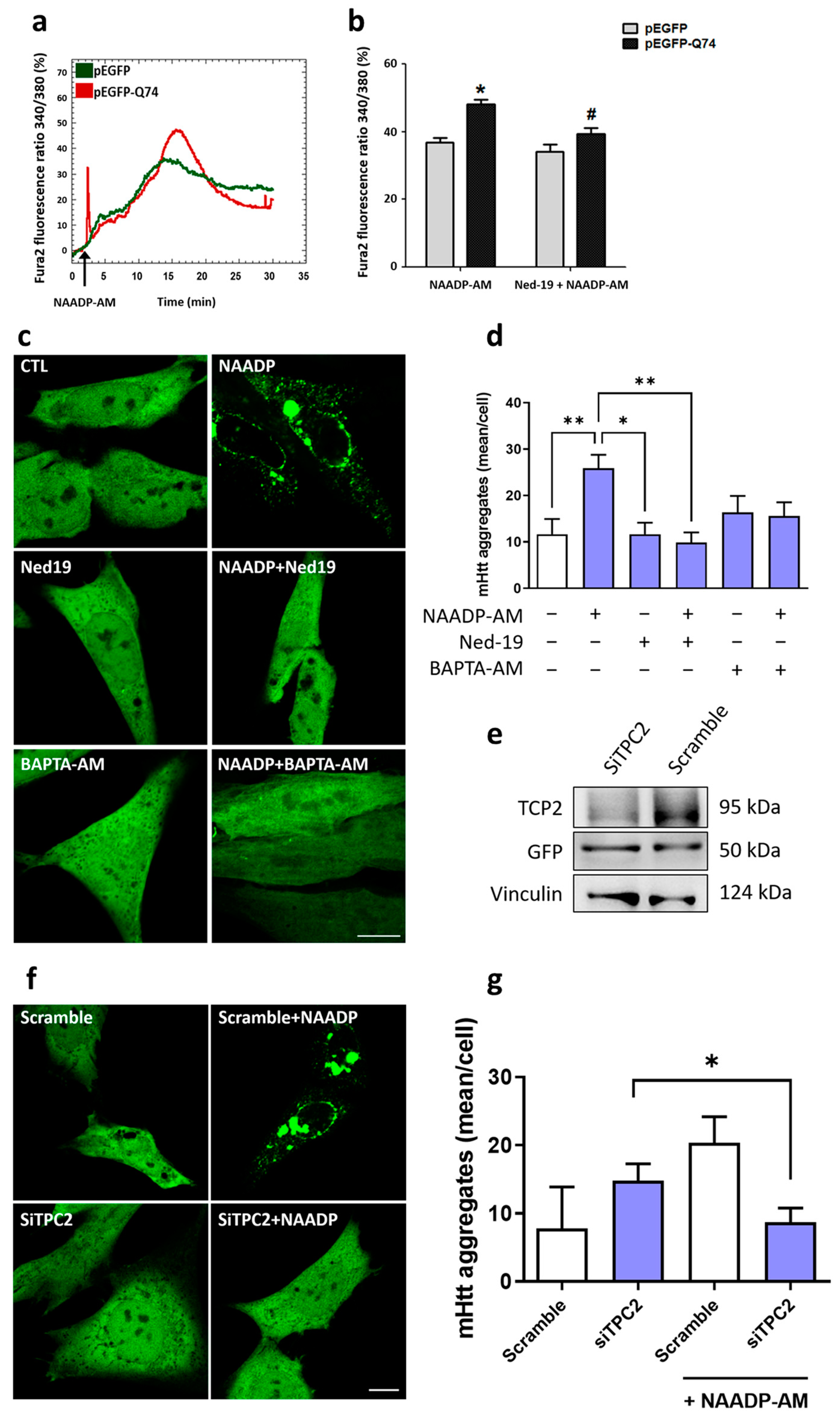
2.1. Overexpression of mHtt-Q74 Enhances NAADP-Mediated Ca2+ Release
2.2. NAADP Induces mHtt-Q74 Aggregation
2.3. mHtt-Q74 Colocalizes with TPC2 Receptor
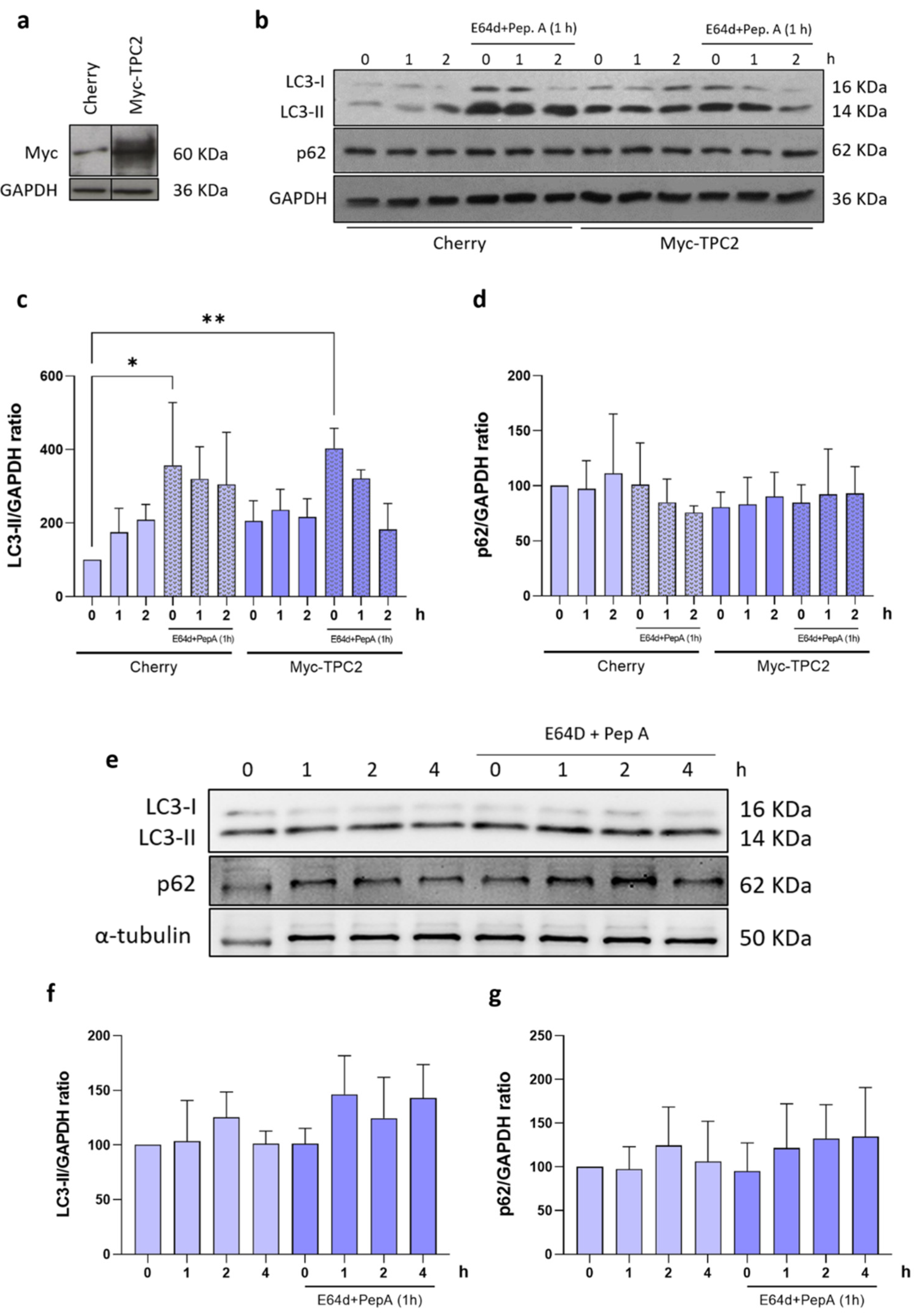
2.4. mHtt-Q74 Inhibits Autophagy in Astrocytes
3. Discussion
4. Materials and Methods
4.1. Cell Culture, Transfections and Retroviral Infection
4.2. Ca2+ Measurements
4.3. Western Blotting
4.4. mHtt Q74 Aggregation Assay
4.5. Colocalization of mHtt-Q74 and TPC2 Receptor
4.6. Statistical Analysis
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Pringsheim, T.; Wiltshire, K.; Day, L.; Dykeman, J.; Steeves, T.; Jette, N. The incidence and prevalence of Huntington’s disease: A systematic review and meta-analysis. Mov. Disord. 2012, 27, 1083–1091. [Google Scholar] [CrossRef] [PubMed]
- Cepeda, C.; Tong, X.P. Huntington’s disease: From basic science to therapeutics. CNS Neurosci. Ther. 2018, 24, 247–249. [Google Scholar] [CrossRef]
- Wyant, K.J.; Ridder, A.J.; Dayalu, P. Huntington’s Disease-Update on Treatments. Curr. Neurol. Neurosci. Rep. 2017, 17, 33. [Google Scholar] [CrossRef]
- Cortes, C.J.; La Spada, A.R. The many faces of autophagy dysfunction in Huntington’s disease: From mechanism to therapy. Drug Discov. Today 2014, 19, 963–971. [Google Scholar] [CrossRef] [PubMed]
- Snowden, J.S. The Neuropsychology of Huntington’s Disease. Arch. Clin. Neuropsychol. 2017, 32, 876–887. [Google Scholar] [CrossRef] [PubMed]
- MacDonald, M.E.; Ambrose, C.M.; Duyao, M.P.; Myers, R.H.; Lin, C.; Srinidhi, L.; Barnes, G.; Taylor, S.A.; James, M.; Groot, N.; et al. A novel gene containing a trinucleotide repeat that is expanded and unstable on Huntington’s disease chromosomes. Cell 1993, 72, 971–983. [Google Scholar] [CrossRef] [PubMed]
- Gusella, J.F.; Wexler, N.S.; Conneally, P.M.; Naylor, S.L.; Anderson, M.A.; Tanzi, R.E.; Watkins, P.C.; Ottina, K.; Wallace, M.R.; Sakaguchi, A.Y.; et al. A polymorphic DNA marker genetically linked to Huntington’s disease. Nature 1983, 306, 234–238. [Google Scholar] [CrossRef]
- Saudou, F.; Humbert, S. The Biology of Huntingtin. Neuron 2016, 89, 910–926. [Google Scholar] [CrossRef]
- Jimenez-Sanchez, M.; Licitra, F.; Underwood, B.R.; Rubinsztein, D.C. Huntington’s Disease: Mechanisms of Pathogenesis and Therapeutic Strategies. Cold Spring Harb. Perspect. Med. 2017, 7, a024240. [Google Scholar] [CrossRef]
- Ross, C.A.; Tabrizi, S.J. Huntington’s disease: From molecular pathogenesis to clinical treatment. Lancet Neurol. 2011, 10, 83–98. [Google Scholar] [CrossRef]
- Jiang, R.; Diaz-Castro, B.; Looger, L.L.; Khakh, B.S. Dysfunctional Calcium and Glutamate Signaling in Striatal Astrocytes from Huntington’s Disease Model Mice. J. Neurosci. 2016, 36, 3453–3470. [Google Scholar] [CrossRef] [PubMed]
- Tang, T.-S.; Tu, H.; Chan, E.Y.; Maximov, A.; Wang, Z.; Wellington, C.L.; Hayden, M.R.; Bezprozvanny, I. Huntingtin and Huntingtin-Associated Protein 1 Influence Neuronal Calcium Signaling Mediated by Inositol-(1,4,5) Triphosphate Receptor Type 1. Neuron 2003, 39, 227–239. [Google Scholar] [CrossRef] [PubMed]
- Suzuki, M.; Nagai, Y.; Wada, K.; Koike, T. Calcium leak through ryanodine receptor is involved in neuronal death induced by mutant huntingtin. Biochem. Biophys. Res. Commun. 2012, 429, 18–23. [Google Scholar] [CrossRef] [PubMed]
- Vigont, V.; Kolobkova, Y.; Skopin, A.; Zimina, O.; Zenin, V.; Glushankova, L.; Kaznacheyeva, E. Both Orai1 and TRPC1 are Involved in Excessive Store-Operated Calcium Entry in Striatal Neurons Expressing Mutant Huntingtin Exon 1. Front. Physiol. 2015, 6, 337. [Google Scholar] [CrossRef] [PubMed]
- Kaltenbach, L.S.; Romero, E.; Becklin, R.R.; Chettier, R.; Bell, R.; Phansalkar, A.; Strand, A.; Torcassi, C.; Savage, J.; Hurlburt, A.; et al. Huntingtin Interacting Proteins Are Genetic Modifiers of Neurodegeneration. PLoS Genet. 2007, 3, e82. [Google Scholar] [CrossRef]
- Carmo, C.; Naia, L.; Lopes, C.; Rego, A.C. Mitochondrial Dysfunction in Huntington’s Disease. In Polyglutamine Disorders; Advances in Experimental Medicine and Biology; Springer: Cham, Switzerland, 2018; Volume 1049, pp. 59–83. [Google Scholar] [CrossRef]
- Rosenstock, T.R.; Bertoncini, C.R.; Teles, A.V.; Hirata, H.; Fernandes, M.J.; Smaili, S.S. Glutamate-induced alterations in Ca2+ signaling are modulated by mitochondrial Ca2+ handling capacity in brain slices of R6/1 transgenic mice. Eur. J. Neurosci. 2010, 32, 60–70. [Google Scholar] [CrossRef]
- Luzio, J.P.; Pryor, P.R.; Bright, N.A. Lysosomes: Fusion and function. Nat. Rev. Mol. Cell Biol. 2007, 8, 622–632. [Google Scholar] [CrossRef]
- Sardiello, M.; Palmieri, M.; Di Ronza, A.; Medina, D.L.; Valenza, M.; Gennarino, V.A.; Di Malta, C.; Donaudy, F.; Embrione, V.; Polishchuk, R.S.; et al. A Gene Network Regulating Lysosomal Biogenesis and Function. Science 2009, 325, 473–477. [Google Scholar] [CrossRef]
- Korolchuk, V.I.; Saiki, S.; Lichtenberg, M.; Siddiqi, F.H.; Roberts, E.A.; Imarisio, S.; Jahreiss, L.; Sarkar, S.; Futter, M.; Menzies, F.M.; et al. Lysosomal positioning coordinates cellular nutrient responses. Nat. Cell Biol. 2011, 13, 453–460. [Google Scholar] [CrossRef]
- Patel, S.; Docampo, R. Acidic calcium stores open for business: Expanding the potential for intracellular Ca2+ signaling. Trends Cell Biol. 2010, 20, 277–286. [Google Scholar] [CrossRef]
- Patel, S. Deviant lysosomal Ca2+ signalling in neurodegeneration. An introduction. Messenger 2016, 5, 24–29. [Google Scholar] [CrossRef]
- Medina, D.L. Lysosomal calcium and autophagy. Int. Rev. Cell Mol. Biol. 2021, 362, 141–170. [Google Scholar] [CrossRef] [PubMed]
- Brailoiu, E.; Churamani, D.; Cai, X.; Schrlau, M.G.; Brailoiu, G.C.; Gao, X.; Hooper, R.; Boulware, M.J.; Dun, N.J.; Marchant, J.S.; et al. Essential requirement for two-pore channel 1 in NAADP-mediated calcium signaling. J. Cell Biol. 2009, 186, 201–209. [Google Scholar] [CrossRef] [PubMed]
- Calcraft, P.J.; Ruas, M.; Pan, Z.; Cheng, X.; Arredouani, A.; Hao, X.; Tang, J.; Rietdorf, K.; Teboul, L.; Chuang, K.-T.; et al. NAADP mobilizes calcium from acidic organelles through two-pore channels. Nature 2009, 459, 596–600. [Google Scholar] [CrossRef] [PubMed]
- Galione, A.; Evans, A.M.; Ma, J.; Parrington, J.; Arredouani, A.; Cheng, X.; Zhu, M.X. The acid test: The discovery of two-pore channels (TPCs) as NAADP-gated endolysosomal Ca2+ release channels. Pflugers Arch. 2009, 458, 869–876. [Google Scholar] [CrossRef] [PubMed]
- Patel, S. Function and dysfunction of two-pore channels. Sci. Signal. 2015, 8, re7. [Google Scholar] [CrossRef]
- Clapper, D.L.; Walseth, T.F.; Dargie, P.J.; Lee, H.C. Pyridine nucleotide metabolites stimulate calcium release from sea urchin egg microsomes desensitized to inositol trisphosphate. J. Biol. Chem. 1987, 262, 9561–9568. [Google Scholar] [CrossRef]
- Lee, H.C.; Walseth, T.F.; Bratt, G.T.; Hayes, R.N.; Clapper, D.L. Structural determination of a cyclic metabolite of NAD+ with intracellular Ca2+-mobilizing activity. J. Biol. Chem. 1989, 264, 1608–1615. [Google Scholar] [CrossRef]
- Zong, X.; Schieder, M.; Cuny, H.; Fenske, S.; Gruner, C.; Rötzer, K.; Griesbeck, O.; Harz, H.; Biel, M.; Wahl-Schott, C. The two-pore channel TPCN2 mediates NAADP-dependent Ca2+-release from lysosomal stores. Pflugers Arch. 2009, 458, 891–899. [Google Scholar] [CrossRef]
- Pereira, G.J.S.; Hirata, H.; Fimia, G.M.; Do Carmo, L.G.; Bincoletto, C.; Han, S.W.; Stilhano, R.S.; Ureshino, R.P.; Bloor-Young, D.; Churchill, G.; et al. Nicotinic Acid Adenine Dinucleotide Phosphate (NAADP) Regulates Autophagy in Cultured Astrocytes. J. Biol. Chem. 2011, 286, 27875–27881. [Google Scholar] [CrossRef]
- Pereira, G.J.S.; Hirata, H.; Do Carmo, L.G.; Stilhano, R.S.; Ureshino, R.P.; Medaglia, N.C.; Han, S.W.; Churchill, G.; Bincoletto, C.; Patel, S.; et al. NAADP-sensitive two-pore channels are present and functional in gastric smooth muscle cells. Cell Calcium 2014, 56, 51–58. [Google Scholar] [CrossRef] [PubMed]
- Pereira, G.J.S.; Antonioli, M.; Hirata, H.; Ureshino, R.P.; Nascimento, A.R.; Bincoletto, C.; Vescovo, T.; Piacentini, M.; Fimia, G.M.; Smaili, S.S. Glutamate induces autophagy via the two-pore channels in neural cells. Oncotarget 2017, 8, 12730–12740. [Google Scholar] [CrossRef] [PubMed]
- Naylor, E.; Arredouani, A.; Vasudevan, S.R.; Lewis, A.M.; Parkesh, R.; Mizote, A.; Rosen, D.; Thomas, J.M.; Izumi, M.; Ganesan, A.; et al. Identification of a chemical probe for NAADP by virtual screening. Nat. Chem. Biol. 2009, 5, 220–226. [Google Scholar] [CrossRef] [PubMed]
- Gómez-Suaga, P.; Luzón-Toro, B.; Churamani, D.; Zhang, L.; Bloor-Young, D.; Patel, S.; Woodman, P.G.; Churchill, G.C.; Hilfiker, S. Leucine-rich repeat kinase 2 regulates autophagy through a calcium-dependent pathway involving NAADP. Hum. Mol. Genet. 2012, 21, 511–525. [Google Scholar] [CrossRef]
- Djajadikerta, A.; Keshri, S.; Pavel, M.; Prestil, R.; Ryan, L.; Rubinsztein, D.C. Autophagy Induction as a Therapeutic Strategy for Neurodegenerative Diseases. J. Mol. Biol. 2020, 432, 2799–2821. [Google Scholar] [CrossRef]
- Smaili, S.S.; Hsu, Y.-T.; Youle, R.J.; Russell, J.T. Mitochondria in Ca2+ signaling and apoptosis. J. Bioenerg. Biomembr. 2000, 32, 35–46. [Google Scholar] [CrossRef]
- Orrenius, S.; Zhivotovsky, B.; Nicotera, P. Regulation of cell death: The calcium–apoptosis link. Nat. Rev. Mol. Cell Biol. 2003, 4, 552–565. [Google Scholar] [CrossRef]
- Walseth, T.F.; Guse, A.H. NAADP: From Discovery to Mechanism. Front. Immunol. 2021, 12, 703326. [Google Scholar] [CrossRef]
- Hockey, L.N.; Kilpatrick, B.S.; Eden, E.R.; Lin-Moshier, Y.; Brailoiu, G.C.; Brailoiu, E.; Futter, C.E.; Schapira, A.H.; Marchant, J.S.; Patel, S. Dysregulation of lysosomal morphology by pathogenic LRRK2 is corrected by TPC2 inhibition. J. Cell Sci. 2015, 128, 232–238. [Google Scholar]
- Lee, J.H.; McBrayer, M.K.; Wolfe, D.M.; Haslett, L.J.; Kumar, A.; Sato, Y.; Lie, P.P.Y.; Mohan, P.; Coffey, E.E.; Kompella, U.; et al. Presenilin 1 Maintains Lysosomal Ca(2+) Homeostasis via TRPML1 by Regulating vATPase-Mediated Lysosome Acidification. Cell Rep. 2015, 12, 1430–1444. [Google Scholar] [CrossRef]
- Pchitskaya, E.; Popugaeva, E.; Bezprozvanny, I. Calcium signaling and molecular mechanisms underlying neurodegenerative diseases. Cell Calcium 2018, 70, 87–94. [Google Scholar] [CrossRef] [PubMed]
- Ureshino, R.P.; Erustes, A.G.; Bassani, T.B.; Wachilewski, P.; Guarache, G.C.; Nascimento, A.C.; Costa, A.J.; Smaili, S.S.; Pereira, G.J.D.S. The Interplay between Ca2+ Signaling Pathways and Neurodegeneration. Int. J. Mol. Sci. 2019, 20, 6004. [Google Scholar] [CrossRef] [PubMed]
- Tang, T.S.; Slow, E.; Lupu, V.; Stavrovskaya, I.G.; Sugimori, M.; Llinás, R.; Kristal, B.S.; Hayden, M.R.; Bezprozvanny, I. Disturbed Ca2+ signaling and apoptosis of medium spiny neurons in Huntington’s disease. Proc. Natl. Acad. Sci. USA 2005, 102, 2602–2607. [Google Scholar] [CrossRef] [PubMed]
- Mackay, J.P.; Nassrallah, W.B.; Raymond, L.A. Cause or compensation?-Altered neuronal Ca2+ handling in Huntington’s disease. CNS Neurosci. Ther. 2018, 24, 301–310. [Google Scholar] [CrossRef]
- Chu, C.T. Mechanisms of selective autophagy and mitophagy: Implications for neurodegenerative diseases. Neurobiol. Dis. 2019, 122, 23–34. [Google Scholar] [CrossRef] [PubMed]
- Cai, Q.; Ganesan, D. Regulation of neuronal autophagy and the implications in neurodegenerative diseases. Neurobiol. Dis. 2022, 162, 105582. [Google Scholar] [CrossRef]
- Sarkar, S.; Rubinsztein, D.C. Huntington’s disease: Degradation of mutant huntingtin by autophagy. FEBS J. 2008, 275, 4263–4270. [Google Scholar] [CrossRef]
- Martin, D.D.; Ladha, S.; Ehrnhoefer, D.E.; Hayden, M.R. Autophagy in Huntington disease and huntingtin in autophagy. Trends Neurosci. 2015, 38, 26–35. [Google Scholar] [CrossRef]
- Croce, K.R.; Yamamoto, A. A role for autophagy in Huntington’s disease. Neurobiol. Dis. 2019, 122, 16–22. [Google Scholar] [CrossRef]
- Wong, Y.C.; Holzbaur, E.L.F. The Regulation of Autophagosome Dynamics by Huntingtin and HAP1 Is Disrupted by Expression of Mutant Huntingtin, Leading to Defective Cargo Degradation. J. Neurosci. 2014, 34, 1293–1305. [Google Scholar] [CrossRef]
- Erustes, A.G.; Stefani, F.Y.; Terashima, J.Y.; Stilhano, R.S.; Monteforte, P.T.; da Silva Pereira, G.J.; Han, S.W.; Calgarotto, A.K.; Hsu, Y.-T.; Ureshino, R.P.; et al. Overexpression of α-synuclein in an astrocyte cell line promotes autophagy inhibition and apoptosis. J. Neurosci. Res. 2018, 96, 160–171. [Google Scholar] [CrossRef] [PubMed]
- Lajoie, P.; Snapp, E.L. Formation and Toxicity of Soluble Polyglutamine Oligomers in Living Cells. PLoS ONE 2010, 5, e15245. [Google Scholar] [CrossRef] [PubMed]
- Karachitos, A.; Grobys, D.; Kulczyńska, K.; Sobusiak, A.; Kmita, H. The Association of VDAC with Cell Viability of PC12 Model of Huntington’s Disease. Front. Oncol. 2016, 6, 238. [Google Scholar] [CrossRef] [PubMed]
- Antonioli, M.; Albiero, F.; Nazio, F.; Vescovo, T.; Perdomo, A.B.; Corazzari, M.; Marsella, C.; Piselli, P.; Gretzmeier, C.; Dengjel, J.; et al. AMBRA1 Interplay with Cullin E3 Ubiquitin Ligases Regulates Autophagy Dynamics. Dev. Cell 2014, 31, 734–746. [Google Scholar] [CrossRef]



Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Pereira, C.A.d.S.; Medaglia, N.d.C.; Ureshino, R.P.; Bincoletto, C.; Antonioli, M.; Fimia, G.M.; Piacentini, M.; Pereira, G.J.d.S.; Erustes, A.G.; Smaili, S.S. NAADP-Evoked Ca2+ Signaling Leads to Mutant Huntingtin Aggregation and Autophagy Impairment in Murine Astrocytes. Int. J. Mol. Sci. 2023, 24, 5593. https://doi.org/10.3390/ijms24065593
Pereira CAdS, Medaglia NdC, Ureshino RP, Bincoletto C, Antonioli M, Fimia GM, Piacentini M, Pereira GJdS, Erustes AG, Smaili SS. NAADP-Evoked Ca2+ Signaling Leads to Mutant Huntingtin Aggregation and Autophagy Impairment in Murine Astrocytes. International Journal of Molecular Sciences. 2023; 24(6):5593. https://doi.org/10.3390/ijms24065593
Chicago/Turabian StylePereira, Cássia Arruda de Souza, Natalia de Castro Medaglia, Rodrigo Portes Ureshino, Claudia Bincoletto, Manuela Antonioli, Gian Maria Fimia, Mauro Piacentini, Gustavo José da Silva Pereira, Adolfo Garcia Erustes, and Soraya Soubhi Smaili. 2023. "NAADP-Evoked Ca2+ Signaling Leads to Mutant Huntingtin Aggregation and Autophagy Impairment in Murine Astrocytes" International Journal of Molecular Sciences 24, no. 6: 5593. https://doi.org/10.3390/ijms24065593
APA StylePereira, C. A. d. S., Medaglia, N. d. C., Ureshino, R. P., Bincoletto, C., Antonioli, M., Fimia, G. M., Piacentini, M., Pereira, G. J. d. S., Erustes, A. G., & Smaili, S. S. (2023). NAADP-Evoked Ca2+ Signaling Leads to Mutant Huntingtin Aggregation and Autophagy Impairment in Murine Astrocytes. International Journal of Molecular Sciences, 24(6), 5593. https://doi.org/10.3390/ijms24065593